

Abstract
Parasitism creates selection for resistance mechanisms in host populations and is hypothesized to promote increased host evolvability. However, the influence of these traits on host evolution when parasites are no longer present is unclear. We used experimental evolution and whole-genome sequencing of Escherichia coli to determine the effects of past and present exposure to parasitic viruses (phages) on the spread of mutator alleles, resistance, and bacterial competitive fitness. We found that mutator alleles spread rapidly during adaptation to any of four different phage species, and this pattern was even more pronounced with multiple phages present simultaneously. However, hypermutability did not detectably accelerate adaptation in the absence of phages and recovery of fitness costs associated with resistance. Several lineages evolved phage resistance through elevated mucoidy, and during subsequent evolution in phage-free conditions they rapidly reverted to nonmucoid, phage-susceptible phenotypes. Genome sequencing revealed that this phenotypic reversion was achieved by additional genetic changes rather than by genotypic reversion of the initial resistance mutations. Insertion sequence (IS) elements played a key role in both the acquisition of resistance and adaptation in the absence of parasites; unlike single nucleotide polymorphisms, IS insertions were not more frequent in mutator lineages. Our results provide a genetic explanation for rapid reversion of mucoidy, a phenotype observed in other bacterial species including human pathogens. Moreover, this demonstrates that the types of genetic change underlying adaptation to fitness costs, and consequently the impact of evolvability mechanisms such as increased point-mutation rates, depend critically on the mechanism of resistance.
Keywords: experimental evolution, coevolution, mutation rate, bacteriophages.
Introduction
Infectious parasites create evolutionary pressure for resistance mechanisms in their hosts (Schmid-Hempel 2011) and indirectly select for mechanisms that generate genetic and phenotypic diversity (Hamilton et al. 1990; Zaman et al. 2014). Bacterial infection by viruses (phages) is the most common host–parasite interaction on Earth, globally estimated at 1025 infections per second (Pedulla et al. 2003), and diverse phage-resistance mechanisms have been characterized (Labrie et al. 2010). Phage selection can also cause alleles that modify bacterial mutation rates to spread through genetic hitchhiking, because resistance mutations are produced more frequently in mutator lineages (Pál et al. 2007; Morgan et al. 2010; O’Brien et al. 2013). Both resistance and mutator alleles have important effects on bacterial evolutionary trajectories (Wielgoss et al. 2013; Scanlan et al. 2015), medical outcomes by altering bacterial growth rate and virulence (Smith and Huggins 1982; Capparelli et al. 2010), and antibiotic resistance (Oliver et al. 2000; Denamur et al. 2002). However, it remains unclear how traits acquired under parasitism, including phage resistance and hypermutability, will influence evolution in the absence of parasites. This question is key to understanding the long-term effects of parasitism: Parasite densities and rates of infection, including those of phages (Weinbauer 2004), are inherently variable in space and time, particularly following the evolution of host resistance (Sasaki 2000). Therefore, a full understanding of how parasitism influences host evolution requires that we account for not only resistance evolution but also subsequent adaptation in parasite-free conditions.
Bacteria can readily adapt to recover fitness defects associated with both phage- (Lenski 1988b) and antibiotic-resistance (Schrag et al. 1997; Maisnier-Patin and Andersson 2004). Experiments with antibiotic-resistant bacteria indicate that mutator alleles can accelerate this process (Perron et al. 2010), but it is not known whether this is also true for phage resistance. Crucially, we can predict that compensatory mechanisms, and potentially the effects of hypermutability, will depend on the original mechanism of resistance. For example, many antibiotics target essential enzymes, and consequently resistance is often due to mutations that alter the structure of these enzymes (Walsh 2000; Blair et al. 2014). When antibiotics are removed, adaptation to recover the cost of resistance is constrained to mutations that maintain the structure and function of the antibiotic target. Typically such mutations occur at specific sites on the enzyme that was originally mutated or subunits that interact with it, and do not restore antibiotic sensitivity (Maisnier-Patin and Andersson 2004; Andersson and Hughes 2011; Brandis et al. 2012). Perron et al. (2010) showed that mutator alleles can accelerate this process for rifampicin-resistant bacteria. In contrast, costs associated with some phage-resistance mechanisms, such as modification of cell surface structures or secretion of extracellular polysaccharides (Labrie et al. 2010), may rely on different types of genetic changes. For example, there are many genes involved in synthesis and secretion of extracellular polysaccharides (Stevenson et al. 1996; Muhammadi and Ahmed 2007). Consequently, there may be a relatively broad range of genetic changes, including reduction- or loss-of-function mutations, that can both downregulate the costly expression of extracellular products and, unlike in the example of rifampicin resistance above, reverse resistance. In this case, if adaptation is not limited by the supply of beneficial mutations, then mutator alleles will not necessarily accelerate this process (Tenaillon et al. 1999). Therefore, the genetic mechanisms of adaptation following exposure to viral parasites and the impact of mutator alleles may deviate from previous observations with antibiotic-resistant bacteria and depend critically on the original mechanism of resistance.
We addressed these questions using experimental populations of E. coli and lytic phages. We manipulated bacterial mutability by use of a mutator genotype that has defective DNA mismatch repair, the same mechanism that confers increased mutability in clinical isolates of several other species including human pathogens (LeClerc et al. 1996; Chopra et al. 2003; Li et al. 2003; Jolivet-Gougeon et al. 2011). We first tested whether increased mutation rate was advantageous in the presence of each of four phages, both alone and in combinations. We then propagated different types of phage-resistant mutants, derived from mutator and nonmutator genetic backgrounds, in the absence of phages for approximately 66 generations. We included phage treatments where the major resistance mechanisms are known to involve cell-surface modification and increased secretion of extracellular polysaccharide (mucoidy), both of which have been observed in several other bacterial species including pathogens (Bohannan and Lenski 2000; Mizoguchi et al. 2003; Labrie et al. 2010; Scanlan and Buckling 2012; Crémet et al. 2013). This strategy, summarized schematically in supplementary figure S1, Supplementary Material online, allowed us to test four hypotheses: 1) Increased mutation rate is advantageous in the presence of different bacteriophages, both alone and in combinations; 2) mutator populations that have evolved resistance to phages adapt more effectively in phage-free conditions than nonmutators that have evolved resistance to the same phage treatment; 3) fitness recovery in the absence of phages depends on the initial mechanism of resistance; and 4) the genetic basis of adaptation depends on both bacterial mutation rate and the initial mechanism of parasite resistance.
Results
Mutator Advantage in the Presence of Parasites
We tested whether an increased mutation rate was advantageous in the presence or absence of parasites by competing a mutator and nonmutator strain (hereafter denoted “Mut” for Mutator and “Ref” for Reference, respectively) in experimental conditions with and without phages (supplementary fig. S1, Supplementary Material online). We included four different phages, both alone and in each pairwise combination, to determine whether mutator alleles have similar effects across different parasite treatments. The likelihood of the mutator spreading varied among phage treatments (F10,33 = 6.26, P < 0.0001; fig. 1): The average selection coefficient for the mutator in each phage treatment was greater than in the phage-free control, and was on average greater in two-phage treatments than single-phage treatments (Welch’s t5.78 = 4.49, P = 0.005). In addition, the mutator was the only genetic background to survive phage selection in 4 of 16 populations from single-phage treatments and 22 of 24 populations from two-phage treatments. In the populations where the mutator did not completely fix, we note that it is possible the Reference would eventually also evolve phage resistance and increase in frequency.
Fig. 1.

Mutator advantage in the presence of phages. Each circle represents a single population (n = 4 in each treatment); bars show the average selection coefficient for the mutator relative to a marked version of the reference strain (K-12 MG1655 Δara).
Mutator advantage was associated with the spread of phage-resistance alleles (supplementary fig. S2, Supplementary Material online). In streaking assays, the colony isolates from two-phage treatments were on average resistant to 2.87 (SE = 0.08) of the four tested phages, whereas colony isolates from one-phage treatments were resistant to only 1.04 (SE = 0.08) phage on average, typically the one they were exposed to during selection (supplementary fig. S2, Supplementary Material online). This can be explained by the frequency of mucoid phage-resistant mutants in the different treatments: 52 of the 196 colony isolates had a mucoid morphology, 48 of which were from two-phage treatments, and every mucoid isolate was resistant to all four phages. No isolates from the phage-free treatment were resistant to any phage.
