

Abstract
Prdm genes encode transcription factors with a subtype of SET domain known as the PRDF1-RIZ (PR) homology domain and a variable number of zinc finger motifs. These genes are involved in a wide variety of functions during animal development. As most Prdm genes have been studied in vertebrates, especially in mice, little is known about the evolution of this gene family. We searched for Prdm genes in the fully sequenced genomes of 93 different species representative of all the main metazoan lineages. A total of 976 Prdm genes were identified in these species. The number of Prdm genes per species ranges from 2 to 19. To better understand how the Prdm gene family has evolved in metazoans, we performed phylogenetic analyses using this large set of identified Prdm genes. These analyses allowed us to define 14 different subfamilies of Prdm genes and to establish, through ancestral state reconstruction, that 11 of them are ancestral to bilaterian animals. Three additional subfamilies were acquired during early vertebrate evolution (Prdm5, Prdm11, and Prdm17). Several gene duplication and gene loss events were identified and mapped onto the metazoan phylogenetic tree. By studying a large number of nonmetazoan genomes, we confirmed that Prdm genes likely constitute a metazoan-specific gene family. Our data also suggest that Prdm genes originated before the diversification of animals through the association of a single ancestral SET domain encoding gene with one or several zinc finger encoding genes.
Keywords: Prdm, transcription factors, evolution, development, metazoans
Introduction
Transcriptional regulation is an important aspect of the development of multicellular organisms, such as animals. Decades of molecular and genetic studies, mainly conducted in bilaterian model organisms, have shown that many key developmental regulators encode transcription factors (TFs) sporting a huge diversity of DNA binding domains. Genomic data from various species belonging to both bilaterians and nonbilaterians, as well as to nonmetazoan opisthokonts, were used to highlight the origin or diversification of several families of animal developmental TFs. These events happened before the divergence of the contemporary animal lineages either in holozoans (a group that includes metazoans and their closest unicellular relatives, such as choanoflagellates and filastereans) or during early metazoan evolution (e.g., Jager et al. 2006; Ryan et al. 2007; Simionato et al. 2007; Degnan et al. 2009; Sebé-Pedrós et al. 2011, 2013; de Mendoza et al. 2013). However, we are still lacking a precise understanding of the evolution of many developmental TF families, such as the Prdm family.
Prdm genes encode proteins that are characterized by the presence of a PR domain originally found to be shared by PRDI-BF1 (positive regulatory domain I-binding factor 1, now known as Prdm1 or Blimp1) and RIZ1 (retinoblastoma protein-interacting zinc finger gene 1, now known as Prdm2) (Fog et al. 2012; Hohenauer and Moore 2012; Di Zazzo et al. 2013). The PR domain corresponds to a subtype of a SET domain, which is found in many histone lysine methyltransferases (HMTs) (Huang 2002; Wu et al. 2010). However, only some Prdm proteins have been shown to display intrinsic HMT activity. Other Prdm proteins are known to mediate indirect epigenetic regulations through binding of histone-modifying enzymes including HMTs, histone deacetylases, and histone acetyltransferases (reviewed in Fog et al. 2012; Hohenauer and Moore 2012). With the exception of vertebrate Prdm11 proteins, all characterized Prdm proteins associate a PR domain to a variable number of Zn fingers (Fumasoni et al. 2007; Kinameri et al. 2008; Sun et al. 2008). Zn fingers motifs mediate sequence-specific DNA binding and protein–protein interactions (Hohenauer and Moore 2012).
The Prdm gene family comprises 17 members in primates (Human, common chimpanzee, and macaque), 16 in rodents (mouse and rat), chick, and Xenopus, whereas only two to three genes have been reported in Caenorhabditis and Drosophila, respectively (Fumasoni et al. 2007; Sun et al. 2008). Prdm genes display a wide variety of expression patterns and functions during development (reviewed in Fog et al. 2012; Hohenauer and Moore 2012; Di Zazzo et al. 2013). This includes germ cell development, neurogenesis, vascular development, brown fat differentiation, hematopoiesis, and insect tracheal development. For example, Prdm1 and Prdm14 are key regulators of primordial germ cell specification in mouse (Ohinata et al. 2005; Kurimoto et al. 2008; Yamaji et al. 2008; Grabole et al. 2013; Magnúsdóttir et al. 2013) and are also important for naive pluripotency in embryonic stem cells (Chu et al. 2011; Grabole et al. 2013; Yamaji et al. 2013). Several Prdm genes are expressed in restricted population of cells of the developing nervous system in vertebrates (e.g., Kinameri et al. 2008) and one of them, Prdm13, has recently been shown to promote GABAergic over glutamatergic neuronal fate in the dorsal spinal cord (Chang et al. 2013; Hanotel et al. 2014). Many Prdm genes are also deregulated in human diseases, in particular hematological and solid tumor cancers where some Prdm genes act as tumor suppressors and others as oncogenes (reviewed in Fog et al. 2012). Several Prdm proteins have also been shown to be modulators of developmental signaling pathways, such as transforming growth factor-β, Notch, and estrogen signaling (reviewed in Hohenauer and Moore 2012; Di Zazzo et al. 2013). One Prdm gene, Prdm9, is a key determinant of sequence-specific recombination hotspots during meiosis in Human and mouse (Baudat et al. 2010; Parvanov et al. 2010) and also behaves as a speciation gene in mouse (Mihola et al. 2009).
Despite the importance of this gene family, only a single study addressed the evolution of Prdm genes in metazoans so far (Fumasoni et al. 2007). The authors proposed that the Prdm genes were specific to animals, that an important expansion of the family occurred in vertebrates, and that one additional duplication took place in primates. However, these conclusions were based on a rather small species sampling (14 among which 8 vertebrates, 2 Drosophila and 2 Caenorhabditis species) and without any lophotrochozoans nor nonbilaterian representatives. Here, we identified Prdm genes in 93 animal species whose genome has been fully sequenced and which are distributed in all the main metazoan lineages. Our phylogenetic analyses allowed the subdivision of the Prdm family into 14 subfamilies and showed that at least 11 of these subfamilies were already present in the last common ancestor (LCA) of bilaterians. Our data indicate that an important diversification of the Prdm family occurred during early metazoan evolution. We were also able to map on the metazoan phylogenetic tree the many gene duplication/loss events in the different Prdm subfamilies that occurred during the metazoan evolutionary history. Based on the examination of a large number of nonmetazoan genomes, we confirmed that the Prdm family is likely specific to metazoans. Finally, we discuss the possibility that different Prdm subfamilies may have originated independently, through the fusion of a single ancestral SET domain encoding gene with several different ancestral Zn finger encoding genes.
Results and Discussion
Genome-Wide Search of Prdm Genes and Definition of Metazoan Prdm Subfamilies
We identified a total number of 976 Prdm genes in the fully sequenced genome of 93 metazoan species that represent a significant sampling of metazoan diversity (table 1). Supplementary tables S1–S6, Supplementary Material online, list all the identified genes (with accession numbers, presence of characteristic domains, sources of the genomic data, and taxonomic information about the studied species). The sequences of all the corresponding proteins can be found in supplementary data set S1, Supplementary Material online. The number of Prdm genes found per species ranges from 2 in the sponge Oscarella carmela, the placozoan Trichoplax adhaerens, and the nematode Caenorhabditis elegans to 19 in teleosteans (table 1). These important differences in Prdm genes number are also found within each main animal lineage. In ecdysozoans, for example, two to seven genes can be found in nematodes, whereas in arthropods the number of Prdm genes ranges from 4 in some insects (such as Apis mellifera) to 13 in the centipede Strigamia maritima (table 1). In deuterostomes, whereas 18 Prdm genes are found in the echinoderm Strongylocentrotus purpuratus, the sea squirt Ciona savignyi possess only five Prdm members. Finally, the interval in vertebrates ranges from 9 to 19 Prdm genes (table 1). Prdm genes encode proteins that are characterized by the presence of a SET domain—this domain is thought to mediate the enzymatic activity of a large number of histone lysine methyltransferases (HMTs). An intrinsic HMT activity has been described for vertebrate Prdm2, Prdm3, Prdm6, Prdm7/9, Prdm8, Prdm13, and Prdm16 proteins (Hohenauer and Moore 2012; Pinheiro et al. 2012; Hanotel et al. 2014). We analyzed a selection of SET domains from a broad range of species and identified several residues that are well conserved in Prdm proteins and could therefore be important for their functions (supplementary text S1 and figs. S1 and S2, Supplementary Material online).
