

abstract
The cancer-testis antigen NY-ESO-1 is expressed by many solid tumors and has limited expression by mature somatic tissues, making it a highly attractive target for tumor immunotherapy. Targeting NY-ESO-1 using engineered T cells has demonstrated clinical efficacy in the treatment of some adult tumors. Neuroblastoma is a significant cause of cancer mortality in children, and is a tumor type shown to be responsive to immunotherapies. We evaluated a large panel of primarily resected neuroblastoma samples and demonstrated that 23% express NY-ESO-1. After confirming antigen-specific activity of T cells genetically engineered to express an NY-ESO-1 directed high-affinity transgenic T cell receptor in vitro, we performed xenograft mouse studies assessing the efficacy of NY-ESO-1-targeted T cells in both localized and disseminated models of neuroblastoma. Disease responses were monitored by tumor volume measurement and in vivo bioluminescence. After delivery of NY-ESO-1 transgenic TCR T cells, we observed significant delay of tumor progression in mice bearing localized and disseminated neuroblastoma, as well as enhanced animal survival. These data demonstrate that NY-ESO-1 is an antigen target in neuroblastoma and that targeted T cells represent a potential therapeutic option for patients with neuroblastoma.
Keywords: Cell therapy, immunotherapy, neuroblastoma, NY-ESO-1, transgenic TCR
Abbreviations
- CAR
chimeric antigen receptor
- CT
cancer testes
- tTCR
transgenic T cell receptor
Introduction
A significant barrier to the development and application of immune-based therapies targeting cancer is the identification of appropriate tumor antigens. Both CD20 and CD19 have proven to be successful antigen targets for B cell malignancies,1,2 however, one of the primary side effects of these therapies is B cell aplasia. While this toxicity can be managed medically, many solid tumor antigens are expressed by normal and indispensible somatic tissues, making the search for appropriate antigen targets difficult. In the late 1990s, analysis of esophageal squamous cell carcinoma led to the identification of NY-ESO-1, a protein normally expressed only in fetal and testicular tissue but aberrantly expressed by some solid malignancies.3 This added NY-ESO-1 to a growing list of molecules expressed appropriately in the germ line and abnormally by some cancers. These molecules were termed “cancer/testis” antigens,4 and are capable of serving as targets for antigen-directed immune therapies.5 Despite initial optimism, trials of vaccination against NY-ESO-1 to induce antitumor immunity have proven largely unsuccessful, and to date have not yet resulted in significant antitumor efficacy.6 Deletion of high-affinity clones directed against NY-ESO-1 during thymic selection and suppression of auto-reactive T cells by peripheral tolerance mechanisms may account for the poor antitumor activity observed in these trials.
One method to overcome the limited antitumor activity of endogenous T cells is infusion of genetically-engineered high-affinity T cells re-directed to tumor antigens. The two primary antigen receptors used to re-target T cells are chimeric antigen receptors (CARs) and transgenic T cell receptors (tTCRs). CAR therapy7 has recently demonstrated significant success in targeting B cell leukemias,2,8-11 and trials targeting solid tumors are underway.12-14 While CARs have great potential as therapeutic agents in cancer immunotherapy, they are limited in their ability to only recognize cell-surface molecules. On the contrary, tTCRs have the ability to identify any processed antigen that is presented by the major histocompatibility complex (MHC), thus greatly expanding the list of possible targets. In vitro studies have demonstrated that cells engineered with endogenously-occurring NY-ESO-1 TCRs have activity against NY-ESO-1-expressing melanoma and non-melanoma cell lines.15 In a recent clinical trial employing NY-ESO-1-directed T cells, engineered cells bearing high-affinity tTCRs were delivered to patients with melanoma and synovial cell carcinoma. Nearly half of patients in this study demonstrated objective clinical responses, highlighting the potential of tTCR T cells in treating established solid tumors.16
Neuroblastoma is the most common extra-cranial pediatric solid tumor. Derived from neuro-endocrine tissue of the sympathetic nervous system, it accounts for 9% of cancer diagnoses and 15% of cancer deaths in children.17 Current standard of care for high-risk disease consists of chemotherapy, surgery, consolidation chemotherapy, stem-cell transplant, tumor-directed radiation, and finally antibody-based therapy. This exhaustive regimen yields a three year event-free survival from diagnosis of only ˜45% of patients.18,19 In addition, the outcome for relapsed neuroblastoma is very poor, with a current achievable goal of short-term disease control and very few patients who achieve longer-term remissions. Improved outcomes for this disease will require incorporation of further innovative therapeutic strategies.
In this study, we established that NY-ESO-1 is a potential antigenic target in neuroblastoma. Our recent clinical experiences2,9 using engineered T cells to target CD19+ tumors have been successful in large part due to a robust, bead-based cell manufacturing process which produces highly effective anti-tumor T cells capable of significant in vivo expansion and persistence for as long as three years.20 Previous data have demonstrated the superiority of high-affinity TCRs in targeting NY-ESO-1,21 and combining our ex vivo cell manufacturing process and a high-affinity HLA-A*02 restricted TCR recognizing the peptide NY-ESO-1157-165 (SLLMWITQC), we demonstrated antigen-specific T-cell activity against NY-ESO-1+ neuroblastomas in vitro. Using a well-characterized bioluminescent xenograft animal model system,22,23 we evaluated the efficacy of these tTCR cells in two in vivo models of neuroblastoma. We demonstrated that these cells were able to slow the progression of both local and disseminated disease, and significantly enhanced animal survival. Together, these data suggest that cells engineered to express tTCRs targeting NY-ESO-1 are a viable therapeutic option for patients with neuroblastoma.
Results
NY-ESO-1 is an antigenic target in neuroblastoma
We first sought to assess NY-ESO-1 expression in tumor biopsies from our patient population at the Children's Hospital of Philadelphia to evaluate this molecule as a relevant immunotherapy target in neuroblastoma. We evaluated a panel of 187 neuroblastoma tumor samples from 165 patients, and of 124 evaluable tumors we found that ˜23% stained positively for NY-ESO-1, with positivity defined as ≥10% of cells expressing target based on immunohistochemical staining, and overall intensity of staining quantified as ≥1 on a 0-3 scale (Table 1) (NY-ESO-1 score was calculated by multiplying % positive with the intensity score). Using immunohistochemical staining, expression of NY-ESO-1 was observed in both the nucleus and cytoplasm. Examining the pathological characteristics of these tumors, 23/28 (82%) NY-ESO-1+ samples were found to be poorly differentiated and/or of unfavorable histology. Additionally, the samples varied from low to high-risk tumors based on the International Neuroblastoma Staging System (INSS) risk score. Similarly, these tumors were found to vary in MYCN amplification status, disease location and patient age at diagnosis.
Table 1.
Profile of NY-ESO-1+ neuroblastoma patient tumors. Resected specimens from the Center for Childhood Cancer Research at the Children's Hospital of Philadelphia were examined histologically, and NY-ESO-1-expressing tumor profiles are represented. These tumors vary widely in histology, grade and overall risk score, and represent ˜23% of all neuroblastomas in our cancer center tumor bank. NY-ESO intensity was graded on a scale of 0-3, and overall score was calculated my multiplying % positive with intensity score.
