

Abstract
Loss of pancreatic β-cell maturity occurs in diabetes and insulinomas. Although both physiological and pathological stresses are known to promote β-cell dedifferentiation, little is known about the molecules involved in this process. Here we demonstrate that activinB, a transforming growth factor β (TGF-β)-related ligand, is upregulated during tumorigenesis and drives the loss of insulin expression and β-cell maturity in a mouse insulinoma model. Our data further identify Pax4 as a previously unknown activinB target and potent contributor to the observed β-cell dedifferentiation. More importantly, using compound mutant mice, we found that deleting activinB expression abolishes tumor β-cell dedifferentiation and, surprisingly, increases survival without significantly affecting tumor growth. Hence, this work reveals an unexpected role for activinB in the loss of β-cell maturity, islet plasticity, and progression of insulinoma through its participation in β-cell dedifferentiation.
INTRODUCTION
Loss of maturity and acquisition of embryonic traits are well-established paradigms that contribute to tumor heterogeneity and metastasis (1, 2). Endocrine tumors that develop from pancreatic islet cells are highly heterogeneous (3). Although poorly differentiated endocrine tumors of the pancreas exist, the cause and contribution of β-cell dedifferentiation in the initiation and progression of those lesions remain undetermined. Loss of insulin expression has been observed in transgenic mouse models of insulinoma, supporting the existence of a mechanism that reverts the differentiated state of mature β cells in β-cell tumors (4, 5). Recently, Landsman et al. demonstrated that elevated Hedgehog/Gli signaling in β cells alters their identity and leads to the development of undifferentiated endocrine pancreatic tumors (6). Therefore, the participation of β-cell dedifferentiation in adult pancreatic pathologies such as islet tumors underscores the need to identify the autocrine factors controlling these mechanisms.
While the characterization of signals that regulate β-cell development and regeneration is the focus of intense work (7), less is known about mechanisms and molecules that control the differentiation state of mature adult β cells under pathological conditions. The phenomenon of β-cell dedifferentiation, characterized by a loss of expression of key β-cell genes, such as those encoding insulin, glucose transporter 2 (Glut2), and transcription factors associated with the cells' mature phenotype, was first reported in mouse and recently confirmed in cultured human islets in the absence of any pathological context (8–10). Further evidence has confirmed that the differentiated state of mature adult β cells is not permanent and is lost in response to signals such as oxidative stress and changes in transcriptional profile (11–13). The contribution of β-cell dedifferentiation to pathological conditions in vivo is also supported by recent work suggesting that β-cell dedifferentiation caused by FoxO1 disruption underlies β-cell failure in type II diabetes (14).
Activins are transforming growth factor β (TGF-β)-related ligands that participate in a wide array of biological processes in development and cancer (15–17). Activins and their receptors control embryonic patterning of foregut-derived organs (18) and are closely associated with the development of the endocrine pancreas (19). Although activinA and activinB are expressed in pancreatic islets, their presence in β cells is still debated (20–22). Nevertheless, transgenic mouse models have confirmed roles for these ligands in adult islets and in β-cell proliferation (23, 24). Interestingly, activinA decreases the expression of mature β-cell genes, highlighting a possible contribution of activins to β-cell dedifferentiation and islet plasticity (25). In contrast, the effect of activinB in pancreatic islets is less clear. Given that activinA and activinB affect the function of islet β cells and are frequently overexpressed in various tumors (26), we hypothesized that activins could contribute to β-cell tumor plasticity. Using a mouse insulinoma model based on the targeted disruption of the Men1 gene (5), we found β-cell tumors to overexpress activinB. Further, our work reveals that activinB mediates β-cell dedifferentiation, causing tumor β cells to lose their mature characteristic while keeping an endocrine identity. The role of activinB in β-cell dedifferentiation was further supported by the absence of dedifferentiated β cells and increased survival in tumors lacking activinB expression.
MATERIALS AND METHODS
Mouse strains and procedure.
βMen1 and InhβB-KO mice have been previously described (5, 27). Control (Men1F/F), βMen1, and InhβB-βMen1 mice were maintained in a mixed 129sv/C57BL6 background. Glucose measurements were done with 6-hour-fasted mice. All animal experiments were performed in accordance with the guidelines of the European Union and French laws and were validated by the local Animal Ethical Committee.
Immunohistological analysis.
Pancreases, collected from 6-h-fasted mice, were fixed in 4% formalin prior to paraffin embedding. Immunohistochemical staining (IHC) was revealed with diaminobenzidine (DAB) (DAB kit; Vector Laboratories, United Kingdom). Immunofluorescence samples were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) (Vector Laboratories, United Kingdom). Lists of antibodies are provided in the supplemental material. The β-cell proliferation index was determined by calculating the percentage of Ki67+ Ins+ double-immunofluorescent cells normalized to Ki67− Ins+ cells. For each genotype, 3 or 4 pancreases were used, and a minimum of 1,000 Ins+ cells were analyzed per animal.
Tumor and β-cell morphometric analyses.
Tumor and β-cell surfaces were evaluated with Histolab software (Alphelys, France) after a visual elimination of artifacts from whole-pancreas scanning acquired on an Eclipse E44 microscope (Nikon, France). Sections were stained with hematoxylin and eosin (H&E) (tumor mass quantification) or for insulin (β-cell mass quantification). Quantifications were performed blind to the genotype on at least 2 randomized sections 30 μm apart. Briefly, for each section, tumor area surfaces were determined based on scanned H&E staining normalized against total pancreas surfaces. Nonpancreatic tissues such as infiltrates, lymph nodes, and sectioning artifacts were manually excluded after visual inspection of all sections. Areas occupied by all pancreatic insulin-positive (insulin+) β cells were subsequently determined on matching serial sections based on insulin immunohistochemical staining. For each section, total insulin+ β-cell surfaces were subsequently normalized against total pancreas surfaces as mentioned above.