Table 1.
Prdm Genes in Metazoans.
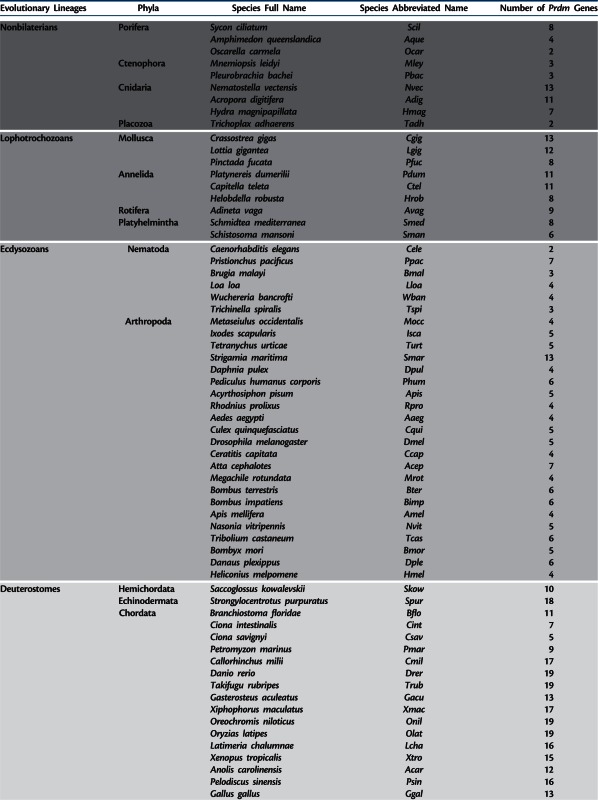

|
Note.—shaded areas discriminate evolutionary lineages
We next performed phylogenetic analyses to assess whether we can define Prdm subfamilies and study their distribution in metazoans. Given the very large number of sequences studied here, we performed these analyses using four different partial sets of sequences: All nonvertebrate sequences + all nonmammalian vertebrates + five mammals (data set 1), all vertebrates + a sampling of nonvertebrate species (data set 2), a sampling of bilaterians only (data set 3), and a sampling of vertebrates and nonvertebrates (data set 4). The use of these reduced data sets allowed performing time-demanding phylogenetic methods, such as bootstrap resamplings and Bayesian analysis, and testing the possible effects of different samplings of species on the robustness of the main nodes of the phylogenetic trees. Supplementary figure S3, Supplementary Material online, shows a representative unrooted tree produced using data set 4. Fourteen different subfamilies of Prdm genes were defined using these phylogenetic analyses. These correspond to monophyletic groups that include sequences from several different species and that are consistently observed in the trees produced by different methods and using the four different data sets. Supplementary table S7, Supplementary Material online, compiles the statistical supports for the nodes defining these groups in the different performed analyses (maximum-likelihood [ML] and Bayesian analysis on the four different data sets). We also tested whether different sequence samplings affected the presence of the different subfamilies in the phylogenetic tree. For this purpose, we conducted several phylogenetic analyses with all Prdm sequences minus the sequences of a given subfamily (e.g., all sequences minus Prdm1 sequences) or minus all orphan genes. The results of these analyses are summarized in supplementary table S7, Supplementary Material online, and show that deleting sequences of any given subfamily do not affect the presence and composition of the other monophyletic groups in the phylogenetic tree.
We named each subfamily using the name of the Human gene(s) included in the group (supplementary fig. S3 and table S1, Supplementary Material online). In most cases, a subfamily contains a single Human member. However, three subfamilies (Prdm3/16, Prdm10/15, and Prdm7/9) contain two Human genes (supplementary table S1, Supplementary Material online) and we obtained evidence that these pairs of genes result from vertebrate- or primate-specific duplications (see below). We were able to confidently assign 931 of the 976 analyzed genes in the 14 defined subgroups. The 45 remaining genes were categorized as “orphans” and were mainly genes that were associated with alternative subfamilies depending on the data set and/or the phylogenetic method used. We also found phylogenetic relationships between some subfamilies, such as between Prdm6 and Prdm12, or between Prdm8 and Prdm13 (supplementary fig. S3, Supplementary Material online), but these groupings were usually not found in all analyses and/or had poor statistical supports (supplementary table S7, Supplementary Material online).
Supplementary table S1, Supplementary Material online, and figure 1 show the distribution of the Prdm genes in the defined subfamilies or in the orphans group in all studied species and the main phylogenetic groups, respectively. Together, these species provide a significant coverage of the main animal evolutionary lineages, including both bilaterians (lophotrochozoans, ecdysozoans, and deuterostomes) and nonbilaterians (poriferans, cnidarians, ctenophores, and placozoans). For most studied phyla, including nonbilaterians, we were able to study Prdm genes from more than one species, an important achievement as hypotheses drawn for lineages represented by single species can be misleading. Our choice of species from various animal lineages allows the study of the evolution of Prdm genes to be studied at the whole metazoan clade scale. In addition, the inclusion in the study of a rich sampling of chordates, arthropods, and to a lesser extent nematodes also allows Prdm gene evolution to be studied in a detailed manner within these three phyla. Although we have tried to be as exhaustive as possible, we must caution that we may have missed some of the Prdm genes in some species, as these genes may lie in unsequenced or badly assembled regions. However, we are confident that our data are sufficient to obtain a qualitatively accurate assessment of the evolution of Prdm genes in metazoans.
Fig. 1.

Prdm subfamilies in the main metazoan groups. The number (or range of numbers) of members of each Prdm subfamily and orphan genes is indicated (none if no member detected). The number of studied species in each phylogenetic group is indicated next to the group name. A final column summarizes the putative ancestral set of Prdm subfamilies in the Bilaterian ancestor.
Ancestral Repertoires of Prdm Subfamilies and Subfamily Losses in Metazoans
We constructed a character matrix (supplementary table S8, Supplementary Material online) that compiles presence/absence states for each Prdm subfamilies in all studied species, coded by 0 or 1 (0 = absence; 1 = presence, regardless of the number of members). This matrix was used to infer the set of Prdm subfamilies that were likely present at the different nodes of the metazoan tree. For this purpose, we used both Dollo-like parsimony and ML approaches as implemented in the ancestral state reconstruction tools of Mesquite (see Materials and Methods for details). Ancestral state reconstruction crucially depends on the established phylogeny of the studied species. Although the relationships between the main bilaterian phyla and lineages are quite consensual (Telford and Copley 2011) (fig. 2), those between bilaterians, cnidarians, placozoans, ctenophores, and poriferans are still very controversial. We considered four recently proposed phylogenies of these groups (Dunn et al. 2008; Philippe et al. 2009; Schierwater et al. 2009; Pick et al. 2010; Ryan et al. 2013) (fig. 3) for ancestral state reconstructions. Results of these analyses obtained by parsimony and ML were very consistent and are shown in supplementary tables S9–S12, Supplementary Material online. By comparing ancestral sets of Prdm subfamilies at different nodes, we were also able to infer the timing of the appearance of the Prdm subfamilies and to position subfamily losses that have occurred in some lineages. The most important inferences are reported on the taxon and species phylogenetic tree shown in figures 2–5 and discussed below.
Fig. 2.

Evolution of Prdm genes in metazoans. Phylogenetic relationships between the studied species are shown (Telford and Copley 2011). A basal polytomy highlights the uncertainties about the relationships between bilaterian and nonbilaterian groups (see fig. 3 for details). Phylogenetic relationships among ecdysozoans and chordates are shown in figures 4 and 5, respectively. The major animal groups and lineages that are discussed in the main text are indicated in blue. The putative ancestral sets of Prdm genes in metazoans and bilaterians are indicated in the dark green boxes. Red boxes indicate subfamily losses that occurred in some lineages. Putative gene losses that have occurred in single species are not shown.
