

Abstract
Atorvastatin calcium is a lipid-lowering agent. It has approximately 15% of bioavailability, remaining amount of drug showed adverse effect which is undesirable for patients. The objective of the study was to enhance the solubility and a dissolution profile of the atorvastatin (AT) calcium. Solid dispersion (SD) is a technique which enhances the solubility and a dissolution profile of poorly soluble drug. Various methods are being used for SDs such as microwave irradiation fusion, kneading, solvent evaporation, fusion, and dropping method. The authors have used here conventional fusion method using PEG 4000 as a hydrophilic carrier. The solubility of pure drug, physical mixture using PEG 4000 (1:3), and SD in phosphate buffer solutions (pH 6.8) was found to be 55.33 ± 0.66, 81.89 ± 2.35, and 93.66 ± 1.35, respectively. Fourier transform infrared and differential scanning calorimetry study showed the significant peak shift of drug in SD. It indicated that the nature of drug had been changed from crystalline form to amorphous form due to conversion into SD formulation. The dissolution rate was significantly increased when the drug polyethylene glycol 4000 ratio was 1:3. The mean cumulative percentage drugs release from pure drug, physical mixture, marketed tablet, and SD at 1 h was 28.92 ± 1.66%, 55.26 ± 0.95%, 72.16 ± 1.33%, and 91.66 ± 1.65%, respectively. It was concluded that the solubility and dissolution profile of SD of AT calcium showed the enhancement of solubility and dissolution when compared with marketed preparations.
Key words: Atorvastatin calcium, bioavailability enhancement, conventional fusion method, solid dispersion
INTRODUCTION
Enhancement of solubility and vis-a-vis bioavailability (BA) of a poorly soluble drug is a challenging job for a drug development scientist. There are many drugs which have good therapeutic value, but absorption is limited to the biological system. A lot of techniques used to overcome the poor aqueous solubility of active pharmaceutical ingredients (API) has been investigated in drug research and development such as salt formation,[1] prodrug formation,[2] particle size reduction,[3] complexation,[4] micelles,[5] microemulsions,[6] nanoemulsions,[7] nanosuspensions,[8] solid lipid nanoparticle,[9] and solid dispersion (SD). SD is considered one of the most successful strategies to improve the dissolution profile of poorly soluble drugs. It is a method to alter the solid state at the particle, or molecular level involves a physical change in the drug and is an attractive option for improving drug solubility.[10] Despite many advantages in improving dissolution profile of poorly soluble drugs, the number of commercial products using SD is limited because of problems in the preparation process and during storage. The most important problem is the recrystallization of drugs from amorphous state during storage which leads to decreased BA of SD.[11] Atorvastatin (AT) is the most preferred drug among statins for hyperlipidemia, used to treat moderate to severe familial or nonfamilial hypercholesterolemia. AT reduces levels of cholesterol (low-density lipoprotein) and triglycerides with increasing levels of “good” cholesterol (high-density lipoprotein). It belongs to Biopharmaceutics Classification System class-II drug and has lower solubility and BA.[12] The drug development scientist can enhance the BA using SD technique. Various group of scientist are working in the field of SD, and good achievements were gained. The API in SDs can be dispersed as separate molecules, amorphous particles, or crystalline particles while the carrier can be in the crystalline or amorphous state. Numerous studies on SD have been published and have showed many advantageous properties of SD in improving the solubility and dissolution rate of poorly soluble drugs. Mohan et al. developed SD of oxcarbazepine using PEG 6000 as hydrophilic carrier with melting technique. They concluded that SDs prepared using this technique was effective in improving drug dissolution. The study also confirmed that PEG 6000 inhibited the crystallization of drugs resulting in the amorphous state form of the drug in SD.[13]
Gorajana et al. developed and characterized cefuroxime axetil SD using hydrophilic carriers PEG 4000. SD formulations were found to have a higher dissolution rate comparatively to pure cefuroxime axetil. The enhancement of dissolution rate of SD by PEG 4000 may be caused by increased wettability, solubility, and reduction in particle size. It may also be caused by amorphization as suggested by differential scanning calorimetry (DSC) and X-ray powder diffraction. Finally concluded as the developed SD of cefuroxime axetil with PEG 4000 will be a better alternative for enhanced therapeutic efficacy.[14]
Sukanya and Kishore developed SD of simvastatin for treatment of hyperlipidemia. They concluded that for increasing the solubility of drug SD was prepared. SD was prepared with PEG 4000 and polyvinylpyrrolidone-K30. They analyzed for their solubility and in vitro dissolution profile, and it is found that SD of the drug with PEG 4000 (1:5) had shown enhanced solubility with improved dissolution profile. Fourier transform infrared, X-ray studies were indicated that amorphous form of SD, which may be also responsible for enhanced the solubility and dissolution profile. Thus showed that dissolution rate of simvastatin can be enhanced to a considerable extent by SD technique with PEG 4000.[15] On the basis of these works, authors have prepared SD of AT and characterized them for various parameters.
