

Abstract
BACKGROUND
Anesthetics cause widespread apoptosis in the developing brain, resulting in neurocognitive abnormalities. However, it is unknown whether anesthesia-induced neurotoxicity occurs in humans because there is currently no modality to assess for neuronal apoptosis in vivo. The retina is unique in that it is the only portion of the central nervous system that can be directly visualized noninvasively. Thus, we aimed to determine whether isoflurane induces apoptosis in the developing retina.
METHODS
CD-1 male mouse pups underwent 1-hour exposure to isoflurane (2%) or air. After exposure, activated caspase-3, caspase-9, and caspase-8 were quantified in the retina, cytochrome c release from retinal mitochondria was assessed, and the number and types of cells undergoing apoptosis were identified. Retinal uptake and the ability of fluorescent-labeled annexin V to bind to cells undergoing natural cell death and anesthesia-induced apoptosis in the retina were determined after systemic injection.
RESULTS
Isoflurane activated the intrinsic apoptosis pathway in the inner nuclear layer (INL) and activated both the intrinsic and extrinsic pathways in the ganglion cell layer. Immunofluorescence demonstrated that bipolar and amacrine neurons within the INL underwent physiologic cell death in both cohorts and that amacrine cells were the likely targets of isoflurane-induced apoptosis. After injection, fluorescent-labeled annexin V was readily detected in the INL of both air-exposed and isoflurane-exposed mice and colocalized with activated caspase-3-positive cells.
CONCLUSIONS
These findings indicate that isoflurane-induced neuronal apoptosis occurs in the developing retina and lays the groundwork for development of a noninvasive imaging technique to detect anesthesia-induced neurotoxicity in infants and children.
Commonly used anesthetics induce widespread neuronal apoptosis in the developing mammalian brain.1–5 Vulnerability coincides with synaptogenesis, and anesthesia-induced neurodegeneration results in significant loss of neurons, cognitive impairment, and behavioral abnormalities in a variety of newborn animal models.6,7 A number of retrospective studies indicate an association between anesthesia exposure and cognitive and behavioral disorders in young children; however, anesthetics have never been shown to cause neurotoxicity in humans.8–13 Furthermore, it is unknown whether anesthesia-induced neurodegeneration occurs in humans because there is currently no modality to noninvasively assess for apoptosis in the brain of infants and children.
Programmed cell death (PCD) is a widespread and natural phenomenon that occurs in 2 waves within the developing central nervous system (CNS).14 The early wave of PCD peaks during midembryogenesis and is important for progenitor cell size, morphogenesis, proliferation, and differentiation, whereas the postnatal wave is critical for synaptogenesis and elimination of aberrant neuronal connections.14 Thus, the prenatal wave regulates progenitor pool size and the later wave ensures proper wiring.15 In the rodent brain, the postnatal peak in developmental PCD occurs within the period of peak synaptogenesis.16–18 Thus, physiologic apoptosis may influence the susceptibility of the developing brain to anesthesia-induced neurotoxicity. The retina is a highly specialized layer of neural tissue within the CNS that enables vision. As in the brain, neurons in the developing retina undergo developmental apoptosis.19 The early embryonic phase of PCD in the retina also coincides with neurogenesis, migration, and differentiation, whereas the postnatal phase is associated with synaptogenesis for the selective elimination of aberrant synapses.19
The retina is unique in that it is the only portion of the CNS that can be directly visualized noninvasively. Recently, high-resolution methods have been developed to image single-cell apoptosis within the retina in vivo.20 Thus, if anesthetics are shown to cause neuronal apoptosis in the developing retina, it may be possible to exploit such neurodegeneration as a surrogate for anesthesia-induced brain neurotoxicity to determine whether the phenomenon occurs in infants and children. However, the effects of anesthetics on the developing retina have not been evaluated.
In this study, we test the hypothesis that anesthetics induce neuronal apoptosis in the developing murine retina. We demonstrate that isoflurane activates caspase-3 in neurons within the inner nuclear layer (INL) of the retina via the intrinsic apoptosis pathway and within the ganglion cell layer (GCL) via both the intrinsic and extrinsic pathways. Furthermore, a systemically injected fluorescent probe readily crossed the blood-retinal barrier to bind to cells undergoing apoptosis in the INL. These findings lay the groundwork for the development of a noninvasive imaging technique to detect anesthesia-induced neuronal degeneration in vivo and indicate that isoflurane-induced neuronal apoptosis occurs in the developing retina.
METHODS
Animal Exposures
The care of the animals in this study was in accordance with the National Institutes of Health (NIH) and Institutional Animal Care and Use Committee guidelines. Study approval was granted by the Children’s National Medical Center. Six- to 8-week-old CD-1 pregnant female mice (20–30 g) were acquired (Charles River, Wilmington, MA) to yield newborn pups. On postnatal day 7 (P7), we exposed male CD-1 mouse pups to either air or isoflurane (2%) in air for 1 hour in a 7-L Plexiglas chamber (25 cm × 20 cm × 14 cm). The chamber had a port for fresh gas inlet and a port for gas outlet which was directed to a fume hood exhaust using standard suction tubing. Air (Air Products, Camden, NJ) was delivered through a variable bypass isoflurane vaporizer and exposure chamber at a flow rate of 8 to 12 L/min. Mice were kept warm with an infrared heating lamp (Cole-Parmer, Court Vernon Hills, IL). P7 was chosen because it is the time point of maximal vulnerability to anesthesia-induced neurodegeneration in the developing brain and because synaptogenesis peaks at day 7 in rodents.16,18 One hour exposure to 2% isoflurane activates brain capsase-3 in 7-day-old mice and represents a brief anesthetic exposure.21,22 Different cohorts of pups were randomly assessed either immediately postexposure or 5 hours post-exposure to isoflurane or air. Pups evaluated at the 5 hours were placed with their respective dams after exposure.