Reversion of Phage Resistance in the Absence of Phages
To determine whether mutator bacteria also adapted relatively rapidly during subsequent evolution in the absence of phages, and whether any effects depended on the original mechanism of phage resistance, we isolated three mucoid and three nonmucoid phage-resistant mutants on both the mutator and nonmutator genetic backgrounds (all from the λ+T4 treatment, because this treatment consistently produced a mixture of mucoid and nonmucoid isolates; supplementary fig. S2, Supplementary Material online). We then propagated these 12 isolates for 10 growth cycles in the absence of phages. Mucoid isolates all rapidly reverted to both phage-sensitivity (quantified in liquid culture; fig. 2) and nonmucoidy (nonmucoid colonies on agar; fig. 3). Reversion was rapid in both mutator and nonmutator genetic backgrounds, and the rate of decline in resistance did not differ on average between mutator and nonmutator selection lines that were initially mucoid (time × mutability interaction in a linear mixed-effects model of resistance averaged across all selection lines derived from each ancestral isolate at each time point, with time as a covariate, mutability [Mut or Ref] as a fixed effect, and ancestral isolate as a random effect: F1,28 = 1.50, P = 0.23). Populations derived from nonmucoid phage-resistant isolates remained resistant to phages on average (effect of time in a mixed model as above: F1,28 = 2.48, P = 0.13; fig. 2), although for one mutator ancestral isolate all three selection lines declined in resistance over time but did not revert to full sensitivity. Colony isolates chosen randomly from evolved populations showed the same qualitative result (supplementary fig. S3, Supplementary Material online), and we confirmed that phage sensitivity measured in liquid culture corresponded to sensitivity in streaking assays as used at the end of competition experiments above.
Fig. 2.
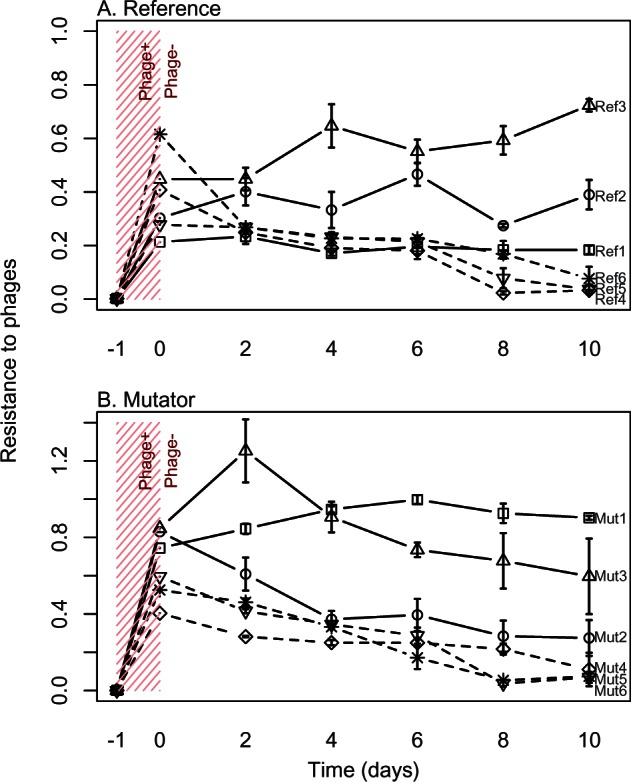
Changes in phage resistance on genetic backgrounds that were initially nonmucoid (solid lines) or mucoid (dashed lines) with (A) normal or (B) elevated mutation rates. Each series shows a single phage-resistant mutant isolated after exposure to phages λ and T4 (T0; phage presence denoted by hatching at left), and then maintained in the absence of phages for ten transfers. Each point shows the average (+SE) resistance score for three independent selection lines. Labels at right denote the six independent phage-resistant isolates sampled from reference (Ref) and mutator (Mut) strains. Note that at transfer 0 each selection line was bottlenecked to a single colony isolate.
Fig. 3.
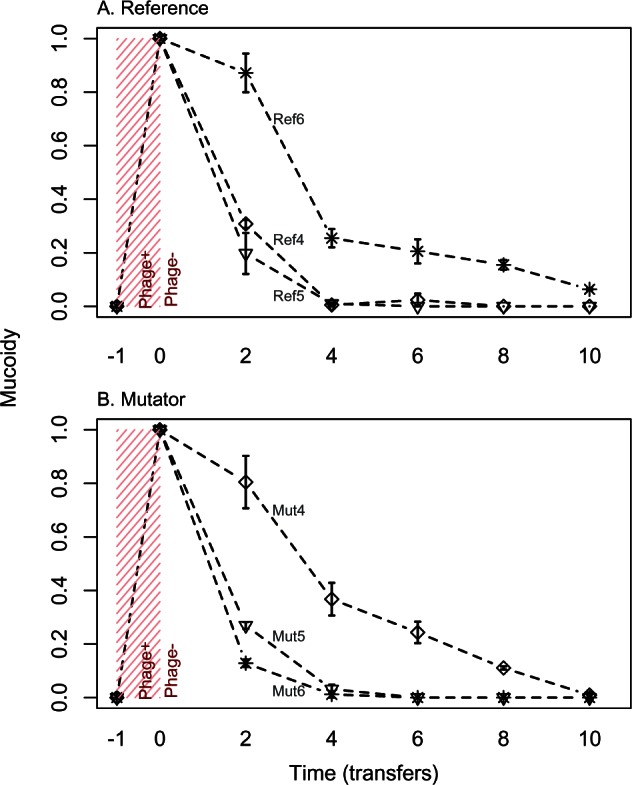
Changes in mucoidy during evolution in the absence of phages for (A) reference and (B) mutator lineages. Mucoidy was scored as the proportion of colonies with a mucoid phenotype in each selection line (three selection lines per mucoid phage-resistant ancestor; each line is labelled with the corresponding ancestral isolate). Each point shows the average ± SE from three independent selection lines.
We also tested whether reversion of mucoidy during evolution in the absence of phages altered the likelihood of re-evolving a mucoid phenotype during subsequent re-exposure to the same phage treatment. Bacteria that were derived from mucoid ancestors but had reverted to nonmucoidy and phage sensitivity during evolution in phage-free conditions did not produce any mucoid mutants upon subsequent re-exposure to phages (supplementary fig. S4, Supplementary Material online). In contrast, three out of six reference populations from the same conditions contained mutants forming mucoid colonies (supplementary fig. S4, Supplementary Material online). Therefore, reversion of mucoidy constrains subsequent evolution of this phenotype.
Recovery of Fitness Costs Associated with Phage Resistance
We measured fitness relative to the ancestor for the 12 phage-resistant colony isolates (three mucoid and three nonmucoid for both Mut and Ref) and a single colony isolate derived from each one after evolution in the absence of phages; we used the same 24 isolates for whole-genome sequencing described below. Our rationale for focusing on single genotypes rather than whole populations was, following Scanlan et al. (2015), to directly link phenotypic data (fitness and resistance) with genomic information (see below); this approach provided multiple independently evolved genotypes in each combination of the categories we were interested in (ancestral/evolved, mucoid/nonmucoid, mutator/nonmutator). In the absence of phages, resistance was costly on average on both Mut and Ref genetic backgrounds prior to experimental evolution in phage-free conditions (mean fitness ± SD = 0.89 ± 0.08 and 0.92 ± 0.15, respectively; fig. 4), although fitness varied among different isolates, including one with no cost. After evolution in the absence of phages, the isolates derived from the 12 ancestral genotypes all showed fitness values larger than or not significantly different from the reference strain (fig. 4), indicating that costs of resistance can be rapidly recovered.
Fig. 4.