These advantages include reducing particle size, possibly to molecular level, enhancing wettability and porosity, as well as changing drug crystalline state, preferably into amorphous state.[16] SD formulation technique is based on formation of solid solutions and eutectic mixtures. The objective of the study was to enhance the solubility and dissolution profile of the AT calcium by preparing SD of AT and compare with marketed formulation.
MATERIALS AND METHODS
Materials
AT was obtained as a gift sample from Jubilant Life Sciences, Noida, Uttar Pradesh (India). PEG 4000, sodium hydroxide, sodium hydrogen phosphate, and HCl, were purchased from S.D. Fine Chemical, Pvt. Ltd., New Delhi, India and other reagents were of analytical grade.
Methods
Preparation of solid dispersion
Conventional fusion method was implied for preparation of SD. PEG 4000 was taken and heated to a molten state at 55–60°C and to this mass added weighed amount of AT with continuous stirring until dissolved. The different ration of drug was added and their optimization was studied on the basis of solubility and dissolution as well as in vitro release. Solidification was allowed to occur at room temperature. The prepared product was stored in a dessicator for 24 h and pulverized using a porcelain mortar and pestle. Then, this pulverized powders were passed through sieve no 80 for uniform size.[17]
Solubility studies
Phase and saturation solubility studies were done as per method described by Higuchi and Connors.[18] The saturation solubility of drug, physical mixtures, and SD with PEG 4000 in distilled water and phosphate buffer (pH 6.8) was determined by adding an excess of drug, physical mixture, and SD to 50 mL distilled water and phosphate buffer in conical flask and were shaken on rotary shaker incubator for 96 h at 37°C ± 0.5°C. The saturated solutions were filtered through a 0.45 μm membrane filter, suitably diluted with water, phosphate buffer and analyzed using Shimadzu UV-1601 spectrophotometer (Kyoto, Japan) at 245 nm.[19]
Fourier transform infrared spectroscopy
Infrared spectroscopy was conducted using PerkinElmer (Massachusetts, USA) Spectrophotometer, and the spectrum was recorded in the wavelength region of 6000–500 cm−1. The procedure consisted of dispersing a sample (drug alone or mixture drug and excipients) in KBr and compressing into discs by applying pressure. The pellet was placed in the light path, and the spectrum was obtained. It is used for confirming any interaction of AT with other excipients.
Differential scanning calorimetry study
The extent of crystallinity was determined using DSC study. The dried sample of AT (about 1 mg) was loaded and sealed into DSC pan with a DSC loading puncher. The sample was scanned between 40°C and 400°C at a heating rate of 5°C/min, under nitrogen atmosphere, using a differential scanning calorimeter, PerkinElmer Pyris 6 DSC (Massachusetts, USA). The thermal behavior of SD of AT-PEG complex was analyzed using DSC to ascertain the formation of complex.
In vitro dissolution studies
Dissolution rate studies were performed in pH 6.8 phosphate buffer at 37°C ± 0.5°C, using 8-station USP type-II apparatus with paddle rotating at 100 rpm. Solid products, SDs as well as physical mixtures, each containing 80 mg of drug were subjected to dissolution. The collected samples were filtered and replaced with fresh media at predetermined time points analyzed spectrophotometrically for the drug content at 245 nm. Each test was performed in triplicate (n = 3).