Fluorophore Injection
In a separate cohort of animals, pups were injected with 25 μL of either fluorescein isothiocyanate (FITC)-conjugated annexin V (BD Biosciences, San Jose, CA) or FITC-conjugated dextran (Sigma-Aldrich, St. Louis, MO) via the intraperitoneal route 4 hours after exposure to isoflurane or air. One hour postinjection, pups were euthanized via intra-peritoneal pentobarbital injection (150 mg/kg). Dose and timing of fluorophore injection were chosen based on previous work and pilot data.23
Cresyl Violet Staining
At the time of euthanasia, after pentobarbital injection (150 mg/kg, intraperitoneal), the animals were systemically perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) via left ventricle injection for 30 minutes. Both eyes were enucleated and then postfixed in additional fixative solution for 24 hours at 4°C. Paraffin-embedded whole eye serial sections were cut at a thickness of 6 μm, and individual sections were slide mounted and stained with cresyl violet for 30 minutes.
Terminal Deoxynucleotidyl Transferase-Mediated UTP Nick End-Labeling Staining
Five hours postexposure, after euthanasia, the animals were systemically perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) via left ventricle injection for 30 minutes. Both eyes were enucleated and then postfixed in additional fixative solution for 24 hours at 4°C. Paraffin-embedded whole eye sections were cut into 6-μm sections, slide mounted, and stained with terminal deoxynucleotidyl transferase (TdT) UTP nick end labeling (TUNEL). Sections were incubated in 0.5% Triton at room temperature, followed by proteinase K at 37°C, then immersed in TdT buffer (30 mmol/L Tris-HCl buffer, pH 7.2, 140 mmol/L sodium cacodylate, and 1 mmol/L cobalt chloride) at room temperature. This was followed by incubation with TdT and biotin-16-dUTP for 60 minutes at 37°C. The reaction was terminated with termination buffer (300 mmol/L sodium chloride with 30 mmol/L sodium citrate) at room temperature, followed by immersion in 3% hydrogen peroxide and 2% fetal bovine serum at room temperature. The sections were then covered with an avidin-biotin complex (1:200 dilution) for 30 minutes at room temperature, incubated with FITC-Avidin D for detection and counterstained with 4′,6-dimidino-2-phenylindole (DAPI). Six to 8 regions of interest were randomly selected under fluorescence microscopy (Olympus BX41 microscope; Olympus America Inc., Melville, NY) in a blinded manner (performed by VP) in 3 to 4 nonserial sections per mouse (15–30 μm from the optic nerve bundle). The numbers of TUNEL-positive nuclei were manually quantified in the outer nuclear layer (ONL), INL, and GCL per square micrometer at 10× magnification and corrected for the calculated area in the region of interest using NIH Image J v. 1.33 in 4 to 8 animals per group (based on sample sizes used previously22).
Immunohistochemistry
In separate cohorts, after euthanasia with pentobarbital injection (150 mg/kg, intraperitoneal), the animals were systemically perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) via left ventricle injection for 30 minutes either immediately after or 5 hours postexposure to isoflurane or air. Both eyes were enucleated and then postfixed in additional fixative solution for 24 hours at 4°C. Paraffin-embedded whole eye serial sections were cut at a thickness of 6 μm, and individual sections were slide mounted. Sections were deparaffinized and rehydrated with xylene, ethanol, and water. Antigen retrieval was performed with 0.01 M sodium citrate (pH 6.0) for 10 minutes at 100°C. Immunohistochemistry was performed using polyclonal rabbit anti-human–activated caspase-3 antibody (9661; Cell Signaling Technology, Beverly, MA), polyclonal rabbit anti-mouse–activated caspase-9 antibody (9509; Cell Signaling Technology), or monoclonal rabbit anti-mouse–activated caspase-8 antibody (8592; Cell Signaling Technology) with biotinylated secondary antibody (goat anti-rabbit; Cell Signaling Technology) and developed with diaminobenzidine. Ten to 12 regions of interest were randomly selected under light microscopy (Olympus BX41 microscope; Olympus America Inc.) in a blinded manner (performed by VP) in 3 to 4 nonserial sections per mouse (15–30 μm from the optic nerve bundle). The numbers of activated caspase-positive cells were manually quantified per square micrometer in the ONL, INL, and GCL at 10× magnification and corrected for the calculated area in the region of interest using NIH Image J v. 1.33 in 3 to 8 animals per group (based on sample sizes used previously22).
Immunoblot Analysis
After euthanasia with pentobarbital injection (150 mg/kg, intraperitoneal), retina mitochondria and cytosol were isolated by differential centrifugation immediately after exposure to isoflurane or air. Retina was harvested and homogenized in ice-cold H medium (70 mM sucrose, 220 mM mannitol, 2.5 mM HEPES, pH 7.4, and 2 mM EDTA). The homogenate was spun at 1500 g for 10 minutes at 4°C. Supernatant was removed and centrifuged at 10,000g for 10 minutes at 4°C. Cytosolic supernatant was collected, and pellet was resuspended in H medium and centrifuged again at 10,000g for 10 minutes at 4°C. Pellet was again resuspended in H medium, and mitochondrial and cytosolic protein concentrations subsequently determined using the method of Lowry.
Ten microgram samples of homogenized retina mitochondrial or cytosolic protein were subjected to SDS-acrylamide gel electrophoresis and immunoblotting. Blots were labeled with a primary rabbit polyclonal anti-equine cytochrome c antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA) or rabbit polyclonal anti-human Bax antibody (Millipore Corporation, Billerica, MA). Mitochondrial blots were labeled with rabbit monoclonal anti-human BCL-xL antibody (Cell Signaling Technology) or rabbit polyclonal anti-human BCL-2 antibody (GeneTex Inc., Irvine, CA). Blots were secondarily exposed to goat anti-rabbit Immunoglobulin G (IgG; Cell Signaling Technology). Mitochondrial protein loading was assessed with a primary monoclonal antibody to mouse Voltage-dependent Anion Channel (Molecular Probes, Eugene, OR) and secondarily exposed to rabbit anti-mouse IgG (Santa Cruz Biotechnology Inc.). Cytosolic protein loading was assessed with a primary monoclonal antibody to mouse glyceraldehyde 3-phosphate dehydrogenase (Thermo Fisher Scientific Inc., Rockford, IL) and secondarily exposed to rabbit anti-mouse IgG (Santa Cruz Biotechnology Inc.). Signal was detected with enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ), and density was measured using scanning densitometry. Five to 6 animals per cohort were evaluated (based on sample sizes used previously24).