Fig. 3.

Inference of Prdm ancestral sets using four recently proposed phylogenies of metazoans. Ancestral sets of genes and gene losses are indicated as in figure 2. In (B), the Prdm subfamily repertoire of the LCA of cnidarians and bilaterians is indicated by a white asterisk; it would be: Prdm6, 7/9, 8, 12, 13, and 14. The phylogeny in (A) is based on Philippe et al. (2009), in (B) on Dunn et al. (2008) and Ryan et al. (2013), in (C) on Pick et al. (2010), and in (D) on Schierwater et al. (2009).
Fig. 4.

Evolution of Prdm genes in nematodes and arthropods. Phylogenetic relationships between the studied species are shown (Meldal et al. 2007; Trautwein et al. 2012). Ancestral sets of genes and gene losses are indicated as in figure 2. Duplications of Prdm3/16 in Pancrustacea/Hexapoda and in Hymenoptera are indicated as light green boxes. Putative gene losses that have occurred in single species are not shown. For sake of simplicity, the names of most of the groups to which belong single species are not indicated. Strigamia maritima belongs to Myriapoda, Daphnia pulex to Crustacea, Pediculus humanus corporis to Phthiraptera, and Tribolium castaneum to Coleoptera. Nematode lineage names are described in the main text.
Fig. 5.

Evolution of Prdm genes in chordates. Phylogenetic relationships between the studied species are shown (based on NBCI taxonomy browser). Ancestral sets of genes and gene losses are indicated as in figure 2. Gene duplications are indicated as light green boxes. Putative gene losses that have occurred in single species are not shown. For sake of simplicity, the name of the group to which belong single species, as well as a few other groups, is not indicated. Branchiostoma floridae belongs to Cephalochordata, Petromyzon marinus to Petromyzontiformes, Callorhinchus milii to Chondrichthyan, Xiphophorus maculatus and Oryzias latipes to Atherinomorphae, Latimeria chalumnae to Coelacanthimorpha, Xenopus tropicalis to Lissamphibia, Ornithorhynchus anatinus to Prototheria, and Tarsius syrichta to Tarsiiformes.
We inferred that the LCA of metazoans possessed at least the Prdm6 subfamily and may have possessed up to five additional ones depending on the considered phylogeny of bilaterians and nonbilaterians phyla (Prdm7/9, Prdm8, Prdm12, Prdm13, and Prdm14; figs. 2 and 3). In the four possible phylogenies, the LCA of cnidarians and bilaterians, wherever this ancestor is positioned, would have possessed at least six Prdm genes (fig. 3). We can infer that cnidarians have lost the Prdm8 subfamily and the placozoan T. adhaerens has lost two to four ancestral Prdm genes (fig. 3). Finally, we conclude that 11 Prdm subfamilies are ancestral to bilaterians (fig. 2). One of these subfamilies, Prdm6 is only found in nonbilaterians and deuterostomes, suggesting that this subfamily was lost during early protostome evolution. Two of the ancestral subfamilies, Prdm2 and Prdm14, are found in deuterostomes and lophotrochozoans, but not in ecdysozoans, suggesting a loss in the ecdysozoan lineage. Although all the bilaterian ancestral subfamilies were also present in the LCA of deuterostomes, one of them, Prdm8, was likely lost in the ambulacrarian lineage (echinoderms and hemichordates; fig. 2). Considering lophotrochozoans, our data suggest that many gene losses have occurred in the platyhelminthe lineage, as Prdm2, Prdm4, Prdm7/9, Prdm10/15, and Prdm12 subfamily members are not found in the two sampled platyhelminthe species (figs. 1 and 2). However, we noticed that there are two to three orphan genes in Schmidtea mediterranea and Schistosoma mansoni respectively, and that these orphans may represent very divergent members of some of the aforementioned Prdm subfamilies. Similar conclusions hold true for the rotifer Adineta vaga which possesses a reduced number of Prdm genes, two of which are orphan genes (supplementary table S1, Supplementary Material online). Our data are also suggestive of a loss of the Prdm12 gene in clitellate annelids (Helobdella robusta and Capitella teleta; fig. 2), although we cannot exclude that the single orphan gene found in both species may be a divergent Prdm12 gene.
In ecdysozoans, from an ancestral situation in which eight Prdm subfamilies were present, three of them (Prdm4, Prdm10/15, and Prdm12) have been lost before the appearance of the LCA of the six studied nematode species (fig. 4). The Prdm13 gene was retained in Trichinella spiralis, but lost in the five remaining species. Additional losses of Prdm3/16 and Prdm8 occurred in the rhabditina (belonging to the so called clade V which includes C. elegans and Pristionchus pacificus) and spirurina (clade III which includes Loa loa, Brugia malayi and Wuchereria bancrofti) lineages, respectively. The Prdm7/9 and Prdm8 genes were lost in Trichi. spiralis. In arthropods, one species, the centipede S. maritima, possesses much more Prdm genes that all the other studied species (table 1). As compared with other arthropods, this large number of Prdm genes in the centipede is due to several reasons: The retention of the ecdysozoan ancestral repertoire of Prdm genes partly lost in the other arthropod lineages, gene duplications in two of the subfamilies and the presence of two orphan genes. The phylogenetic relationships between the main arthropod lineages (pancrustaceans, chelicerates, and myriapods) are still a matter of controversy (Telford et al. 2008; Budd and Telford 2009): One hypothesis is that pancrustaceans and myriapods form a monophyletic group (Mandibulata), whereas another hypothesis suggests a monophyletic group composed of myriapods and chelicerates (Myriochelata). This controversy does not hinder our interpretations as in light of both hypotheses, Prdm4 and Prdm7/9 genes appear to have been independently lost in chelicerates and pancrustaceans, the latter having also lost the Prdm12 subfamily (fig. 4). Finally, the Prdm10/15 subfamily was lost in the Mecopterida clade that includes lepidopterans and dipterans (fig. 4).
In chordates, the putative ancestral set of bilaterian Prdm genes is composed of the same 11 subfamilies present in the LCA of bilaterians (fig. 5). The cephalochordate Branchiostoma floridae (Amphioxus) has retained most of these genes (only Prdm8 is missing; supplementary table S1, Supplementary Material online), whereas the urochordates Ci. intestinalis and Ci. savignyi (ascidians) have lost six of these ancestral subfamilies (fig. 5). In vertebrates, only few gene losses are observed, such as the loss of Prdm11 in most teleosteans, Prdm17 in marsupial mammals and birds, and Prdm7/9 in birds (fig. 5). In contrast, the Prdm gene family has significantly expanded in vertebrates. Three subfamilies (Prdm5, Prdm11, and Prdm17) arose during the course of vertebrate evolution. We determined that Prdm5 was ancestral to vertebrates, Prdm17 to gnathostomes, and Prdm11 to euteleostomes (fig. 5). However, how these new subfamilies have been produced remains unclear. An obvious possibility is that the genes belonging to these subfamilies have been produced through gene duplications of ancestral genes followed by extensive divergence of one of the paralogs (supplementary fig. S4A, Supplementary Material online). Under this hypothesis, one could expect the vertebrate-specific subfamilies to group with some other subfamilies in the phylogenetic trees (supplementary fig. S4B, Supplementary Material online). As mentioned earlier, we failed to detect robust groupings of subfamilies, even if we found an association of Prdm5 and Prdm14 subfamilies in one phylogenetic analysis (supplementary table S7, Supplementary Material online). Consequently, this deprives us of conclusive arguments in favor of the duplication hypothesis.