Cell culture.
Min6 and βTC3 cells were cultured as described previously (25). Cells were subjected to 24 h of serum starvation prior to 72 h of stimulation with a subphysiological concentration (i.e., 1 nM) of activinB (Peprotech, USA). When mentioned, 10 nM SB431542 (Sigma, France) was added to the stimulation medium.
Real-time reverse transcriptase PCR (RT-PCR), Western blotting, and ELISA analysis.
RNAs were extracted using RNeasy kits (Qiagen, Valencia, CA). Real-time PCR analyses were carried out on a Step-One RT system (Applied Biosystems, France) using SYBR green (Life Technology, France) and results for each sample normalized to Tpb. Primer sequences are provided in the supplemental material. Antibodies for Western blots are listed in the supplemental material. Quantification of activinB intraislet production was done with a commercial enzyme-linked immunosorbent assay (ELISA) kit (mouse INHbB kit; USCNK, Houston, TX) according to the manufacturer's instructions.
Statistical analyses.
Statistical analyses were performed as described in the figure legends; unpaired Student t tests were used unless otherwise indicated. All analyses were done using Prism5 software (GraphPad, USA); a P value of ≤0.05 was considered significant. Results are given as means ± standard errors of the means (SEM).
RESULTS
Men1 advanced β-cell tumors show a heterogenic loss of insulin expression.
Expression of insulin is a hallmark of mature differentiated β cells. Having previously reported reduced insulin immunoreactivity in insulinomas that develop from Men1F/F; RipCre mice (βMen1) (5), we hypothesized that insulinomas undergo cellular dedifferentiation. Using an immunohistochemical approach to detect insulin, we first confirmed a robust reduction of insulin expression in advanced βMen1 tumors. Although insulin expression was barely detectable in several tumor cells (termed Ins−) within 14-month-old tumors, we found a few β cells scattered within islet structures to maintain a strong insulin immunoreactivity (termed Ins+) (Fig. 1A; see Fig. S1 in the supplemental material). Quantification of the respective surface areas occupied by Ins+ and Ins− tumor cells revealed that 7.45% ± 1.508% of the pancreas was occupied by islet tumors, whereas Ins+ tumor β cells accounted for only for 1.35% ± 0.581% of this surface in βMen1 tumors (Fig. 1B). Analysis of serum glucose concentrations revealed that βMen1 animals had became severely hypoglycemic at the age of 14 months (Fig. 1C), suggesting that the remaining Ins+ β-cell lesions are capable of producing an overload of insulin that impacts glycemia.
FIG 1.
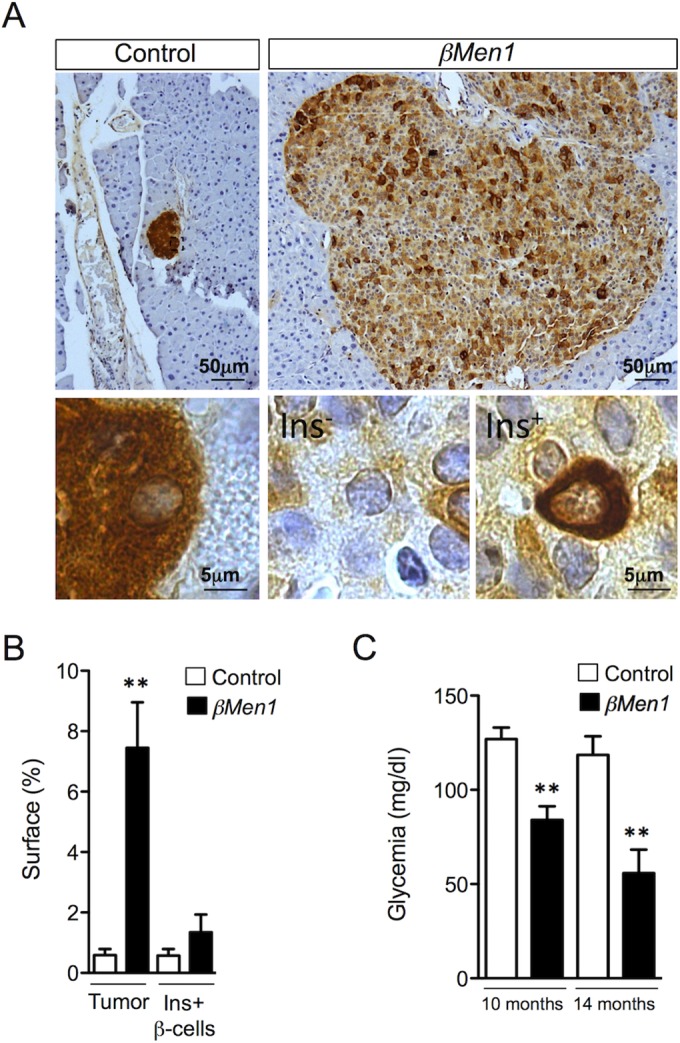
Analysis of insulin expression in βMen1 β-cell tumors reveals heterogeneity in intratumoral insulin expression. (A) Representative insulin staining heterogeneity seen in 14-month-old control and βMen1 tumors. Lower panels represent magnified views of upper panels. The Ins− and Ins+ panels depict β cells expressing low and high insulin levels, respectively, in tumors. (B) Morphometric measurements of tumor versus insulin-immunoreactive (Ins+) β-cell surfaces observed in 14-month-old βMen1 and control mice. Data represent the means of surface ± SEM. **, P < 0.01 by the Student t test. (C) Mean glycemia ± SEM; **, P < 0.01 by the Student t test.