Immunofluorescence
After euthanasia with pentobarbital injection (150 mg/kg, intraperitoneal), the animals were systemically perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) via left ventricle injection for 30 minutes 5 hours after exposure to isoflurane or air. Both eyes were enucleated and then postfixed in additional fixative solution for 24 hours at 4°C. Paraffin-embedded whole eye serial sections were cut at a thickness of 6 μm, and individual sections were slide mounted. Sections were deparaffinized and rehydrated with xylene, ethanol, and water. Antigen retrieval was performed with 0.01 M sodium citrate (pH 6.0) for 10 minutes at 100°C. Immunofluorescence was performed using monoclonal mouse anti-human PKC α antibody, monoclonal mouse anti-rat HPC-1 antibody, polyclonal goat anti-human calbindin antibody, or polyclonal goat anti-human Glutamate Aspartate Transporter (GLAST) antibody (Santa Cruz Biotechnology Inc.). Trimethylrhoadmine isothiocyanate (TRITC)-conjugated goat anti-mouse IgG secondary antibody (Jackson Immunoresearch Laboratories, Inc., West Grove, PA) was used for PKC and HPC-1, and donkey anti-goat IgG secondary antibody (Jackson Immunoresearch Laboratories, Inc.) for calbindin and GLAST. Double immunofluorescence was performed with polyclonal rabbit anti-human–activated caspase-3 antibody (9661; Cell Signaling Technology) and either FITC-conjugated goat anti-rabbit IgG secondary antibody (Jackson Immunoresearch Laboratories, Inc.) (for PKC and HPC-1) or FITC-conjugated donkey anti-rabbit IgG secondary antibody (Jackson Immunoresearch Laboratories, Inc.) (for calbindin and GLAST). Sections were counter-stained with DAPI, and colocalization of fluorescence was assessed at 40× and 100× magnification (oil immersion) using inverted confocal microscopy (Olympus Fluoview FV1000 confocal laser scanning microscope; Olympus America Inc.) in 3 to 4 animals per group (based on sample sizes used previously for immunohistochemistry22). Excitation/emission settings were 488/515 to 700 nm for FITC, 559/515 to 700 nm for TRITC, and 405/420 to 700 nm for DAPI. Six to 8 regions of interest were randomly selected in a blinded manner (performed by VP) in 3 to 4 nonserial sections per mouse (15–30 μm from the optic nerve bundle). The number of activated caspase-3-positive cells was manually quantified per square micrometer in INL at 40× magnification and corrected for the calculated area in the region of interest using NIH Image J v. 1.33. The number and percentage of cells with colocalized fluorescence were then manually quantified.
For cohorts injected with FITC-conjugated annexin V or dextran, pups were euthanized with pentobarbital injection (150 mg/kg, intraperitoneal) 1 hour after intraperitoneal injection. The animals were systemically perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) via left ventricle injection for 30 minutes, and eyes were postfixed in additional fixative solution for 24 hours at 4°C. Paraffin-embedded whole eye serial sections were cut at a thickness of 6 μm, and individual sections were slide mounted. Immunofluorescence was performed with polyclonal rabbit anti-human–activated caspase-3 antibody (9661; Cell Signaling Technology) and TRITC-conjugated goat anti-rabbit IgG secondary antibody (Jackson Immunoresearch Laboratories, Inc.). Sections were counterstained with DAPI, and colocalization of fluorescence was assessed at 40× and 100× magnification (oil immersion) using inverted confocal microscopy in 5 to 6 animals per group (based on sample sizes used previously for immunohistochemistry22). Six to 8 regions of interest were randomly selected in a blinded manner (performed by V.P.) in 3 to 4 nonserial sections per mouse (15–30 μm from the optic nerve bundle). The number of activated caspase-3-positive cells and FITC-positive cells was manually quantified per square micrometer at 40× magnification and corrected for the calculated area in the region of interest using NIH Image J v. 1.33. The number and percentage of cells with colocalized fluorescence was then quantified.
Statistical Analysis
Although we did not perform a power analysis for this investigation, we used sample sizes from previous work for each end point.22,24 Data are presented in bar plots as mean ± SD.
Statistical significance was assessed using a 2-sample t test for the 2-group comparisons for immunohistochemistry, immunofluorescence, immunoblot, and TUNEL data. One-sided t test was used for TUNEL, immunohistochemistry, and immunoblot data, given that only unidirectional change for each parameter would be considered relevant after isoflurane exposure. This is based on evidence from previous work indicating that anesthetics induce apoptosis and do not decrease it.1–5,22 Furthermore, natural apoptosis occurs in 2% (or less) of neurons at any given time during development.18 Because this value is physiologically close to 0, a reduction <0 would not be likely. In addition, anesthetics are known to induce apoptosis in developing neurons by activating caspases (not inactivating them) and causing cytochrome c release from mitochondria (not enhancing cytochrome c levels in mitochondria).6 Thus, with regard to TUNEL, immunohistochemistry, and immunoblot analyses, results from the experimental group could not be any less than that of the controls. Therefore, 1-tailed t test was used. However, 2-sided t test was used for immunofluorescence. Significance for each t test was set at P < 0.01. Statistical analyses were conducted using SAS 9.3 (SAS Institute, Inc.).
RESULTS
Isoflurane Induces Apoptosis and Activates Caspase-3 in the Developing Retina
Isoflurane has previously been shown to induce widespread neuronal apoptosis in the developing rodent brain coincident with the peak in synaptogenesis.25 Thus, we determined whether isoflurane induces PCD and activates caspase-3 in the developing retina with TUNEL staining and immunohistochemistry on slide-mounted retinal sections. Different cohorts of pups were evaluated immediately after 1-hour exposure to isoflurane in air (activated caspase-3) or 5 hours after exposure (both TUNEL and activated caspase-3) on P7. Air-exposed cohorts served as controls.