| Patient ID | Age (days) | Histology | Grade | INSS Risk | % of cells NY-ESO-1+ | NY-ESO-1 intensity | NY-ESO-1 score |
|---|---|---|---|---|---|---|---|
| UPN 6 | 1747 | Unfavorable | Poorly differentiated | Low | 20 | 1 | 20 |
| UPN 7 | 154 | Unfavorable | Poorly differentiated | Low | 20 | 1 | 20 |
| UPN 9 | 804 | Unfavorable | Poorly differentiated | Low | 50 | 3 | 150 |
| UPN 10 | 355 | Unfavorable | Poorly differentiated | Low | 60 | 3 | 180 |
| UPN 16 | 317 | Favorable | Poorly differentiated | Low | 20 | 1 | 20 |
| UPN 30 | 532 | Favorable | Differentiating | Low | 10 | 1 | 10 |
| UPN 31 | 124 | Favorable | Poorly differentiated | Intermediate | 20 | 2 | 40 |
| UPN 41 | 573 | Favorable | Differentiating | Low | 20 | 1 | 20 |
| UPN 120 | 401 | Unfavorable | Poorly differentiated | High | 50 | 1 | 50 |
| UPN 121 | 1936 | Unfavorable | N/A | Low | 70 | 3 | 210 |
| UPN 123 | 709 | Unfavorable | N/A | Low | 60 | 2 | 120 |
| UPN 129 | 10 | Unfavorable | Undifferentiated | High | 20 | 1 | 20 |
| UPN 131 | 1467 | Unfavorable | Poorly differentiated | High | 20 | 1 | 20 |
| UPN 133 | 3624 | Unfavorable | Differentiating | High | 20 | 1 | 20 |
| UPN 153 | 3871 | Unfavorable | Differentiating | High | 40 | 1 | 40 |
| UPN 169 | 4233 | Unfavorable | Differentiating | High | 90 | 2 | 180 |
| UPN 170 | 342 | Favorable | Poorly differentiated | Low | 40 | 2 | 80 |
| UPN 173 | 19 | Favorable | Poorly differentiated | Low | 10 | 1 | 10 |
| UPN 185 | 101 | Favorable | Poorly differentiated | Intermediate | 80 | 1 | 80 |
| UPN 187 | 1155 | Favorable | Differentiating | Low | 70 | 2 | 140 |
| UPN 241 | 75 | Favorable | Poorly differentiated | Low | 50 | 1 | 50 |
| UPN 244 | 5229 | N/A | Differentiating | Intermediate | 60 | 1 | 60 |
| UPN 245 | 173 | Favorable | Poorly differentiated | Intermediate | 10 | 1 | 10 |
| UPN 247 | 611 | Favorable | Differentiating | Low | 80 | 1 | 80 |
| UPN 267 | 40 | Favorable | Poorly differentiated | Low | 10 | 1 | 10 |
| UPN 288 | 1064 | Unfavorable | Poorly differentiated | High | 20 | 1 | 20 |
| UPN 292 | 533 | Unfavorable | Poorly differentiated | Low | 10 | 1 | 10 |
| UPN 299 | 32 | Favorable | Poorly differentiated | Intermediate | 10 | 1 | 10 |
NY-ESO-1+ neuroblastomas stimulate T cell cytotoxicity
With a validated antigenic target in our pediatric cancer population, we then assessed the efficacy of NY-ESO-1-directed T cells against neuroblastoma in vitro. While transgenic expression of NY-ESO-1 by genetic engineering of neuroblastoma cell lines would create viable antigen targets for assay development, this method is likely to result in supra-physiologic target expression, and thus risk potential over-presentation of antigen as compared to endogenous presentation seen in actual tumor cells. To more closely capture physiologic expression and presentation, we examined the endogenous expression of NY-ESO-1 in our neuroblastoma cancer cell lines. Evaluation of several cell lines as assessed by qRT-PCR revealed one with robust NY-ESO-1 expression (NB16), one with moderate expression (SK-NAS), and one with undetectable NY-ESO-1 levels (SY5Y) (Table 2), thus giving us neuroblastoma lines expressing a range of NY-ESO-1 to test. Our tTCR targeting NY-ESO-1 is HLA-A*02-restricted. Thus, to allow for TCR recognition of presented antigen, these neuroblastoma cell lines were engineered to express HLA-A2 using lentiviral vector engineering, creating sublines NB16-A2, SK-NAS-A2 and SY5Y-A2. This engineering strategy allowed unhindered antigen presentation, without driving over-presentation.
Table 2.
NY-ESO-1 expression profile of neuroblastoma cell lines. Quantitative RT-PCR evaluation of NY-ESO-1 expression in neuroblastoma cell lines is demonstrated. Harvested mRNA was evaluated by RT-PCR, and relative expression quotients (RQ) calculated using an internal control gene (GUS B) and the ΔΔCt method for expression standardization. As shown, SY5Y exhibit undetectable (ND, not detected) NY-ESO-1 transcript levels, whereas SKNAS and NB16 cell lines have progressively higher levels of transcript
| Sample | GUS B Ct | NY-ESO-1 Ct | NY-ESO-1 RQ |
|---|---|---|---|
| SY5Y | 22.91 | ND | ND |
| SKNAS | 23.10 | 23.50 | 0.3839 |
| NB16 | 22.57 | 22.28 | 0.6217 |
T cells were engineered to express tTCRs and were combined in vitro with HLA-A2+ neuroblastoma cell lines. T-cell activation and degranulation was assessed by expression of CD107a, a marker of cytotoxic T-cell function,24 after 4 hours in co-culture with target cells (Fig. 1). Not all T cells in the co-culture expressed the tTCR, and thus the tTCR-negative cells served as controls to evaluate antigen-driven degranulation. To quantify the degree of antigen-specific degranulation, we developed a metric to assess the specificity of surface CD107a expression, termed the “degranulation ratio.” This is a ratio of cells expressing the transgenic receptor that demonstrated degranulation (tTCR+CD107a+, antigen-dependent degranulation) compared to cells that didn't express the transgenic receptor that demonstrated degranulation (tTCR−CD107a+, antigen-independent degranulation), and provides a method to assess tTCR-dependent T-cell activation while controlling for non-specific activation across groups.
Figure 1.
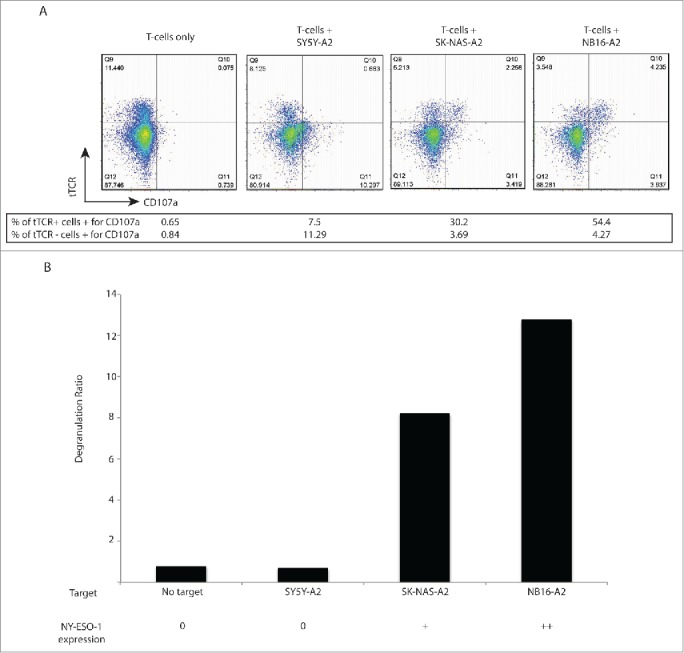
NY-ESO-1 tTCR cells degranulate in response to NY-ESO-1+ tumors. T cells transduced with engineered transgenic T cell receptor (tTCR) targeting NY-ESO-1 were incubated with target neuroblastoma cells and activation was measured by CD107a upregulation. (A) Flow cytometry plots demonstrating increasing degranulation with increasing NY-ESO-1 expression. Percentages shown are percent of tTCR+ cells expressing CD107a / total tTCR+ cells (upper number, reflecting antigen-specific degranulation) and percent of tTCR− cells expressing CD107a / total tTCR− cells (bottom number, reflecting non-specific degranulation). (B) Bar graph representing the data shown in Figure 1A. The degranulation ratio represents degranulation of tTCR+ cells relative to tTCR− cells after incubation with neuroblastoma cell lines, thus controlling for non-specific degranulation.