Our data therefore imply that an important part of the diversification of Prdm genes occurred before the emergence of present-day bilaterian main lineages. This is at odds with the previous study of Prdm gene evolution, which suggested that only very few Prdm genes were ancestral to bilaterians (Fumasoni et al. 2007). The discrepancy between our study and the previous one is due to our inclusion of a large number of nonvertebrate sequences in contrast to the few species included in the previous study. Moreover, this earlier work taxonomic sampling included species such as Drosophila melanogaster and C. elegans which were characterized here to have lost several ancestral Prdm genes. Our findings are however not surprising, as similar important early metazoan diversifications have been observed for many genes families involved in development including TFs and cell signaling molecules (e.g., Kusserow et al. 2005; Jager et al. 2006; Ryan et al. 2006; Simionato et al. 2007; Larroux et al. 2008; Degnan et al. 2009). The determination of the exact timing of the early diversification of Prdm genes is rendered difficult, in part by the still relative paucity of sequenced nonbilaterian genomes, and even more importantly by the uncertainties about the phylogenetic relationships of nonbilaterian phyla between each other and bilaterians. Our data nevertheless suggest a two-step increase in the number of Prdm genes: A first one at the origin of metazoans and a second one in the cnidarian/bilaterian lineage. A similar timing has been proposed for other developmental gene families, such as basic Helix–Loop–Helix TFs (Simionato et al. 2007), and may hold true for many other TF families (Degnan et al. 2009).
Interestingly, though the groupings between some subfamilies (Prdm6+Prdm12, Prdm8+Prdm13, Prdm2+Prdm3/16, Prdm4+10/15, and Prdm5+Prdm14) were not statistically significant, it still matched a trend of paralog retention/loss in that if a lineage lost a subfamily, it retained its putative paralog subfamily most of the time. This holds true for loss of Prdm 6 and retention of Prdm12 in Protostomes (at least ancestrally), loss of Prdm 8 and retention of Prdm13 in cnidarians and ambulacrarians, loss of Prdm2 and retention of Prdm3/16 in ecdysozoans, planarians and bivalves. This trend is less clear for the grouping Prdm4 and 10/15. Because Prdm5 is never lost after its emergence in vertebrates, that trend is not tested in that case, Prdm14 being also always present.
Gene Duplications within Prdm Subfamilies
Ten of the 14 identified Prdm subfamilies possess more than one member in at least one of the studied species (supplementary table S1, Supplementary Material online), suggesting the occurrence of gene duplications during their evolutionary history. Consequently, no genes duplications were detected in the four remaining Prdm subfamilies, Prdm4, Prdm5, Prdm11, and Prdm17. We studied the evolution of all the Prdm subfamilies by performing phylogenetic analyses (supplementary text S2, Supplementary Material online; figs. 6 and 7, supplementary figs. S5–S16, Supplementary Material online). Many duplications seem to have occurred in lineages that lead to single studied species. Nevertheless, seven subfamilies (Prdm 1, Prdm2, Prdm3/16, Prdm7/9, Prdm 8, Prdm10/15, and Prdm12) are likely to display more ancient duplications as several broadly related species possess more than one gene for these subfamilies.
Fig. 6.

Phylogenetic analysis of the Prdm3/16 subfamily. An ML tree is shown. Midpoint rooting has been used. A similar tree topology has been obtained by BI. Statistical supports (aLRT and aBayes values for ML; posterior probabilities for BI) are indicated on the nodes by color circles (color code is indicated in the figure). Nodes without color circles are not statistically supported and/or not congruent between ML and BI methods. Species names are abbreviated using the first letter of the genus name followed by the three first letters of the species name. All abbreviations can be found in table 1. Black arrows indicate pairs of paralogs that are closely related in the phylogenetic tree, whereas asterisks denote paralogs that are not closely associated in the phylogenetic tree. Duplications that likely occurred in Gnathostomata, Pancrustacea, and Hymenoptera are indicated. Nodes that define the monophyletic groups that allowed to position these duplications are also highlighted (Prdm3/Prdm16; Prdm3_16a/Prdm3_16b; Prdm3_16a1/Prdm3_16a2). These monophyletic groups are also found in ML trees constructed with several different species samplings (sampling 1: only deuterostome genes; sampling 2: only chordate genes; sampling 3: only vertebrate genes; sampling 4: only deuterostomes and ecdysozoans; sampling 5: only protostomes; sampling 6: only ecdysozoans; sampling 7: only arthropods).
Fig. 7.

Phylogenetic analysis of the Prdm10/15 subfamily. An ML tree is shown. Midpoint rooting has been used. A similar tree topology has been obtained by BI. Legend is as in figure 6. The duplication that likely took place during early vertebrate evolution is indicated. Nodes that define the monophyletic groups that allowed to position this duplication are also highlighted (Prdm10/Prdm15). These monophyletic groups are also found in ML trees constructed with several different species samplings (sampling 1: only deuterostome genes; sampling 2: only chordate genes; sampling 3: only vertebrate genes).
We mapped these duplications onto the species phylogenetic tree using both ancestral state reconstruction and phylogenetic analyses. We constructed a character matrix (supplementary table S13, Supplementary Material online) that compiles the number of members for the seven aforementioned Prdm subfamilies in all studied species, with a multistate code (0 = no member, 1 = one member, 2 = two members, 3 = three or more members). This matrix was used to infer the number of members of each of these subfamilies at the different nodes of the metazoan tree. For this purpose, we used both unbiased parsimony and ML approaches as implemented in the ancestral state reconstruction tools of Mesquite (see Materials and Methods for details). Results of these analyses obtained by parsimony and ML were very consistent and are shown in supplementary tables S14 and S15, Supplementary Material online. By comparing the number of members at different nodes, we can both infer the position of gene duplications and identify potential secondary gene losses. These inferences were confronted to the phylogenetic analysis of the corresponding subfamilies. The robustness of the obtained phylogenetic trees was further assessed using species resampling and approximately unbiased (AU) topology tests (see Materials and Methods).
Prdm1 Subfamily
Ancestral state reconstruction indicates one duplication having occurred in the Gnathostomata lineage, followed by the secondary loss of one of the duplicates in the Tetrapoda lineage and a second duplication in the Euteleostei lineage (fig. 5). Both hypotheses are at first sight supported by the phylogenetic tree of this subfamily (supplementary fig. S5, Supplementary Material online). However, the nodes defining the crucial monophyletic groups for the hypothesis of the duplication in the Gnathostomata lineage (the Prdm1b group in particular) are not statistically supported and are not robust over species resampling (supplementary fig. S5, Supplementary Material online). In addition, alternative tree topologies that are not consistent with this hypothesis cannot be rejected by the AU test (supplementary table S16, Supplementary Material online). In contrast, the node that defines a monophyletic group made of euteleost Prdm1a and Prdm1c genes (and therefore supporting a duplication in the Euteleostei lineage) is statistically supported and found with different species samplings (supplementary fig. S5, Supplementary Material online). Alternative topologies not consistent with this hypothesis are rejected by the AU test (supplementary table S16, Supplementary Material online). The phylogenetic analyses provide therefore strong support for the hypothesis of a duplication having occurred in the Euteleostei lineage, but are not helpful for positioning the other duplication which may have occurred in the Gnathostomata lineage.
Prdm2 Subfamily
Ancestral state reconstruction indicates that one duplication has occurred in the lineage leading to the Teleostei ancestor. This hypothesis is supported by the phylogenetic analysis as we observed a monophyletic group that includes the teleost Prdm2a and Prdm2b genes (supplementary fig. S6, Supplementary Material online). This monophyletic group is also found in trees constructed with different species samplings and alternative topologies in which the teleost Prdm genes do not form a monophyletic group are rejected by the AU test (supplementary table S16, Supplementary Material online). The phylogenetic analyses therefore provide support for the hypothesis of a duplication in the Teleostei lineage.