Isoflurane significantly increased the number of TUNEL-positive nuclei in the INL (P < 0.0001), perhaps with a trend toward significance in the GCL and ONL (Fig. 1). The number of activated caspase-3-positive cells significantly increased in the INL after isoflurane exposure at the 5-hour time point versus air-exposed controls (P = 0.008) (Fig. 2). Given our sample size, we could not establish whether activated caspase-3 levels were increased in INL immediately after isoflurane exposure compared with air exposure (P = 0.138; 95% confidence interval [CI], −1.51 to 4.40). Isoflurane may have also increased the number of activated caspase-3-positive cells in the GCL at both time points after exposure compared with air-exposed controls, based on a trend toward significance (Fig. 2). Activated caspase-3 levels remained relatively unchanged in the ONL after isoflurane exposure (0 hour versus air: P = 0.765; 95% CI, −0.43 to 0.57; 5 hours versus air: P = 0.076; 95% CI, −0.43 to 2.05).
Figure 1.
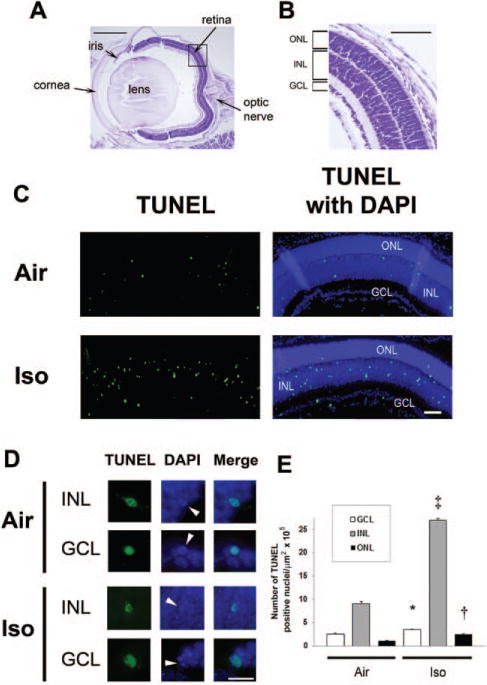
Isoflurane induces apoptosis in the developing retina. Terminal deoxynucleotidyl transferase–mediated UTP nick end-labeling (TUNEL) assays were performed on retinal sections 5 hours postexposure to isoflurane or air. A, For orientation, a cresyl violet– stained section from a mouse eye imaged at 4× is provided. The major structures including the retina and optic nerve are labeled. Scale bar, 500 μm. A magnified image of the retina within the boxed section is shown at 10× (B). The outer nuclear layer (ONL), inner nuclear layer (INL), and ganglion cell layer (GCL) are labeled. Scale bar, 250 μm. C, Representative sections of TUNEL assays with and without 4′,6-dimidino-2-phenylindole (DAPI) after exposure to either air or isoflurane (Iso) at 10× are depicted. Green TUNEL-positive nuclei are visible. Scale bar, 100 μm. D, Representative images of individual TUNEL-positive nuclei within the INL and GCL magnified at 60× are demonstrated. DAPI staining of TUNEL-positive nuclei undergoing apoptosis are indicated (white arrowheads), and colocalization of fluorescence is shown in the merged images. Scale bar, 25 μm. E, Quantification of TUNEL-positive nuclei is demonstrated. Values are expressed as means + SD. N = 4–8 animals per cohort. *P = 0.022 versus air-exposed cohort, 95% confidence interval of the difference between groups (0.03–1.99). †P = 0.011 versus air-exposed cohort, 95% confidence interval (0.38–3.18). ‡P < 0.0001 versus air-exposed cohort.
Figure 2.
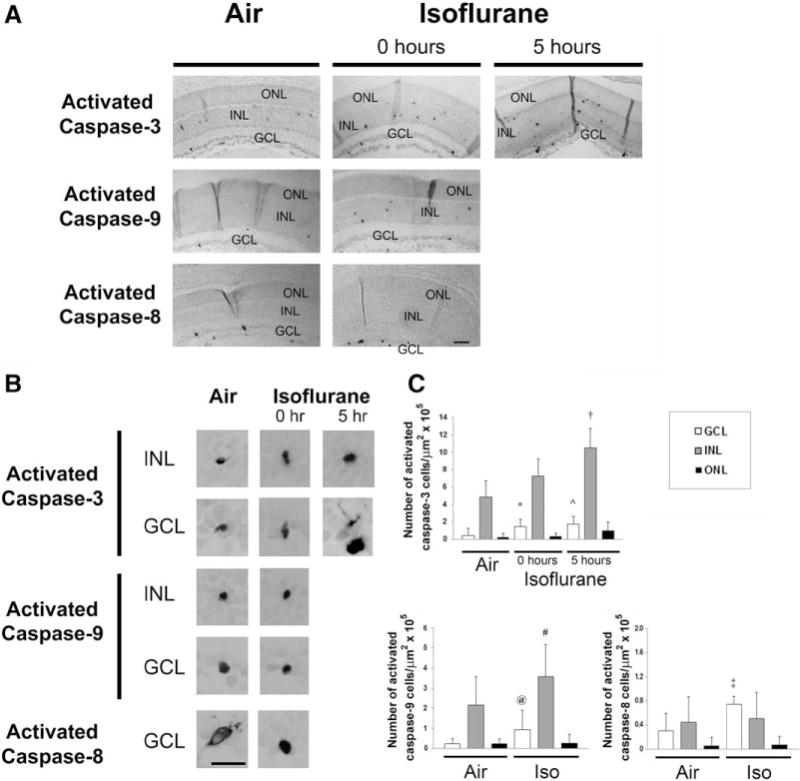
Isoflurane induces caspase activation in the developing retina. Immunohistochemistry for activated caspase-3, caspase-9, and caspase-8 was performed on retinal sections immediately after (0 hour) and 5 hours postexposure to isoflurane (Iso). A, Representative sections imaged at 10× after 1-hour exposure to air or isoflurane are depicted. Activated caspase-3-, caspase-9-, and caspase-8-stained cells are visible within the retinal layers. The outer nuclear layer (ONL), inner nuclear layer (INL), and ganglion cell layer (GCL) are labeled. Scale bar, 100 μm. B, Representative images of activated caspase-positive cells undergoing degeneration within the INL and GCL are magnified at 60×. Scale bar, 25 μm. C, Quantification of activated caspase-3-, caspase-9-, and caspase-8-stained cells is demonstrated. Values are expressed as means + SD. N = 3–8 animals per cohort. †P = 0.008 versus air-exposed cohort. *P = 0.029 versus air-exposed cohort; 95% confidence interval (CI), −0.04 to 2.00. ˆP = 0.025 versus air-exposed cohort; 95% CI, −0.01 to 2.56. @P = 0.005 versus air-exposed cohort. #P = 0.046 versus air-exposed cohort; 95% CI, −0.31 to 3.43. ‡P = 0.001 versus air-exposed cohort.
