

Abstract
A brain consists of numerous distinct neurons arising from a limited number of progenitors, called neuroblasts in Drosophila. Each neuroblast produces a specific neuronal lineage. To unravel the transcriptional networks that underlie the development of distinct neuroblast lineages, we marked and isolated lineage-specific neuroblasts for RNA sequencing. We labeled particular neuroblasts throughout neurogenesis by activating a conditional neuroblast driver in specific lineages using various intersection strategies. The targeted neuroblasts were efficiently recovered using a custom-built device for robotic single-cell picking. Transcriptome analysis of mushroom body, antennal lobe and type II neuroblasts compared with non-selective neuroblasts, neurons and glia revealed a rich repertoire of transcription factors expressed among neuroblasts in diverse patterns. Besides transcription factors that are likely to be pan-neuroblast, many transcription factors exist that are selectively enriched or repressed in certain neuroblasts. The unique combinations of transcription factors present in different neuroblasts may govern the diverse lineage-specific neuron fates.
KEY WORDS: Neural stem cell, RNA-seq, Transcription factor, Type II neuroblast, Antennal lobe, Mushroom body
Summary: Transcriptome analysis of lineage-specific neural progenitors reveals molecular heterogeneity at the single-cell level and the origin for the numerous distinct neurons present in a complex brain.
INTRODUCTION
The complex brain consists of numerous distinct types of neuron. Generating such cellular diversity involves heterogeneous neural stem cells (NSCs) (Greig et al., 2013). A mouse brain consists of ∼75 million neurons (Oh et al., 2014) that arise from an undefined number of NSCs. Conserved neurogenic programs with stem cell-like progenitors underlie the generation of ∼30,000 neurons from ∼200 neuroblasts (NBs) in the much smaller Drosophila cerebrum (Urbach and Technau, 2004; Yu et al., 2013). Moreover, Drosophila NBs, like mammalian NSCs, exhibit both basic and complex patterns of neurogenesis (Homem and Knoblich, 2012). In the basic type I lineages, one progenitor buds off a series of intermediate precursors, called ganglion mother cells (GMCs), that either divide once to produce two neurons or directly differentiate into a single mature neuron. By contrast, the complex type II lineages are further amplified through intermediate neural progenitors (INPs) that, like the founding progenitor, self-renew to produce an independent series of offspring. Given the genetic, cellular and molecular conservations, studying Drosophila NBs should shed light on the development of the brains of more complex organisms.
Drosophila NBs are fated early in development to produce specific lineages of post-mitotic neurons and/or glia. Approximately 100 cerebral NBs arise per hemisphere, with each expressing a unique combination of early patterning genes (Urbach and Technau, 2003). Labeling the offspring made by individual cerebral NBs has revealed the composition of ∼100 discrete NB clones per hemisphere with characteristic morphologies (Yu et al., 2013). Mapping individual neurons serially made by one NB provides a detailed description of the developmental fate of that NB. Such single-cell lineage analyses, enabled by twin-spot MARCM (Yu et al., 2009), have not only substantiated the hypothesis that specific neuron sets are produced by specific NBs, but also revealed the distinct patterns of neuronal diversification characteristic of different NB lineages. Striking distinctions are observed among the extensively studied cerebral lineages, which include, per hemisphere, the four ‘equivalent’ lineages of mushroom body (MB) intrinsic neurons (Ito et al., 1997), four of the five known antennal lobe (AL) lineages (Jefferis et al., 2001; Lai et al., 2008), and the eight complex type II NB lineages (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008). The MB is the insect learning and memory center, and the AL is the primary olfactory center. Notably, the MB NBs, as the sole NBs that divide incessantly until fly eclosion (Truman and Bate, 1988), yield only three major classes of MB neurons, with no sister fate diversification in the paired neurons made by one GMC (Lee et al., 1999). By contrast, the AL NBs end proliferation around pupation but can generate ∼40 neuron types from a single hemilineage, as their GMCs make daughter cells with distinct A/B fates due to a Notch-mediated binary sister fate decision (Lin et al., 2010, 2012; Yu et al., 2010). Additional offspring diversities arise in the complex type II lineages through the production of variant INP sublineages by each type II NB and the derivation of distinct neuron/glia types from each INP (Wang et al., 2014). Transcriptome analysis of such diverse NBs should reveal the transcriptional networks governing distinct NB developmental fates as opposed to general NB programs.
Molecular profiling of distinct NBs requires means for targeting the NB(s) of interest throughout neurogenesis. Despite extensive lineage analysis, one cannot recognize specific NBs without markers. Probably owing to the combinatorial and dynamic nature of cell fate-determining gene expression, it is rare to see drivers that label a small number of specific NBs exclusively and persistently in the developing nervous system. Even with such drivers in hand, it is challenging to obtain enough pure lineage-specific NBs for genome-wide molecular profiling. Bulk fluorescently labeled Drosophila NBs have been successfully collected by fluorescence-activated cell sorting (FACS) for RNA sequencing (RNA-seq) (Berger et al., 2012). However, it is very challenging to find an appropriate gating condition for sorting rare cell populations with FACS. Manual picking of dissociated cells under the microscope was therefore adopted to recover rare target cells without contamination (Nagoshi et al., 2010; Okaty et al., 2011). However, such procedures are labor intensive, require very fine motor control, and become unreliable in the detection of dim cells. These technical constraints limit systematic efforts in the molecular characterization of NSC heterogeneity.
Here we report effective strategies for engineering lineage-specific NB drivers, and describe a custom-built, single-cell picking device for robotically recovering rare target cells from dissociated tissues. Transcriptome analyses of various NB subsets compared with non-selective NBs revealed novel pan-NB transcription factors and distinct sets of lineage-specific transcription factors expressed in different NB subsets. The lineage-specific transcription factors may govern the specification of lineage-characteristic offspring. The robotic single-cell picking device we describe should allow efficient purification of any cell type that can be uniquely labeled. The same strategy can be readily adopted for cell type-specific molecular profiling in diverse tissues.
RESULTS
Lineage-specific NB drivers
The first challenge in molecular profiling of NB heterogeneity and dynamics was to mark various NB subsets specifically and persistently through development. To achieve permanent NB labeling we engineered recombinase-dependent NB drivers in which a chimeric dpnEE promoter (active in all cerebral NBs) is separated from an exogenous transcriptional activator (GAL4 or LexA::P65) (Awasaki et al., 2014) by a site-specific recombination cassette carrying transcription/translation stop signals (Pfeiffer et al., 2010). Only in dpnEE-active cells can excision of the intervening cassette lead to activation of the driver. Thus, one can transform complex, often dynamic, patterns of recombinase expression, driven by various cis-regulatory elements, into clean, permanent NB drivers covering distinct NB subsets (Fig. 1). However, most isolated cis-regulatory elements show not only high activities in some restricted patterns but also weaker expression in other, often larger, domains (Awasaki et al., 2014). Such low-level ‘background’ activities could activate the recombinase-dependent NB driver in many more NBs at low frequencies, and drastically reduce the targeting specificity of otherwise sparse NB drivers.
Fig. 1.