Prdm3/16 Subfamily
Ancestral state reconstruction indicates that two duplications have occurred, one in vertebrates in the lineage leading to the Gnathostomata ancestor (this duplication gave rise to the Prdm3 and Prdm16 genes; fig. 5) and another in arthropods in the lineage leading to the Hexapoda ancestor (Prdm3/16a and Prdm3/16b; fig. 4). The phylogenetic analysis supports the positioning of a duplication in the Gnathostomata lineage, as the gnathostome sequences form a monophyletic group and the single gene from the nongnathostome vertebrate Petromyzon (Pmar_prdm3_16) is found as outgroup of this monophyletic group (fig. 6). This topology is robust over species resampling and alternative topologies that are not consistent with the hypothesis of a duplication in the Gnathostomata lineage are rejected by the AU test (supplementary table S16, Supplementary Material online). The phylogenetic tree also shows that the single gene found in the crustacean Daphnia (Dpul_Prdm3_16) clusters with hexapod Prdm3_16a genes, whereas the single genes that are found in other nonhexapod species are found as outgroup of the Prdm3a + Prdm3b clade (fig. 6). This suggests that the duplication may have occurred before the divergence between crustaceans and hexapods within the Pancrustacea lineage and that Daphnia has lost the Prdm3b gene. In addition, the two paralogs that are found in some hymenopterans clustered together within the Prdm3_16a group, suggesting an additional duplication and the loss of the Prdm3_16b genes in hymenopterans (fig. 6). These groups are found in trees constructed with other species samplings (fig. 6) and alternative groupings are rejected by the AU test (supplementary table S16, Supplementary Material online). Phylogenetic analyses therefore support the occurrence of three duplications, in the Gnathostomata, Pancrustacea, and Hymenoptera lineages (figs. 4 and 5).
Prdm7/9 Subfamily
Ancestral state reconstruction indicates that in Primates, a duplication may have occurred in the lineage that leads to the ancestor of either Haplorrhini or Simiiformes (fig. 5). The phylogenetic tree does not provide support for this hypothesis as the Primate Prdm7 and Prdm9 genes do not form separate monophyletic groups (supplementary fig. S10, Supplementary Material online). More generally, this gene tree shows many incongruences with the species tree, which could be due to the rapid and unusual evolution of these genes in mammals, and in primates in particular (Oliver et al. 2009; Thomas et al. 2009). Strikingly different topologies were found when the trees were constructed with different species samplings, but we never found separate Prdm7 and Prdm9 monophyletic groups. We tested alternative topologies in which the Prdm7 and Prdm9 genes form separate monophyletic groups and found that these alternative topologies are nevertheless not rejected by the AU test (supplementary table S16, Supplementary Material online). We therefore conclude that the phylogenetic analysis of the Prdm7/9 subfamily is unreliable for positioning the duplication that has occurred.
Prdm8 Subfamily
Ancestral state reconstruction indicates that duplications may have independently occurred in the lineages leading to Danio, Oryzias, and Callorhinchus. The phylogenetic analysis in contrast suggests a duplication having occurred in the lineage leading to the Gnathostomata ancestor, followed by gene losses in the Sarcopterygia lineage and in the lineages leading to several teleosts (fig. 5 and supplementary fig. S11, Supplementary Material online). Indeed, one of the paralogs found in Danio, Oryzias, and Callorhinchus (Prdm8b sequences) forms a monophyletic group (that also includes the single gene found in Gasterosteus) that is found as the sister group to a large monophyletic group that includes all sarcopterygian Prdm8 sequences, the single genes found in some teleosts and the second Danio, Oryzias, and Callorhinchus paralogs (Prdm8a genes). This topology is robust over species resamplings and alternative topologies are rejected by the AU test (supplementary fig. S11 and table S16, Supplementary Material online). Phylogenetic analyses therefore suggest a duplication in Gnathostomata.
Prdm10/15 Subfamily
Ancestral state reconstruction indicates that a duplication, which gave rise to Prdm10 and Prdm15 genes, has occurred in the lineage leading to the Gnathostomata ancestor. Accordingly, Prdm10 and Prdm15 genes form well-supported monophyletic groups in the phylogenetic tree of this subfamily (fig. 7). However, the single gene found in the nongnathostome Petromyzon is found within the Prdm15 group. Gnathostome Prdm15 proteins display a few stretches of amino acids that are not found in gnathostome Prdm10 or nonvertebrate Prdm10/15 proteins (not shown). The single Petromyzon protein displays these stretches, further supporting that it is encoded by a bona fide Prdm15 gene. This therefore suggests that the duplication has occurred at the root of the vertebrates (fig. 5) and that the Prdm10 paralog has been lost in Petromyzon (or is located in a nonsequenced part of the genome). A same tree topology than in trees constructed with different species samplings (fig. 7) and alternative topologies in which the Petromyzon gene clusters with the Prdm10 genes are rejected by the AU test (supplementary table S16, Supplementary Material online). We therefore conclude that the duplication has likely occurred in the lineage leading to the Vertebrata ancestor.
Prdm12 Subfamily
Ancestral state reconstruction indicates that either one duplication occurred in the Euteleostei lineage or one duplication occurred in the Ovalentariae lineage and possibly a second in the Percomorpharia lineage (fig. 5). Phylogenetic analyses strongly support the hypothesis of a duplication in the Euteleostei lineage. Indeed, we can observe in the phylogenetic tree two monophyletic groups that each includes one of the two paralogs (Prdm12a and Prdm12b) found in Euteleostei species, with the single gene from Danio as outgroup, this topology is robust over species resamplings and alternative topologies not consistent with the duplication in Euteleostei are rejected by the AU test (supplementary fig. S13 and table S16, Supplementary Material online). We therefore conclude that the duplication has likely occurred in the lineage leading to the Euteleostei ancestor.
In conclusion, the combination of ancestral state reconstruction and phylogenetic analyses allowed us to determine the timing of several duplications that occurred during the evolution of some Prdm subfamilies. Two duplications occurred during early evolution of vertebrates, giving rise to Prdm10 and Prdm15 genes, on one hand, and Prdm3 and Prdm16 genes, on the other hand. Together with the origination of the three vertebrate-specific subfamilies (Prdm5, Prdm11, and Prdm17), these duplications contribute to the overall expansion of the Prdm gene repertoire in vertebrates. Two other duplications likely took place in the Gnathostomata lineage (Prdm1 and Prdm8 subfamilies), but only one paralog was retained in most vertebrates. Several duplications (Prdm1, Prdm2, and Prdm12 subfamilies) occurred in teleosts and one duplication took place in primates, giving rise to Prdm7 and Prdm9 genes.
Evolutionary Origin of Prdm Genes
So far we only identified and studied Prdm genes in metazoans. To determine whether Prdm genes can be found in nonmetazoan species, we made Basic Local Alignment Search Tool (BLAST) searches against the fully sequenced genomes of 40 species belonging to several eukaryotic lineages, with a special focus on opisthokonts, the group to which metazoans belong (supplementary table S17, Supplementary Material online). We used the same methodology than for the searches in metazoans: The 17 Human Prdm genes were used as queries, the best BLAST hits for each species were used to make BLAST against Human Refseq genes, and only those that allow retrieving Prdm genes as best BLAST hits were retained. One or a few such sequences were found in 25 of the 40 studied species (supplementary table S17 and data set S2, Supplementary Material online). In most cases, sequence similarity to Prdm genes was low. None of the encoded proteins contains both a SET domain and Zn fingers, whereas many of the proteins lack these domains entirely. Three of the proteins contain a SET domain (supplementary table S17, Supplementary Material online)—however, in a phylogenetic analysis with a larger sample of SET domain proteins (see below), these sequences do not cluster with Prdm proteins (not shown). We determine that it is unlikely that bona fide Prdm genes exist outside metazoans and therefore conclude that the Prdm genes constitute a metazoan-specific family of TFs.