Men1 β-cell tumors progressively lose insulin expression, while they retain their endocrine character.
We next analyzed the loss of insulin expression at various tumor stages. Dysplastic lesions that develop at 6 months showed insulin staining comparable to that for controls (Fig. 2A). The earliest insulin reduced expression was evident in 10-month-old adenomas and was even more pronounced at 14 months (Fig. 2A). We subsequently used chromograninA, a marker of neuroendocrine tumors, to address whether Ins− lesions retain their endocrine character. We found chromograninA to be expressed in all tumors analyzed and to be colocalized with insulin in only a subset of tumor β cells (Fig. 2B). Next, we thought to determine menin expression in β cells that had lost insulin expression. First, we checked menin expression at 1 month of age and found that menin disruption was almost complete in β cells, whereas all α cells and Ins− chromogranin+ cells were expressing menin at that age (see Fig. S2 and S3 in the supplemental material). More interestingly, at 6 and 14 months of age, we found menin expression to be lost in all Ins+ and Ins− chromograninA+ islet tumors that developed in βMen1 mice (see Fig. S3 in the supplemental material), supporting the idea that Ins− chromograninA+ tumors may originate from Ins+ menin-disrupted β cells. Given the known α- to β-tumor-cell transdifferentiation that occurs in Men1 glucagonomas (28), we analyzed whether Ins− lesions expressed glucagon, somatostatin, or pancreatic polypeptide and failed to detect those hormones in Ins− β-cell lesions (Fig. 2C). This observation suggests that the loss of insulin expression observed in tumor β cells reflects a dedifferentiation rather than a transdifferentiation. Finally, we found dedifferentiated lesions to lack the expression of Sox9 and Ngn3, supporting the idea that tumor β cells had lost their maturity rather than acquired a progenitor-like phenotype (Fig. 2D and E).
FIG 2.

Progressive loss of insulin expression in βMen1 tumor β cells. (A) Immunohistochemical analysis of insulin expression at the indicated ages. (B) Analysis of chromograninA and insulin coexpression. Representative pictures of double-immunofluorescent staining from 14-month-old βMen1 and control mice are shown. (C) Immunohistochemical staining of insulin, glucagon, pancreatic (Panc.) polypeptide, and somatostatin in 14-month-old pancreas serial sections. (D and E) Analysis of Sox9 and Ngn3 expression in 14-month-old Ins− βMen1 tumor lesions. Note that pancreatic ducts and intestinal tissues present on the same pancreatic sections were used as internal controls expressing Sox9 and Ngn3, respectively.
ActivinB expression is selectively increased during β-cell tumorigenesis.
We next speculated that an autocrine factor produced by tumor cells promotes the loss of insulin expression. We explored transcriptomic analyses of Men1-KO β-cell and insulinoma models (29, 30), looking for candidates known to inhibit insulin expression. Given the importance of TGF-β signaling in insulin synthesis and maintenance of β-cell maturity (24, 31), we paid particular attention to related ligands. In this way, we identified the InhβB gene, coding for activinB, to be upregulated in insulinomas depleted of Men1. Histological analyses performed with 2 independent activinB antibodies confirmed the selective induction of activinB expression in β-cell tumors and the near absence of activinB in control β cells (Fig. 3A; see Fig. S4 in the supplemental material). Knowing the importance of both activinA and activinB in islet β-cell functions (25, 32, 33), we further checked whether activinA expression was altered, but we did not find this ligand to be increased in Men1 β-cell tumors (data not shown). Next, we sought to delineate the onset of activinB overexpression from aging βMen1 mice. Whereas activinB was absent in 4-month-old hyperplastic islets, we detected this ligand within a group of clustered cells randomly located within 6-month-old dysplastic islets. At 10 and 14 months of age, the number of cells expressing activinB was robustly increased (Fig. 3A). Islet content analysis confirmed that activinB was induced in 9-month-old βMen1 isolated islets compared to age-matched controls (Fig. 3B). Using a coimmunofluorescence approach, we further found lesions with reduced levels of insulin or a lack of insulin to express high levels of activinB, while activinB was absent in Ins+ tumor cells and control islets (Fig. 3C). Taken together, these observations support that activinB induced-expression could represent a triggering signal that reduces insulin expression in βMen1 tumor β cells.
FIG 3.

Induced activinB expression in β-tumor cells leads to a progressive downregulation of insulin expression. (A) Immunohistochemical analysis of activinB expression in βMen1 and control pancreas. Islets are outlined by dashed lines. (B) Quantification of intraislet activinB content. Size-matched hand-picket islets from 9-month-old βMen1 (n = 4) and control (n = 4) animals were used to extract protein and quantify activinB content by ELISA. Data are represented as means ± SEM of triplicate measurements of 4 independent samples per genotype. *, P < 0.05 by the Student t test. (C) Immunofluorescent colocalization of activinB and insulin within 10- and 14-month-old tumor lesions and control islets. Right panels are higher magnifications of dashed areas.