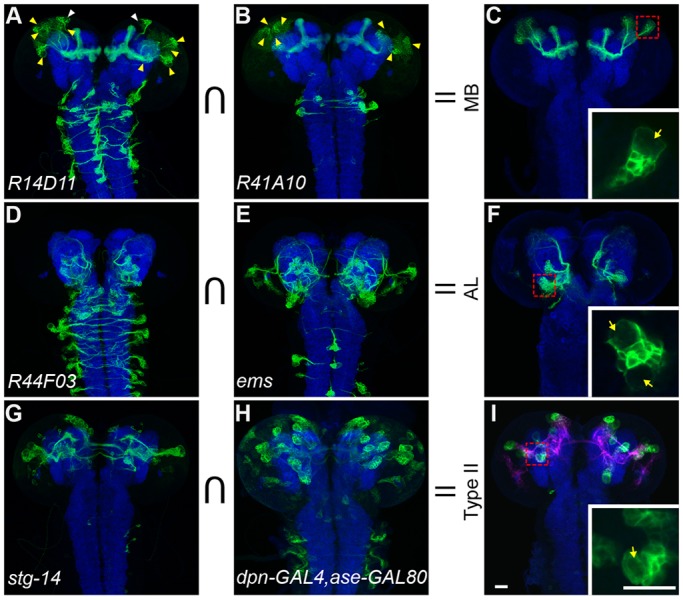
Refining NB drivers by intersection. (A-C) Via activation of dpnEE>KDRTs-stop-KDRTs>LexA::P65, the drivers R14D11-GAL4 (A) and R41A10-GAL4 (B) show activities (green) in MB NBs (yellow arrowheads) plus various non-overlapping NBs (white arrowheads). Intersection with the split GAL4 components, driven respectively by R14D11 and R41A10, achieves exclusive targeting of MB NBs (C). The CNS of 50 h ALH larvae was counterstained for Bruchpilot with nc82 mAb (blue). (D-F) Analogously, combining R44F03 with ems allows specific targeting of AL NBs. (G-I) stg-KD selectively activates dpn>KDRTs-stop-KDRTs>GAL4 in type II NBs plus various type I NBs (G). asense-GAL80 can repress GAL4 activity in type I NBs (H). Combining stg-KD with asense-GAL80 achieves exclusive targeting of type II NBs, which lie next to their derived INPs marked with R9D11-LexA::P65 (magenta) (I). Insets show that NBs (arrows) are much larger than their offspring. Scale bars: 20 µm.
Therefore, to target NBs of interest in small subsets with minimal background, we further incorporated various intersectional strategies to refine driver patterns and eliminate all ‘off-target’ expression. We successfully made three clean NB drivers that specifically label the four equivalent MB NBs, four of the five diverse AL NBs, and the eight complex type II NBs, respectively (Fig. 1, Fig. S1). We achieved unique and permanent MB NB labeling by restricting recombinase induction using split GAL4 (Luan et al., 2006; Pfeiffer et al., 2010) with R14D11 and R41D10 (Jenett et al., 2012), which show common MB NB activities and non-overlapping backgrounds (Fig. 1A-C). To exclusively label the four major AL NBs (ALad1, ALl1, ALv1 and ALv2), we first drove a recombinase with R44F03 (Awasaki et al., 2014), which confined the pan-NB driver primarily to the AL. Then we used another characterized AL promoter, ems (Lichtneckert et al., 2008; Lin et al., 2013), to drive a second recombinase for activation of the GFP reporter only in the AL NBs (Fig. 1D-F). For targeting type II NBs, we started with the stg14 enhancer (Wang et al., 2014) that selectively labels type II NBs but also occasionally labels type I NBs and INPs. We further restricted the stg14-patterned dpnEE-GAL4 activity by inhibiting GAL4 activity in type I NBs with asense-GAL80 (Fig. 1G-I).
A custom-built, single-cell picking device
The next challenge was to isolate a sufficient number of GFP-marked NBs from many developing brains of the same age. We accomplished this by building an integrated microscopy system for rapid target cell detection and robotic single-cell picking. The hardware of our custom-built, single-cell picking device includes three main components (Fig. 2A). The imaging component was built with the Leica M205 FA fluorescence stereomicroscope, a motorized XY stage for scanning a slide and tracking xy coordinates automatically, and an electron-multiplying CCD camera/sensor for capturing both fluorescence and bright-field images with enhanced sensitivity. The picking component was assembled with a metal capillary needle, a syringe pump and a micromanipulator. The stainless steel needle of internal diameter 35 µm was mounted via a holder onto a piezo micromanipulator with a positioning range of 20×20×20 mm3, 0.5 μm resolution and 3 μm repeatability (Fig. 2B). The end of the capillary needle was connected to a 100 µl syringe via a PEEK tube and tubing fitting. The syringe was mounted on a standard infuse or withdraw pump that drives fluid flow through the metal capillary needle. The control component was built upon a desktop computer with essential input/output peripherals.
Fig. 2.

Robotic sorting of NBs for RNA-seq. (A) The integrated microscopy system for rapid target cell detection and robotic single-cell picking. (B) The metal capillary needle connected to a syringe pump via a PEEK tube and moved by a micromanipulator. (C) Screen shot of the user interface for operating the single-cell picking device. The large window (left) shows real-time fluorescence and/or bright-field imaging of a 1×1 mm2 visual field. The metal capillary tip is automatically positioned next to the target cell with GFP fluorescence and a diameter of 16 µm. The small windows (right) show magnified views of the bright-field/fluorescence overlay and fluorescence-only images in the selected area. The functional keys arranged beneath are grouped into multiple categories. (D) Average size distribution of GFP-positive MB NB clonal cells from ∼30 dissociated larval CNS of four trials. Note the presence of a small population of cells (14-20 µm in diameter; likely NBs) that are distinct from the remaining smaller cells (presumably GMCs and post-mitotic neurons). (E) Relative abundance of transcript levels in isolated MB NBs following different collection procedures: a two-step 2 h collection scheme (normal), the normal condition with an additional wash step (2wash), and the normal condition plus a 2 h incubation (+2hr). Transcript levels were determined by qPCR and normalized to RNA from larval CNS. Data are represented as mean±s.d. of three trials. Note the enrichment of known pan-NB transcripts (dpn and mira) and reduction of neuronal-specific (nSyb) and glial-specific (repo) transcripts. One-way ANOVA analysis showed no significant difference among the three picking conditions. Note that the y-axis of nSyb and repo is presented as log10 value. (F) Correlation coefficients (mean±s.d.) between the transcriptomes of the MB NBs obtained following the different collection procedures.
The software that monitors and controls the imaging and picking components was coded with LabView. The user interface provides control buttons for operating (1) the motorized XY stage to scan through the slide or move to specific xy coordinates, (2) the microscope to adjust fluorescence versus bright-field imaging, (3) the micromanipulator to align and place the capillary tip at the selected position, and (4) the syringe pump to control the fluid flow direction, duration and rate for collecting, releasing or separating objects (Fig. 2C). In addition, distinct views of real-time images are shown in three windows. The CCD camera/sensor continuously reads both bright-field and green or red fluorescence images. The overlay image with a field of view of 1×1 mm is shown in the left large window, while enlarged views of the overlay and fluorescence images around the ‘red cross’ (positioned by the user's mouse click) are displayed in the other two windows (Fig. 2C). Moreover, the system automatically calculates independent object diameters based on their fluorescence signals and selectively highlights those objects that fall within a predefined size range. This enabled us to select the larger NBs of interest and not the daughter cells that had perdurance of the fluorescent label.
To eliminate background fluorescence, which is crucial for locating target cells expressing modest levels of fluorescent proteins (to avoid toxicity), we placed dissociated cells on glass slides without ‘autofluorescent’ plastic substrates. Moreover, the slide surface was prepared with PLL-PEG to reduce adhesiveness. We created two chambers on the microscope slide using transparent silicon clear rubber carved into two 30×30 mm2 chambers. Each chamber was pre-wet with 1300 µl medium (AHS buffer containing 2% FBS). The dissociated cells were concentrated via centrifugation in 150 µl medium and then loaded into one chamber for the initial cell picking; the other chamber was reserved for cleaning the isolated cells. We picked the cells using flexible metal capillaries, as opposed to fragile glass micropipettes.
Prompt recovery of Drosophila NBs for RNA-seq
The engineered MB NB driver stochastically labels about four of the eight MB NBs that make the bilaterally symmetric adult MBs throughout larval and pupal development (Ito et al., 1997; Lee et al., 1999). We practiced picking the GFP-labeled MB NBs, ranging from 14 to 22 μm in size (Fig. 2D), dissociated from larval CNS using the custom-built, single-cell picking device. We established a two-step procedure that allows efficient, clean recovery of most of the 60 or so targeted NBs that can be reliably dissociated from 30 larvae carrying ∼120 marked MB NBs in total (Fig. S2, Movie 1).