Stability studies
Stability studies were carried out as per the International Conference on Harmonization guidelines. The samples were stored at 40°C ± 0.5°C/75 ± 5% RH for 3 months, Thermolab Scientifific Equipments Pvt. Ltd., Thane, India. The samples were withdrawn and evaluated for the content and in vitro release at predetermined time intervals. The variations are analyzed and compared with the freshly prepared formulations.
Statistical analysis
All the results are represented as a mean ± standard deviation and analyzed by Student's t-test for difference between the groups. More than two groups were compared using ANOVA and difference greater at P < 0.05 was considered as significant.
RESULTS AND DISCUSSION
Solubility studies
The results of saturation solubility studies are given in Figure 1. The solubility of pure drug in water and in phosphate buffer solutions (PBS) (pH 6.8) was found to be 26.08 ± 0.37 and 55.33 ± 0.66 μg/mL. The solubility of physical mixture prepared using PEG 4000 in the ratio (1:1, 1:2, and 1:3) was 35.59 ± 1.12, 46.87 ± 1.24, 57.79 ± 1.35 μg/mL in water and 63.78 ± 1.19, 72.76 ± 1.21, 81.89 ± 2.35 μg/mL in PBS. The solubility of SD using PEG 4000 (1:1, 1:2, and 1:3) in water were found to be 40.33 ± 2.18, 51.33 ± 1.16, 76.50 ± 2.66 μg/mL and in PBS (pH 6.8) 65.36 ± 1.28, 77.56 ± 1.24, and 93.66 ± 1.35, respectively. These results showed that after preparation of SD using PEG solubility was enhanced. It is concurrent to the study done by other group of researchers.
Figure 1.
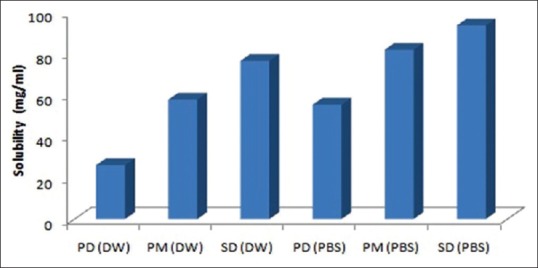
Saturation solubility studies of atorvastatin in distilled water and phosphate buffer saline
Fourier transform infrared study
Fourier transform infrared study was done, and results were analyzed. The pure drug had shown a characteristics peak at 3364 and 3275 cm−1. Other O-H stretching was also observed. However, SD with PEG 4000 was analyzed the typical peaks were shifted, broadened, some have low intensity, and some are missed. It indicated that the formation of SD with carriers. It is a common technique used to investigate the intermolecular interaction and drug–carrier compatibility because it can detect the physical and chemical reaction between drug and carriers. H-bonding between drug and carriers is an important characterization method to explain physical state and the stability of API in SD.[20]
Differential scanning calorimetry study
It is the most preferred thermal technique for SD characterization, provides accurate information about melting point, glass transition temperature (Tg) as well as the energy, changes associated with the phase transitions including crystallization and fusion process. The lack of a drug melting peak in the DSC thermogram of an SD indicated that the drug exists in an amorphous form or encapsulated form as seen in Figure 2. The previous studies showed that PEG was the most suitable excipient which provide single phase SD. The molecular miscibility, recrystallization, and phase separation of SD after preparation and under accelerated stability conditions were characterized by DSC. In this study, another peak was observed with SD showed molecular miscibility. The SD was physically stable if no recrystallization peaks were observed.[21]
Figure 2.