ActivinB expression induces the downregulation of insulin expression in β cells in vitro.
Given that Smad3 has the capacity to bind and mediate repression of the insulin gene promoter (34), we next tested the direct effect of activinB stimulation on transcription of insulin genes in vitro. Using cultured Min6, we found that activinB significantly inhibited both Ins1 and Ins2 gene expression (Fig. 4). Use of the Psmad2/3-SB431542 inhibitor demonstrated a significant rescue of the Ins1 and Ins2 gene inhibition by activinB, confirming that part of this inhibition was taking place through Smad2/3, as previously reported (34). Interestingly Ins2 gene inhibition was partly rescued by SB431542, suggesting that a Smad2/3 noncanonical mechanism was also involved.
FIG 4.
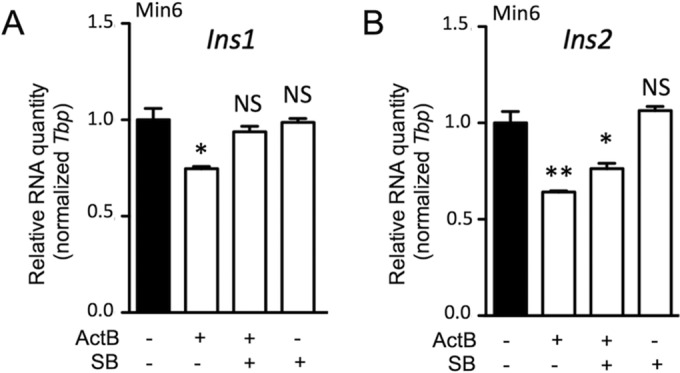
ActivinB inhibits insulin expression in vitro. Quantitative RT-PCR analysis of Ins1 (A) and Ins2 (B) gene expression in Min6 cells following 72 h of activinB (ActB) treatment is shown. SB, SB431542 inhibitor. Graphs are representative examples of 3 independent experiments. Data represent means ± SEM for triplicate samples per experiment. *, P < 0.05; **, P < 0.01; NS, not significant (by the Student t test).
ActivinB disruption in βMen1 mice rescues the loss of insulin expression in tumor β cells.
We next examined the consequences of an activinB loss for β-cell tumorigenesis. Taking advantage of the viability of the homozygous InhβB-KO mouse (27), we generated InhβBKO/KO; Men1F/F; RipCre compound mutants (termed InhβB-βMen1) to investigate whether the lack of activinB impacted the progressive loss of insulin expression in tumor β cells. Although quantification of the tumor cell mass progression did not revealed differences between βMen1 and InhβB-βMen1 compound mutant mice, we found that the surface of the compound-mutant lesions plateaued at 6 months supporting the fact that activinB may impact tumor growth (Fig. 5A). Analysis of activinB immunoreactivity further confirmed the lack of activinB in InhβB-βMen1 tumors (see Fig. S5 in the supplemental material). Interestingly, we found that islet tumors of all analyzed 10- and 14-month-old InhβB-βMen1 mice stained homogeneously for insulin, yet the intensity of the observed staining was slightly weaker than in control or InhβB-KO mutant islets (Fig. 5B and C; see Fig. S1 in the supplemental material). Moreover, we found that βMen1 animals lacking activinB demonstrated a prolonged survival after 10 months of age (Fig. 5D), which appears to be consistent with the slight, not significant, reduction of the Ki67 proliferation index of InhβB-βMen1 lesions at 14 months of age (Fig. 5E). Taken together, these observations support the notion that the loss of insulin observed in the βMen1 lesions results from an autocrine effect of activinB and, more importantly, that the lack of activinB could negatively impact the progression of βMen1 tumors.
FIG 5.

Inactivation of activinB restores insulin expression in βMen1 insulinomas and increases survival. (A) Measurements of islet cell surface during the course of tumorigenesis. Endocrine surface were calculated using H&E-stained whole pancreatic sections at the indicated ages. The graph shows the percentage of endocrine surface normalized to exocrine surfaces. Numbers of analyzed pancreases per genotype and age are indicated (n). Data are represented as mean of mouse surface ± SEM. (B) Immunohistochemical analysis of insulin expression in 14-month-old pancreas from animals of the indicated genotype. (C) Morphometric measurements of tumor versus insulin-immunoreactive (Ins+) β-cell surfaces observed in 14-month-old InhβB-βMen1 and control mice. Data represent the mean of surface ± SEM. **, P < 0.01 by the Student t test. (D) Survival curve of βMen1 (n = 16), InhβB-βMen1 (n = 14), and control (n = 14) mice. **, P < 0.01; ***, P < 0.001 (by the Mantel-Cox test). (E) Ki67 proliferative index of Ins+ lesions and control β cells at 14 months of age. Data represent the mean of the number of Ki67+/Ins+ cells found in mutant and control animals ± SEM. NS, not significant the Student t test.
ActivinB decreases the expression of mature β-cell genes and contributes to the dedifferentiation of β-cell tumors.