We collected over 300 MB NBs isolated ∼50 h after larval hatching (ALH). We explored whether an extra wash step can enhance the purity, and we collected another 300 MB NBs following two rounds of cleaning. We also examined how critical the timing of the procedure is, and we collected another 300 MB NBs that had been left in medium for an additional 2 h after cleaning, a total of 4 h after tissue disruption. We used 100 NBs for each RNA/cDNA preparation, and three replicates were executed for each condition. Using qPCR, we assessed the expression of various genes characteristic of NBs and of potential neuronal and glial contaminants. Compared with the RNA of late larval CNS, we observed substantial enrichment of various known NB genes, including deadpan (dpn) (Bier et al., 1992) and miranda (mira) (Ikeshima-Kataoka et al., 1997; Shen et al., 1997), and minimal expression of neuronal Synaptobrevin (nSyb) (Südhof et al., 1989) and glial reversed polarity (repo) (Xiong et al., 1994) in all conditions of isolated MB NBs (Fig. 2E). This lends support to the purity of the picked NBs. In addition, both qPCR and RNA-seq results showed no detectable effect on the transcriptomes of the isolated MB NBs due to additional cleaning or prolonged incubation (Fig. 2E,F).
Taken together, our 2 h, two-step protocol of single-cell picking is adequate to recover a pure population of rare NBs, freshly dissociated from highly heterogeneous neural tissues, that are apparently maintained in a stable state through the procedure.
Distinguishable transcriptomes of different NBs
Distinct NBs carry out analogous neurogenic programs but yield different lineage-specific neuron types in diverse lineage-characteristic patterns. Transcriptome analysis of discrete NB subsets should reveal the transcriptional networks governing individual NB developmental fates as opposed to general NB programs. Using the lineage-specific NB drivers, we isolated 300 AL and 300 type II NBs at 50 h ALH. We also picked 300 non-selective NBs marked with a pan-NB driver (Awasaki et al., 2014), as a reference pool. Three replicates (100 NBs per RNA/cDNA preparation) were executed for each NB group. We confirmed the fidelity of picking different sets of NBs by qPCR. The qPCR analysis showed selective enrichment of the type I NB-specific gene asense (ase) (Bowman et al., 2008) in the MB and AL NBs and of the type II-specific gene pointed (pnt) (Zhu et al., 2011) in type II NBs, as compared with late larval CNS (Fig. 3A). In further support of minimal contamination or sampling bias, we obtained high replicate consistency in the NB RNA-seq data that unambiguously showed analogous, yet distinguishable, transcriptomes for different NB collections, highlighting their clear distinctions from the transcriptomes of post-mitotic neurons and glia (Fig. 3B, Table S1).
Fig. 3.

Profiling lineage-specific NBs. (A) Enrichment of known NB transcripts in various NB subsets. Note selective expression of asense (ase) and pointed (pnt) in the type I and type II NBs, respectively. Transcript levels were determined by qPCR and normalized to RNA from total larval CNS. Data are represented as mean±s.d. of three trials. One-way ANOVA analysis showed that in type II NBs ase was significantly reduced (*P<0.05) and pnt was significantly enriched (***P<0.001). (B) Correlation analysis of the overall transcriptomes of the MB, AL, type II and all-NBs with neuron and glia data as outliers. Each replicate is shown. (C) Venn diagram showing the numbers of differentially expressed genes (average TPM>10, q-value <0.05) recovered from pairwise transcriptome comparisons of the MB, AL and type II NBs.
Pairwise comparisons among the transcriptomes of the age-matched MB, AL or type II NBs uncovered 585 differentially expressed genes with average transcripts per million (TPM) greater than 10 and q-value <0.05 (see Materials and Methods) (Fig. 3C). About 12% of the differentially expressed genes were annotated as transcription factors (TFs), making the TF class the most over-represented gene class with differential expression among distinct NB subsets (Fig. S3). Nonetheless, only 68 out of 445 TFs with average TPM>10 were recovered as differentially expressed TFs, supporting the presence of shared neurogenic programs.
Abundantly expressed TFs common to diverse NBs
Universal NB TFs should be expressed in the three distinct NB subsets as well as in the non-selective NB pool. A Venn diagram of the expressed TFs among these four groups of NB collections revealed 348 common TFs with average TPM>10 and 70 common TFs with average TPM>100 (Fig. 4A). Sixty-one of the 70 strong common TFs are comparably expressed with less than a 2-fold change (FC<2) in the expression level between different collections (Fig. 4B). However, nine abundant NB TFs (TPM>100) show varied expression levels (FC>2) in the sampled NBs (Fig. 4C). These include: HmgZ, E(spl)mγ-HLH, dm (Myc), apt, E(spl)m8-HLH, dj-1β, E(spl)m7-HLH, CG12911 and tai. The three E(spl) transcripts are selectively lower in MB NBs, which might reflect substantially weaker Notch activities in the MB NBs, as E(spl) genes are well-known Notch downstream targets (Delidakis et al., 2014). Fifty-two of the 70 abundant NB TFs (TPM>100 in all four NB groups) were independently recovered by comparing NB samples with neuron/glia samples using the limma-voom package with the q-value <0.05 to identify 195 NB-enriched TFs in total (Fig. S4).
Fig. 4.
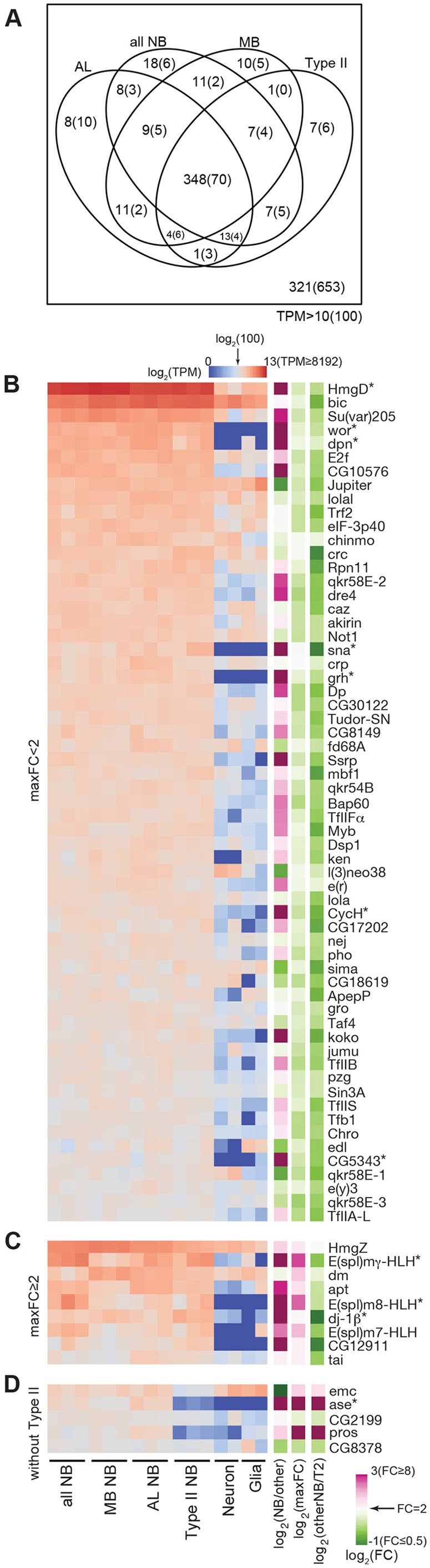
Transcription factors (TFs) abundantly expressed in diverse NBs. (A) Venn diagram showing the numbers of TFs with groupwise average TPM>10 or 100 (parentheses) in the transcriptomes of MB, AL, type II and all NBs. Note that 321 of 784 annotated TFs (combining PANTHER10 and GO) were weakly or not expressed (average TPM<10) in all examined NB pools. (B) Sixty-one TFs with a groupwise average larger than 100 TPM for all groups (all NB, MB NB, AL NB and type II NB) and maximum fold change between groups of <2. TPM values in log2 scale are shown from low (blue) to high (red) with TPM=100 as the middle (white). Shown on the side are maximum fold change (maxFC) in log2 scale and fold change between NBs and neuron/glia (NB/other) or between MB/AL/all NB and type II NB (otherNB/T2) in log2 scale. (C) Similar to A, but with maxFC >2. (D) Similar to A,B, but with type II removed from the calculation. Only additional genes not included in A,B are shown. TFs greatly enriched in NB samples as compared with neuron/glia data (NB/other >10) are indicated by asterisks.