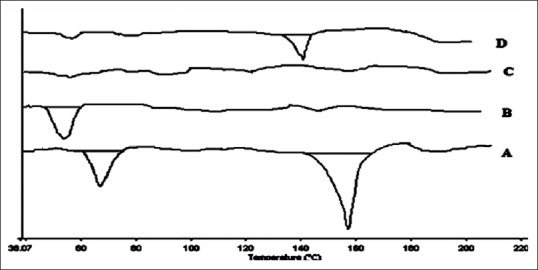
Differential scanning calorimetry thermograms of various preparations; A: Pure drug, B: Carrier-PEG 4000, C: Atorvastatin solid dispersion, and D: Physical mixture of PEG 4000 and atorvastatin
In vitro drug release
The dissolution profiles of AT for SD and physical mixture performed in 6.8 phosphate buffer were as shown in Figure 3. The dissolution rate was significantly increased when the AT: PEG 4000 ratio was at 1:3. The mean cumulative percentage drugs release from physical mixture at 60 min was 56.34 ± 1.93%, 69.64 ± 1.62%, 72.16 ± 1.33% for 1:1, 1:2, 1:3, respectively as shown in Figure 3 and 28.92 ± 1.66% and 55.26 ± 0.95% for pure drug and marketed tablet. However in the case of SD, release rate was observed as 91.66 ± 1.65%. These results are concurrent to the previous study performed by Choudhary et al. also.[22] The drug release from all the formulations followed zero order kinetics, as the plot observed in between amount of drug released versus time was found to be linear. To analyze the mechanism of drug release from these formulations, the data were followed Hixson–Crowell equation. The slope values (n) obtained to decline between 0.4756 and 0.5895 for all formulations for the release of AT indicating nonFickian diffusion.
Figure 3.
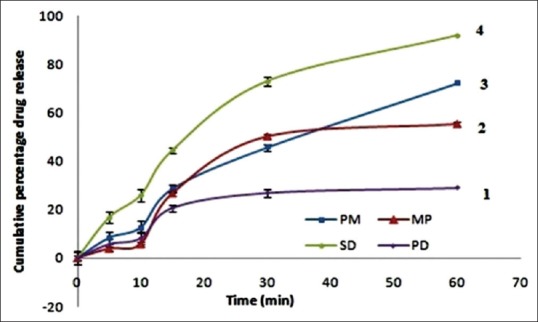
In vitro release study of atorvastatin in phosphate buffer saline; 1: Pure drug, 2: Marketed preparation, 3: Physical mixture, 4: Solid dispersion
Stability studies
After removal of predetermined time interval, samples were analyzed for the content and in vitro release study and results were compared with the original data. There are no significant (P < 0.05) variations were found in results. It is indicated that the prepared SD is stable under accelerated storage conditions.
CONCLUSION
The SD of AT was prepared successfully using conventional fusion method. Solubility was increased significantly as compared to pure drug and physical mixture with PEG 4000. In vitro release study was performed and it showed a marked increase in cumulative percentage release and it showed a better alternative to the AT conventional formulation. PEG 4000 is a better inert carrier for SD. DSC study showed the absence of sharp drug peak in SD indicated that the drug physical property has been changed which is a proof of molecular interaction of PEG with AT. This is why the increased solubility, as well as BA of AT, has been found with SD. The stability study also indicated that the prepared SD was stable, and there is no significant changes were observed. This current study suggested that it will be an alternative approach for the treatment of hyperlipidemia in near future, but the clinical study have been warranted before its marketing.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Berge SM, Bighley LD, Monkhouse DC. Pharmaceutical salts. J Pharm Sci. 1977;66:1–19. doi: 10.1002/jps.2600660104. [DOI] [PubMed] [Google Scholar]
- 2.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T, et al. Prodrugs: Design and clinical applications. Nat Rev Drug Discov. 2008;7:255–70. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 3.Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3:785–96. doi: 10.1038/nrd1494. [DOI] [PubMed] [Google Scholar]
- 4.Loftsson T, Duchêne D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329:1–11. doi: 10.1016/j.ijpharm.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 5.Lavasanifar A, Samuel J, Kwon GS. Poly (ethylene oxide) -block-poly (L-amino acid) micelles for drug delivery. Adv Drug Deliv Rev. 2002;54:169–90. doi: 10.1016/s0169-409x(02)00015-7. [DOI] [PubMed] [Google Scholar]