Isoflurane Activates the Intrinsic Apoptosis Pathway in the INL and Both the Intrinsic and Extrinsic Pathways in the GCL
Although the exact upstream mechanisms that initiate anesthesia-induced neurodegeneration are not completely known, the downstream mechanism of neurotoxicity appears to be caspase activation via the mitochondrial pathway of apoptosis followed by activation of the death receptor pathway and neurotrophic factor-activated pathways.6,25 Isoflurane-induced activation of the intrinsic pathway is mediated by downregulation of the antiapoptotic proteins, such as BCL-xL and BCL-2, permeabilization of the outer mitochondrial membrane after Bax translocation, and subsequent release of cytochrome c from mitochondria into the cytosol.6,25 Release of cytochrome c results in formation of the apoptosome and autoactivation of caspase-9, which, in turn, activates the effector caspase, caspase-3.26 The extrinsic apoptosis pathway is activated after binding of various apoptosis-inducing ligands to death receptors which then recruit and activate caspase-8.27 Caspase-8 then activates caspase-3.27 To determine the downstream mechanism of isoflurane-induced activation of caspase-3 in the developing retina, we performed immunohistochemistry for activated caspase-9 and caspase-8 immediately after 1-hour exposure to isoflurane or air on slide-mounted retinal sections. We isolated mitochondria and cytosol from retina and performed immunoblot analyses for the various proapoptotic and antiapoptotic mediators.
Consistent with the activation of the mitochondrial apoptosis pathway, isoflurane significantly increased the number of activated caspase-9-positive cells in the GCL (P = 0.005) (Fig. 2). There may have also been an increase in activated caspase-9 levels in the INL, based on a trend toward significance immediately after exposure (Fig. 2). However, isoflurane had no effect on the levels of activated caspase-9 in the ONL (P = 0.500; 95% CI, −0.35 to 0.35) (Fig. 2). Steady-state levels of Bax were relatively unchanged in the cytosolic and mitochondrial fractions after isoflurane exposure compared with controls (P = 0.101; 95% CI, −0.03 to 0.12 and P = 0.217; 95% CI, −0.19 to 0.41, respectively) suggesting that Bax translocation remained intact (Fig. 3). However, there may have been a decrease in steady-state levels of the antiapoptotic mediator, BCL-xL, in mitochondria after isoflurane exposure, based on a trend toward significance (Fig. 3). This was associated with a significant decrease in steady-state levels of mitochondrial cytochrome c after isoflurane exposure (P = 0.004) and, perhaps, an increase in cytosolic levels with a trend toward a significance indicating the release of cytochrome c from mitochondria (Fig. 3). Although steady-state levels of the antiapoptotic protein, BCL-2, slightly decreased after isoflurane exposure, this difference was not statistically significant (P = 0.073; 95% CI, −0.07 to 0.38) (Fig. 3).
Figure 3.
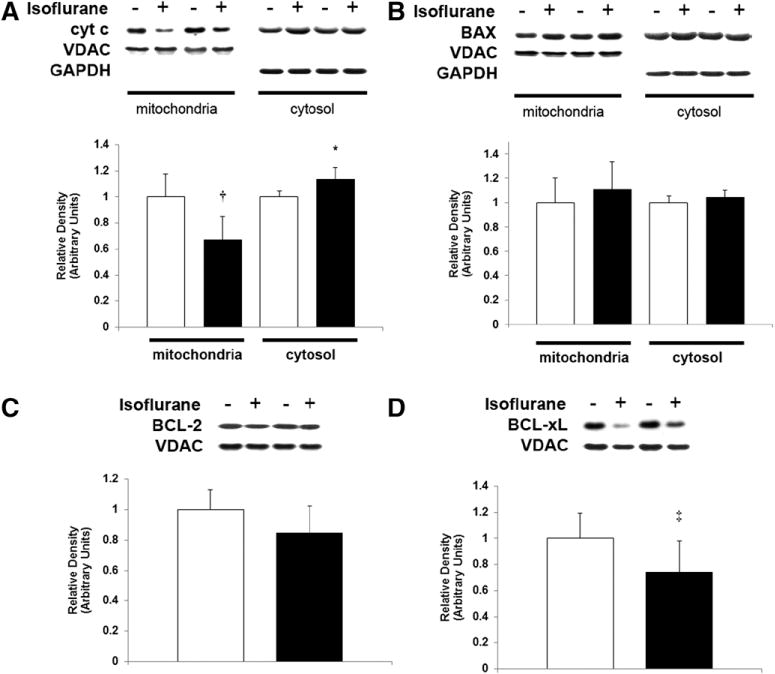
Isoflurane induces cytochrome c release from mitochondria in the developing retina. Mitochondria and cytosol were isolated from retina immediately after 1-hour exposure to air (−) or isoflurane (+). Representative immunoblots for (A) cytochrome c (cyt c), (B) Bax, (C) BCL-2, and (D) BCL-xL are depicted. Voltage-dependent Anion Channel and glyceraldehyde 3-phosphate dehydrogenase were used as loading controls for mitochondrial and cytosolic protein, respectively. Graphical representations are shown. White bars are air-exposed controls and black bars are isoflurane-exposed cohorts. Air-exposed values were arbitrarily set to 1. Values are expressed as means + SD. N = 5–6. *P = 0.015 versus air-exposed cohort; 95% CI, 0.02–0.25. †P = 0.004 versus air-exposed cohort. ‡P = 0.033 versus air-exposed cohort; 95% CI, −0.54 to 0.02.