ase and pros, two well-known type I NB genes, were not recovered as universal NB TFs due to minimal expression in type II NBs. Notably, there are five TFs with TPM>100 in all, except type II, NB groups. These additional potentially pan-type I NB TFs include emc, ase, CG2199, pros and CG8378 (Smyd4-4) (Fig. 4C). Of all 75 TFs abundantly expressed in most, if not all, NBs (Fig. 4), 11 [CG5343 (Bug22), CycH, E(spl)m8-HLH, E(spl)mγ-HLH, HmgD, ase, dj-1β, dpn, grh, sna and wor] are greatly enriched in NBs (FC>10) as compared with post-mitotic neurons and glia (Fig. 4, asterisks). Given the known involvement of wor, dpn, grh, sna, ase and pros (not recovered as NB-specific TFs due to significant glia expression) in NB proliferation versus differentiation (Ashraf and Ip, 2001; Cai et al., 2001; Almeida and Bray, 2005; Maurange et al., 2008; Lai et al., 2012; Zhu et al., 2012; Lai and Doe, 2014; Yasugi et al., 2014), these general NB-specific TFs are likely to constitute the transcriptional networks that orchestrate the expression of gene batteries required for NB-characteristic programs, including asymmetric cell division and neuron/glia production.
TFs differentially expressed in distinct NBs
In addition to pan-NB TFs, distinct NBs express different combinations of lineage-specific TFs, as evidenced by the recovery of 68 differentially expressed TFs from the above pairwise transcriptome comparisons among the MB, AL and type II NBs. We do not know the expression patterns of those differentially expressed TFs in the remaining, highly heterogeneous NBs. To reveal TFs that are more specifically enriched or repressed in particular NBs, we next compared a given NB subset with all other NB collections, including the non-selective NB pool. We also compared the non-selective NB pool with the three distinct NB subsets that we sampled. We used the limma-voom package with q-value <0.05 to recover TFs specifically enriched or repressed in the MB NBs, the AL NBs, the type II NBs, and the non-selective NBs (Fig. 5).
Fig. 5.

TFs characteristic of specific NB subsets. TFs selected via comparisons of MB with other NBs (A), AL with other NBs (B), type II with other NBs (C), and all NB with other NBs (D) (q-value <0.05, using limma-voom package and Benjamini-Hochberg correction). TPM values normalized by the average of the most strongly expressed group are shown as a heatmap. Side bars: average TPM and fold change (FC) values in log2 scale. Asterisks indicate the genes for which protein expression pattern was analyzed by immunostaining.
We uncovered seven TFs that were specifically enriched in the MB group comprising a homogenous population of NBs (Fig. 5A). Among the heterogeneous populations of NBs, we identified six different TFs enriched in the AL group (Fig. 5B) and another six TFs enriched in the type II group (Fig. 5C). Furthermore, we detected seven TFs that stood out in the non-selective NB pool (Fig. 5D) despite modest to low TPM values, apparently due to their minimal expressions in the MB, AL and Type II NBs. It is unclear how restricted their expression is in the remaining NBs. We also uncovered eight, two and three TFs that are specifically repressed in the MB, AL and type II NBs, respectively. We did not recover any TF specifically repressed in the non-selective NB pool, consistent with its composition of all diverse NBs.
We examined the expression patterns of five differentially expressed TFs by immunostaining (Fig. 6). We detected Rx expression in all MB NBs (Fig. 6A′, arrows), Mid expression in one of four AL NBs (Fig. 6D′, arrow), and Dr expression in another AL NB (Fig. 6E′, arrow). Notably, Dr is abundant in the progeny of two additional AL lineages (Fig. 6E′, yellow asterisks). We also confirmed that there is no detectable expression of SoxN in the MB NBs (Fig. 6B′, asterisk) but that there is strong SoxN expression in many brain cells, including the progeny of type II lineages (outlined in Fig. 6B′). By contrast, Tll is abundant in the offspring of MB NBs (Kurusu et al., 2009) (Fig. 6C, arrows) and the developing optic lobe (Li et al., 2013), but could not be detected within the AL lineages (Fig. 6C,C′, green). These immunostaining results substantiate the expression of Rx, and not SoxN, in the MB NBs, the absence of Tll in AL NBs, and the expression of Mid and Dr in various AL NBs, which further attests to the molecular heterogeneity of the AL NBs.
Fig. 6.

Endogenous Rx, SoxN, Tll, Mid and Dr expression patterns and effect of SoxN misexpression on Tll expression. Composite (A-F) or single focal plane (A′-F′) confocal images of 50 h ALH larval brains. Green: GAL4-OK107-positive cells in A,B,F or AL lineages in C-E. Magenta: immunostaining for Rx, SoxN, Tll, Mid or Dr. Blue: Dpn in all NBs and INPs of type II lineages. (A,A′) Rx can be detected in MB NBs (arrows), but not in some other NBs (asterisk). (B,B′) SoxN can be detected in type II lineages (dashed outline), but not in MB NBs (asterisk). (C,C′) Tll exists abundantly in MB neurogenic areas (arrows) and the optic lobe region, and weakly in many NBs of unknown identity (dashed outlined), but could not be detected in AL lineages (asterisks). (D,D′) Mid exists in only one AL lineage (arrow). Inset shows another focal plane of the boxed region. (E,E′) Dr can be detected in one AL NB (arrow) and in the offspring of two Dr-negative AL NBs (yellow asterisks). (F,F′) Ectopic SoxN, driven by GAL4-OK107, caused the loss of MB Tll expression, except for just one MB NB area (arrow), as compared with strong Tll expression in all wild-type MB NB areas (arrows in inset). Scale bar: 20 µm.
The repression of otherwise broadly expressed SoxN in the MB NBs is crucial for normal MB neurogenesis. Ectopic expression of SoxN altered MB NB fate as evidenced by gradual loss of Tll expression (Fig. 6F). Consistent with the known requirement of Tll for MB neurogenesis (Kurusu et al., 2009), the SoxN-positive MBs showed reduced neuron numbers and aberrant morphologies (data not shown). This exemplifies the importance of lineage-specific expression or repression of various TFs in programming distinct NBs and their production of lineage-characteristic progenies.
DISCUSSION
Single-cell analysis has been shedding much new light on the development and function of complex tissues. Molecular profiling of single cells will further revolutionize biomedical research. For instance, a large-scale, single-cell RNA-seq study has allowed the molecular classification of cell types in the mouse cortex and hippocampus and revealed that TFs form complex layered codes in diverse cell types (Zeisel et al., 2015). However, it is challenging to capture a particular cell for a single-cell RNA-seq reaction. Moreover, single-cell RNA-seq techniques remain exploratory. One practical solution is RNA-seq of a few ‘identical’ cells obtained from different individuals. In addition to the reliability or otherwise of RNA/cDNA preparation, there are two major concerns over the quality of the sample that can affect the consistency and interpretation of the data. One concern is the purity of the collected cells, since even trace contamination with cells of other identities could have a significant impact when the sample size is small. The other concern is the complexity of sample composition, as heterogeneous target cells might not be adequately sampled if only a few cells are picked. Additional concerns include the well-being of the collected cells, cell type/status-dependent variations in RNA content, and possible contamination with RNAs released from damaged cells during tissue disruption. In summary, absolute accuracy becomes important when dealing with a small number of target cells.