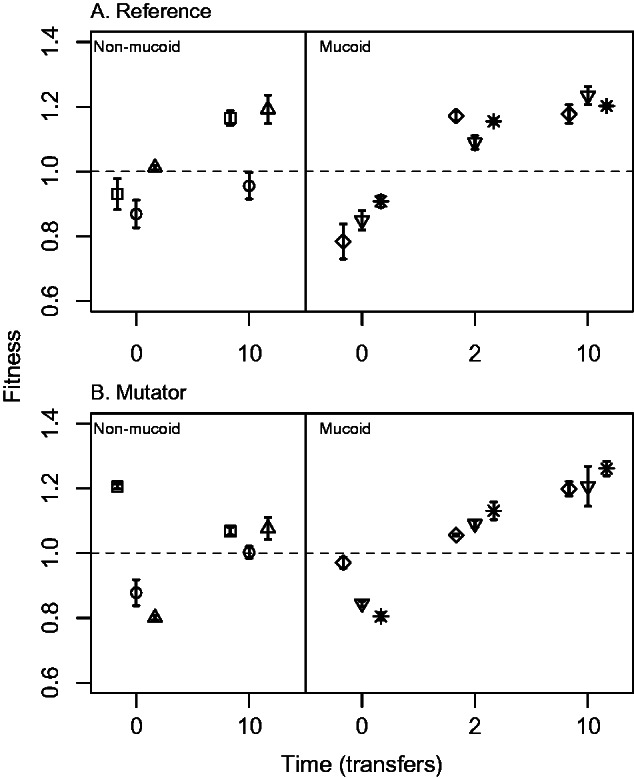
Changes in competitive fitness during evolution in the absence of phages. Competitive fitness was measured in the absence of phages for three nonmucoid (left panels) and three mucoid (right panels) phage-resistant isolates derived from (A) the reference and (B) mutator strains. Symbols represent each of the three independent isolates sampled in each case (as in figs. 2 and 3). Each point shows the average ± SE of three independent assays for a single colony isolate.
We found no difference on average between the fitness of isolates derived from mutator and nonmutator genetic backgrounds (F1,9 = 0.21, P = 0.67 in a mixed model with fitness as the response, mucoidy and mutability as fixed effects, and ancestral isolate as a random effect). However, average fitness of evolved isolates that were initially mucoid was higher on average than that of isolates derived from nonmucoid ancestors (F1,9 = 11.31, P = 0.008). In other words, mutation rate did not strongly impact the outcome of evolution in terms of changes in resistance and competitive fitness, but the original phage-resistance phenotype (mucoid or nonmucoid) did.
Note that the two ancestral phage-sensitive genetic backgrounds from which phage-resistant mutants were derived can adapt to these experimental conditions over this time-scale (supplementary fig. S5, Supplementary Material online). Therefore, the increased fitness of evolved isolates may reflect a combination of adaptation to recover the cost of phage resistance and general adaptation to the experimental environment. The response to selection (change in fitness) was greater on average for phage-resistant genetic backgrounds than that observed for control selection lines (mean ± SD = 0.24 ± 0.16 compared with 0.11 ± 0.09; one-sample t11 = 2.76, P < 0.02), consistent with compensatory evolution (Moore et al. 2000; Hall et al. 2010).
Parallel Genetic Changes Acquired in the Presence of Phages
Whole-genome sequencing showed that every mucoid phage-resistant isolate had a genetic change in a locus involved in colanic acid biosynthesis (figs. 5 and 6; supplementary table S1, Supplementary Material online, at time point T0): Two had single nucleotide polymorphisms (SNPs) in rcsC, encoding a sensory kinase controlling capsule biosynthesis, and four had SNPs or insertion sequence (IS) insertions in or upstream of yrfF, encoding a putative response attenuator of the RcsCDB system in which this type of genetic change is known to cause mucoidy (Miskinyte et al. 2013). We also detected strong parallelism among nonmucoid phage-resistant mutants: Five out of six had mutations affecting the coding region or transcription activation site of ompC, encoding the outer membrane protein C (OmpC), known to confer phage resistance (Yu and Mizushima 1982). One of these mutations was an IS5-mediated 23-kb deletion (supplementary table S1, Supplementary Material online; lineage Mut3), which included both ompC and rcsD. The other nonmucoid resistant mutant had two mutations that may also be linked to phage resistance by the same mechanism (supplementary table S1, Supplementary Material online; lineage Ref3): A nonsynonymous SNP in the coding region of envZ, which encodes the sensor EnvZ known to alter the expression of OmpC through the response regulator OmpR (Wandersman et al. 1980; Garrett et al. 1983; Slauch et al. 1988), and a frameshift mutation after an insertion of G into the coding region of manZ. The manZ gene encodes a subunit of the maltose permease, a gateway for phage λ to enter the host cytosol from the periplasm (Erni et al. 1987; Esquinas-Rychen and Erni 2001), and is known to be involved in phage-resistance evolution (Meyer et al. 2012). We tested for altered phage susceptibility in knockouts of these genes from the Keio collection (Baba et al. 2006), finding that, as expected from earlier studies (Wandersman et al. 1980; Yu and Mizushima 1982), the OmpC knockout was relatively resistant to T4 and the T4+λ combination (supplementary fig. S6, Supplementary Material online); the EnvZ knockout was relatively resistant to T4, but deleting ManZ did not have a strong effect on phage resistance in liquid culture on its own. Thus, OmpC and EnvZ mutations can be linked to phage resistance, whereas any benefit of ManZ mutation in liquid may be contingent on the EnvZ mutation that we found on the same genome.
Fig. 5.

Representation of evolved mutations along the whole genomes of single colony isolates sampled at different time points (T) in reference (Ref) and mutator (Mut) lineages and derived from both (A; green symbols) nonmucoid and (B; red symbols) mucoid ancestors. Gray areas highlight regions of parallel evolution as described in the main text. Symbols depict the six mutational categories.
Fig. 6.

Position of genetic changes associated with phage resistance and reversion of mucoidy in genes for which high parallelism was detected.
Parallel Genetic Changes Driving Adaptation and Reversion of Mucoidy
All six isolates derived from mucoid ancestors, which reverted to nonmucoid phage-sensitive phenotypes in the absence of phages, had acquired additional changes in rcsD, rcsB or rcsC and retained their original mutations (figs. 5B and 6B; supplementary table S1, Supplementary Material online). For two of these genotypes, clonal isolates from the same populations at an earlier time point (transfer 2) had the same genetic change in this region (Ref6: IS insertion in rcsC; Mut5: SNP in rcsB), indicating that these changes appeared early and were maintained. For two other genotypes, isolates sampled from the same populations at transfer 2 had genetic changes in regions that were subsequently deleted in the isolates sampled at transfer 10 (Ref4: IS insertion in rcsD followed by large deletion; Mut6: small deletion [64 bp] in rcsC followed by a 14-kb deletion spanning the entire region including all three rcs-genes). In these populations the later genetic change could have occurred on a genome that already had the first genetic change, or on a competing lineage derived from the same ancestor. The latter scenario is consistent with our observations in the remaining two populations derived from mucoid ancestors: The genetic change observed at transfer 2 was not present at transfer 10 (Ref5: insertion in rcsD at transfer 2 and deletion in rcsD at transfer 10; Mut4: insertion in rcsB at transfer 2 and deletion in rcsC at transfer 10). Thus, reversion of mucoidy in mutator and nonmutator lineages was always associated with alterations of rcs (table 1), and in some populations we detected independent revertant lineages at different time points.
Table 1.