T cells incubated without target cells demonstrated <1% CD107a expression, independent of tTCR expression (degranulation ratio 0.774). tTCR+ cells incubated with SY5Y-A2 (no NY-ESO-1) showed 7.5% degranulation, with a degranulation ratio of 0.664. tTCR+ cells incubated with SK-NAS-A2 (moderate NY-ESO-1) showed 30.2% degranulation, and a degranulation ratio of 8.18. Finally, tTCR+ cells incubated with NB16-A2 (high NY-ESO-1) showed 54.4% degranulation, with a degranulation ratio of 12.74 (Fig. 1). The results of this sensitive assay demonstrate that varying levels of NY-ESO-1 expression may result in varying levels of T-cell activation in this short time period. Additionally, we demonstrate that tTCR T cells exhibit cytolytic activity against only NY-ESO-1-positive targets, and not against NY-ESO-1-negative HLA-A2+ targets, confirming selective antigen-driven T-cell activity.
NY-ESO-1 directed T cells control localized tumors in mice
After confirming in vitro activity, we next assessed the immunologic activity of these anticancer immune cells in vivo. We first assessed antigen expression in SY5Y-A2 and NB16-A2 cells using immunohistochemistry (as described previously) and found that SY5Y-A2 tumors had an NY-ESO-1 score of 0 (0% cells positive) and NB16-A2 cells had an NY-ESO-1 score of 270-285 (90-95% cells positive, intensity grading 3 – data not shown), consistent with our RT-PCR findings. We next injected 2 × 107 neuroblastoma cells, either SY5Y-A2 or NB16-A2, subcutaneously into immunodeficient NOD/SCID/cγ−/− (NSG) mice. When large tumors were established and reached a mean volume of 500 mm3, we injected 107 T cells, either tTCR+ (73% tTCR+) or untransduced (NTD) directly into tumor sites. In order to control for MHC-restriction of the T cells injected, all T cells were harvested from HLA-A2+ normal human donors. To control for the non-specific activity of untransduced T cells (derived from HLA-A2+ donors) in response to HLA-A2+ tumors, each study included a group of animals injected with saline only (PBS). Animals bearing SY5Y-A2 (i.e., NY-ESO-1 negative) tumors were injected with T cells on day 22 after tumor injection, and demonstrated rapid tumor growth independent of treatment modality (Fig. 2A, p = 0.75). Animals with NB16-A2 (NY-ESO-1 positive) tumors were injected on day 10 after tumor injection, and those animals that were treated with untransduced cells or PBS also demonstrated rapid growth. In contrast, those animals injected with a single dose of NY-ESO-1-directed tTCR T cells (˜7.3 × 106 tTCR+ cells) demonstrated significant control of disease and delay of disease progression (Fig. 2B, P < 0.001). NB16-A2 tumors were excised from several mice at day 7 and day 14 after T-cell injection and examined histologically. Tumors injected with tTCR cells demonstrated extensive T-cell infiltration and expansion within tumor sites by day 7, while tumors treated with untransduced T cells lacked significant T-cell infiltration. Examination on day 14 revealed necrosis of tumors treated with tTCR cells, while untransduced cells did not alter tumor architecture, and T cells were found at the periphery of tumor tissue (Fig. S1).
Figure 2.
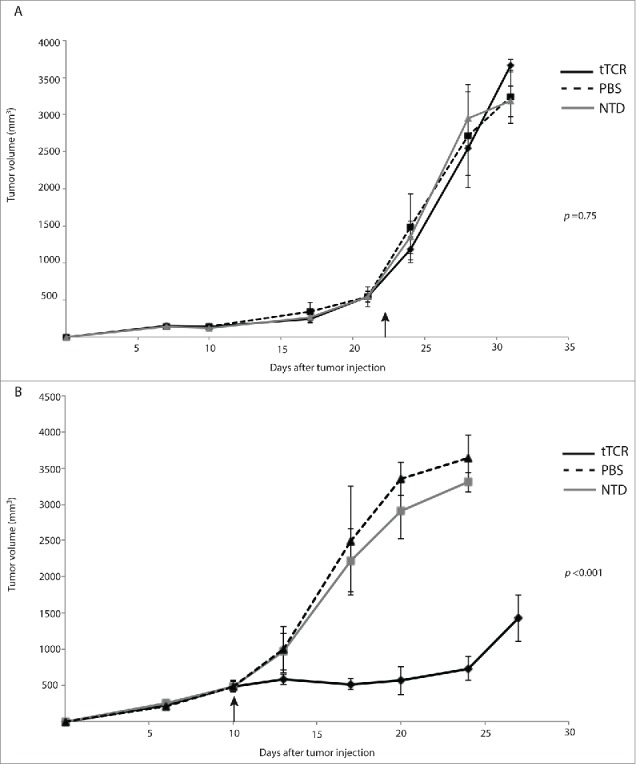
NY-ESO-1 tTCR cells control growth of localized neuroblastoma. Host mice were injected s.c. with 2 × 107 neuroblastoma xenograft cells followed by treatment with human donor-derived T cells. After large tumors were established, 1 × 107 NY-ESO-1 tTCR cells (73% tTCR+) or untransduced cells (NTD) were injected intratumorally (black arrow). (A) NY-ESO-1 tTCR cells have no effect on NY-ESO-1 negative SY5Y-A2 tumor growth. (B) NY-ESO-1 tTCR cells restrict NY-ESO-1-expressing NB16-A2 tumor growth. (n = 6 animals in each group).
Survival of animals harboring SY5Y-A2 tumors was similar among all treatment groups, with median survival ranging from 28-31 days (Fig. 3A, p = 0.84). In contrast, survival of animals with NB16-A2 tumors treated with tTCR T cells was significantly enhanced (median survival 42 days) as compared to those treated with untransduced T cells (median survival 20 days) or PBS (median survival 17 days) (Fig. 3B, p = 0.0019). Of note, comparison of animals treated with PBS demonstrates an impressive difference in median survival of animals with SY5Y-A2 (28-31 days) compared with NB16-A2 (17-20 days), highlighting the aggressive nature of this tumor cell line.
Figure 3.
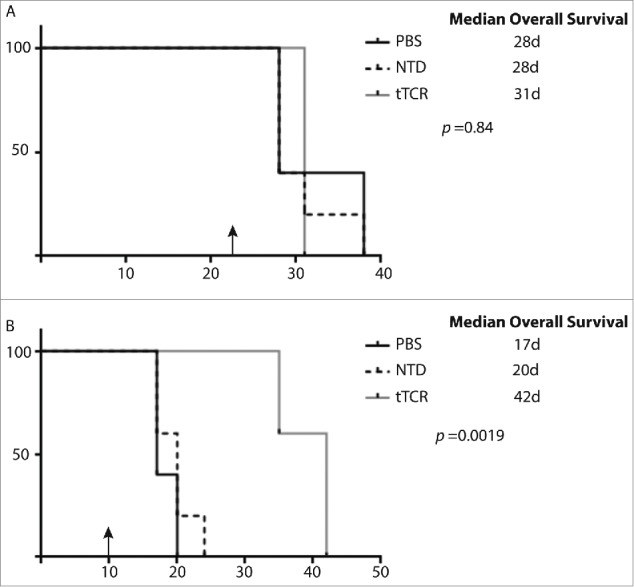
NY-ESO-1 tTCR cells extend survival of mice bearing localized NY-ESO-1+ neuroblastoma. NOD-SCID-yc-/- mice with established flank tumors were injected with T cells or PBS at the time indicated (black arrows). (A) Mice with SY5Y-A2 tumors treated with tTCR cells, untransduced cells (NTD) and PBS had equivalent overall survival. (B) Intratumoral injection of NB16-A2 tumors with tTCR cells resulted in significantly enhanced survival as compared to injection with NTD cells and PBS treated animals. Shown are the median (n D 6 animals per group) survival. Statistical analysis was performed by log-rank test with Bonferroni correction.