We addressed these concerns by sampling a small number of specific NBs using robotic single-cell picking. Remarkable consistency exists in the RNA-seq data of all MB NB samples collected at the same developmental time (50 h ALH) with three replicates per collection scheme. The high reproducibility (correlation coefficient of 0.928±0.0039, mean±s.d.) demonstrates the fidelity of our single-cell picking device as well as the homogeneity of larval MB NBs. Only six genes (RpL18, RpL23, RpLP1, RpLP2, sna, CG34300) differed significantly in the extra wash condition, and no gene showed a significant difference in the extra 2 h incubation condition based on the criteria of FC>2, TPM>10, q-value <0.05. This suggests that the established 2 h one-wash protocol is well within the time limits for recovering pure target cells by single-cell picking without progressive fate or viability changes. Even the replicates of heterogeneous AL or type II NBs show excellent reproducibility in the transcriptome of the same group (correlation coefficients of 0.94 and 0.95, respectively), indicating no sampling bias in the coverage of the four or eight targeted NBs with collections of 100 NBs.
Pairwise comparisons among various NB subsets revealed 585 genes with average TPM>10 and q-value <0.05 (Fig. 3C). TFs account for ∼12% of the differentially expressed genes. We uncovered a small number of TFs that were strongly enriched in, or absent from, a particular NB subset as compared with other NB subsets and the non-selective NBs. The selective expression or repression of such lineage-specific TFs may modify core NB programs and/or govern the specification of lineage-characteristic offspring. For instance, in the type II lineages, the expression of pnt, but not ase, maintains the NB fate, and the expression of btd, but not pros, promotes the INP cell fate (Bowman et al., 2008; Bayraktar et al., 2010; Zhu et al., 2011; Komori et al., 2014; Xie et al., 2014). In addition, tll is essential for efficient and extended production of MB neurons (Kurusu et al., 2009), and lack of SoxN ensures proper Tll expression in MB NBs and young progenies (Fig. 6F). Moreover, retn and ems have been shown to govern MB and AL neuronal differentiation, respectively (Ditch et al., 2005; Lichtneckert et al., 2008). The expression of multiple lineage-specific TFs in a given NB, possibly as a combinatorial TF code, contrasts with the recent report of otd (oc) as a master lineage cell fate-determining gene in a particular NB lineage (Sen et al., 2014).
Our RNA-seq data also reveal 75 TFs abundantly expressed in most, if not all, NBs. A putative transcriptional network of 28 TFs enriched in ase-positive type I NBs, as compared with their neuronal offspring, has been depicted (Berger et al., 2012). Only seven of the TFs [E(spl)mγ-HLH, wor, dpn, crc, Ssrp, grh and ken] appear on our list of abundantly expressed NB TFs. Six additional genes (mod, Ssb-c31a, klu, CG4570, CG15715 and CG10565) are not annotated as TFs and would otherwise have been included in our list. The remaining 15 previously identified type I NB-characteristic TFs fail to pass our arbitrary abundance threshold of average TPM>100 in the non-selective NB group (Fig. S5); five of these TFs show an average TPM of less than 30. Although they might be significantly enriched in NBs as compared with post-mitotic neurons, these weakly expressed TFs show similarly low levels of expression in all our sampled NB groups and are thus unlikely to play central roles in core NB programs. By contrast, the ∼80 strongly expressed, pan-NB TFs reported here, especially the seven uncharacterized NB-enriched TFs [CG5343, CycH, E(spl)m8-HLH, E(spl)mγ-HLH, HmgD, dj-1β and mod], along with the six well-known NB-specific TFs (ase, dpn, grh, sna, wor and pros), are likely to govern various aspects of core NB programs.
In conclusion, we have established an integrated microscopy system for robotic single-cell picking and demonstrated how it facilitates the identification and prompt recovery of rare target cells from dissociated tissues. The system is ideal for picking genetically marked cells. Three specific subsets of Drosophila NBs, targeted via various intersectional transgenic tactics, were collected for RNA-seq, which revealed the molecular heterogeneities of neural progenitors. Transcriptome analysis of lineage-specific precursors promises to elucidate the gene regulatory networks underlying the development of complex tissues.
MATERIALS AND METHODS
Genetic models for picking lineage-specific NBs
Transgenes used in this study are listed in Table 1. To label MB NBs specifically, we crossed dpnEE>KDRTs-stop-KDRTs>LexA::P65; lexAop-myrGFP female flies with UAS-KD; R41A10-ZpGDBD, R14D11-P65ADZ male flies. To label the four AL (ALad1, ALl1, ALv1 and ALv2) NBs specifically, we crossed dpnEE>KDRTs-stop-KDRTs>LexA::P65; lexAop>FRT-stop-FRT>myrGFP; R44F03-KD female flies with lexAop>FRT-stop-FRT>myrGFP; EMS-GAL4 2.6D, UAS-FLP male flies. To label the eight type II NBs specifically, we crossed dpn>KDRTs-stop-KDRTs>GAL4; UAS-mCD8::GFP (46), UAS-mCD8::GFP (48); stg14-KD, lexAop-myr::tdTomato female flies with R9D11-LexA::P65, 20XUAS-mCD8::GFP; R9D11-GAL80, asense-GAL80 male flies.
Table 1.
Transgenic flies

For ectopic expression of SoxN, GAL4-OK107 was used to drive UAS-SoxN (Overton et al., 2002) throughout MB neurogenesis.
Dissection and trituration of larval CNS
Drosophila larvae hatched within a 2 h time window were cultured at 25°C for 50 h. We dissected out the synchronized CNS in freshly prepared adult hemolymph saline (AHS) buffer (108 mM NaCl, 5 mM KCl, 2 mM CaCl2, 8.2 mM MgCl2, 4 mM NaHCO3, 1 mM NaH2PO4, 5 mM trehalose, 10 mM sucrose, 5 mM HEPES, pH 7.0). As much as possible of the ventral nerve cord was removed. A collection of ∼30 larval brain samples was immediately transferred to an Eppendorf tube (1.5 ml) containing 200 µl AHS with Pronase (1 mg/ml; P5147, Sigma-Aldrich) and Dispase (1 mg/ml; LS02104, Worthington Biochemical Corporation). After tissue digestion for 30 min at 25°C, the enzyme medium was removed, followed by two brief, gentle washes with fresh AHS containing 50 μM AP-5 (76326-31-3, Sigma-Aldrich), 20 μM DNQX (2379-57-9, Sigma-Aldrich), 0.1 μM TTX citrate (1069, Tocris) and 2% FBS. The brain samples were then triturated by gently pipetting in 1 ml AHS through four fire-polished Pasteur pipettes (13-678-20B, Fisher Scientific) with descending pore sizes for ∼20 min. Next, the dissociated cells were centrifuged for 5 min at 49 RCF and the AHS was replaced to minimize any RNA content released from damaged cells during the trituration process. Finally, the dissociated cells were concentrated by centrifuging 5 min at 49 RCF before single-cell picking.