- 6.Mueller EA, Kovarik JM, van Bree JB, Tetzloff W, Grevel J, Kutz K. Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion formulation. Pharm Res. 1994;11:301–4. doi: 10.1023/a:1018923912135. [DOI] [PubMed] [Google Scholar]
- 7.Fatouros DG, Deen GR, Arleth L, Bergenstahl B, Nielsen FS, Pedersen JS, et al. Structural development of self nano emulsifying drug delivery systems (SNEDDS) during in vitro lipid digestion monitored by small-angle X-ray scattering. Pharm Res. 2007;24:1844–53. doi: 10.1007/s11095-007-9304-6. [DOI] [PubMed] [Google Scholar]
- 8.Muller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs: I. preparation by a size-reduction technique. Int J Pharm. 1998;160:229–37. [Google Scholar]
- 9.Potta SG, Minemi S, Nukala RK, Peinado C, Lamprou DA, Urquhart A, et al. Development of solid lipid nanoparticles for enhanced solubility of poorly soluble drugs. J Biomed Nanotechnol. 2010;6:634–40. doi: 10.1166/jbn.2010.1169. [DOI] [PubMed] [Google Scholar]
- 10.Narasaiah VL, Reddy BK, Kishore K, Kumar MR, Srinivasa Rao P, Reddy V. Enhanced dissolution rate of atorvastatin calcium using solid dispersion with PEG 6000 by dropping method. J Pharm Sci Res. 2010;2:484–91. [Google Scholar]
- 11.Baird JA, Taylor LS. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv Drug Deliv Rev. 2012;64:396–421. doi: 10.1016/j.addr.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 12.Khan FN, Dehghan MH. Enhanced bioavailability of atorvastatin calcium from stabilized gastric resident formulation. AAPS Pharm Sci Tech. 2011;12:1077–86. doi: 10.1208/s12249-011-9673-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohan A, Madhavi M, Jyosthna P. Preparation, in vitro and in vivo characterization of solid dispersions of oxcarbazepine using melting technique. Pharm Innov J. 2015;3:99–103. [Google Scholar]
- 14.Gorajana A, Rajendran A, Yew LM, Dua K. Preparation and characterization of cefuroxime axetil solid dispersions using hydrophilic carriers. Int J Pharm Investig. 2015;5:171–8. doi: 10.4103/2230-973X.160857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sukanya M, Kishore VS. Design and development of solid dispersions of simvastatin for enhancing the solubility. Am J PharmTech Res. 2012;2:733–40. [Google Scholar]
- 16.Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12:1068–75. doi: 10.1016/j.drudis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Sharma M, Garg R, Gupta GD. Formulation and evaluation of solid dispersion of atorvastatin calcium. J Pharm Sci Innov. 2013;2:73–81. [Google Scholar]
- 18.Arunkumar N, Deecaraman M, Rani C, Mohanraj KP, Kumar VK. Preparation and solid state characterization of atrovastatin nanosuspension for enhanced solubility and dissolution. Int J Pharm Tech Res. 2009;1:1725–30. [Google Scholar]
- 19.Narasaiah VL, Reddy BK, Kumar MR, Kiran Kumar A, Raju C, Kumar AS, et al. Improved dissolution rate of atorvastatin calcium using solid dispersion with PEG 4000. J Chem Pharm Res. 2010;2:304–11. [Google Scholar]
- 20.Miyazaki T, Aso Y, Yoshioka S, Kawanishi T. Differences in crystallization rate of nitrendipine enantiomers in amorphous solid dispersions with HPMC and HPMCP. Int J Pharm. 2011;407:111–8. doi: 10.1016/j.ijpharm.2011.01.035. [DOI] [PubMed] [Google Scholar]
- 21.Vo CL, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85(3 Pt B):799–813. doi: 10.1016/j.ejpb.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Choudhary A, Rana AC, Aggarwal G, Kumar V, Zakir F. Development and characterization of an atorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharm Sin B. 2012;2:421–8. [Google Scholar]