NY-ESO-1 tTCR T cells significantly delay progression of a rapidly progressive disseminated neuroblastoma in vivo
After demonstrating efficacy of tTCR T cells in vitro and in a localized model of neuroblastoma in vivo, we next sought to investigate their potential in a model that more closely mimicked the clinical scenario in which genetically engineered T cells are likely to be used. We have developed a disseminated model of neuroblastoma in which neuroblastoma cells are genetically engineered to express click beetle green (CBG) luciferase and then delivered systemically to NSG mice.25 Using an in vivo imaging bioluminescent system, we were able to monitor disease progression over time. We have previously demonstrated that neuroblastoma delivered in this manner establishes significant disease burdens in the liver and lymph nodes, both relevant sites of neuroblastoma metastasis.25
We engineered both SY5Y-A2 cells and NB16-A2 cells to express CBG, and injected 2 × 106 cells i.v. via tail vein. We monitored the hosts for the establishment of systemic disease as demonstrated by bioluminescent signal of >0.5 log10 over background. Animals were then injected with 107 T cells (again, engineered cells were 73% tTCR+). Animals with disseminated SY5Y-A2 tumors demonstrated rapid disease progression (Fig. 4A, p = 0.99), again independent of therapy delivered. Infusion with tTCR transduced T cells significantly delayed disease progression in animals with disseminated NB16-A2 tumor cells as compared to those animals treated with untransduced cells or PBS (Fig. 4B, P < 0.001), and a ˜2.5 log10 difference in radiance (reflecting tumor burden) at day 30. These therapeutic effects again translated to animal survival, with no difference in survival of animals with SY5Y-A2 tumors (median survival of 63 days in all groups, Fig. 5A, p = 0.31) and significantly enhanced survival in animals with NB16-A2 tumors receiving tTCR cells (median survival of animals treated with PBS 50.5 days, untransduced cells 47 days and tTCR cells 92 days, Fig. 5B, p = 0.0077). As in the localized model, all animals in this disseminated model eventually succumbed to disease.
Figure 4.
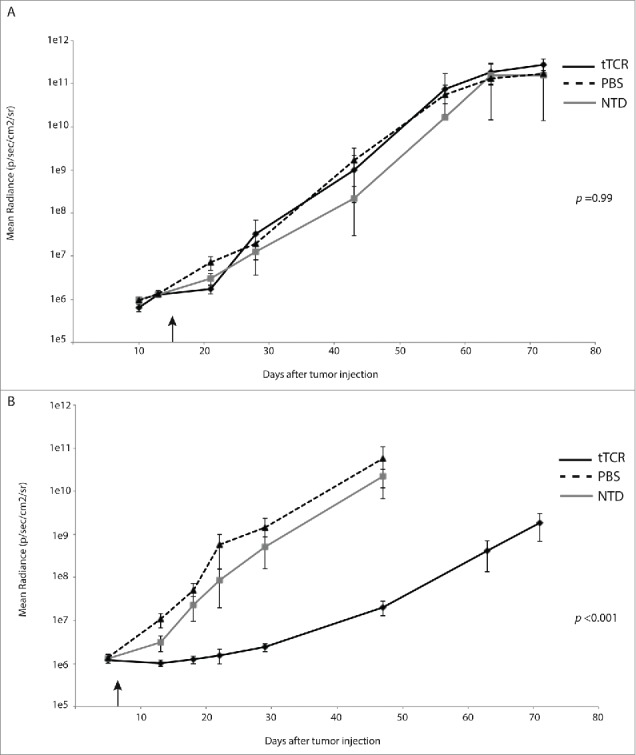
NY-ESO-1 tTCR cells delay progression of disseminated neuroblastoma. To establish disseminated neuroblastoma, 2 × 106 neuroblastoma cells were injected i.v. into immunocompromised host mice. After tumor burdens reached mean radiance of 1 × 106 p/sec/cm2/sr, animals were injected with 1 × 107 transgenic T cell receptor (tTCR) expressing T cells i.v. (black arrows). (A) NY-ESO-1 tTCR cells have no impact on SY5Y-A2 tumor progression. (B) NY-ESO-1 tTCR cells significantly delayed progression of NY-ESO-1+ NB16-A2 tumors. (n = 7 animals in each group).
Figure 5.
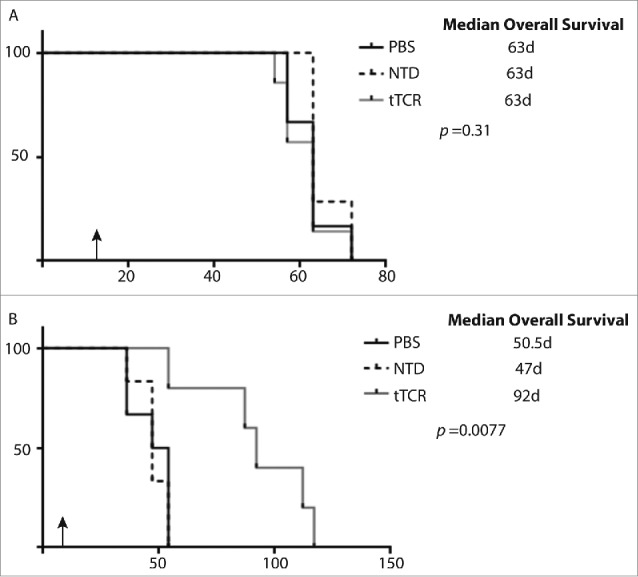
NY-ESO-1 tTCR cells extend survival of mice bearing disseminated NY-ESO-1+ neuroblastoma. NOD-SCID-yc-/- mice with established disseminated tumors were injected with 1 x 107 T cells (black arrows). (A) Animals with SY5Y-A2 tumors have no survival benefit when treated with tTCR cells. (B) tTCR cells significantly enhanced survival of mice with disseminated NB16-A2 tumors.
Visual representation of animals over time (Fig. 6) highlights the effects of these tTCR cells. Initial disease suppression is followed by extended tumor growth delay. Several animals demonstrated transient disease progression (day 29), which was temporarily controlled (Day 47) and likely serves as a harbor site of eventual disease progression (day 63).
Figure 6.

NY-ESO-1+ tTCR cells delay progression of neuroblastoma. NY-ESO-1+ NB16-A2 disseminated tumors were established, followed by delivery to 1 x 107 T-cells on day 6 after tumor injection (black arrow). Untransduced T-cells (NTD) have no impact on tumor progression, while tTCR T-cells suppress and control established neuroblastoma long-term. At shown, several animals had disease progression (here present at day 29), which was subsequently controlled, but then reappeared after day 60.
Discussion
Identification of clinically-viable tumor antigens is one of the largest hurdles facing rapid development of targeted immunotherapies for cancer, specifically solid tumors. Currently, tumor antigen identification methods focus on defined groups of targetable antigens: novel peptides produced by somatic mutations,26 antigens expressed by expendable tissues,9 and cancer-testes antigens.16 Several immunotherapy platforms have now demonstrated clinical efficacy in re-targeting immune tissue to cancer cells by manipulating the endogenous immune response, including antibody-based and cytokine-based therapies. Novel therapies like bi-specific antibodies are demonstrating early clinical success,27,28 and methods that bypass the endogenous immune system, such as genetically-engineered T cells, have also drawn a great deal of attention for the successes demonstrated using chimeric antigen receptors.2,8,9,29
While several adult cancer have been evaluated for NY-ESO-1 expression and its ability to serve as an immunotherapeutic target,15,16,30 pediatric cancers have been largely overlooked. A trial targeting NY-ESO-1+ synovial cell carcinoma using the T cells investigated in this report is currently enrolling; however, this clinical trial is, to our knowledge, the only such pediatric trial targeting NY-ESO-1 (Clinicaltrials.gov Identifier NCT01343043). While pediatric cancer diagnoses certainly represent a small proportion of the worldwide cancer burden, outcomes have largely plateaued since the mid-1990s31 and further progress will require alternate therapeutic platforms. In this study we show that T cells engineered to target NY-ESO-1 using a transgenic TCR demonstrate immunotherapeutic efficacy against NY-ESO-1+ neuroblastoma.