Seventeen of the retrieved nonmetazoan genes encode proteins with one or more Zn fingers and we further studied these genes. We noticed that, when making BLAST searches of Prdm genes in animals, we also often retrieved non-Prdm Zn finger genes with some similarity to Prdm genes. We therefore made phylogenetic analyses using a multiple alignment of the Zn fingers of a few Prdm proteins (from two deuterostomes [Human and Saccoglossus], one ecdysozoan Strigamia, and one lophotrochozoan Platynereis), a large set of Human Zn finger proteins retrieved in the BLAST searches (supplementary data set S3, Supplementary Material online), and the 17 aforementioned nonmetazoan proteins. We found a monophyletic group that includes all Prdm proteins (except Human Prdm9), the 17 nonmetazoan Zn finger proteins, and 11 Human Zn finger proteins (supplementary fig. S17, Supplementary Material online). Within this group, the Prdm proteins do not form a monophyletic group and are intermixed with Human and nonmetazoan Zn finger proteins, suggesting that some Prdm Zn fingers are more closely related to some non-Prdm Zn fingers than to other Prdm Zn Fingers. One simple interpretation of this phylogenetic tree would therefore be that the Prdm Zn fingers originated from several different ancestral genes. The phylogenetic tree has however to be taken with much caution, as 1) many nodes of the tree have low to very low statistical support, 2) different groupings were found in a few instances in the ML and Bayesian inference (BI) trees, and 3) some phylogenetic relationships between Prdm proteins were at odds with our other analyses (e.g., Human Prdm3 and Prdm16 do not cluster together nor with Platynereis Prdm3/16). Therefore, we cannot exclude that some of the phylogenetic relationships displayed in this tree could be artefactual.
We next wondered whether a similar situation can be observed for the modified SET domain of Prdm proteins: The PR domain. We started from the SET domain proteins identified by Sun et al. (2008) in Human, Drosophila, Saccharomyces, and Schizosaccharomyces and retrieved their putative orthologs by BLAST searches in 14 additional species belonging to Unikonts (supplementary table S18 and data set S4, Supplementary Material online). Orthology relationships were assessed by reciprocal best BLAST hit and only the putative orthologs that encode a protein with a recognizable SET domain were retained for further analysis. We then performed phylogenetic analyses using a multiple alignment of the SET domain of the 419 retrieved proteins. We found a well-supported monophyletic and exclusive group of Prdm proteins, in addition to several other monophyletic groups (fig. 8). This phylogenetic tree is largely consistent with previous analyses of SET domain proteins made with different species samples (Sun et al. 2008; Zhang and Ma 2012; Lhuillier-Akakpo et al. 2014). Therefore, this phylogenetic analysis strongly suggests that the PR-modified SET domain of all Prdm proteins originates from a single ancestral SET domain. Interestingly, we found that SET domain sequences from three nonmetazoan opisthokont species (Capsaspora owczarzaki, Sphaeroforma arctica, and Spizellomyces punctatus) cluster with the Prdm proteins in the phylogenetic tree (fig. 8). The corresponding genes, which are found in three different opisthokont lineages (filastereans, ichthyosporeans, and fungi), might therefore derive from the same ancestral gene that gave rise to Prdm genes. However, we have to note that the three aforementioned nonmetazoan proteins, when blasted against animal genomes, do not allow retrieving Prdm proteins among the best BLAST hits. In addition, the PR domain of Prdm proteins is considered to be a very divergent type of SET domain (Sun et al. 2008; Wu et al. 2010). Therefore, we cannot rule out that the association of these three proteins with Prdm ones in the phylogenetic tree may be an artefactual grouping of very divergent SET domains. In addition, if the three nonmetazoan genes do indeed derive from the ancestral gene from which the Prdm genes originated, we have to assume that this ancestral gene has been lost in most studied fungi and choanoflagellates. This type of differential loss/retention of ancestral genes in different lineages is not uncommon, as it has been observed for other TF families and RNA-binding proteins (Kerner et al. 2011; de Mendoza et al. 2013), and has been proposed as one of the reasons behind the existence of taxonomically restricted genes (Forêt et al. 2010).
Fig. 8.

Phylogenetic analysis of SET domain proteins. An unrooted ML tree is shown. Statistical supports for the different highlighted groups are as in figure 6. The different groups were named according to Sun et al. (2008) and Zhang and Ma (2012).
Taken together, our data suggest that Prdm genes appeared alongside early animal evolution through a genome rearrangement event that put together Zn fingers and SET domain-encoding regions from different genes (supplementary fig. S18, Supplementary Material online). Although the SET domain-encoding region of all Prdm genes would have originated from a single ancestral gene, Zn fingers would have derived from several different ancestral genes (supplementary fig. S18A, Supplementary Material online). We cannot however rule out an alternative scenario in which after gene duplications from a single ancestral Prdm gene, genome rearrangement events, such as exon shuffling or gene conversion, may have brought different or additional Zn finger-encoding regions to separate Prdm genes (supplementary fig. S18B, Supplementary Material online).
Conclusions
Our data suggest that Prdm genes originated at the dawn of the animal kingdom through the association of a single ancestral SET domain-encoding gene with one or several Zn finger-encoding genes. It has been often proposed that appearance of “new” genes may be correlated to the appearance of new traits (Khalturin et al. 2009; Forêt et al. 2010) and it is therefore tempting to speculate that Prdm genes may have had an importance for the acquisition of some metazoan traits. Given the involvement of many Prdm genes in development in present-day bilaterians, one obvious possibility would be implications in the evolution of developmental processes associated with the acquisition of multicellularity in metazoans. Our study also sheds new light on the evolution of Prdm genes in animals. We defined four probable phases of Prdm gene family diversification: 1) one phase prior to metazoan cladogenesis and which gave rise to two to three different Prdm genes (this phase may in fact correspond to the appearance of Prdm genes as discussed above); 2) a second phase before cnidarians and bilaterians diverged and which gave rise to six different genes; 3) a third phase before the divergence of the main bilaterian lineages, giving rise to 11 different genes; and 4) a fourth phase during early vertebrate evolution leading to the presence of three additional Prdm subfamilies in most vertebrates lineages. Finally, by identifying Prdm genes in many species in which these genes were so far unknown, including species amenable to gene expression and functional studies, our study also paves the way toward a better understanding of the evolution of the expression patterns and functions of the Prdm genes in animals.
Materials and Methods
Whole-Genome Analysis
For all the studied species, genomic sequences (genome contigs and/or gene predictions and/or peptide predictions) were retrieved from the various sources listed in supplementary tables S2–S6, Supplementary Material online. BLAST searches (Altschul et al. 1997) were performed using the KoriBlast and ngKLAST softwares (Korilog Company, Muzillac, France) on local databases constructed from the downloaded genomic sequences. We first used the 17 described Prdm genes from Homo sapiens (Fumasoni et al. 2007) as queries for these BLAST searches. To enhance the comprehensiveness of our searches, we next used the Prdm sequences of some additional species as queries in BLAST searches against the genome of species from the same taxonomic group. We used the Prdm sequences from the well-assembled and annotated Drosophila and Tribolium genomes as queries to search the Prdm genes in the other arthropod genomes; the Danio Prdm sequences were used to screen the genomes of the other teleosts; and the Prdm genes from Amphimedon and Nematostella were used for searches in the nonbilaterian and nonmetazoan genomes. Sequences were recognized as Prdm genes if they 1) show high sequence similarity to Human Prdm genes, 2) display the characteristic Prdm domains (PR domain and/or Zn fingers), and 3) allow retrieving Prdm genes as best BLAST hits when used as query to make BLAST against the Human genome. Conserved domains were identified using NCBI batch CD-search (Marchler-Bauer et al. 2015). To test the extensiveness of our approach, we chose a sample of 11 species considered as having the best-characterized species genomes and allowing a fair coverage of the diversity of the studied animals. The species are H. sapiens, B. floridae, Stro. purpuratus, D. melanogaster, S. maritima, C. elegans, Cap. teleta, Lottia gigantea, Nematostella vectensis, Pleurobrachia bachei, and Amphimedon queenslandica. We took the identified Prdm sequences from each of these 11 species and used them as queries for BLAST searches against the genome of the remaining ten species (110 different BLASTs in total). To identify Prdm genes in these BLAST searches, we retrieved all hits and 1) blasted them against the genome from which came the query, 2) blasted them against the Human genome, and 3) identified the putative domains present in the sequences (with CD-search). We considered a given sequence to be part of a Prdm gene if this sequence matches a known Prdm genes in a reciprocal BLAST and/or a Human genome BLAST, and if it contains a least one Zinc finger or a PR domain. Strikingly, these BLAST searches only recovered already identified Prdm sequences and did not allow the identification of any additional ones, providing good evidence that our identification scheme was working very well. We also used all the “orphan” genes for BLAST searches against the genomes of the aforementioned 11 species. The rationale behind these searches was the idea that orphan genes may correspond to very divergent genes, not found in vertebrates or Drosophila, which would nonetheless allow BLAST retrieval of other divergent genes that cannot be found using Human Prdms. Eleven performed BLAST searches were performed and analyzed as mentioned above, but did not allow the identification of any additional Prdm genes in the 11 studied species, further supporting the comprehensiveness of our approach.