Maintenance of β-cell maturity is orchestrated by a subset of transcription factors that are essential for adult β cells (11, 23); therefore, we analyzed the impact of activinB on mature β-cell gene expression. Using Min6 and βTC3 cells derived from mouse insulinomas, we found the expression of Ins1, Ins2, Slc2a2, Nkx6.1, and Pdx1 to be significantly reduced in those cells following activinB treatment (Fig. 6A and B). The inhibition of Mafa and Nkx2.2 by activinB appeared to be mutually exclusive in βTC3 and Min6 cells, while the expression of Mafb remained unchanged in both cell lines. The downregulation of MafA, Nkx6.1, Nkx2.2, and Pdx1 following activinB stimulation was also validated at the protein level (Fig. 6C). Interestingly, the expression of the paired box 4 (Pax4), a transcription factor involved in pancreatic β-cell differentiation and more recently shown to promote β-cell dedifferentiation in mature β cells (35), was strongly induced by activinB treatment in both βTC3 and Min6 cells (Fig. 6A and B). Use of the SB431542 inhibitor further demonstrated the specificity of the activinB-mediated induction through Smad2/3 signaling in βTC3 cells (Fig. 6D and E) as well as in Min6 cells (data not shown).
FIG 6.

ActivinB decreases the expression of mature β-cell genes while promoting Pax4 expression. (A and B) Quantitative RT-PCR analysis of mature β-cell gene expression in Min6 (A) or βTC3 (B) cells following 72 h of activinB stimulation. (C) Analysis of MafA, Nkx6.1, Nkx,2.2 and Pdx1 protein expression in Min6 cells subjected to 72 h of activinB stimulation (+). Tubulin was used as a loading control. (D and E) Analysis of Pax4 gene (D) and protein (E) expression in βTC3 cells subjected to 72 h of activinB (ActB) stimulation in the presence or absence of SB431542 (SB). Graphs are representative examples of 3 independent experiments. Data are presented as mean ± SEM for triplicate samples per experiment. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant (by the Student t test).
ActivinB disruption rescues the loss of tumor β-cell maturity in βMen1 tumors.
We next investigated Ins− lesions, which do express activinB. We were unable to detect MafA in Ins− tumors, whereas we found Pdx1 and Nkx6.1 expression to be reduced yet still present in those lesions compared to control islets (Fig. 7A to C). Membrane expression of the glucose transporter Glut2 (encoded by the Slc2a2 gene) was absent at the surface of the Ins− tumor β cells, suggesting that those cells had lost their glucose-sensing capacity (Fig. 7D). A marked increase in Pax4 expression was also evident in Ins− lesions that overexpressed activinB, as seen by the strong staining observed in the lesions lacking insulin expression (Fig. 7E). Lesions that developed in the InhβB-βMen1 mice were found to be well differentiated and to express MafA, Nkx6.1, Pdx1, and Pax4 levels comparable to those in age-matched control islets (Fig. 7). As in normal pancreas, the expression of Glut2 was obvious at the surface of β cells in InhβB-βMen1 tumors (Fig. 7D). Together these observations confirm that βMen1 tumors lacking activinB were protected against β-cell dedifferentiation and loss of maturity.
FIG 7.

Inactivation of activinB rescues the loss of maturity seen in βMen1 insulinomas. Immunofluorescent colocalization of insulin and mature β-cell markers MafA (A), Nkx6.1 (B), Pdx1 (C), Glut2 (D), and Pax4 (E) in Ins− βMen1 and InhβB-βMen1 tumors developed at the age of 14 months is shown. Lower panels are magnified views of each separate marker fluorescent channel.
DISCUSSION
Ample evidence from clinical studies and work with mice shows that pancreatic cells demonstrate a high degree of cellular plasticity in response to various stresses and pathological conditions. Here, we report that islet β-cell tumors induced by the loss of the Men1 suppressor gene are subjected to β-cell dedifferentiation. More importantly, we demonstrate that activinB, which is barely detectable in normal β cells, is markedly induced in dysplastic lesions and causes tumor cells to progressively lose their insulin expression and their capacity to express mature markers essential for β-cell function, such as MafA, Pdx1, Nkx6.1, and Glut2.
The identification of activinB as an endogenous modulator of β-cell plasticity brings a new twist to its previously reported role in inhibiting insulin secretion (32, 33). This role is consistent with the emerging functions of the TGF-β/bone morphogenic protein (BMP) superfamilies in the suppression of adult β-cell maturation (31). Smad-BMP signaling prevents β-cell differentiation in stem cells, zebrafish, and mice (36), and TGF-β, activinA, and Smad3 are capable of repressing insulin and a subset of mature β-cell genes in islet β-cell lines and isolated mouse islets (25, 34). Using Min6 and βTC3 cells, we found that activinB represses the expression of several genes associated with β-cell maturity. Interestingly, Szabat et al. reported that activinA stimulation of Min6 cells has the same effects (25). Thus, both activinA and activinB can promote β-cell dedifferentiation in vitro, through the same target genes. This observation conflicts somewhat with the acute, antagonistic roles previously reported for activinA and activinB in β-cell insulin secretion (32, 33). Nevertheless, both activins have the capacity to activate Smad3 phosphorylation through the use of Alk4/Alk7 type I receptors, which may explain their common in vitro effects on β-cell maturity. Wu et al. recently demonstrated that activinB is a more potent inducer of Smad3 phosphorylation than activinA in isolated islets and β-cell lines (33). Given that Smad3 has the capacity to bind and mediate repression of the insulin gene promoter (34), our data suggest that activinA may not induce β-cell dedifferentiation in vivo as seen for activinB. Indeed, we did not detect an increase in activinA expression in Men1 β-cell tumors, confirming that the observed β-cell dedifferentiation results solely from the autocrine effects of activinB. This conclusion is supported by the lack of β-cell dedifferentiation in the InhβB-βMen1 compound mutant mice. While the inactivation of activinB convincingly blocks tumor β-cell dedifferentiation, we cannot exclude the possibility that other factors also contribute to this process. Interestingly, some target candidates, such as MafA, seem to be partly rescued in tumors developed by compound mutant mice (Fig. 7A). Whereas the origin of this observation remains elusive, it supports the participation of complementary pathways and molecules that maintain the dedifferentiation process beyond activinB. Among possible candidates, menin itself could have an important role, as it was previously shown to contribute to decreased MafA expression in insulinoma cell lines (37). It will therefore be important to determine to what extent menin contributes to β-cell dedifferentiation through an activinB-independent mechanism.