Using the Children's Hospital of Philadelphia tumor bank, we were able to analyze the expression of NY-ESO-1 among a large number of resected primary neuroblastoma samples. We found that ˜23% of our samples express NY-ESO-1, approximately the same percentage of malignant melanomas that have been shown to express NY-ESO-1.32 When considering potential clinical translation, it is important to remember that this therapy is restricted by expression of the appropriate MHC Class I (HLA-A2). Approximately 40% of the Caucasian population of the United States is positive for HLA-A2, and from this we can predict that 10% of all neuroblastoma patients will be eligible for this therapy. If shown to be promising among HLA-A2+ patients, this will spur the development of NY-ESO-1 targeting TCRs with other common HLA restrictions, a key step to broaden application of engineered TCRs. The therapeutic effect combined with the poor outcomes for this disease warrant clinical evaluation of this therapy.
We posit that the successful targeting and inhibition of tumor growth demonstrated in this study primarily relies on two issues: targeting the immunodominant epitope of NY-ESO-1 (peptides 157-165)33 and constraining the long-term cytotoxicity of these engineered T cells. We have previously demonstrated that ex vivo expansion using CD3/CD28 beads produces a T-cell population that is more effective in vivo, at least in part due to the phenotype of cells produced using this stimulation method.20 These cells have a greater proliferative capacity in vivo, and in our CD19 CAR model they demonstrated enhanced antitumor activity as compared to cells produced using other methods of expansion.
As assessed by CD107a degranulation, we demonstrated that lower levels of NY-ESO-1 expression are sufficient to induce an antigen-driven response, highlighting the sensitivity of our high-affinity tTCR cells. Interestingly, the level of degranulation seemed to correlate with the level of antigen expression. Whether this finding is an artifact of a 4-hour incubation period such that cells expressing lower levels of NY-ESO-1 would have demonstrated further degranulation over time, or if this is a sustained phenomenon, is unclear. NY-ESO-1 expression was found to be variable among our primary patient samples, and the observed degranulation with lower-levels of NY-ESO-1 expression suggests that tumors with lower levels of antigen production would also be targetable, although this assumption was not evaluated in our in vivo xenograft model. To date, clinical trials targeting NY-ESO-1 have excluded patients with lower levels of NY-ESO-1 expression, limiting enrollment to those with moderate or high levels of expression.34 We thus have no clinical data to suggest that NY-ESO-1 transgenic TCR expressing T cells are capable of successfully targeting low expressing tumors. These in vitro data suggest that tTCR cells can recognize and degranulate in response to lower levels of antigen, but they do not answer the question of what the “threshold” for recognition and resultant cytotoxicity is required in vivo. We can hypothesize 2 response models: (1) the degree of antigen production and presentation correlates to T-cell activity, and thus “lower-expressers” will have a more limited T cell-driven anti-tumor response (a gradient-response model), or (2) T-cell activity is driven by a threshold effect, in that a minimal antigen burden is needed to initiate robust T-cell activity that is “on” or “off” (a binary-response model). Further clinical studies are necessary to determine the role of antigen intensity on response and outcome.
In both the subcutaneous model and the disseminated tumor model, we observed that tTCR cells were able to slow disease progression of NY-ESO-1+ neuroblastoma but were unable to eradicate disease. The majority of this effect is observed in the first 30-40 days after T-cell infusion, after which time tumor growth rate accelerates. Several mechanisms may account for this observation. Immune escape resulting from selective pressure may take the form of downregulation of antigen presenting machinery or antigen expression itself. MHC downregulation is a well-described method of tumor immune escape.35 Indeed, loss of HLA-A*02 in a murine model of multiple myeloma was confirmed as a mechanism of immunoevasion when animals were treated with NY-ESO-1 tTCR cells.36 While we have not confirmed this either histologically or molecularly, HLA-A2 downregulation is less likely to account for tumor progression in our study. The NB16 neuroblastoma cell line used was genetically engineered to express HLA-A2, and silencing of this engineered locus driven by the Ef-1α promoter, while possible, is unlikely and has not been observed in our experience using lentivirally-engineered cells. Deletion or silencing of target antigens is also a well-described mechanism of immune escape, and our group has reported immunoevasion of CD19-negative ALL observed in our trial of CD19 CAR T cells.9 Preliminary data have demonstrated that subcutaneous NB16 tumors retain similar levels of NY-ESO-1 expression after treatment with NY-ESO-1 tTCR cells, even in the setting of disease response followed by progression (equivalent NY-ESO-1 score prior to infusion and at time of animal death, data not shown). These data suggest that neither NY-ESO-1 silencing nor outgrowth of endogenously NY-ESO-1-negative cells are contributing mechanisms of disease progression in this setting. Larger studies to compare the expression levels of both HLA-A2 and NY-ESO-1 in tumors from animals treated with control cells and those under selective pressure from NY-ESO-1 tTCR cells should be pursued to confirm this finding.
We have observed that T-cell persistence is an essential component of a maintained antitumor response in our CAR T cell therapy trials8 and our CAR T cells have been successful in large part due to their maintained persistence in vivo. While persistence was not directly evaluated in our current study, previous data from prior studies using the same NY-ESO tTCR T cells in patients with sarcoma and myeloma demonstrated maintained persistence up to 2 years after cell infusion.37 Evaluation of persistence will be an essential component of our studies moving forward. If indeed persistence is not maintained, a multiple infusion strategy may be of value to enhance the antitumor responses observed with a single infusion.
We hypothesize that one specific mechanism that greatly contributes to the lack of disease control long-term is the differential rate of cell division between our T cells and the tumor cells. As demonstrated in our study, the NB16 cell line is an extremely aggressive cancer. In vitro it is the fastest growing neuroblastoma line that we have evaluated, and this malignancy results in remarkably rapid animal death in vivo (as quickly as 17 days after tumor injection in the case of subcutaneous disease). Prior to T-cell infusion, animals with both subcutaneous and disseminated disease were permitted to establish significant disease burdens. While it is clear that an adequate effector:target cell ratio is achieved acutely as reflected by the stabilization of disease growth immediately following T cell infusion, the timing of treatment may have been too late to achieve an adequate effector:target cell ratio over the long-term. Tumor cell division may have simply outpaced T-cell division within the tumor site. It is indeed true that in our clinical experience with acute lymphoblastic leukemia (ALL), an aggressive and rapidly dividing tumor, we do observe the ability of CAR T cells to achieve adequate effector:target cell ratios long term. Solid tumors, however, present different barriers to achieve effective antitumor activity. Leukemia occupies the same anatomical niche as the transferred T cells, and thus obviates the need for effective tumor infiltration dependent on an unstable vascular supply.25,38 These barriers may be rate-limiting with regards to achieving successful antitumor responses in large, rapidly dividing solid tumors. When considering clinical translation, both disease burden and rate of tumor growth will be relevant parameters when determining patient eligibility for engineered T-cell therapy for solid tumors.
We have previously reported long-term control of disseminated neuroblastoma using CAR T cells targeting the antigen GD2.25 In this prior study, we observed disease eradication in animals treated with GD2 CAR T cells whereas the animals in this study demonstrated attenuation of disease progression, but with animals eventually died of disease. Although several factors may contribute to the observed difference in outcome, a key component is likely the role of integrated vs accessory co-stimulatory signals. Initial T-cell activity may be enhanced with the integrated 4-1BB activation that is present in CAR-mediated activity, as may long-term T-cell persistence, and further studies are necessary to illuminate this and other potential differences in signaling that may result from disintegrated vs integrated co-stimulation. These distinct outcomes highlight an opportunity to establish different therapeutic goals for patients receiving engineered T-cell therapy based on their unique clinical characteristics. Many neuroblastoma patients are ineligible for autologous stem cell transplant due to large disease burdens, rapidly progressive disease or other co-morbidities, and the goal for these patients may be simply disease stabilization prior to proceeding with standard therapies. Recent advances and a large influx of clinical trial data now allow us to identify the different clinical settings in which these novel therapies may be applied, and that the goals for all diseases may not be uniform.