Multiple Alignments, Phylogenetic Analyses, and Topology Tests
Multiple alignments were obtained using MUSCLE 3.5 (Edgar 2004), available on the Bioinformatics toolkit platform of the Max Planck Institute for Developmental Biology at Tübingen (http://toolkit.tuebingen.mpg.de, last accessed November 25, 2015). The resulting multiple alignments were manually improved. Multiple alignments were handled using SEAVIEW 4 (Gouy et al. 2010). BoxShade (http://www.ch.embnet.org/software/BOX_form.html, last accessed November 25, 2015) was used to generate printouts of some multiple alignments. WebLogo (http://weblogo.berkeley.edu, last accessed November 25, 2015) was used to generate sequence logos. ML analyses were performed using the online available PHYML software (http://www.atgc-montpellier.fr/phyml/, last accessed November 25, 2015; Guindon and Gascuel 2003; Guindon et al. 2005). Le and Gascuel amino acid substitution model was used (Le and Gascuel 2008). Equilibrium frequencies, proportion of invariable sites, and gamma shape parameter were estimated from the data. We used four substitution rate categories. The starting trees were generated using BIONJ and the NNI type of tree improvement was used. Statistical supports for the different internal branches were determined by approximate likelihood-ratio test (aLRT), a Bayesian-like transformation of aLRT (aBayes) and in some cases bootstrap resampling (Anisimova and Gascuel 2006; Anisimova et al. 2011). BI was performed using MRBAYES 3.2 (Huelsenbeck and Ronquist 2001; Ronquist et al. 2012) with JTT + G model (Jones et al. 1992). Two independent Markov chains were sampled every 200 generations. The trees obtained in the two runs were mixed and the first 25% of the trees were discarded as “burn-in.” Convergence was assessed by looking at the average standard deviation split frequencies and potential scale reduction factor values, following the software’s instructions. Phylogenetic trees were visualized and rooted using FigTree 1.4 developed by Andrew Rambaud (http://tree.bio.ed.ac.uk/software/figtree/, last accessed November 25, 2015). We also performed AU test (Shimodaira 2002). We generated trees with different topologies by rearranging the branching order of the ML trees produced by PhyML. Likelihoods of these test trees and best PhyML tree were compared by the AU test using CONSEL (Shimodaira and Hasegawa 2001). A given topology was considered as rejected if the P value of this tree was inferior to 0.05.
Ancestral State Reconstructions
Character matrices were constructed with Mesquite (Version 3.03; http://mesquiteproject.org, last accessed November 25, 2015). Species trees were manually constructed (using the nexus format) and then visualized with Mesquite. Ancestral state reconstructions were performed using the “Trace all characters” tool of Mesquite, using both parsimony and ML methods. To reconstruct the ancestral states of the presence/absence of the different Prdm subfamilies with the ML method, the asymmetrical two-parameter Markov-k Model (AsymmMk) was used. We tested a range of forward and backward rates—the ancestral rates shown in the article were consistently obtained when the forward rate is less than 0.05 and the backward rate greater than 0.3. Values shown in supplementary tables S11 and S12, Supplementary Material online, were obtained with a forward rate of 0.00340136 and a backward rate of 0.59183673. Likelihoods were calculated assuming that root state frequencies are same as equilibrium. To reconstruct the ancestral states of the number of members of the Prdm1, 2, 3/16, 7/9, 8, 10/15, and 12 subfamilies with the ML method, the symmetrical one-parameter Markov-k Model (Mk1) was used.
Supplementary Material
Supplementary texts S1 and S2, tables S1–S18, figures S1–S18, and data sets S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors are grateful to Maja Adamska for giving them access to the Sycon ciliatum genomic data. They also thank the Genoscope and the EMBL for giving access to unpublished Platynereis expressed sequence tags and genomic sequences. They also thank Eric Bellefroid for stimulating discussions about Prdm gene functions and their evolution. They thank Eve Gazave and B. Duygu Ozpolat for critical reading of the manuscript. They are grateful to Adrien Demilly for help with the figures. This work was funded by the CNRS, the Agence Nationale de la Recherche (France) (ANR grant Blanc METAMERE), the Institut Universitaire de France and the Who am I? laboratory of excellence (No.ANR-11-LABX-0071) funded by the French Government.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Gascuel O. 2006. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 55:539–552. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Gil M, Dufayard J-F, Dessimoz C, Gascuel O. 2011. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol. 60:685–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327:836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd GE, Telford MJ. 2009. The origin and evolution of arthropods. Nature 457:812–817. [DOI] [PubMed] [Google Scholar]
- Chang JC, Meredith DM, Mayer PR, Borromeo MD, Lai HC, Ou Y-H, Johnson JE. 2013. Prdm13 mediates the balance of inhibitory and excitatory neurons in somatosensory circuits. Dev Cell. 25:182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu L-F, Surani MA, Jaenisch R, Zwaka TP. 2011. Blimp1 expression predicts embryonic stem cell development in vitro. Curr Biol. 21:1759–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mendoza A, Sebé-Pedrós A, Sestak MS, Matejcic M, Torruella G, Domazet-Loso T, Ruiz-Trillo I. 2013. Transcription factor evolution in eukaryotes and the assembly of the regulatory toolkit in multicellular lineages. Proc Natl Acad Sci U S A. 110:E4858–E4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan BM, Vervoort M, Larroux C, Richards GS. 2009. Early evolution of metazoan transcription factors. Curr Opin Genet Dev. 19:591–599. [DOI] [PubMed] [Google Scholar]
- Di Zazzo E, De Rosa C, Abbondanza C, Moncharmont B. 2013. PRDM proteins: molecular mechanisms in signal transduction and transcriptional regulation. Biology 2:107–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, Seaver E, Rouse GW, Obst M, Edgecombe GD, et al. 2008. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452:745–749. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fog CK, Galli GG, Lund AH. 2012. PRDM proteins: important players in differentiation and disease. Bioessays 34:50–60. [DOI] [PubMed] [Google Scholar]
- Forêt S, Knack B, Houliston E, Momose T, Manuel M, Quéinnec E, Hayward DC, Ball EE, Miller DJ. 2010. New tricks with old genes: the genetic bases of novel cnidarian traits. Trends Genet. 26:154–158. [DOI] [PubMed] [Google Scholar]
- Fumasoni I, Meani N, Rambaldi D, Scafetta G, Alcalay M, Ciccarelli FD. 2007. Family expansion and gene rearrangements contributed to the functional specialization of PRDM genes in vertebrates. BMC Evol Biol. 7:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 27:221–224. [DOI] [PubMed] [Google Scholar]
- Grabole N, Tischler J, Hackett JA, Kim S, Tang F, Leitch HG, Magnúsdóttir E, Surani MA. 2013. Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep. 14:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704. [DOI] [PubMed] [Google Scholar]
- Guindon S, Lethiec F, Duroux P, Gascuel O. 2005. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33:W557–W559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanotel J, Bessodes N, Thélie A, Hedderich M, Parain K, Van Driessche B, Brandao Kde O, Kricha S, Jorgensen MC, Grapin-Botton A, et al. 2014. The Prdm13 histone methyltransferase encoding gene is a Ptf1a-Rbpj downstream target that suppresses glutamatergic and promotes GABAergic neuronal fate in the dorsal neural tube. Dev Biol. 386:340–357. [DOI] [PubMed] [Google Scholar]
- Hohenauer T, Moore AW. 2012. The Prdm family: expanding roles in stem cells and development. Development 139:2267–2282. [DOI] [PubMed] [Google Scholar]