Besides inducing the loss of β-cell maturity, activinB strongly upregulates Pax4, both in a β-cell line and in Ins− dedifferentiated lesions. While Pax4 is essential for the development and differentiation of insulin-producing β cells in the mammalian pancreas (38), its functions in adult β cells have been proposed to orchestrate a network of genes that govern β-cell expansion and survival under physiological and pathological conditions (39). Examining the consequences of increased Pax4 expression in activinB-expressing tumor cells is therefore important. However, the low proliferative index of dedifferentiated tumors does not corroborate the previously described role of Pax4 in β-cell-induced proliferation in rat and human islets (40). Interestingly, long-term conditional overexpression of Pax4 in mature islet β cells represses the expression of insulin, MafA, and Glut2, consistent with the occurrence of β-cell dedifferentiation (35). This observation suggests that activinB-mediated induction of Pax4 may contribute to triggering or maintaining β-cell dedifferentiation in the pathophysiological context of β-cell tumors, a hypothesis supported by the lack of Pax4 overexpression and β-cell dedifferentiation in tumors in InhβB-βMen1 mice lacking activinB expression.
While we initially demonstrated that activinB inhibits insulin secretion (32), work by Wu et al. confirmed that transient stimulation of isolated pancreatic islets with exogenous activinB suppresses glucose-stimulated induced secretion (GSIS) and ATP production (33). In the diabetic state, loss of acute GSIS is associated with a loss of the β-cell mature phenotype (41). Therefore, it will be important to determine whether a sustained inhibition of GSIS and ATP production mediated by upregulation of activinB in β cells could contribute to the observed plasticity of tumor β cells. Our observations suggest that tumor β cells overexpress activinB as a means of protecting against the overproduction of insulin in animals developing insulinomas. Although physiological protection against insulin overload may occur through limitation of GSIS in the early stages of tumor progression, the loss of β-cell functionality through dedifferentiation is likely to be the major protective mechanism operating in these tumors.
While in the field of pancreatic endocrine tumors much is still unknown about islet tumors due to their functional complexity and cellular diversity, our results identify activinB as an intrinsic β-cell candidate that contributes to the plasticity of islet tumors. Unlike previous insulinomas mouse models, which develop aggressively proliferative, poorly dedifferentiated carcinomas upon forced activation of Hedgehog/Gli signaling or expression of the simian virus 40 (SV40) large T antigen in β cells (6, 42), the dedifferentiated tumors in the βMen1 mouse model have a low proliferative potential (data not shown) and lack expression of the Sox9 and Ngn3 markers of early pancreatic progenitors, similar to the case for the pancreatic endocrine tumors found in MEN1 patients. Such observations highlight the complexity and subtlety of the mechanism that may control β-cell tumor dedifferentiation in vivo. Published work from Anlauf et al. and Perren et al. (43, 44) has shown that most of the early islet lesions seen in MEN1 patients are glucagon positive, whereas the advanced hormone-secreting islet tumors detected in these same patients are mainly insulinomas or nonsecreting tumors. These observations suggest that glucagon-positive early lesions may be replaced or overtaken by insulinomas and mixed islet tumors during tumorigenesis, an observation consistent with our previous published work (28). To that extent, we believe that the identification of activinB-induced expression in βMen1 insulinomas and its role in their dedifferentiation/loss of mature characteristics sheds light on an important issue regarding the mechanism of origin of the insulinomas and nonfunctioning tumors seen in MEN1 patients. Therefore, activinB has potential as a biomarker and drug target that could refine the clinical classification of pancreatic endocrine tumors. More importantly, our work uncovers a previously unknown physiological role of activinB in the maintenance of β-cell maturity. Identifying the in vivo signals that trigger the expression of activinB in pancreatic islet cells will be crucial to further understanding the cellular contexts that promote β-cell dedifferentiation.
Supplementary Material
ACKNOWLEDGMENTS
We thank the animal care staff of ALECs-SFP (Lyon, France) and ANICAN (CRCL-Lyon, France) for maintenance of mouse strains. We are grateful to B. Sosa-Pineda (St. Jude Children's Research Hospital, Memphis, TN) for the anti-Pax4 antibody. We thank Tamsin Lindström (Stockholm, Sweden) for critical reading and editing of the manuscript.
This work was supported by La Ligue contre le Cancer (CD42 and CD69), Lyon I University, l'Agence Nationale de Recherche (grant SVSE2 ANR10BLAN124004), and the European Foundation for the Study of Diabetes (EFSD/AZ cellular plasticity program 2014). D.R. was supported by fellowships from the French Ministry of Education and Research and La Ligue.
D.R. and P.B. conceived and designed the experiments. D.R., J.C., A.H., R.J., R.B., D.G., R.T., M.C.-B., and P.B. performed the experiments and analyzed the data. O.A., O.R., and C.X.Z. contributed reagents and provided advice on experiments. D.R., C.X.Z., O.R., O.A., and P.B. discussed the results. P.B. wrote the manuscript.