Summary of Genetic Changes in Mutator and Nonmutator Lineages.
| Time (transfer) | Mutator | Mucoid Ancestor | SNPs | Insertions | Deletions | Big Deletions | IS Insertions | Duplications | Total |
|---|---|---|---|---|---|---|---|---|---|
| 0 | No | No | 1 ± 0 | 0.3 ± 0.6 | 0 ± 0 | 0.7 ± 1.2 | 0.7 ± 0.6 | 0.3 ± 0.6 | 3 ± 1 |
| 0 | No | Yes | 0.7 ± 0.6 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.3 ± 0.6 | 0 ± 0 | 1 ± 0 |
| 0 | Yes | No | 5 ± 4.6 | 0 ± 0 | 0.7 ± 0.6 | 0.3 ± 0.6 | 0 ± 0 | 0 ± 0 | 6 ± 4.6 |
| 0 | Yes | Yes | 6.3 ± 1.5 | 1.7 ± 1.5 | 0.3 ± 0.6 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 8.3 ± 1.2 |
| 10 | No | No | 1 ± 0 | 0.3 ± 0.6 | 0 ± 0 | 1 ± 1 | 1.7 ± 0.6 | 0.7 ± 0.6 | 4.7 ± 0.6 |
| 10 | No | Yes | 0.7 ± 0.6 | 0 ± 0 | 0 ± 0 | 0.7 ± 0.6 | 1.7 ± 0.6 | 0 ± 0 | 3 ± 0 |
| 10 | Yes | No | 6.3 ± 4.9 | 0.7 ± 1.2 | 0.7 ± 0.6 | 0.3 ± 0.6 | 1.3 ± 0.6 | 0 ± 0 | 9.3 ± 4.5 |
| 10 | Yes | Yes | 8.3 ± 0.6 | 2 ± 1.7 | 0.3 ± 0.6 | 0.7 ± 0.6 | 1 ± 1 | 0.3 ± 0.6 | 13 ± 2.5 |
NOTE.—Each value is the mean number of changes ± SD for three independent genotypes.
Isolates derived from nonmucoid resistant genotypes also retained their original mutations, in genes affecting ompC or manZ (supplementary table S1, Supplementary Material online), and acquired additional genetic changes. Unlike for isolates derived from mucoid genotypes, there was little evidence for parallelism, except for IS movements in the coding sequence of fimE, a regulator involved in phase variation of type 1 fimbriation, and either of its two flanking intergenic regions, which we found in four out of six genotypes. We also found IS mutations in this region in five out of six genotypes derived from mucoid ancestors, indicating that they had a beneficial function across the different genetic backgrounds in our experimental conditions. In support, when we sequenced this region for six independently evolved control isolates, which had never been exposed to phages but adapted to the same phage-free conditions (3 × Ref; 3 × Mut; supplementary fig. S5, Supplementary Material online), we found genetic changes in this region in all six isolates (supplementary table S3, Supplementary Material online), three of which were IS movements. Mutations in genes regulating the expression of fimbrial proteins, including IS insertions into fimA and fimB genes, have also been previously detected during adaptation to laboratory media in E. coli (Barrick et al. 2009; Winkler et al. 2014).
Mutator Effect Is Limited to SNPs
The presence of the mutator allele had a strong impact on genetic diversity during the initial period of phage-resistance evolution (table 1; Welch’s t-test on total number of genetic changes in mutator and reference genotypes: t6.5 = 3.6, P = 0.01). This effect was driven by a difference in the average number of SNPs (mutator: 5.7, SD = 3.1; reference: 0.8, SD = 0.4; P = 0.01), and is not significant when SNPs are excluded (P = 0.6). Genotypes derived from the mutator also had more SNPs after evolution in phage-free conditions (7.3, SD = 3.3 compared with 0.83, SD = 0.4 for the reference; P = 0.005), but there was no significant difference in the total number of other types of genetic changes (P = 0.45). Of all SNPs in coding regions, four out of four were nonsynonymous in Reference isolates and 26 out of 37 were nonsynonymous in Mutator isolates (supplementary table S4, Supplementary Material online). In summary, lineages with defective DNA mismatch repair acquired more SNPs, and this was advantageous in the presence of phages. However, during subsequent evolution in the absence of phages other types of genetic changes, mostly IS insertions (table 1), were relatively more important and our phenotypic analyses showed that mutator lineages did not adapt or revert costly phenotypes more rapidly.
Discussion
Returning to our four hypotheses, we found that 1) elevated bacterial mutation rate was highly advantageous in the presence of parasitic viruses, and particularly in multiparasite treatments; 2) elevated mutation rate did not detectably accelerate adaptation to or reversion of costly parasite-resistance phenotypes in the absence of phages, unlike observations with costly antibiotic-resistance phenotypes (Perron et al. 2010); 3) changes in bacterial fitness and the likelihood of reversion to phage sensitivity were dependent on the initial mechanism of phage resistance. Phage resistance here, as in other experiments with E. coli (Hancock and Reeves 1975; Bohannan and Lenski 2000; Mizoguchi et al. 2003; Kim et al. 2015) and other bacterial species (Ohshima et al. 1988; Labrie et al. 2010; Scanlan and Buckling 2012), typically resulted from modification of cell surface receptors (OmpC) or mucoidy owing to mutations in genes involved in extracellular polysaccharide biosynthesis (rcs genes). Both mucoid and nonmucoid phage-resistance mutants paid a cost in competitive fitness in the absence of phages, but rapidly recovered during evolution in phage-free conditions, attaining similar fitness to phage-sensitive controls. Nonmucoid resistant mutants maintained resistance in the absence of phages, as observed with E. coli B (Lenski 1988b). However, for mucoid mutants, which do not form in E. coli B (Lenski 1988a), adaptation was associated with rapid reversion to nonmucoidy and phage sensitivity. The lack of mucoid mutants in E. coli B probably results from the complete lack of rcsA-D, attributable to IS-mediated deletions (Studier et al. 2009). 4) Our sequence data showed that the genetic basis of adaptation was strongly related to the initial mechanism of phage resistance: Reversion of mucoidy occurred through diverse SNPs, IS insertions and deletions, but always in rcs genes; mutations in this region were not observed in lineages derived from nonmucoid ancestors.
Why is mucoidy so reversible? Mucoidy evolves in the presence of phages and is associated with a large mutational target including many genes involved in colanic acid biosynthesis and regulation (Gottesman et al. 1985; Costa and Anton 2001; Ionescu and Belkin 2009). As such, there are many mutational pathways to a mucoid phenotype (Lieberman and Markovitz 1970; Ophir and Gutnick 1994). In agreement with our findings, Costa and Anton (2001) and Scanlan and Buckling (2012) noted that mucoidy is unstable: Revertants are often observed during routine laboratory culture. Our results suggest that such reversibility may be due to a large mutational target for reversion of mucoidy: Although there was strong parallelism in that every revertant genotype had acquired a mutation in an rcs gene, this included SNPs, IS insertions and deletions across three different genes. Consistent with this, in some populations we detected independent revertant genotypes derived from the same mucoid ancestor at different time points. This large mutational target may also partially explain why mutator lineages did not revert faster than nonmutator lineages. Evidently the supply rate of mutations affecting mucoidy is sufficient for nonmutator populations of this size (∼109 at stationary phase) to contain at least one reversion mutation at the start of the experiment. In agreement, our own and previous estimates for rates of spontaneous mutation and IS movements suggest that genetic changes conferring and reversing mucoidy were abundant, respectively, during and after phage exposure (Wielgoss et al. 2011; Sousa et al. 2013; Supplementary data, Supplementary Material online). Moreover, the fact that reversion was often due to IS mutations or deletions which, unlike SNPs, were not more frequent on the mutator genetic background compared with the reference, suggests that the increased genetic diversity in mutator lineages is limited to SNVs (single nucleotide variants). When adaptation relies on other types of genetic changes an SNV mutator, such as our mutS-defective strain, does not gain an indirect fitness advantage due to increased evolvability.
The rapid reversibility of mucoidy is also relevant in other contexts. Mucoid isolates are often recovered from chronic infections caused by key pathogens including E. coli and Pseudomonas aeruginosa (Doggett 1969; Ciofu et al. 2001; Crémet et al. 2013). In P. aeruginosa, where the genetic basis of mucoidy is different to E. coli, mutations resulting in constitutive mucoidy appear to be involved in adaptation to host immune responses during early stages of chronic infection (Yang et al. 2012; Marvig et al. 2015). However, mucoidy can subsequently be repressed by mutations at other sites, hypothesized to result from adaptation to regulate costly expression of this phenotype (Marvig et al. 2015). More generally, mucoidy can also alter bacterial adaptation to other diverse stresses including antibiotics (Piña and Mattingly 1997), macrophages (Miskinyte et al. 2013), menthol (Landau and Shapira 2012), and dessication (Ophir and Gutnick 1994). Therefore, we speculate that acquisition of mucoidy and regulation of associated costs, as observed in our experiments, may be common features of bacterial adaptation.