With regard to the extrinsic pathway, activated caspase-8 levels significantly increased in the GCL after isoflurane exposure (P = 0.001) (Fig. 2). Although activated caspase-8 was detectable in both the INL and ONL, there was no change in levels after exposure to isoflurane compared with air-exposed controls (P = 0.486; 95% CI, −0.61 to 0.63 and P = 0.496; 95% CI, −0.24 to 0.24, respectively).
Isoflurane Induces Apoptosis in Amacrine Neurons in the Developing Retina
The retina is made up of several different types of neurons and glia. Thus, we attempted to determine which specific retinal cells undergo apoptosis after isoflurane exposure with double-labeling immunofluorescence on slide-mounted retinal sections 5 hours postexposure. We assessed for colocalization of activated caspase-3 with various retina cell–specific protein markers in isoflurane-exposed and air-exposed cohorts. Activated caspase-3 fluorescence in the INL colocalized with that of the bipolar neuron-specific cytosolic protein, PKC, with the amacrine neuron-specific cytosolic protein, HPC-1, and with the horizontal cell-specific protein, calbindin, in both air-exposed and isoflurane-exposed cohorts (Fig. 4). There may have been an increase in the number and percentage of cells positive for both activated caspase-3 and HPC-1 after isoflurane exposure compared with air-exposed controls, based on a trend toward significance (Fig. 4). It should be noted that we only focused on the INL and did not assess for displaced amacrine cell staining in the GCL. There was no significant effect of isoflurane versus air exposure on the number or percentage of cells positive for both activated caspase-3 and PKC (P = 0.123; 95% CI, −1.78 to 11.02 and P = 0.721; 95% CI, −16.11 to 21.67, respectively) or calbindin (P = 0.749; 95% CI, −14.00 to 18.44 and P = 0.670; 95% CI, −22.36 to 32.36, respectively) (Fig. 4). Our findings suggest that caspase-3 was activated in the cytosol of amacrine cells within the INL after exposure to isoflurane and that caspase activation in bipolar neurons likely represents natural PCD.
Figure 4.
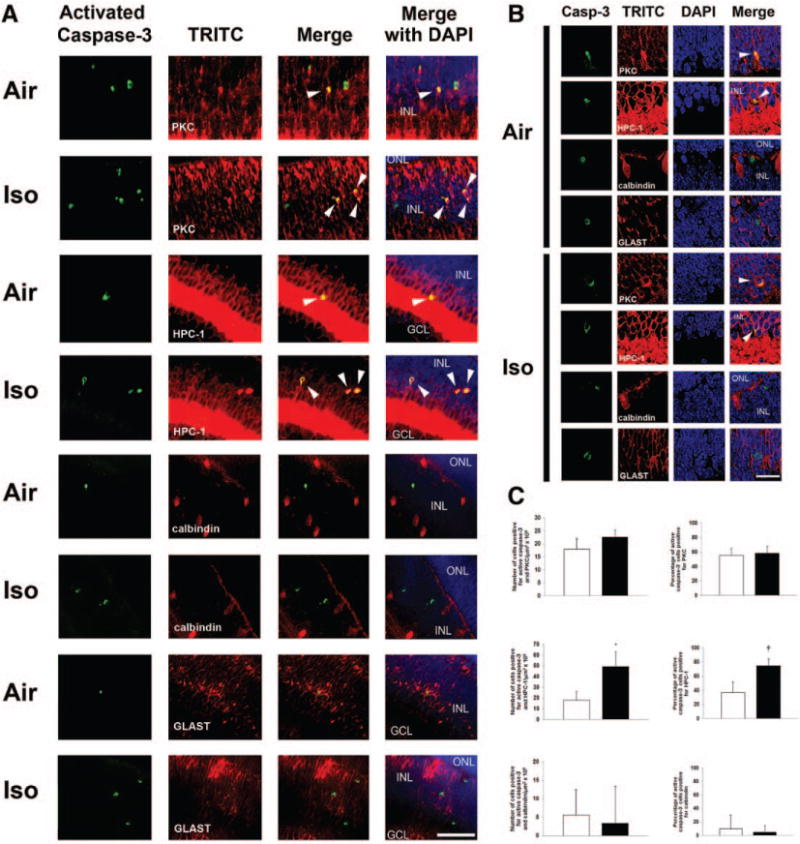
Bipolar neurons and amacrine cells undergo developmental programmed cell death within the inner nuclear layer (INL) on P7 while isoflurane induces apoptosis only in amacrine cells. Double-labeling immunofluorescence was performed on retinal sections 5 hours after exposure to air or isoflurane (Iso). A, Representative INL sections imaged at 40× are depicted. Bipolar neurons were labeled with PKC. Amacrine cells were labeled with HPC-1. Horizontal cells were labeled with calbindin. Müller glia were labeled with GLAST. Green activated capsase-3 staining indicates cells undergoing apoptosis. Arrowheads indicate cells with colocalization of fluorescence with activated caspase-3. Activated caspase-3 staining was rarely associated with calbindin (horizontal cells) and did not colocalize with GLAST in the cytosol of Müller glia. Scale bar, 50 μm. The outer nuclear layer (ONL), INL, and ganglion cell layer (GCL) are labeled. Sections are oriented with ONL at the top of the image and GCL at the bottom. B, Representative images magnified at 100× demonstrating colocalization (arrowheads) of activated caspase-3 (casp-3) green fluorescence with trimethylrhoadmine isothiocyanate (TRITC) (red) fluorescence of PKC in the cytosol of bipolar neurons and HPC-1 of amacrine neurons in air-exposed and isoflurane-exposed cohorts. There was rare colocalization of fluorescence of activated caspase-3 with calbindin of horizontal cells and no colocalization with GLAST of Müller glia. Scale bar, 20 μm. C, Quantification of cells positive for both activated caspase-3 and TRITC within the INL of air-exposed (white bars) and isoflurane-exposed (black bars) cohorts is demonstrated. Percentage of activated caspase-3-positive cells demonstrating colocalization is also depicted. Values are expressed as means + SD. *P = 0.028 versus air-exposed cohort; 95% CI, 5.5–57.2. †P = 0.022 versus air-exposed cohort; 95% CI, 8.8–67.1. PKC = bipolar neuron-specific cytosolic protein; HPC-1 = amacrine neuron-specific cytosolic protein; DAPI = 4′,6-dimidino-2-phenylindole.