We have no conflicts to disclose.
Funding Statement
Olov Andersson thanks the Ragnar Söderbergs foundation, Swedish Research Council and Strategic Research Initiative in Stem Cells and Regenerative Medicine, for support.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00930-15.
REFERENCES
- 1.Marusyk A, Almendro V, Polyak K. 2012. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 2.Brabletz T. 2012. To differentiate or not—routes towards metastasis. Nat Rev Cancer 12:425–436. doi: 10.1038/nrc3265. [DOI] [PubMed] [Google Scholar]
- 3.Asa SL. 2011. Pancreatic endocrine tumors. Mod Pathol 24(Suppl 2):S66–S77. doi: 10.1038/modpathol.2010.127. [DOI] [PubMed] [Google Scholar]
- 4.Pelengaris S, Khan M, Evan GI. 2002. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 109:321–334. doi: 10.1016/S0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 5.Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. 2003. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res 63:4836–4841. [PubMed] [Google Scholar]
- 6.Landsman L, Parent A, Hebrok M. 2011. Elevated Hedgehog/Gli signaling causes beta-cell dedifferentiation in mice. Proc Natl Acad Sci U S A 108:17010–17015. doi: 10.1073/pnas.1105404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziv O, Glaser B, Dor Y. 2013. The plastic pancreas. Dev Cell 26:3–7. doi: 10.1016/j.devcel.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 8.Weinberg N, Ouziel-Yahalom L, Knoller S, Efrat S, Dor Y. 2007. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes 56:1299–1304. doi: 10.2337/db06-1654. [DOI] [PubMed] [Google Scholar]
- 9.Russ HA, Bar Y, Ravassard P, Efrat S. 2008. In vitro proliferation of cells derived from adult human beta-cells revealed by cell-lineage tracing. Diabetes 57:1575–1583. doi: 10.2337/db07-1283. [DOI] [PubMed] [Google Scholar]
- 10.Spijker HS, Ravelli RB, Mommaas-Kienhuis AM, van Apeldoorn AA, Engelse MA, Zaldumbide A, Bonner-Weir S, Rabelink TJ, Hoeben RC, Clevers H, Mummery CL, Carlotti F, de Koning EJ. 2013. Conversion of mature human beta-cells into glucagon-producing alpha-cells. Diabetes 62:2471–2480. doi: 10.2337/db12-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernardo AS, Hay CW, Docherty K. 2008. Pancreatic transcription factors and their role in the birth, life and survival of the pancreatic beta cell. Mol Cell Endocrinol 294:1–9. doi: 10.1016/j.mce.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. 2009. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab 10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. 2011. Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell 20:419–429. doi: 10.1016/j.devcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. 2012. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sulyok S, Wankell M, Alzheimer C, Werner S. 2004. Activin: an important regulator of wound repair, fibrosis, and neuroprotection. Mol Cell Endocrinol 225:127–132. doi: 10.1016/j.mce.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Xia Y, Schneyer AL. 2009. The biology of activin: recent advances in structure, regulation and function. J Endocrinol 202:1–12. doi: 10.1677/JOE-08-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ. 2010. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Kim SK, Hebrok M, Li E, Oh SP, Schrewe H, Harmon EB, Lee JS, Melton DA. 2000. Activin receptor patterning of foregut organogenesis. Genes Dev 14:1866–1871. [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang YQ, Cleary MM, Si Y, Liu G, Eto Y, Kritzik M, Dabernat S, Kayali AG, Sarvetnick N. 2004. Inhibition of activin signaling induces pancreatic epithelial cell expansion and diminishes terminal differentiation of pancreatic beta-cells. Diabetes 53:2024–2033. doi: 10.2337/diabetes.53.8.2024. [DOI] [PubMed] [Google Scholar]
- 20.Ogawa K, Abe K, Kurosawa N, Kurohmaru M, Sugino H, Takahashi M, Hayashi Y. 1993. Expression of alpha, beta A and beta B subunits of inhibin or activin and follistatin in rat pancreatic islets. FEBS Lett 319:217–220. doi: 10.1016/0014-5793(93)80549-A. [DOI] [PubMed] [Google Scholar]
- 21.La Rosa S, Uccella S, Marchet S, Capella C, Lloyd RV. 2004. Localization of inhibins and activins in normal endocrine cells and endocrine tumors of the gut and pancreas: an immunohistochemical and in situ hybridization study. J Histochem Cytochem 52:217–225. doi: 10.1177/002215540405200210. [DOI] [PubMed] [Google Scholar]
- 22.Brown ML, Kimura F, Bonomi LM, Ungerleider NA, Schneyer AL. 2011. Differential synthesis and action of TGFss superfamily ligands in mouse and rat islets. Islets 3:367–375. doi: 10.4161/isl.3.6.18013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliver-Krasinski JM, Stoffers DA. 2008. On the origin of the beta cell. Genes Dev 22:1998–2021. doi: 10.1101/gad.1670808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown ML, Schneyer AL. 2010. Emerging roles for the TGFbeta family in pancreatic beta-cell homeostasis. Trends Endocrinol Metab 21:441–448. doi: 10.1016/j.tem.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabat M, Johnson JD, Piret JM. 2010. Reciprocal modulation of adult beta cell maturity by activin A and follistatin. Diabetologia 53:1680–1689. doi: 10.1007/s00125-010-1758-0. [DOI] [PubMed] [Google Scholar]