We found strong parallelism in the types of mutations responsible for acquiring mucoidy and phage resistance, reversing mucoidy, and adaptation to the experimental abiotic environment. This is consistent with previous experimental studies showing variable but pervasive genetic parallelism in adaptation to novel environments (Tenaillon et al. 2012; Bailey et al. 2015) and stressors such as antibiotics (Vogwill et al. 2014), as well as comparative studies of clinical isolates of human pathogens (Lieberman et al. 2011; Marvig et al. 2015). This indicates that genetic parallelism can result from adaptation of independent populations to identical, strong selection pressures, such as antibiotics and parasites, as well as selection for improved growth in a benign abiotic environment, although in many cases parallelism is manifest at the level of genes, rather than individual genetic changes (Woods et al. 2006; Barrick et al. 2009).
Why are other types of resistance less reversible? Resistance through modification of OmpC, or phage receptors in general, can probably only be reverted by regaining the original and functional three-dimensional structure of the phage receptor, which may only be possible by a reversion to the ancestral sequence of this gene. Thus, the mutational target size for reversion probably equals 1 when resistance is due to receptor modification. In a more extreme scenario, when the receptor is deleted it may be impossible to restore receptor function in a single step (or without horizontal gene transfer). Consistent with the low reversibility of resistance to T4 in nonmucoid E. coli, Lenski (1988b) found that resistance was maintained in the absence of phages for 400 generations in E. coli B. The same rationale has also been used to explain why compensatory adaptation to costs of antibiotic resistance is more likely to occur through mutations at other loci than by reversion of the initial resistance mutation (Levin et al. 2000; Maisnier-Patin and Andersson 2004): Many mutations can restore or compensate for the function impaired by resistance mutations, but only one mutation may restore antibiotic-target binding specificity. In contrast, mucoidy can be reversed by any mutation that reduces biosynthesis or secretion of extracellular polysaccharide, including large deletions as observed in our experiment. Therefore, the mutational target size is much larger than one. This raises the possibility that, in general, resistance resulting from mutations on drug targets or phage receptors will have very low rates of reversion compared with resistance due to mutations that up- or downregulate expression of existing proteins such as those involved in EPS biosynthesis.
Active IS elements are known to be rapid drivers of genome evolution (Papadopoulos et al. 1999; Schneider et al. 2000; Cooper et al. 2001). Recently, and in line with our findings, it has been shown that IS elements can also lead to large genome rearrangements, including deletions, owing to homologous recombination events between identical elements during experimental evolution (Raeside et al. 2014). Our results suggest that such events can contribute to recovery of fitness costs associated with resistance mechanisms in the absence of phages. Thus, although IS elements may impose a genetic burden by causing deleterious changes (Touchon and Rocha 2007), they can also cause adaptive changes in some scenarios (Chou et al. 2009; Stoebel and Dorman 2010; Gaffé et al. 2011), in this case by allowing relatively rapid adjustment of expression of energetically costly proteins. Together with our other results, this suggests bacterial adaptation to changing environments can be accelerated by a range of diversity-generating mechanisms. These include 1) mutator alleles as identified in clinical and natural isolates (LeClerc et al. 1996; Matic et al. 1997; Turrientes et al. 2010) and shown here to be advantageous in single- and multiple-parasite environments, which are likely to be relevant in nature (Koskella et al. 2012); and 2) IS elements that our study showed to play a key role when parasites were no longer present.
Mutator and nonmutator phage-resistant mutants in our evolution experiment had different phage-resistance mutations. In fact, although there was very high parallelism at the level of genes, individual putative phage-resistance mutations within rcsCDB, yrfF, ompC or manZ were each found in a single isolate. Therefore, one may argue that the failure of mutators to adapt or revert phage resistance more rapidly than nonmutators was due to their having “hard-to-recover-from” phage-resistance mutations in the first place. Although we cannot exclude the possibility that a mutator and nonmutator with identical phage-resistance mutations would adapt at different rates, our aim was instead to test for a difference in the outcomes of evolution in mutator and nonmutator lineages with phage-resistance mutations acquired during evolution in the presence of phages; that is, realistic random draws from the spectrum of possible phage-resistance mutations on each genetic background. Additionally, resistance mutations were in the same genes and had similar fitness costs, a key determinant of the rate of adaptation (Moore et al. 2000; Sousa et al. 2012), suggesting that our experimental design nevertheless captured similar evolutionary processes in mutator and nonmutator lineages, with multiple independent replicates for each.
Materials and Methods
Organisms and Growth Conditions
We used E. coli K-12 MG1655 grown with orbital shaking at 37 °C in all experiments. Unless otherwise stated, bacteria and phages were grown in lysogeny broth (LB) supplemented with 10 mM MgSO4. In competition experiments in the presence of phages and for experimental evolution in the absence of phages, we compared the reference strain with a conditional mutator strain that has the native mutS promoter replaced by the ParaBAD promoter. Thus, ParaBAD-mutS is repressed in the absence of arabinose, leading to an approximately 50-fold increase in mutation rate (supplementary fig. S7, Supplementary Material online). The reference mutation rate can be restored by adding 0.2% (w/v) arabinose, which induces the ParaBAD promoter, thereby activating the expression of mutS. We exploited this feature when growing phage-resistant colony isolates of the mutator for freezer storage or DNA extraction, to minimize the likelihood of additional mutations influencing our analysis of mutations occurring during the experiment. We denote the mutator and reference genetic backgrounds and lineages derived from them by the prefixes “Mut” and “Ref” respectively. We used four different lytic phages: λ, T4, T5, and T6. The λ strain we used is an obligate lytic strain called cI26 (Meyer et al. 2012).
Competition Experiments in the Presence of Phages
We tested whether the mutator was at an evolutionary advantage over the reference strain in the presence of each phage (supplementary fig. S1, Supplementary Material online), both on its own (n = 4) and in each pairwise combination (n = 6). We competed the mutator against a marked Δara version of the reference. This strain is otherwise isogenic but is distinguished by red colonies on Tetrazolium–Arabinose (TA) agar plates. For a given competition, we grew independent overnight cultures of the mutator and reference, before mixing them 1:1 (v/v) and diluting 100-fold to a final volume of 1 ml in 96-deep-well plates (ABgene, Thermo Fisher Scientific, Waltham, MA). We then added either approximately 5 × 106 phage particles suspended in 10 µl phosphate-buffered solution (PBS), or 10 µl sterile PBS for phage-free treatments. After 24 h of growth, we transferred an aliquot to fresh media (100-fold dilution) and incubated for a further 24 h. We estimated the frequency of each bacterial genetic background at 0 and 48 h by dilution and plating on TA agar. Every competition was replicated four times independently. We estimated the selection coefficient for the mutator relative to the Δara reference strain as s = ln(R48h/R0h)/2, where R48h and R0h are the ratios of the two genetic backgrounds at the start and end of the competition, respectively (Lenski et al. 1991; Travisano and Lenski 1996), giving the difference in Malthusian growth parameters per day (Travisano and Lenski 1996; Lopez-Pascua and Buckling 2008; Gómez and Buckling 2013); a per-generation-estimate in the presence of phages is precluded by the difficulty of estimating the number of bacterial generations in these conditions. In cases where only one genetic background was present at the end of the assay, we conservatively took the final frequency to be 0.99.
Qualitative Phage-Resistance Assays
To determine whether phage-resistant mutants appeared and spread during competition experiments, we picked four independent colony isolates from each of the two genetic backgrounds in every replicate population. In cases where only one genetic background was present at the end, we took four colony isolates from that genetic background. We then grew each colony isolate in 100 µl of liquid LB+MgSO4 for 3 h, before streaking it across a line of each phage that had been previously dried onto an agar plate. Isolates were scored resistant if there was no visible growth inhibition after 24-h incubation (Buckling and Rainey 2002; Brockhurst et al. 2007). Because phage-resistant mutants frequently have a mucoid colony phenotype in these conditions (Tazzyman and Hall 2015), we also scored whether each colony isolate was mucoid or not.