Single-cell picking
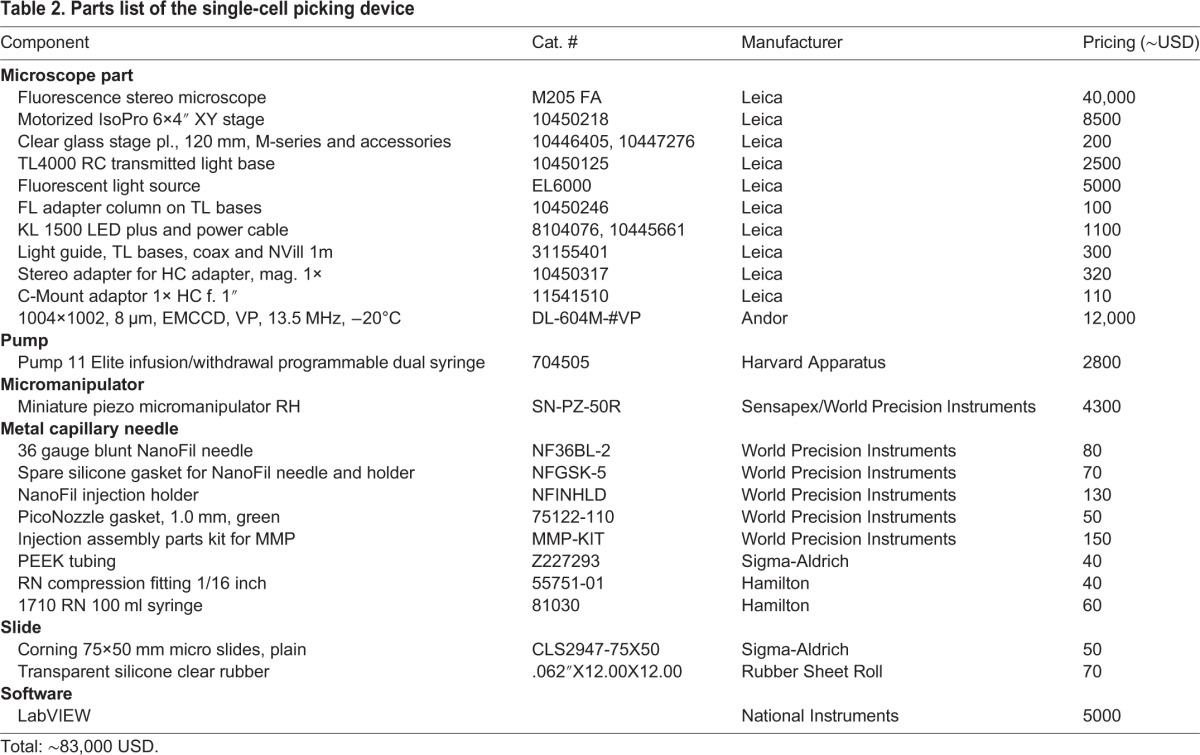
Parts of our custom-built single-cell picking device, including specifications and sources, are listed in Table 2.
Table 2.
Parts list of the single-cell picking device
The 75×50 mm microscope slide was cleaned with double-distilled water and the surface was dried with compressed air (Air'it; 23-022523, Fisher Scientific). A 75×40 mm piece of transparent silicone clear rubber (thickness ∼1.6 mm), carved into two 30×30 mm chambers, was placed on the center of the slide. To reduce adhesion, chamber surfaces were treated with 500 μl 300 μg/ml PLL-PEG [PLL(20)-g[3.5]- PEG(2); SuSoS] for 2 h at room temperature. Each chamber was rinsed twice with AHS then pre-wet by adding 400 μl AHS. Concentrated cells were resuspended in 150 μl AHS then gently added into the center of the pre-wet left chamber. A final volume of 1300 μl AHS was added to each chamber. The two-step picking procedure was able to collect ∼60 NBs in 2 h (Fig. S2). Picked cells were released directly into a PCR tube (PCR-02-C, Corning Life Sciences-Axygen Scientific), and 9.5 μl PicoPure extraction buffer (12317-03, Life Technologies) was immediately added. The mixture was incubated at 42°C for 30 min before storing at −80°C.
For the extra wash experiment, pure target cells were transferred to a fresh chamber, ten cells at a time, using a new capillary needle. After all the pure target cells were transferred, ten target cells were collected using another capillary needle and then released directly into a PCR tube for RNA extraction and storage. For the extra 2 h incubation experiment, pure target cells were incubated in the collection chamber for 2 h after removing non-target cells.
RNA/cDNA preparation and qPCR
Each PCR tube containing approximately ten cells was stored at −80°C until RNA preparation and amplification. Approximately 100 NBs of the identical experimental condition were combined from ten tubes and used as an input for RNA extraction with the PicoPure RNA Isolation Kit (KIT0204, Life Technologies). The RNA solution in 11 μl elution buffer (Life Technologies) was concentrated to 4 μl with a SpeedVac. The quality of RNA extracted from ∼100 NBs was examined on a bioanalyzer using the RNA 6000 Pico Kit (5067-1513, Agilent), with a yield of ∼500 pg (Fig. S6). After adding 1 μl ERCC RNA Spike-in Mix (1/100,000 of the original concentration; 4456740, Life Technologies), the RNA was immediately converted to cDNA and underwent further amplification with the Ovation RNA-Seq System V2 (7102-A01, NuGEN). The final cDNA was dissolved in 30 μl TE buffer (75793, Affymetrix). Total RNA from larval CNS was isolated by TRIzol purification (15596-026, Invitrogen).
Before cDNA fragmentation, qPCR was performed to determine the expression levels of known NB-specific, neuron-specific and glia-specific genes, as a preliminary quality control. For each gene, at least three qPCR primers, either as suggested in http://www.flyrnai.org/FlyPrimerBank or as used in previous publications, were screened through standard curve analysis and the best primers were chosen based on their efficiency and specificity. For qPCR, 2.5 ng cDNA was used for inputs, which is within the best range of DNA input suggested by the standard curve analysis.
After confirming cDNA quality, ∼2.25 μg cDNA from each sample was fragmented by ultrasonicator (LE220, Covaris), followed by final library construction with the Ovation Rapid DR Multiplex System 1-96 kit (0328-96, NuGEN). Each of the final fragmented cDNA libraries was dissolved in 9 μl buffer EB (19086, Qiagen) and sequenced (single-end 100 bp read) using the HiSeq 2500 system (Illumina).
RNA-seq and transcriptome analysis
A total of 778 million reads (average 43.2 million reads per sample) were obtained. The FASTQ data were first processed with cutadapt (Martin, 2011) to remove Illumina adaptor and NuGEN SPIA adaptor. Then, reads mapping to ribosomal and other abundant or low complexity sequences (polyA, polyC, phiX, mitochondrial sequences) were removed using Bowtie 2 (Langmead and Salzberg, 2012). On average, 77.2% of the reads were removed at this stage. The remaining reads were then mapped to UCSC dm3 genome using STAR (Dobin et al., 2013). On average, 22.3% of the initial reads were mapped to dm3.
CPM (counts per million fragments mapped) and FPKM (fragments per kilobase of exon per million fragments mapped) were calculated from the mapped reads using HTSeq (Anders et al., 2015) supplied with Illumina iGenome UCSC dm3 gene annotation gtf. FPKM values were converted to TPM (transcripts per million) for downstream analysis (Table S1).
For differential expression analysis (Fig. 3C, Fig. 5, Fig. S4), the limma-voom package (Law et al., 2014) was used as supplied with CPM filtered with the criteria that at least four samples should have CPM>1 (9968 selected out of 15340). q-values were calculated using the Benjamini-Hochberg method.
As TFs we used a combined list of genes annotated in Gene Ontology (term: GO:0001071) of QuickGo (Binns et al., 2009) and genes annotated as ‘transcription factor' in PANTHER (Mi et al., 2013). PANTHER classification was used for the enrichment analysis in Fig. S3.
RNA-seq data are available in NCBI Gene Expression Omnibus with accession number GSE71104.