- 26.Antsiferova M, Werner S. 2012. The bright and the dark sides of activin in wound healing and cancer. J Cell Sci 125:3929–3937. doi: 10.1242/jcs.094789. [DOI] [PubMed] [Google Scholar]
- 27.Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. 1994. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev 8:414–427. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- 28.Lu J, Herrera PL, Carreira C, Bonnavion R, Seigne C, Calender A, Bertolino P, Zhang CX. 2010. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 138:1954–1965. doi: 10.1053/j.gastro.2010.01.046. [DOI] [PubMed] [Google Scholar]
- 29.Serewko-Auret MM, Mould AW, Loffler KA, Duncan R, Kay GF, Hayward NK. 2010. Alterations in gene expression in MEN1-associated insulinoma development. Pancreas 39:1140–1146. doi: 10.1097/MPA.0b013e3181dc67fc. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y, Gurung B, Wu T, Wang H, Stoffers DA, Hua X. 2010. Reversal of preexisting hyperglycemia in diabetic mice by acute deletion of the Men1 gene. Proc Natl Acad Sci U S A 107:20358–20363. doi: 10.1073/pnas.1012257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, Davis JC, Peterson QP, Rubin LL, Melton DA. 2014. Reversal of beta cell de-differentiation by a small molecule inhibitor of the TGFbeta pathway. eLife 3:e02809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertolino P, Holmberg R, Reissmann E, Andersson O, Berggren PO, Ibanez CF. 2008. Activin B receptor ALK7 is a negative regulator of pancreatic beta-cell function. Proc Natl Acad Sci U S A 105:7246–7251. doi: 10.1073/pnas.0801285105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H, Mezghenna K, Marmol P, Guo T, Moliner A, Yang SN, Berggren PO, Ibanez CF. 2014. Differential regulation of mouse pancreatic islet insulin secretion and Smad proteins by activin ligands. Diabetologia 57:148–156. doi: 10.1007/s00125-013-3079-6. [DOI] [PubMed] [Google Scholar]
- 34.Lin HM, Lee JH, Yadav H, Kamaraju AK, Liu E, Zhigang D, Vieira A, Kim SJ, Collins H, Matschinsky F, Harlan DM, Roberts AB, Rane SG. 2009. Transforming growth factor-beta/Smad3 signaling regulates insulin gene transcription and pancreatic islet beta-cell function. J Biol Chem 284:12246–12257. doi: 10.1074/jbc.M805379200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu He KH, Lorenzo PI, Brun T, Jimenez Moreno CM, Aeberhard D, Vallejo Ortega J, Cornu M, Thorel F, Gjinovci A, Thorens B, Herrera PL, Meda P, Wollheim CB, Gauthier BR. 2011. In vivo conditional Pax4 overexpression in mature islet beta-cells prevents stress-induced hyperglycemia in mice. Diabetes 60:1705–1715. doi: 10.2337/db10-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung WS, Andersson O, Row R, Kimelman D, Stainier DY. 2010. Suppression of Alk8-mediated Bmp signaling cell-autonomously induces pancreatic beta-cells in zebrafish. Proc Natl Acad Sci U S A 107:1142–1147. doi: 10.1073/pnas.0910205107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamze Z, Vercherat C, Bernigaud-Lacheretz A, Bazzi W, Bonnavion R, Lu J, Calender A, Pouponnot C, Bertolino P, Roche C, Stein R, Scoazec JY, Zhang CX, Cordier-Bussat M. 2013. Altered MENIN expression disrupts the MAFA differentiation pathway in insulinoma. Endocr Relat Cancer 20:833–848. doi: 10.1530/ERC-13-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. 1997. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- 39.Brun T, Gauthier BR. 2008. A focus on the role of Pax4 in mature pancreatic islet beta-cell expansion and survival in health and disease. J Mol Endocrinol 40:37–45. doi: 10.1677/JME-07-0134. [DOI] [PubMed] [Google Scholar]
- 40.Brun T, Franklin I, St-Onge L, Biason-Lauber A, Schoenle EJ, Wollheim CB, Gauthier BR. 2004. The diabetes-linked transcription factor PAX4 promotes β-cell proliferation and survival in rat and human islets. J Cell Biol 167:1123–1135. doi: 10.1083/jcb.200405148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weir GC, Bonner-Weir S. 2004. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53(Suppl 3):S16–S21. doi: 10.2337/diabetes.53.suppl_3.S16. [DOI] [PubMed] [Google Scholar]
- 42.Hunter KE, Quick ML, Sadanandam A, Hanahan D, Joyce JA. 2013. Identification and characterization of poorly differentiated invasive carcinomas in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS One 8:e64472. doi: 10.1371/journal.pone.0064472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anlauf M, Schlenger R, Perren A, Bauersfeld J, Koch CA, Dralle H, Raffel A, Knoefel WT, Weihe E, Ruszniewski P, Couvelard A, Komminoth P, Heitz PU, Kloppel G. 2006. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol 30:560–574. doi: 10.1097/01.pas.0000194044.01104.25. [DOI] [PubMed] [Google Scholar]
- 44.Perren A, Anlauf M, Henopp T, Rudolph T, Schmitt A, Raffel A, Gimm O, Weihe E, Knoefel WT, Dralle H, Heitz PU, Komminoth P, Kloppel G. 2007. Multiple endocrine neoplasia type 1 (MEN1): loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J Clin Endocrinol Metab 92:1118–1128. doi: 10.1210/jc.2006-1944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.