Experimental Evolution in the Absence of Phages
We independently isolated three mucoid and three nonmucoid phage-resistant mutants from both the reference and mutator genetic backgrounds, before propagating them in the absence of phages (supplementary fig. S1, Supplementary Material online). To do this we grew replicate cultures of each genetic background in the λ+T4 treatment before streaking them on agar, picking a single colony from each population and testing for phage resistance by streaking assays as above. Colony isolates that were resistant to one or both phages were purified by restreaking them twice, and three mucoid and nonmucoid isolates from each genetic background were randomly chosen. Each was then grown for 2 h in phage-free liquid culture and frozen at −80 °C. We experimentally evolved these 12 phage-resistant mutants in the absence of phages in the same conditions as above, with three replicate selection lines for each ancestral genotype (n = 36), for ten growth cycles in batch culture with 100-fold dilution at each transfer (∼66 generations), freezing samples of each population every second transfer. We also included three replicate selection lines each for the ancestral phage-sensitive reference and mutator strains (n = 42 total). At the end of the experiment, we tested for changes in phage resistance, mucoidy, and competitive fitness as described below.
Quantifying Phage Resistance after Evolution in Phage-Free Conditions
We quantified changes in phage resistance after ten growth cycles in the absence of phages by measuring the reduction in bacterial growth, RBG (Poullain et al. 2008; Hall et al. 2011), attributable to phages in liquid cultures of each ancestral genotype and evolved population. For a given assay, independent cultures were grown in the presence and absence of phages, and initial and final bacterial population densities were estimated by taking OD at 600 nm using an M2 SpectraMax spectrophotometer (Molecular Devices, Sunnyvale, CA). RBG was then calculated as ΔODwith phage/ΔODwithout phage, so that RBG = 1 if phages had no effect on bacterial growth, and RBG < 1 if bacterial growth was reduced by phages. We assayed RBG three times independently for samples of every population taken every second transfer. We also tested for changes in mucoidy by streaking each population from each time point on agar and counting the proportion of mucoid colonies.
Changes in Competitive Fitness due to Phage Resistance and Evolution in the Absence of Phages
To test both whether phage resistance was associated with a pleiotropic cost in terms of competitive fitness, and whether that cost was ameliorated during evolution in the absence of phages, we did pairwise competition assays for each phage-resistant ancestor (n = 12), as well as a single clone derived from each at the end of ten transfers of experimental evolution in the absence of phages (n = 12). We also assayed fitness for the ancestral, phage-sensitive reference and mutator, plus three clones derived from each that we isolated from independently evolved populations at the end of the experiment. These assays were done as above, except that they lasted a single transfer and selection coefficients were estimated per generation, taking the number of generations in each assay as log2(Nfinal/Ninitial), where N is the total population size, allowing us to estimate fitness w as 1+s relative to the reference strain, after correction for the cost of the ara-marker (Trindade et al. 2009). Additionally, we measured phage resistance for each of these genotypes by RBG assays to test for reversion of phage resistance. Finally, having observed reversion of both mucoidy and phage resistance as early as the second transfer of our experiment, we also isolated six revertants (colony isolates that were mucoid at the beginning but nonmucoid after two transfers) and measured competitive fitness and resistance alongside the other isolates.
Testing for Re-Evolution of Mucoidy
We hypothesized that, following reversion of mucoidy in the absence of phages, bacteria may be less likely to re-evolve mucoidy upon subsequent re-exposure to the same phage treatment that originally selected for mucoid mutants, compared with control populations that had never encountered the phages previously. We tested this by growing six replicate populations of each of the six revertant genotypes from transfer 10 (phage-sensitive nonmucoid isolates derived from phage-resistant mucoid ancestors). We also included six replicate populations of the reference strain in the same phage treatment as used above to isolate resistant mutants. We then plated each population and scored the fraction of mucoid colonies.
Whole-Genome Resequencing
We sequenced the genomes of 32 bacterial strains: The ancestral reference and mutator strains, 12 phage-resistant mutants used to initiate our evolution experiment (time point 0; T0), 12 evolved clones from the end of the experiment (T10), and 6 revertants (nonmucoid isolates derived from mucoid phage-resistant clones) isolated after two transfers (T2) to investigate in more detail the genetic basis of reversion of phage resistance. Genomic DNA was extracted using Qiagen’s Genomic DNA extraction buffers with 20/G Genomic-tips. We typically obtained greater than 10 µg of high quality DNA (average fragment sizes exceeding 20 kb). DNA extracts were prepared for sequencing using NEBNext Library Prep Kit for Illumina sequencers according to the manufacturer’s recommendations with insert sizes of 400–600 bp. Sequencing was performed on a Illumina HiSeq 2500 machines in paired-end mode of either 2× 100 bp or 2× 125 bp sequence lengths. Sequences were trimmed to remove both Illumina specific adapters/primers and bad quality bases in single reads using Trimmomatic v0.32 (Bolger et al. 2014); with these basic parameters ILLUMINACLIP: TruSeq3-PE-2.fa:2:25:10 CROP:99 LEADING:30 TRAILING:25 SLIDINGWINDOW:4:25 MINLEN:36. Quality was assessed before and after trimming using FastQC v0.11.2 (Andrews 2015). Finally, both trimmed paired and nonpaired reads were mapped to the E. coli str. K-12 substr. MG1655 reference genome (Blattner et al. 1997) using breseq v0.24rc5 (Barrick et al. 2014; Deatherage and Barrick 2014). The ancestral sequences had five extra mutations compared with the K-12 reference, which served as internal positive controls and indicated reliable resequencing results (supplementary table S2, Supplementary Material online). These were all removed prior to downstream statistical analyses. Given the strengths and limitations of short resequencing we focus our analyses on the following mutation categories: SNPs, insertions and deletions, with the latter spanning regions up to several 10 kb, IS movements, and finally, small duplications, with the maximum close to the read lengths, that is, <125 bp.
Supplementary Material
Supplementary data, figures S1–S7 and tables S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank three anonymous reviewers and the editor for helpful comments on the manuscript, as well as Dominique Schneider for feedback on an earlier draft, Jenna Gallie for lytic λ and Julien Capelle for T5 and T6. This work was supported by the Swiss National Science Foundation (PZ00P3_148255 to A.H.) and an EU Marie Curie PEOPLE Postdoctoral Fellowship for Career Development (FP7-PEOPLE-2012-IEF-331824 to S.W.).