A potential hurdle to the application of this therapy is the fact that neuroblastoma is known to have low levels of MHC Class I expression in vivo.39 This low level of Class I expression has been correlated with a low degree of T-cell infiltration, and thus it has been proposed that reduced MHC expression levels may confer “protection” from cell-mediated immunity in some tumors. In contradistinction, CAR T cells targeting neuroblastoma via an MHC-independent mechanism demonstrate robust tumor infiltration and antitumor responses.25,38 The cell lines used in our in vitro and in vivo experiments were engineered to express high levels of MHC Class I, thus removing the potential for low levels of antigen presentation at the time of tTCR T-cell infusion. Previous work from our group and others has demonstrated that exogenous interferon γ (IFNγ) results in upregulation of MHC Class I on previously MHC Class I-low/negative tumors, and that this upregulation can result in increased T-cell infiltration and enhanced tumor killing in vivo.39,40 Combining this observation with the robust antitumor activity of the cells described in this study, one could envision a clinical trial design in which high-risk neuroblastoma patients are treated with IFNγ surrounding the time of tTCR T-cell infusion to enhance tumor infiltration and antitumor activity. There is also the possibility that not all tumor cells will have undetectable levels of Class I, and some tTCR T cells will be able to engage and activate. As mentioned, these cells are cultured and engineered in the same manner as our CAR T cells, which we have recently reported secrete large amounts of IFNγ.8 This alone may be sufficient to induce MHC Class I upregulation within the tumor microenvironment, without the need for exogenous IFNγ.
Low MHC Class I expression is most often associated with high-grade neuroblastomas, and has been shown to be biochemically driven by MYCN expression.41 Examination of our neuroblastoma tissue microarray (TMA) demonstrates that approximately 31% of these all resected neuroblastomas are classified as high-risk, as assessed by INSS staging. Of these high-risk tumors, 47% are MYCN amplified, reflecting the poor prognosis of this genetic amplification. Interestingly, while 30% of the NY-ESO-1+ tumors are high-risk (a similar to the proportion seen in the TMA as a whole) only 14% were found to display MYCN amplification. Similarly, whereas ˜16% of all neuroblastomas were found to express MYCN (independent of INSS risk), only 3.7% of all NY-ESO-1+ tumors were MYCN amplified. While this may simply represent sampling error from our group of tumors, NY-ESO-1+ tumors may represent a subgroup of neuroblastomas less likely to express MYCN, and thus perhaps be less likely to downregulate MHC molecules. Further evaluation is necessary to confirm this correlation.
Previously published data has suggested that the hypo-methylating agent decitabine may induce increased CT antigen expression.42 Although we were not able to reproduce this finding in our own cell lines (data not shown), it is possible that this effect may be observed in some primary neuroblastomas, thus increasing the number of patients eligible for this T-cell immunotherapy. Mechanisms to enhance antigen expression certainly warrant further investigation. Another approach to improve efficacy of engineered T cells against solid malignancies is combination with checkpoint blockade inhibitors. The recent clinical success of these molecules in the treatment of a variety of solid tumors43,44 has led to investigation of combining this immunotherapy with adoptive cellular therapy and some initial success has been demonstrated.45 Addition of PD-1 or CTLA4 inhibition may result in enhanced antitumor responses using our engineered tTCR cells.
To date, successful trials of engineered T cells have largely targeted B cell malignancies using CD19. The on-target off-tissue loss of normal B cells is a clinically tolerable side effect. Previous studies targeting solid tumors using CAR T cells have resulted in numerous adverse events mediated by multiple mechanisms.46-48 These events further drive the search for improved antigen selection. Previously published data using tTCR cells targeted to NY-ESO-1 demonstrated efficacy and, importantly, did not demonstrate on-target toxicity.16 The limited expression restricted to germ-line tissues that do not express MHC Class I molecules likely accounts for this limited toxicity and confirms that CT antigens are promising immunotherapeutic targets.
To our knowledge, this is the first assessment of a transgenic TCR targeting pediatric neuroblastoma. We have found that NY-ESO-1 is a relevant immunotherapy target in this disease, and T cells engineered to target this molecule using engineered tTCRs demonstrate antitumor efficacy and significantly improve animal survival. Given these findings, clinical translation to investigate the efficacy of these cells in patients with refractory neuroblastoma is both justified and needed.
Methods
Neuroblastoma tissue microarray and immunohistochemistry
The Center for Childhood Cancer Research at the Children's Hospital of Philadelphia has developed a tissue microarray of 187 neuroblastoma tumor cores collected from 165 patients. Flank tumors from our xenograft animal studies were excised and preserved in 4% paraformaldehyde. Evaluation of antigen expression, tumor grade, histologic profile, Mycn amplification status, and International Neuroblastoma Staging System (INSS) stage was done for each sample. Staining was performed on a Bond Max automated staining system, using the Bond Refine polymer staining kit (Leica Microsystems, Buffalo Grove, IL). Standard staining protocol was followed, with the exception of the primary antibody incubation, which was extended to 1 h at room temperature. NY-ESO-1 antibody (Life Technologies, Grand Island, NY, Catalog #35-6200) and anti-human CD3 antibody (Dako, Carpinteria, CA, Catalog #M7254) were used at 1:50 dilution and antigen retrieval was performed with E1 retrieval solution for 20 min (Leica Microsystems). Slides were rinsed and dehydrated through a series of ascending concentrations of ethanol and xylene. Stained slides were then digitally scanned at 20x magnification on an Aperio OS slide scanner and analyzed using ImageScope Software (both Aperio,Vista, CA). NY-ESO-1 expression was evaluated by both percentage of cells staining positively for NY-ESO-1 (graded 0-100%), as well as intensity of staining (graded 0-3). These two numbers were multiplied to calculate a combined “NY-ESO-1 score,” ranging from 0-300. Only samples that had >10% of cells stain positively and had a combined NY-ESO-1 score of >10 were considered positive.
NY-ESO-1 mRNA expression in neuroblastoma cell lines
Neuroblastoma cell lines were evaluated for expression of NY-ESO-1 using quantitative reverse-transcription polymerase chain reaction (RT-PCR). RNA extraction was performed using Ambion RNAqueous or RiboPure kits (Life Technologies, Grand Island, NY), and cDNA was produced using iScript DNA Synthesis kits (Bio-Rad Laboratories, Life Technologies, Grand Island, NY). Quantitation was performed on an ABI 7500 Fast Real-Time PCR System (Life Technologies, Grand Island, NY). Relative quantities (RQs) were calculated using the comparative Ct method on ABI Data Assist software.
Lentiviral vector production
High-titer, replication-defective lentiviral vectors were produced using 293T human embryonic kidney cells as previously described.49 Briefly, HEK293T cells were seeded at 107 cells per T150 tissue culture flask 24 h before transfection. On the day of transfection, cells were treated with 7 μg of pMDG.1, 18 μg of pRSV.rev, 18 μg of pMDLg/p.RRE packaging plasmids and 15 μg of transfer plasmid in the presence of Lipofectamine 2000 transfection reagent (Life Technologies, Grand Island, NY, Catalog #11668019). Transfer plasmids containing TCR constructs were modified so that expression of the TCR was under control of the EF-1α promoter as previously described.50 Viral supernatants were harvested 24 h and 48 h after transfection and concentrated by ultracentrifugation overnight at 10,500xg.
T cell engineering
Primary human T cells from normal donors were procured through the University of Pennsylvania Human Immunology Core. T cells were combined at a ratio of 1:1 CD4:CD8 cells at a concentration of 106 cells/mL in T cell culture media with stimulatory microbeads coated with antibodies directed against CD3 and CD28 (Life Technologies, Grand Island, NY, Catalog #111.32D) at a concentration of 3 beads/cell, as reported previously.51 For each experiment, cells were derived from the same donor and split into 2 large cultures. Following 24 h after initial stimulation, one culture was exposed to lentiviral vector at a multiplicity of infection (MOI) of 5-10 particles/cell, and the other culture was left undisturbed. Cells were counted and volumes measured serially until growth and size trends indicated cells were rested down, at which time they were frozen. Cells were then thawed 12-18 hours prior to in vivo injection.