- Huang S. 2002. Histone methyltransferases, diet nutrients and tumour suppressors. Nat Rev Cancer. 2:469–476. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755. [DOI] [PubMed] [Google Scholar]
- Jager M, Queinnec E, Houliston E, Manuel M. 2006. Expansion of the SOX gene family predated the emergence of the Bilateria. Mol Phylogenet Evol. 39:468–477. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 8:275–282. [DOI] [PubMed] [Google Scholar]
- Kerner P, Degnan SM, Marchand L, Degnan BM, Vervoort M. 2011. Evolution of RNA-binding proteins in animals: insights from genome-wide analysis in the sponge Amphimedon queenslandica. Mol Biol Evol. 28:2289–2303. [DOI] [PubMed] [Google Scholar]
- Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TCG. 2009. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet. 25:404–413. [DOI] [PubMed] [Google Scholar]
- Kinameri E, Inoue T, Aruga J, Imayoshi I, Kageyama R, Shimogori T, Moore AW. 2008. Prdm proto-oncogene transcription factor family expression and interaction with the Notch-Hes pathway in mouse neurogenesis. PLoS One 3:e3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto K, Yabuta Y, Ohinata Y, Shigeta M, Yamanaka K, Saitou M. 2008. Complex genome-wide transcription dynamics orchestrated by Blimp1 for the specification of the germ cell lineage in mice. Genes Dev. 22:1617–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusserow A, Pang K, Sturm C, Hrouda M, Lentfer J, Schmidt HA, Technau U, von Haeseler A, Hobmayer B, Martindale MQ, et al. 2005. Unexpected complexity of the Wnt gene family in a sea anemone. Nature 433:156–160. [DOI] [PubMed] [Google Scholar]
- Larroux C, Luke GN, Koopman P, Rokhsar DS, Shimeld SM, Degnan BM. 2008. Genesis and expansion of metazoan transcription factor gene classes. Mol Biol Evol. 25:980–996. [DOI] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol. 25:1307–1320. [DOI] [PubMed] [Google Scholar]
- Lhuillier-Akakpo M, Frapporti A, Denby Wilkes C, Matelot M, Vervoort M, Sperling L, Duharcourt S. 2014. Local effect of enhancer of zeste-like reveals cooperation of epigenetic and cis-acting determinants for zygotic genome rearrangements. PLoS Genet. 10:e1004665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir E, Dietmann S, Murakami K, Gunesdogan U, Tang F, Bao S, Diamanti E, Lao K, Gottgens B, Azim Surani M. 2013. A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat Cell Biol. 15:905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. 2015. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43:D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldal BHM, Debenham NJ, De Ley P, De Ley IT, Vanfleteren JR, Vierstraete AR, Bert W, Borgonie G, Moens T, Tyler PA, et al. 2007. An improved molecular phylogeny of the Nematoda with special emphasis on marine taxa. Mol Phylogenet Evol. 42:622–636. [DOI] [PubMed] [Google Scholar]
- Mihola O, Trachtulec Z, Vlcek C, Schimenti JC, Forejt J. 2009. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323:373–375. [DOI] [PubMed] [Google Scholar]
- Ohinata Y, Payer B, O’Carroll D, Ancelin K, Ono Y, Sano M, Barton SC, Obukhanych T, Nussenzweig M, Tarakhovsky A, et al. 2005. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 436:207–213. [DOI] [PubMed] [Google Scholar]
- Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N, Beatson SA, Lunter G, Malik HS, Ponting CP. 2009. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 5:e1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. 2010. Prdm9 controls activation of mammalian recombination hotspots. Science 327:835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Derelle R, Lopez P, Pick K, Borchiellini C, Boury-Esnault N, Vacelet J, Renard E, Houliston E, Queinnec E, et al. 2009. Phylogenomics revives traditional views on deep animal relationships. Curr Biol. 19:706–712. [DOI] [PubMed] [Google Scholar]
- Pick KS, Philippe H, Schreiber F, Erpenbeck D, Jackson DJ, Wrede P, Wiens M, Alie A, Morgenstern B, Manuel M, et al. 2010. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Mol Biol Evol. 27:1983–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M, et al. 2012. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150:948–960. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JF, Burton PM, Mazza ME, Kwong GK, Mullikin JC, Finnerty JR. 2006. The cnidarian-bilaterian ancestor possessed at least 56 homeoboxes: evidence from the starlet sea anemone, Nematostella vectensis. Genome Biol. 7:R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JF, Mazza ME, Pang K, Matus DQ, Baxevanis AD, Martindale MQ, Finnerty JR. 2007. Pre-bilaterian origins of the Hox cluster and the Hox code: evidence from the sea anemone, Nematostella vectensis. PLoS One 2:e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JF, Pang K, Schnitzler CE, Nguyen AD, Moreland RT, Simmons DK, Koch BJ, Francis WR, Havlak P, Program NCS, et al. 2013. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342:1242592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierwater B, Eitel M, Jakob W, Osigus H-J, Hadrys H, Dellaporta SL, Kolokotronis S-O, Desalle R. 2009. Concatenated analysis sheds light on early metazoan evolution and fuels a modern “urmetazoon” hypothesis. PLoS Biol. 7:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebé-Pedrós A, Ariza-Cosano A, Weirauch MT, Leininger S, Yang A, Torruella G, Adamski M, Adamska M, Hughes TR, Gomez-Skarmeta JL, et al. 2013. Early evolution of the T-box transcription factor family. Proc Natl Acad Sci U S A. 110:16050–16055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebé-Pedrós A, de Mendoza A, Lang BF, Degnan BM, Ruiz-Trillo I. 2011. Unexpected repertoire of metazoan transcription factors in the unicellular holozoan Capsaspora owczarzaki. Mol Biol Evol. 28:1241–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H. 2002. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 51:492–508. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. 2001. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17:1246–1247. [DOI] [PubMed] [Google Scholar]
- Simionato E, Ledent V, Richards G, Thomas-Chollier M, Kerner P, Coornaert D, Degnan BM, Vervoort M. 2007. Origin and diversification of the basic helix-loop-helix gene family in metazoans: insights from comparative genomics. BMC Evol Biol. 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X-J, Xu P-F, Zhou T, Hu M, Fu CT, Zhang Y, Jin Y, Chen Y, Chen SJ, Huang QH, et al. 2008. Genome-wide survey and developmental expression mapping of zebrafish SET domain-containing genes. PLoS One 3:e1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford MJ, Bourlat SJ, Economou A, Papillon D, Rota-Stabelli O. 2008. The evolution of the Ecdysozoa. Philos Trans R Soc Lond B Biol Sci. 363:1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford MJ, Copley RR. 2011. Improving animal phylogenies with genomic data. Trends Genet. 27:186–195. [DOI] [PubMed] [Google Scholar]
- Thomas JH, Emerson RO, Shendure J. 2009. Extraordinary molecular evolution in the PRDM9 fertility gene. PLoS One 4:e8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein MD, Wiegmann BM, Beutel R, Kjer KM, Yeates DK. 2012. Advances in insect phylogeny at the dawn of the postgenomic era. Annu Rev Entomol. 57:449–468. [DOI] [PubMed] [Google Scholar]
- Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali-Hassani A, Campagna-Slater V, Vedadi M, Arrowsmith CH, et al. 2010. Structural biology of human H3K9 methyltransferases. PLoS One 5:e8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji M, Seki Y, Kurimoto K, Yabuta Y, Yuasa M, Shigeta M, Yamanaka K, Ohinata Y, Saitou M. 2008. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat Genet. 40:1016–1022. [DOI] [PubMed] [Google Scholar]
- Yamaji M, Ueda J, Hayashi K, Ohta H, Yabuta Y, Kurimoto K, Nakato R, Yamada Y, Shirahige K, Saitou M. 2013. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell 12:368–382. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ma H. 2012. Complex evolutionary history and diverse domain organization of SET proteins suggest divergent regulatory interactions. New Phytol. 195:248–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.