References
- Andersson DI, Hughes D. 2011. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol Rev. 35:901–911. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2015. FastQC: a quality control tool for high throughput sequence data. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed on 5 December 2014
- Baba T, Ara T, Hasegawa M, Takai Y, Yoshiko O, Baba M, Datsenko KA, Tomita M, Wanner BL, Morio H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SF, Rodrigue N, Kassen R. 2015. The effect of selection environment on the probability of parallel evolution. Mol Biol Evol. 32:1436–1448 [DOI] [PubMed] [Google Scholar]
- Barrick JE, Colburn G, Deatherage DE, Traverse CC, Strand MD, Borges JJ, Knoester DB, Reba A, Meyer AG. 2014. Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genomics 15:1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF. 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461:1243–1247 [DOI] [PubMed] [Google Scholar]
- Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. 2014. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 13:42–51 [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462 [DOI] [PubMed] [Google Scholar]
- Bohannan BJM, Lenski RE. 2000. Linking genetic change to community evolution: insights from studies of bacteria and bacteriophage. Ecol Lett. 3:362–377 [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandis G, Wrande M, Liljas L, Hughes D. 2012. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol Microbiol. 85:142–151 [DOI] [PubMed] [Google Scholar]
- Brockhurst MA, Morgan AD, Fenton A, Buckling A. 2007. Experimental coevolution with bacteria and phage: the Pseudomonas fluorescens—Φ2 model system. Infect Genet Evol. 7:547–552 [DOI] [PubMed] [Google Scholar]
- Buckling A, Rainey PB. 2002. Antagonistic coevolution between a bacterium and a bacteriophage. Proc R Soc Lond B Biol Sci. 269:931–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparelli R, Nocerino N, Lanzetta R, Silipo A, Amoresano A, Giangrande C, Becker K, Blaiotta G, Evidente A, Cimmino A, et al. 2010. Bacteriophage-resistant Staphylococcus aureus mutant confers broad immunity against staphylococcal infection in mice. PLoS One 5:e11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra I, O’Neill AJ, Miller K. 2003. The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist Updat. 6:137–145 [DOI] [PubMed] [Google Scholar]
- Chou HH, Berthet J, Marx CJ. 2009. Fast growth increases the selective advantage of a mutation arising recurrently during evolution under metal limitation. PLoS Genet. 5:e1000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofu O, Fussing V, Bagge N, Koch C, Hoiby N. 2001. Characterization of paired mucoid/non-mucoid Pseudomonas aeruginosa isolates from Danish cystic fibrosis patients: antibiotic resistance, beta-lactamase activity and RiboPrinting. J Antimicrob Chemother. 48:391–396 [DOI] [PubMed] [Google Scholar]
- Cooper VS, Schneider D, Blot M, Lenski RE. 2001. Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of Escherichia coli B. J Bacteriol. 183:2834–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa CS, Anton DN. 2001. Role of the ftsAlp promoter in the resistance of mucoid mutants of Salmonella enterica to mecillinam: characterization of a new type of mucoid mutant. FEMS Microbiol Lett. 200:201–205 [DOI] [PubMed] [Google Scholar]
- Crémet L, Caroff N, Giraudeau C, Reynaud A, Caillon J, Corvec S. 2013. Detection of clonally related Escherichia coli isolates producing different CMY beta-lactamases from a cystic fibrosis patient. J Antimicrob Chemother. 68:1032–1035 [DOI] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 1151:165–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denamur E, Bonacorsi S, Giraud A, Duriez P, Hilali F, Amorin C, Bingen E, Andremont A, Picard B, Taddei F, et al. 2002. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J Bacteriol. 184:605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett RG. 1969. Incidence of mucoid Pseudomonas aeruginosa from clinical sources. Appl Microbiol. 18:936–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erni B, Zanolari B, Kocher HP. 1987. The mannose permease of Escherichia coli consists of three different proteins. Amino acid sequence and function in sugar transport, sugar phosphorylation, and penetration of phage λ DNA. J Biol Chem. 262:5238–5247 [PubMed] [Google Scholar]
- Esquinas-Rychen M, Erni B. 2001. Facilitation of bacteriophage lambda DNA injection by inner membrane proteins of the bacterial phosphoenol-pyruvate: carbohydrate phosphotransferase system (PTS). J Mol Microbiol Biotechnol. 3:361–370 [PubMed] [Google Scholar]
- Gaffé J, McKenzie C, Maharjan RP, Coursange E, Ferenci T, Schneider D. 2011. Insertion sequence-driven evolution of Escherichia coli in chemostats. J Mol Evol. 72:398–412 [DOI] [PubMed] [Google Scholar]
- Garrett S, Taylor RK, Silhavy TJ. 1983. Isolation and characterization of chain-terminating nonsense mutations in a porin regulator gene, Envz. J Bacteriol. 156:62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez P, Buckling A. 2013. Coevolution with phages does not influence the evolution of bacterial mutation rates in soil. ISME J. 7:2242–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, Trisler P, Torres-Cabassa A. 1985. Regulation of capsular polysaccharide synthesis in Escherichia coli K-12: characterization of three regulatory genes. J Bacteriol. 162:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AR, Griffiths VF, MacLean RC, Colegrave N. 2010. Mutational neighbourhood and mutation supply rate constrain adaptation in Pseudomonas aeruginosa. Proc R Soc Lond B Biol Sci. 277:643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AR, Scanlan PD, Lopez-Pascua LDC, Buckling A. 2011. Bacteria-phage coevolution and the emergence of generalist pathogens. Am Nat. 177:44–53 [DOI] [PubMed] [Google Scholar]
- Hamilton WD, Axelrod R, Tanese R. 1990. Sexual reproduction as an adaptation to resist parasites (a review). Proc Natl Acad Sci U S A. 87:3566–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock RE, Reeves P. 1975. Bacteriophage resistance in Escherichia coli K-12: general pattern of resistance. J Bacteriol. 121:983–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu M, Belkin S. 2009. Overproduction of exopolysaccharides by an Escherichia coli K-12 rpoS mutant in response to osmotic stress. Appl Environ Microbiol. 75:483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivet-Gougeon A, Kovacs B, Le Gall-David S, Le Bars H, Bousarghin L, Bonnaure-Mallet M, Lobel B, Guillé F, Soussy CJ, Tenke P. 2011. Bacterial hypermutation: clinical implications. J Med Microbiol. 60:563–573 [DOI] [PubMed] [Google Scholar]
- Kim MS, Kim YD, Hong SS, Park K, Ko KS, Myung H. 2015. Phage-encoded colanic acid-degrading enzyme permits lytic phage infection of a capsule-forming resistant mutant Escherichia coli strain. Appl Environ Microbiol. 81:900–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella B, Lin DM, Buckling A, Thompson JN. 2012. The costs of evolving resistance in heterogeneous parasite environments. Proc R Soc Lond B Biol Sci. 279:1896–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 8:317–327 [DOI] [PubMed] [Google Scholar]
- Landau E, Shapira R. 2012. Effects of subinhibitory concentrations of menthol on adaptation, morphological, and gene expression changes in enterohemorrhagic Escherichia coli. Appl Environ Microbiol. 78:5361–5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeClerc JE, Li BG, Payne WL, Cebula TA. 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208–1211 [DOI] [PubMed] [Google Scholar]
- Lenski RE. 1988a. Experimental studies of pleiotropy and epistasis in Escherichia coli 1. Variation in competitive fitness among mutants resistant to virus T4. Evolution 42:425–432 [DOI] [PubMed] [Google Scholar]
- Lenski RE. 1988b. Experimental studies of pleiotropy and epistasis in Escherichia coli 2. Compensation for maladaptive effects associated with resistance to virus T4. Evolution 42:433–440 [DOI] [PubMed] [Google Scholar]
- Lenski RE, Rose MR, Simpson SC, Tadler SC. 1991. Long-term experimental evolution in Escherichia coli 1. Adaptation and divergence during 2,000 generations. Am Nat. 138:1315–1341 [Google Scholar]
- Levin BR, Perrot V, Walker N. 2000. Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 154:985–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tsui HC, LeClerc JE, Dey M, Winkler ME, Cebula TA. 2003. Molecular analysis of mutS expression and mutation in natural isolates of pathogenic Escherichia coli. Microbiology 149:1323–1331 [DOI] [PubMed] [Google Scholar]
- Lieberman MM, Markovitz A. 1970. Depression of guanosine diphosphate-mannose pyrophosphorylase by mutations in two different regulator genes involved in capsular polysaccharide synthesis in Escherichia coli K-12. J Bacteriol. 101:965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman TD, Michel JB, Aingaran M, Potter-Bynoe G, Roux D, Davis MR, Jr, Skurnik D, Leiby N, LiPuma JJ, Goldberg JB, et al. 2011. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 43:1275–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pascua LDC, Buckling A. 2008. Increasing productivity accelerates host-parasite coevolution. J Evol Biol. 21:853–860 [DOI] [PubMed] [Google Scholar]
- Maisnier-Patin S, Andersson DI. 2004. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res Microbiol. 155:360–369 [DOI] [PubMed] [Google Scholar]
- Marvig RL, Sommer LM, Molin S, Johansen HK. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet. 47:57–64 [DOI] [PubMed] [Google Scholar]
- Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J. 1997. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277:1833–1834 [DOI] [PubMed] [Google Scholar]
- Meyer JR, Dobias DT, Weitz JS, Barrick JE, Quick RT, Lenski RE. 2012. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335:428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskinyte M, Sousa A, Ramiro RS, de Sousa JAM, Kotlinowski J, Caramalho I, Magalhães S, Soares MP, Gordo I. 2013. The genetic basis of Escherichia coli pathoadaptation to macrophages. PLoS Pathog. 9:e1003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi K, Morita M, Fischer CR, Yoichi M, Tanji Y, Unno H. 2003. Coevolution of bacteriophage PP01 and Escherichia coli O157:H7 in continuous culture. Appl Environ Microbiol. 69:170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore FBG, Rozen DE, Lenski RE. 2000. Pervasive compensatory adaptation in Escherichia coli. Proc R Soc Lond B Biol Sci. 267:515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AD, Bonsall MB, Buckling A. 2010. Impact of bacterial mutation rate on coevolutionary dynamics between bacteria and phages. Evolution 64:2980–2987 [DOI] [PubMed] [Google Scholar]
- Muhammadi Ahmed N. 2007. Genetics of bacterial alginate: alginate genes distribution, organization and biosynthesis in bacteria. Curr Genomics. 8:191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien S, Rodrigues AM, Buckling A. 2013. The evolution of bacterial mutation rates under simultaneous selection by interspecific and social parasitism. Proc R Soc Lond B Biol Sci. 280:20131913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima Y, Schumacher-Perdreau F, Peters G, Pulverer G. 1988. The role of capsule as a barrier to bacteriophage adsorption in an encapsulated Staphylococcus simulans strain. Med Microbiol Immunol. 177:229–233 [DOI] [PubMed] [Google Scholar]
- Oliver A, Canton R, Campo P, Baquero F, Blázquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1253 [DOI] [PubMed] [Google Scholar]
- Ophir T, Gutnick DL. 1994. A role for exopolysaccharides in the protection of microorganisms from desiccation. Appl Environ Microbiol. 60:740–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal C, Maciá MD, Oliver A, Schachar I, Buckling A. 2007. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 450:1079–1081 [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Schneider D, Meier-Eiss J, Arber W, Lenski RE, Blot M. 1999. Genomic evolution during a 10,000-generation experiment with bacteria. Proc Natl Acad Sci U S A. 96:3807–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, et al. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182 [DOI] [PubMed] [Google Scholar]
- Perron GG, Hall AR, Buckling A. 2010. Hypermutability and compensatory adaptation in antibiotic-resistant bacteria. Am Nat. 176:303–311 [DOI] [PubMed] [Google Scholar]
- Piña SE, Mattingly SJ. 1997. The role of fluoroquinolones in the promotion of alginate synthesis and antibiotic resistance in Pseudomonas aeruginosa. Curr Microbiol. 35:103–108 [DOI] [PubMed] [Google Scholar]
- Poullain V, Gandon S, Brockhurst MA, Buckling A, Hochberg ME. 2008. The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 62:1–11 [DOI] [PubMed] [Google Scholar]
- Raeside C, Gaffé J, Deatherage DE, Tenaillon O, Briska AM, Ptashkin RN, Cruveiller S, Médigue C, Lenski RE, Barrick JE, et al. 2014. Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. MBio 5:e01377–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A. 2000. Host-parasite coevolution in a multilocus gene-for-gene system. Proc R Soc Lond B Biol Sci. 267:2183–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan PD, Buckling A. 2012. Co-evolution with lytic phage selects for the mucoid phenotype of Pseudomonas fluorescens SBW25. ISME J. 6:1148–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan PD, Hall AR, Blackshields G, Friman VP, Davis MR, Jr, Goldberg JB, Buckling A. 2015. Coevolution with bacteriophages drives genome-wide host evolution and constrains the acquisition of abiotic-beneficial mutations. Mol Biol Evol. 32:1425–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Hempel P. 2011. Evolutionary parasitology. Oxford (United Kingdom): Oxford University Press. [Google Scholar]
- Schneider D, Duperchy E, Coursange E, Lenski RE, Blot M. 2000. Long-term experimental evolution in Escherichia coli IX. Characterization of insertion sequence-mediated mutations and rearrangements. Genetics 156:477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag SJ, Perrot V, Levin BR. 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc R Soc Lond B Biol Sci. 264:1287–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slauch JM, Garrett S, Jackson DE, Silhavy TJ. 1988. EnvZ functions through OmpR to control porin gene expression in Escherichia coli K12. J Bacteriol. 170:439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HW, Huggins MB. 1982. Successful treatment of experimental Escherichia coli infections in mice using phage: its general superiority over antibiotics. J Gen Microbiol. 128:307–318 [DOI] [PubMed] [Google Scholar]
- Sousa A, Bourgard C, Wahl LM, Gordo I. 2013. Rates of transposition in Escherichia coli. Biol Lett. 9:20130838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa A, Magalhães S, Gordo I. 2012. Cost of antibiotic resistance and the geometry of adaptation. Mol Biol Evol. 29:1417–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson G, Andrianopoulos K, Hobbs M, Reeves PR. 1996. Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J Bacteriol. 178:4885–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoebel DM, Dorman CJ. 2010. The effect of mobile element IS10 on experimental regulatory evolution in Escherichia coli. Mol Biol Evol. 27:2105–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Daegelen P, Lenski RE, Maslov S, Kim JF. 2009. Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E. coli B and K-12 genomes. J Mol Biol. 394:653–680 [DOI] [PubMed] [Google Scholar]
- Tazzyman SJ, Hall AR. 2015. Lytic phages obscure the cost of antibiotic resistance in Escherichia coli. ISME J. 9:809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Rodríguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS. 2012. The molecular diversity of adaptive convergence. Science 335:457–461 [DOI] [PubMed] [Google Scholar]
- Tenaillon O, Toupance B, Le Nagard H, Taddei F, Godelle B. 1999. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152:485–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M, Rocha EP. 2007. Causes of insertion sequences abundance in prokaryotic genomes. Mol Biol Evol. 24:969–981 [DOI] [PubMed] [Google Scholar]
- Travisano M, Lenski RE. 1996. Long-term experimental evolution in Escherichia coli. IV. Targets of selection and the specificity of adaptation. Genetics 143:15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I. 2009. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. 5:e1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrientes MC, Baquero MR, Sánchez MB, Valdezate S, Escudero E, Berg G, Cantón R, Baquero F, Galán JC, Martínez JL. 2010. Polymorphic mutation frequencies of clinical and environmental Stenotrophomonas maltophilia populations. Appl Environ Microbiol. 76:1746–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogwill T, Kojadinovic M, Furió V, MacLean RC. 2014. Testing the role of genetic background in parallel evolution using the comparative experimental evolution of antibiotic resistance. Mol Biol Evol. 31:3314–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. 2000. Molecular mechanisms that confer antibacterial drug resistance. Nature 406:775–781 [DOI] [PubMed] [Google Scholar]
- Wandersman C, Moreno F, Schwartz M. 1980. Pleiotropic mutations rendering Escherichia coli K12 resistant to bacteriophage TP1. J Bacteriol. 143:1374–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbauer MG. 2004. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 28:127–181 [DOI] [PubMed] [Google Scholar]
- Wielgoss S, Barrick JE, Tenaillon O, Cruveiller S, Chane-Woon-Ming B, Medigue C, Lenski RE, Schneider D. 2011. Mutation rate inferred from synonymous substitutions in a long-term evolution experiment with Escherichia coli. G3 1:183–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielgoss S, Barrick JE, Tenaillon O, Wiser MJ, Dittmar WJ, Cruveiller S, Chane-Woon-Ming B, Médigue C, Lenski RE, Schneider D. 2013. Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc Natl Acad Sci U S A. 110:222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler JD, Garcia C, Olson M, Callaway E, Kao KC. 2014. Evolved osmotolerant Escherichia coli mutants frequently exhibit defective N-acetylglucosamine catabolism and point mutations in cell shape-regulating protein MreB. Appl Environ Microbiol. 80:3729–3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods R, Schneider D, Winkworth CL, Riley MA, Lenski RE. 2006. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc Natl Acad Sci U S A. 103:9107–9112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Hengzhuang W, Wu H, Damkiæer S, Jochumsen N, Song Z, Givskov M, Høiby N, Molin S. 2012. Polysaccharides serve as scaffold of biofilms formed by mucoid Pseudomonas aeruginosa. FEMS Immunol Med Microbiol. 65:366–376 [DOI] [PubMed] [Google Scholar]
- Yu F, Mizushima S. 1982. Roles of lipopolysaccharide and outer membrane protein OmpC of Escherichia coli K-12 in the receptor function for bacteriophage T4. J Bacteriol. 151:718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman L, Meyer JR, Devangam S, Bryson DM, Lenski RE, Ofria C. 2014. Coevolution drives the emergence of complex traits and promotes evolvability. PLoS Biol. 12:e1002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.