T cells were transduced with a lentiviral vector encoding a high-affinity NY-ESO-1157-165-directed transgenic T cell receptor (AdaptImmune, Abington, United Kingdom) at a concentration of 7 infectious particles per T cell using protocols previously described.52 Using an HLA-A*0201-restricted MHC molecule loaded with a dextramer specific for this tTCR (SLLMWITQV) conjugated to either phycoerythrin (PE, Catalog #WB2696-PE) or allophycocyanin (APC, Catalog #WB2696-APC) fluorophores (Immudex USA, Fairfax, Virginia), transduced cells were stained to assess tTCR expression. Fluorescence was assessed using an Accuri C6 Flow Cytometer (BD Biosciences, Franklin Lakes, NJ).
Modification of neuroblastoma cell lines
Lentiviral vectors encoding HLA-A2-restricted Class I MHC molecules and Click Beetle Luciferase – GFP (CBG/GFP) were manufactured as described above. Neuroblastoma cell lines were plated and given 24 h to adhere to culture vessels, and then exposed to lentiviral vectors encoding HLA-A2 at multiplicity of infection (MOI) of 8-10. HLA-A2 expression was then assessed by flow cytometry after incubation of neuroblastoma cells with antibodies directed against HLA-A2 (BD Biosciences, Franklin Lakes, NJ). Both SY5Y and NB16 cell lines had >95% HLA-A2 expression. Cells were then re-plated and exposed to vectors encoding CBG/GFP, after which expression of GFP was assessed by flow cytometry. Again, all cells demonstrated >95% CBG/GFP expression.
CD107a degranulation assay
T cells modified to express the NY-ESO-1 tTCR were co-incubated with target neuroblastoma cells engineered to express HLA-A2 at a ratio of 5:1 target:T cell. This co-culture was combined with an antibody cocktail consisting of anti-CD107a-e660 (eBiosciences, San Diego, CA, Catalog #50-1079), and stimulatory antibodies directed against CD28 (clone 9.3, generously provided by Dr. Bruce Levine, University of Pennsylvania) and CD49d (BD Biosciences, Franklin Lakes, NJ, Catalog #555051) for one hour. Intracellular protein transport was halted by addition of GolgiStop (BD Biosciences, Franklin Lakes, NJ, Catalog #554724) and cells were incubated for an additional three hours. Cells were then harvested and stained for CD3 (BD Biosciences, Franklin Lakes, NJ, Catalog #554832) and tTCR as described and analyzed on an Accuri C6 Flow Cytometer. Specificity of degranulation was controlled for by calculation of the Degranulation Ratio. This number was calculated using the formula ((%tTCR+CD107a+)/(%tTCR+))/((%tTCR-CD107a+)/(%tTCR-)), which controlled for non-specific degranulation of tTCR-negative cells.
Mouse xenograft studies
6-16 week old NOD-SCID-γc−/− (NSG) were obtained from the Jackson Laboratory (Bar Harbor, ME) or bred in house under an approved Institutional Animal Care and Use Committee (IACUC) protocol and maintained in pathogen-free conditions. For flank tumor studies, animals were injected with 2 × 107 neuroblastoma cells suspended in 0.2 mL Matrigel (BD Biosciences, San Jose, CA, Catalog #354234) subcutaneously. For disseminated tumor studies, animals were injected via tail vein with 2 × 106 cells in 0.1 mL sterile PBS. T cells were injected in 0.1 mL sterile PBS either directly intratumoral for flank tumor studies or intravenously for disseminated studies at the indicated times. Animals were monitored for signs of disease progression and overt toxicity, such as xenogeneic graft-vs.-host disease, as evinced by >20% loss in body weight, loss of fur, diarrhea, conjunctivitis and disease-related hind limb paralysis. Histological studies presented in Figure S1 were performed on flank tumors excised from animals sacrificed at the indicated times.
Measurement of flank tumors
Flank tumor measurements were made bi-weekly using electronic calipers (Fowler-Sylvac, Boston, MA, Catalog #54-200-777). Longest length and width measurements were recorded and tumor volume was calculated according to the formula ((width + length)/2)3)/2. Mice were sacrificed when tumors reached 3 cm3, or when tumor burden inhibited activity.
Bioluminescent imaging
Disease burdens were monitored over time using the Xenogen sensitive bioluminescent imaging system, as described previously.22,23 Animals were sacrificed when bioluminescent signal reached > 1011 photons/sec/cm2/steridian.
Cell line identity testing
Parent cell lines were genotyped by short tandem repeat (STR) analysis.53 Cell lines and samples were verified every 6 months, or after any genetic modification to ensure identity.
Statistical considerations
All statistical analysis was performed with Prism 4 (Graphpad Software, La Jolla, CA). For comparison among multiple groups, Kruskal-Wallis analysis was performed with Dunn Multiple Comparison tests to compare individual groups. Survival curves were compared using the log-rank test with Bonferroni correction for multiple comparisons.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
The authors wish to thank Junior Hall and Jessica Hulitt for technical assistance with the animal studies described.
Author contributions
N.S., M.K., G.B.S., B.J., D.M.B., C.H.J, and S.A.G. designed the research. N.S., I.K., D.M., B.P. performed the research.
Funding
Supported by grants from the Jeffrey Jay Weinberg Memorial Foundation, Cookies for Kids Cancer, Solving Kids Cancer, and a Stand Up to Cancer–St. Baldrick's Pediatric Dream Team translational research grant (SU2C-AACR-DT1113). Carl H. June (University of Pennsylvania) and Michael Kalos (Eli Lilly) have patent applications in some of the technology described in this manuscript. Gwen Binder-Scholl and Bent Jakobsen are employees of AdaptImmune.