Immunohistochemistry and confocal imaging
Brain tissues dissected from larvae at a specific stage were fixed with 2% paraformaldehyde and immunostained as described previously (Lin et al., 2012). The following primary antibodies were used: rabbit anti-GFP 1:1500 (A11122, Life Technologies), rat anti-GFP 1:500 (GF090R, Nacalai), rabbit anti-DsRed 1:500 (632496, Clontech), mouse nc82 mAb 1:100 (Developmental Studies Hybridoma Bank), rabbit anti-SoxN 1:400 (gift from S. Crews, University of North Carolina at Chapel Hill), guinea pig anti-Tll 1:500 (gift from C. Desplan, New York University), rabbit anti-Rx 1:300, rabbit anti-Mid 1:1000 and rabbit anti-Dr 1:500 (gifts from H. Lacin, HHMI). Corresponding fluorescent secondary antibodies (1:500) were purchased from Life Technologies. Images were collected on a Zeiss LSM 710 confocal microscope.
Acknowledgements
We thank the Janelia Neuro-Seq Project Team for essential technical support; Dr Haluk Lacin for Rx, Mid and Dr antibodies; Dr Steven Russell for UAS-SoxN and SoxN antibody; Dr Stephen Crews for SoxN antibody; Dr Claude Desplan for Tll antibody; R. Miyares for input and critical reading of the manuscript; and C. Sullivan for administrative support.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
C.-P.Y., Z.L. and T.L. designed the study. C.-P.Y., K.S. and Z.L. performed experiments. C.-C.F. constructed the cell-picking device with suggestions from L.P.L. and K.S. L.-Y.L. contributed to fly brain dissection and cell dissociation. Q.R. engineered the MB NB-specific driver. X.Y. made various DNA constructs. C.-P.Y., Z.L., K.S. and T.L. analyzed the data and wrote the manuscript. C.-P.Y., C.-C.F., K.S. and Z.L. contributed equally to this work.
Funding
This work was supported by the Howard Hughes Medical Institute. Deposited in PMC for release after 6 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.129163/-/DC1
References
- Almeida M. S. and Bray S. J. (2005). Regulation of post-embryonic neuroblasts by Drosophila Grainyhead. Mech. Dev. 122, 1282-1293. 10.1016/j.mod.2005.08.004 [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P. T. and Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166-169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf S. I. and Ip Y. T. (2001). The Snail protein family regulates neuroblast expression of inscuteable and string, genes involved in asymmetry and cell division in Drosophila. Development 128, 4757-4767. [DOI] [PubMed] [Google Scholar]
- Awasaki T., Kao C.-F., Lee Y.-J., Yang C.-P., Huang Y., Pfeiffer B. D., Luan H., Jing X., Huang Y.-F., He Y. et al. (2014). Making Drosophila lineage–restricted drivers via patterned recombination in neuroblasts. Nat. Neurosci. 17, 631-637. 10.1038/nn.3654 [DOI] [PubMed] [Google Scholar]
- Bayraktar O., Boone J. Q., Drummond M. L. and Doe C. Q. (2010). Drosophila type II neuroblast lineages keep Prospero levels low to generate large clones that contribute to the adult brain central complex. Neural Dev. 5, 26 10.1186/1749-8104-5-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello B. C., Izergina N., Caussinus E. and Reichert H. (2008). Amplification of neural stem cell proliferation by intermediate progenitor cells in Drosophila brain development. Neural Dev. 3, 5 10.1186/1749-8104-3-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C., Harzer H., Burkard T. R., Steinmann J., van der Horst S., Laurenson A.-S., Novatchkova M., Reichert H. and Knoblich J. A. (2012). FACS purification and transcriptome analysis of drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep. 2, 407-418. 10.1016/j.celrep.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E., Vaessin H., Younger-Shepherd S., Jan L. Y. and Jan Y. N. (1992). deadpan, an essential pan-neural gene in Drosophila, encodes a helix-loop-helix protein similar to the hairy gene product. Genes Dev. 6, 2137-2151. 10.1101/gad.6.11.2137 [DOI] [PubMed] [Google Scholar]
- Binns D., Dimmer E., Huntley R., Barrell D., O'Donovan C. and Apweiler R. (2009). QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics 25, 3045-3046. 10.1093/bioinformatics/btp536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone J. Q. and Doe C. Q. (2008). Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev. Neurobiol. 68, 1185-1195. 10.1002/dneu.20648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman S. K., Rolland V., Betschinger J., Kinsey K. A., Emery G. and Knoblich J. A. (2008). The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell 14, 535-546. 10.1016/j.devcel.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Chia W. and Yang X. (2001). A family of snail-related zinc finger proteins regulates two distinct and parallel mechanisms that mediate Drosophila neuroblast asymmetric divisions. EMBO J. 20, 1704-1714. 10.1093/emboj/20.7.1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delidakis C., Monastirioti M. and Magadi S. S. (2014). E(spl): genetic, developmental, and evolutionary aspects of a group of invertebrate Hes proteins with close ties to Notch signaling. Curr. Top. Dev. Biol. 110, 217-262. 10.1016/B978-0-12-405943-6.00006-3 [DOI] [PubMed] [Google Scholar]
- Ditch L. M., Shirangi T., Pitman J. L., Latham K. L., Finley K. D., Edeen P. T., Taylor B. J. and McKeown M. (2005). Drosophila retained/dead ringer is necessary for neuronal pathfinding, female receptivity and repression of fruitless independent male courtship behaviors. Development 132, 155-164. 10.1242/dev.01568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15-21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig L. C., Woodworth M. B., Galazo M. J., Padmanabhan H. and Macklis J. D. (2013). Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 14, 755-769. 10.1038/nrn3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem C. C. F. and Knoblich J. A. (2012). Drosophila neuroblasts: a model for stem cell biology. Development 139, 4297-4310. 10.1242/dev.080515 [DOI] [PubMed] [Google Scholar]
- Ikeshima-Kataoka H., Skeath J. B., Nabeshima Y.-i., Doe C. Q. and Matsuzaki F. (1997). Miranda directs Prospero to a daughter cell during Drosophila asymmetric divisions. Nature 390, 625-629. 10.1038/37641 [DOI] [PubMed] [Google Scholar]
- Ito K., Awano W., Suzuki K., Hiromi Y. and Yamamoto D. (1997). The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development 124, 761-771. [DOI] [PubMed] [Google Scholar]
- Jefferis G. S. E., Marin E. C., Stocker R. F. and Luo L. (2001). Target neuron prespecification in the olfactory map of Drosophila. Nature 414, 204-208. 10.1038/35102574 [DOI] [PubMed] [Google Scholar]
- Jenett A., Rubin G. M., Ngo T.-T., Shepherd D., Murphy C., Dionne H., Pfeiffer B. D., Cavallaro A., Hall D., Jeter J. et al. (2012). A GAL4-driver line resource for Drosophila neurobiology. Cell Rep. 2, 991-1001. 10.1016/j.celrep.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori H., Xiao Q., Janssens D. H., Dou Y. and Lee C.-Y. (2014). Trithorax maintains the functional heterogeneity of neural stem cells through the transcription factor buttonhead. eLife 3, e03502 10.7554/eLife.03502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurusu M., Maruyama Y., Adachi Y., Okabe M., Suzuki E. and Furukubo-Tokunaga K. (2009). A conserved nuclear receptor, Tailless, is required for efficient proliferation and prolonged maintenance of mushroom body progenitors in the Drosophila brain. Dev. Biol. 326, 224-236. 10.1016/j.ydbio.2008.11.013 [DOI] [PubMed] [Google Scholar]
- Lai S.-L. and Doe C. Q. (2014). Transient nuclear Prospero induces neural progenitor quiescence. eLife 3, e03363 10.7554/eLife.03363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai S.-L., Awasaki T., Ito K. and Lee T. (2008). Clonal analysis of Drosophila antennal lobe neurons: diverse neuronal architectures in the lateral neuroblast lineage. Development 135, 2883-2893. 10.1242/dev.024380 [DOI] [PubMed] [Google Scholar]