FITC-Annexin V Binds to Cells Undergoing Apoptosis in the Developing Retina
Translocation of phosphatidylserine (PS) from the inner leaflet of the plasma membrane to the outer leaflet is one of the earliest features of apoptosis.28 Annexin V, a 35-kD calcium-dependent phospholipid-binding protein, binds to PS with high affinity and has been used to detect apoptosis in different cell and tissue types.28 As a first step in developing a potential fluorescent probe, we aimed to determine whether the fluorophore, FITC-annexin V, could cross the blood-retinal barrier and bind to cells undergoing apoptosis in the retina. We injected FITC-annexin V via the intraperitoneal route 4 hours postexposure to isoflurane or air. One hour later, we assessed for FITC fluorescence and colocalization with activated caspase-3 on slide-mounted retinal sections. FITC-dextran served as a control fluorophore. After intra-peritoneal injection, FITC-annexin V was readily detected in the INL of both air-exposed and isoflurane-exposed mice and colocalized with activated caspase-3-positive cells (Fig. 5). However, FITC-dextran was not visible (Fig. 5). The number of FITC-annexin V–positive cells significantly increased in the retina after isoflurane exposure compared with air-exposed controls (P = 0.002) (Fig. 5C). Importantly, all FITC-annexin V–positive cells were positive for activated caspase-3. However, not all caspase-3-positive cells were FITC-annexin V positive. Only about 20% of cells that were positive for activated caspase-3 also demonstrated FITC-annexin V fluorescence in the air-exposed cohort (95% CI, 17.70–25.29, calculated for the single proportion using the Newcombe-Wilson test) (Fig. 5D). However, isoflurane exposure significantly increased the percentage of cells in the retina with colocalized fluorescence (activated caspase-3 and FITC-annexin V) approximately 2-fold versus air-exposed controls (P = 0.002) (Fig. 5D). This suggests that PS translocation occurred later than activation of caspase-3 in the apoptosis pathway but that FITC-annexin V was able to gain access to the retina after systemic injection and bind specifically to cells undergoing PCD.
Figure 5.
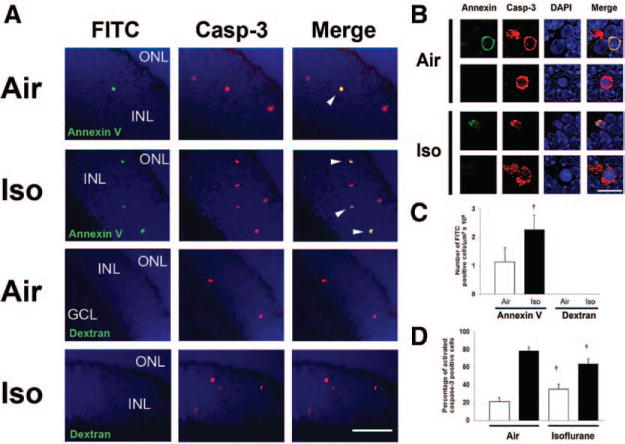
Systemically injected fluorescein isothiocyanate (FITC)-conjugated annexin V labels cells undergoing apoptosis in the developing retina. A, Mouse pups were injected with either FITC-annexin V or FITC-dextran 4 hours postexposure to either isoflurane or air. Immunohistochemistry for activated caspase-3 was performed on retinal sections 1 hour postinjection. Representative sections imaged at 40× are depicted. Scale bar, 50 μm. Green annexin V–positive cells and red activated caspase-3 (casp-3)-positive cells are visible within the inner nuclear layer (INL) of air-exposed and isoflurane (Iso)-exposed animals. Merged images demonstrate colocalization of FITC-annexin V with activated caspase-3 in the INL of both air-exposed and isofluane-exposed cohorts (arrowheads). B, Representative images of cells fluorescent for both activated caspase-3 and annexin V magnified at 100× are demonstrated. Merged images demonstrate colocalization of fluorescence. All FITC-annexin V–positive cells demonstrated colocalized fluorescence with activated caspase-3. Activated caspase-3-positive cells lacking annexin V fluorescence are also shown. Scale bar, 10 μm. C, Quantification of FITC-labeled cells after annexin V or dextran injection in air-exposed and isoflurane (Iso)-exposed cohorts is demonstrated. Values are expressed as means + SD. N = 5–6 animals per cohort. †P = 0.002 versus air-exposed cohort. D, Percentage of activated caspase-3-positive cells that demonstrated FITC-annexin V fluorescence (white bars) and that lacked FITC fluorescence (activated caspase-3 only; black bars) in both air-exposed and isoflurane-exposed cohorts is demonstrated. Values are expressed as means + SD. N = 5–6 animals per cohort. †P = 0.002 versus air-exposed cohort. GCL = ganglion cell layer; ONL = outer nuclear layer; DAPI = 4′,6-dimidino-2-phenylindole.
DISCUSSION
In this study, we demonstrate that isoflurane induces apoptosis in the developing murine retina on P7. Our findings indicate that, as with the developing brain, one of the downstream mechanisms of anesthesia-induced neurotoxicity in the retina is the activation of the intrinsic apoptosis pathway. This is evidenced by the isoflurane-induced release of cytochrome c from retinal mitochondria, caspase-9 activation in the GCL immediately after exposure, and a trend toward caspase-9 activation in the INL with subsequent activation of caspase-3.
Importantly, the number of TUNEL-positive nuclei exceeded the number of activated caspase-3-positive cells in all 3 layers of the retina in both air-exposed and isoflurane-exposed cohorts. This likely relates to the temporal profile of the activation of the various components of the intrinsic apoptosis pathway.29 Caspase-3 is activated at about 3 hours postrelease of cytochrome c and peaks at around 8 hours. TUNEL-positive staining begins between 3 to 8 hours post-stimulus and peaks at 24 hours. Because apoptosis can occur from 24 to 48 hours in some cells, TUNEL assays will identify cells that begin DNA fragmentation due to isoflurane exposure as well as cells that were already in the process of undergoing natural PCD 24 or even 48 hours before exposure. Thus, the number of TUNEL-positive cells will be greater than activated caspase-3. Furthermore, because developmental PCD is also mediated by the intrinsic apoptosis pathway, it is unknown whether cell death results from an isoflurane-induced acceleration of the natural physiologic process within the retina or if cells destined to survive are targeted by the anesthetic. Such a question will need to be further explored.