References
- 1.Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med 2012; 366:2008-16; PMID:22621628; http://dx.doi.org/ 10.1056/NEJMct1114348 [DOI] [PubMed] [Google Scholar]
- 2.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725-33; PMID:21830940; http://dx.doi.org/ 10.1056/NEJMoa1103849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen YT, Scanlan MJ, Sahin U, Türeci O, Gure AO, Tsang S, Williamson B, Stockert E, Pfreundschuh M, Old LJ. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A 1997; 94:1914-8; PMID:9050879; http://dx.doi.org/ 10.1073/pnas.94.5.1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen YT, Gure AO, Tsang S, Stockert E, Jäger E, Knuth A, Old LJ. Identification of multiple cancer/testis antigens by allogeneic antibody screening of a melanoma cell line library. Proc Natl Acad Sci U S A 1998; 95:6919-23; PMID:9618514; http://dx.doi.org/ 10.1073/pnas.95.12.6919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev 2002; 188:22-32; PMID:12445278; http://dx.doi.org/ 10.1034/j.1600-065X.2002.18803.x [DOI] [PubMed] [Google Scholar]
- 6.Jager E, Gnjatic S, Nagata Y, Stockert E, Jäger D, Karbach J, Neumann A, Rieckenberg J, Chen YT, Ritter G, et al.. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci U S A 2000; 97:12198-203; PMID:11027314; http://dx.doi.org/ 10.1073/pnas.220413497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med 2014; 65:333-47; PMID:24274181; http://dx.doi.org/ 10.1146/annurev-med-060512-150254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al.. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507-17; PMID:25317870; http://dx.doi.org/ 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al.. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509-18; PMID:23527958; http://dx.doi.org/ 10.1056/NEJMoa1215134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3:95ra73; PMID:21832238; http://dx.doi.org/ 10.1126/scitranslmed.3002842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al.. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5:177ra38; PMID:23515080; http://dx.doi.org/ 10.1126/scitranslmed.3005930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al.. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014; 2:112-20; PMID:24579088; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al.. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011; 118:6050-6; PMID:21984804; http://dx.doi.org/ 10.1182/blood-2011-05-354449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al.. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14:1264-70; PMID:18978797; http://dx.doi.org/ 10.1038/nm.1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol 2005; 174:4415-23; PMID:15778407; http://dx.doi.org/ 10.4049/jimmunol.174.7.4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al.. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011; 29:917-24; PMID:21282551; http://dx.doi.org/ 10.1200/JCO.2010.32.2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet 2007;369:2106-20; PMID:17586306; http://dx.doi.org/ 10.1016/S0140-6736(07)60983-0 [DOI] [PubMed] [Google Scholar]
- 18.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, et al.. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med 1999; 341:1165-73; PMID:10519894; http://dx.doi.org/ 10.1056/NEJM199910143411601 [DOI] [PubMed] [Google Scholar]
- 19.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al.. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010; 363:1324-34; PMID:20879881; http://dx.doi.org/ 10.1056/NEJMoa0911123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett DM, Singh N, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy 2014; 16:619-30; PMID:24439255; http://dx.doi.org/ 10.1016/j.jcyt.2013.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, Karbach J, Jäger E, Cameron BJ, Lissin N, et al.. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol 2006; 176:7308-16; PMID:16751374; http://dx.doi.org/ 10.4049/jimmunol.176.12.7308 [DOI] [PubMed] [Google Scholar]
- 22.Barrett DM, Seif AE, Carpenito C, Teachey DT, Fish JD, June CH, Grupp SA, Reid GS. Noninvasive bioluminescent imaging of primary patient acute lymphoblastic leukemia: a strategy for preclinical modeling. Blood 2011; 118:e112–7; PMID:21856863; http://dx.doi.org/ 10.1182/blood-2011-04-346528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther 2011; 22:1575-86; PMID:21838572; http://dx.doi.org/ 10.1089/hum.2011.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burkett MW, Shafer-Weaver KA, Strobl S, Baseler M, Malyguine A. A novel flow cytometric assay for evaluating cell-mediated cytotoxicity. J Immunother 2005; 28:396-402; PMID:16000959; http://dx.doi.org/ 10.1097/01.cji.0000165357.11548.6d [DOI] [PubMed] [Google Scholar]
- 25.Singh N, Liu X, Hulitt J, Jiang S, June CH, Grupp SA, Barrett DM, Zhao Y. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol Res 2014; 2:1059-70; PMID:25104548; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al.. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344:641-5; PMID:24812403; http://dx.doi.org/ 10.1126/science.1251102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, et al.. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013; 121:5154-7; PMID:23678006; http://dx.doi.org/ 10.1182/blood-2013-02-485623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, Gökbuget N, Neumann S, Goebeler M, Viardot A, et al.. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012; 119:6226-33; PMID:22592608; http://dx.doi.org/ 10.1182/blood-2012-01-400515 [DOI] [PubMed] [Google Scholar]
- 29.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al.. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra25; PMID:24553386; http://dx.doi.org/ 10.1126/scitranslmed.3008226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuberth PC, Jakka G, Jensen SM, Wadle A, Gautschi F, Haley D, Haile S, Mischo A, Held G, Thiel M, et al.. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther 2013; 20:386-95; PMID:22739387; http://dx.doi.org/ 10.1038/gt.2012.48 [DOI] [PubMed] [Google Scholar]
- 31.Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O'Leary M, Smith FO, Reaman GH. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 2010; 28:2625-34; PMID:20404250; http://dx.doi.org/ 10.1200/JCO.2009.27.0421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aung PP, Liu YC, Ballester LY, Robbins PF, Rosenberg SA, Lee CC. Expression of New York esophageal squamous cell carcinoma-1 in primary and metastatic melanoma. Hum Pathol 2014;. 45(2):259-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Held G, Matsuo M, Epel M, Gnjatic S, Ritter G, Lee SY, Tai TY, Cohen CJ, Old LJ, Pfreundschuh M, et al.. Dissecting cytotoxic T cell responses towards the NY-ESO-1 protein by peptide/MHC-specific antibody fragments. Eur J Immunol 2004; 34:2919-29; PMID:15368308; http://dx.doi.org/ 10.1002/eji.200425297 [DOI] [PubMed] [Google Scholar]
- 34.Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, et al.. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-Reactive T-cell receptor: Long-term follow-up and correlates with response. Clin Cancer Res 2015;21:1019-27; PMID:25538264; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol 2003; 195:346-55; PMID:12704644; http://dx.doi.org/ 10.1002/jcp.10290 [DOI] [PubMed] [Google Scholar]
- 36.Klippel ZK, Chou J, Towlerton AM, Voong LN, Robbins P, Bensinger WI, Warren EH. Immune escape from NY-ESO-1-specific T-cell therapy via loss of heterozygosity in the MHC. Gene Ther 2014; 21:337-42; PMID:24451117; http://dx.doi.org/ 10.1038/gt.2013.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merchant M, Cristea M, Stadtmauer E, Tap W, D'Angelo S, Grupp SA, Holdich T, Binder-Scholl G, Jakobsen B, Odunsi K, et al.. Genetically engineered NY-ESO-1 Specific T cells in HLA-A201+ Patients with Advanced Cancers. In: ASCO 2015 Annual Meeting 2015. J Clin Oncol 33, 2015 (suppl; abstr TPS3102) [Google Scholar]
- 38.Singh N, Barrett DM, Grupp SA. Roadblocks to success for RNA CARs in solid tumors. OncoImmunology 2015; 3(12):e962974; PMID:25964863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reid GS, Shan X, Coughlin CM, Lassoued W, Pawel BR, Wexler LH, Thiele CJ, Tsokos M, Pinkus JL, Pinkus GS, et al.. Interferon-gamma-dependent infiltration of human T cells into neuroblastoma tumors in vivo. Clin Cancer Res 2009; 15:6602-8; PMID:19825945; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Merchant MS, Romero ME, Tsokos M, Wexler LH, Kontny U, Mackall CL, Thiele CJ. Induction of caspase 8 by interferon gamma renders some neuroblastoma (NB) cells sensitive to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) but reveals that a lack of membrane TR1/TR2 also contributes to TRAIL resistance in NB. Cancer Res 2003; 63:1122-9; PMID:12615731 [PubMed] [Google Scholar]
- 41.Bernards R, Dessain SK, Weinberg RA. N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell 1986; 47:667-74; PMID:3096575; http://dx.doi.org/ 10.1016/0092-8674(86)90509-X [DOI] [PubMed] [Google Scholar]
- 42.Bao L, Dunham K, Lucas K. MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells to facilitate cytotoxic T lymphocyte-mediated tumor cell killing. Cancer Immunol Immunother 2011; 60:1299-307; PMID:21626030; http://dx.doi.org/ 10.1007/s00262-011-1037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/ 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res 2013; 19:5636-46; PMID:23873688; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0458 [DOI] [PubMed] [Google Scholar]
- 46.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 2010; 18:666-8; PMID:20357779; http://dx.doi.org/ 10.1038/mt.2010.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18:843-51; PMID:20179677; http://dx.doi.org/ 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 2013; 1:26-31; PMID:24777247; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol 2003; 171:166-74; PMID:12816995; http://dx.doi.org/ 10.4049/jimmunol.171.1.166 [DOI] [PubMed] [Google Scholar]
- 50.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al.. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17:1453-64; PMID:19384291; http://dx.doi.org/ 10.1038/mt.2009.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laport GG, Levine BL, Stadtmauer EA, Schuster SJ, Luger SM, Grupp S, Bunin N, Strobl FJ, Cotte J, Zheng Z, et al.. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood 2003; 102:2004-13; PMID:12763934; http://dx.doi.org/ 10.1182/blood-2003-01-0095 [DOI] [PubMed] [Google Scholar]
- 52.Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, Binder GK, Slepushkin V, Lemiale F, Mascola JR, et al.. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A 2006; 103:17372-7; PMID:17090675; http://dx.doi.org/ 10.1073/pnas.0608138103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, Toji LH, Ohno T, Tanabe H, Arlett CF, et al.. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci U S A 2001; 98:8012-7; PMID:11416159; http://dx.doi.org/ 10.1073/pnas.121616198 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.