- Lai S.-L., Miller M. R., Robinson K. J. and Doe C. Q. (2012). The Snail family member Worniu is continuously required in neuroblasts to prevent Elav-induced premature differentiation. Dev. Cell 23, 849-857. 10.1016/j.devcel.2012.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. and Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C. W., Chen Y., Shi W. and Smyth G. K. (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T., Lee A. and Luo L. (1999). Development of the Drosophila mushroom bodies: sequential generation of three distinct types of neurons from a neuroblast. Development 126, 4065-4076. [DOI] [PubMed] [Google Scholar]
- Li X., Erclik T., Bertet C., Chen Z., Voutev R., Venkatesh S., Morante J., Celik A. and Desplan C. (2013). Temporal patterning of Drosophila medulla neuroblasts controls neural fates. Nature 498, 456-462. 10.1038/nature12319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtneckert R., Nobs L. and Reichert H. (2008). Empty spiracles is required for the development of olfactory projection neuron circuitry in Drosophila. Development 135, 2415-2424. 10.1242/dev.022210 [DOI] [PubMed] [Google Scholar]
- Lin S., Lai S.-L., Yu H.-H., Chihara T., Luo L. and Lee T. (2010). Lineage-specific effects of Notch/Numb signaling in post-embryonic development of the Drosophila brain. Development 137, 43-51. 10.1242/dev.041699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Kao C.-F., Yu H.-H., Huang Y. and Lee T. (2012). Lineage analysis of Drosophila lateral antennal lobe neurons reveals notch-dependent binary temporal fate decisions. PLoS Biol. 10, e1001425 10.1371/journal.pbio.1001425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Marin E. C., Yang C.-P., Kao C.-F., Apenteng B. A., Huang Y., O'Connor M. B., Truman J. W. and Lee T. (2013). Extremes of lineage plasticity in the Drosophila brain. Curr. Biol. 23, 1908-1913. 10.1016/j.cub.2013.07.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan H., Peabody N. C., Vinson C. R. and White B. H. (2006). Refined spatial manipulation of neuronal function by combinatorial restriction of transgene expression. Neuron 52, 425-436. 10.1016/j.neuron.2006.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17.1, 10-12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Maurange C., Cheng L. and Gould A. P. (2008). Temporal transcription factors and their targets schedule the end of neural proliferation in Drosophila. Cell 133, 891-902. 10.1016/j.cell.2008.03.034 [DOI] [PubMed] [Google Scholar]
- Mi H., Muruganujan A. and Thomas P. D. (2013). PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377-D386. 10.1093/nar/gks1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagoshi E., Sugino K., Kula E., Okazaki E., Tachibana T., Nelson S. and Rosbash M. (2010). Dissecting differential gene expression within the circadian neuronal circuit of Drosophila. Nat. Neurosci. 13, 60-68. 10.1038/nn.2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. W., Harris J. A., Ng L., Winslow B., Cain N., Mihalas S., Wang Q., Lau C., Kuan L., Henry A. M. et al. (2014). A mesoscale connectome of the mouse brain. Nature 508, 207-214. 10.1038/nature13186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaty B. W., Sugino K. and Nelson S. B. (2011). Cell type-specific transcriptomics in the brain. J. Neurosci. 31, 6939-6943. 10.1523/JNEUROSCI.0626-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton P. M., Meadows L. A., Urban J. and Russell S. (2002). Evidence for differential and redundant function of the Sox genes Dichaete and SoxN during CNS development in Drosophila. Development 129, 4219-4228. [DOI] [PubMed] [Google Scholar]
- Pfeiffer B. D., Ngo T.-T. B., Hibbard K. L., Murphy C., Jenett A., Truman J. W. and Rubin G. M. (2010). Refinement of tools for targeted gene expression in Drosophila. Genetics 186, 735-755. 10.1534/genetics.110.119917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S., Cao D., Choudhary R., Biagini S., Wang J. W., Reichert H. and VijayRaghavan K. (2014). Genetic transformation of structural and functional circuitry rewires the Drosophila brain. eLife 3, e04407 10.7554/eLife.04407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C.-P., Jan L. Y. and Jan Y. N. (1997). Miranda is required for the asymmetric localization of Prospero during mitosis in Drosophila. Cell 90, 449-458. 10.1016/S0092-8674(00)80505-X [DOI] [PubMed] [Google Scholar]
- Südhof T. C., Baumert M., Perin M. S. and Jahn R. (1989). A synaptic vesicle membrane protein is conserved from mammals to Drosophila. Neuron 2, 1475-1481. 10.1016/0896-6273(89)90193-1 [DOI] [PubMed] [Google Scholar]
- Truman J. W. and Bate M. (1988). Spatial and temporal patterns of neurogenesis in the central nervous system of Drosophila melanogaster. Dev. Biol. 125, 145-157. 10.1016/0012-1606(88)90067-X [DOI] [PubMed] [Google Scholar]
- Urbach R. and Technau G. M. (2003). Molecular markers for identified neuroblasts in the developing brain of Drosophila. Development 130, 3621-3637. 10.1242/dev.00533 [DOI] [PubMed] [Google Scholar]
- Urbach R. and Technau G. M. (2004). Neuroblast formation and patterning during early brain development in Drosophila. Bioessays 26, 739-751. 10.1002/bies.20062 [DOI] [PubMed] [Google Scholar]
- Wang Y.-C., Yang J. S., Johnston R., Ren Q., Lee Y.-J., Luan H., Brody T., Odenwald W. F. and Lee T. (2014). Drosophila intermediate neural progenitors produce lineage-dependent related series of diverse neurons. Development 141, 253-258. 10.1242/dev.103069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Li X., Zhang X., Mei S., Li H., Urso A. and Zhu S. (2014). The Drosophila Sp8 transcription factor Buttonhead prevents premature differentiation of intermediate neural progenitors. eLife 3, e03596 10.7554/eLife.03596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W. C., Okano H., Patel N. H., Blendy J. A. and Montell C. (1994). repo encodes a glial-specific homeo domain protein required in the Drosophila nervous system. Genes Dev. 8, 981-994. 10.1101/gad.8.8.981 [DOI] [PubMed] [Google Scholar]
- Yasugi T., Fischer A., Jiang Y., Reichert H. and Knoblich J. A. (2014). A regulatory transcriptional loop controls proliferation and differentiation in Drosophila neural stem cells. PLoS ONE 9, e97034 10.1371/journal.pone.0097034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.-H., Chen C.-H., Shi L., Huang Y. and Lee T. (2009). Twin-spot MARCM to reveal the developmental origin and identity of neurons. Nat. Neurosci. 12, 947-953. 10.1038/nn.2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.-H., Kao C.-F., He Y., Ding P., Kao J.-C. and Lee T. (2010). A complete developmental sequence of a Drosophila neuronal lineage as revealed by twin-spot MARCM. PLoS Biol. 8, e1000461 10.1371/journal.pbio.1000461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.-H., Awasaki T., Schroeder M. D., Long F., Yang J. S., He Y., Ding P., Kao J.-C., Wu G. Y.-Y., Peng H. et al. (2013). Clonal development and organization of the adult Drosophila central brain. Curr. Biol. 23, 633-643. 10.1016/j.cub.2013.02.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A., Munoz-Manchado A. B., Codeluppi S., Lonnerberg P., La Manno G., Jureus A., Marques S., Munguba H., He L., Betsholtz C. et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138-1142. 10.1126/science.aaa1934 [DOI] [PubMed] [Google Scholar]
- Zhu S., Barshow S., Wildonger J., Jan L. Y. and Jan Y. N. (2011). Ets transcription factor Pointed promotes the generation of intermediate neural progenitors in Drosophila larval brains. Proc. Natl. Acad. Sci. USA 108, 20615-20620. 10.1073/pnas.1118595109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Wildonger J., Barshow S., Younger S., Huang Y. and Lee T. (2012). The bHLH repressor Deadpan regulates the self-renewal and specification of Drosophila larval neural stem cells independently of Notch. PLoS ONE 7, e46724 10.1371/journal.pone.0046724 [DOI] [PMC free article] [PubMed] [Google Scholar]