In addition to activating the intrinsic pathway, isoflurane also activated caspase-8 in the GCL after exposure, suggesting activation of the extrinsic pathway. Although activated caspase-8 was detectable within the INL, the GCL was the only layer to demonstrate a significant increase in levels after anesthetic exposure. It is unknown why caspase-8 became activated specifically in the GCL; however, the finding suggests that isoflurane may activate the death receptor pathway in addition to the intrinsic pathway in retinal ganglion cells. Region-specific activation of the extrinsic apoptosis pathway has been demonstrated in the developing brain after anesthetic exposure.25 For example, caspase-8 became activated in neurons within the parietal and occipital cortex of 7-day-old rats after isoflurane exposure while nuclei of the anterior thalamus showed minimal activation.25 In addition, the phenomenon of GCL-specific caspase-8 activation has been demonstrated in the retina after optic nerve transection.30 Thus, retinal ganglion cells may be susceptible to activation of both pathways after exposure to proapoptotic stimuli.
Tumor necrosis factor (TNF)-α is a proinflammatory cytokine that initiates the extrinsic apoptosis pathway.27 Retinal ganglion cells express the R1 receptor of TNF-α.31 Thus, these neurons may be uniquely susceptible to death receptor ligands. Isoflurane has previously been shown to increase TNF-α levels in the brain after exposure.32 Furthermore, in experimental glaucoma, Müller glia– derived TNF-α has been shown to be a potent mediator of bystander retinal ganglion cell death.31 Thus, although we did not measure TNF-α production in this study, it is possible that isoflurane directly induced TNF-α production in the GCL, leading to activation of the death receptor pathway in ganglion cells. Alternatively, since Müller cells can become secondarily activated after primary ganglion cell death, it is possible that isoflurane directly activated the intrinsic pathway within retinal ganglion cells, leading to Müller glia–derived TNF-α production and secondary ganglion cell death via activation of the extrinsic apoptosis pathway.31
As with the developing brain, the cells in the retina undergoing natural PCD, and those that demonstrate susceptibility to the proapoptotic effect of isoflurane on P7 appear to be neurons.33 This is evidenced by colocalization of fluorescence of activated caspase-3 with that of neuron-specific proteins in both air-exposed and isoflurane-exposed cohorts. Activated caspase-3 labeling in the cytosol of retinal cells undergoing apoptosis colocalized with a bipolar neuron-specific cytosolic protein (PKC), an amacrine neuron-specific cytosolic protein (HPC-1), and rarely with a horizontal neuron-specific cytosolic protein (calbindin) within the INL of both experimental cohorts. Only the number and percentage of cells positive for both HPC-1 and activated caspase-3 increased after isoflurane exposure. These findings indicate that bipolar and amacrine neurons undergo natural PCD on P7 and that amacrine cells are the potential targets of isoflurane-induced apoptosis in the INL of the developing retina. This is consistent with the work demonstrating that most neurons undergoing physiologic apoptosis in the developing rodent retina on P7 are amacrine and bipolar cells within the INL and ganglion cells within the GCL.19,34–36 It is unclear why neurons within the ONL were relatively spared from isoflurane-induced neurodegeneration, but this may relate to the fact that photoreceptor cell death does not peak until later in development.19,35,36 Thus, developmental PCD may play a role in the susceptibility of neurons to anesthesia-induced neurotoxicity. Furthermore, the lack of anesthesia-induced photoreceptor cell death may indicate some level of protection and may provide mechanistic insights for future therapies with further investigation.
Because the retina can be directly visualized noninvasively, the opportunity exists to develop a technique to image anesthesia-induced retinal neurotoxicity in vivo. After intraperitoneal injection, FITC-annexin V was readily visible in the INL of the retina and colocalized with activated caspase-3-positive cells in both air-exposed and isoflurane-exposed mice. Our findings indicate that systemically injected FITC-annexin V crosses the blood-retina barrier and binds to cells undergoing apoptosis, providing a potential opportunity to image natural PCD and isoflurane-induced apoptosis in the developing retina. A method has recently been developed using fluorescent-labeled annexin V to detect neuronal apoptosis in the retina in vivo with noninvasive confocal laser scanning ophthalmoscopy.23 This approach has been shown to enable visualization of single-cell apoptosis within the retina in animal models of glaucoma, optic nerve transection, and after intravitreal injection of staurosporine and is currently in clinical trials in adults.23 In order for such a technique to be of value in the context of anesthesia-induced toxicity, however, a method will need to be developed to differentiate cells dying from natural PCD versus those dying from anesthesia-induced apoptosis. Furthermore, FITC-annexin V labeled only 40% of activated caspase-3-positive cells after isoflurane exposure. Differences in fluorescent-labeled annexin V staining after isoflurane exposure versus models of ocular hypertension and optic nerve transection, for example, likely relate to the route of fluorophore injection, the degree of apoptosis induced by the stimulus, and the retinal tissue layer affected. We injected FITC-annexin V systemically, whereas others have injected annexin V directly into the intravitreal fluid.23 Furthermore, isoflurane exposure on P7 affected neurons mostly within the INL, whereas experimental glaucoma and other models of retinal neurodegeneration appear to affect primarily retinal ganglion cells within the GCL.23 Intravitreal injection permits annexin V to access retinal ganglion cells directly, whereas intraperitoneal injection requires that the fluorophore cross the blood-retinal barrier and gain access to the inner retinal layers. Thus, further development and refinement of this approach will likely be necessary. However, in time, we may be able to translate this novel approach to the human condition to determine whether neuronal apoptosis occurs in the retina after anesthesia exposure.
It should be noted that attempting to extrapolate experimental findings in rodents to humans has major limitations.37 It has been suggested by some that the structural maturity of the murine brain on P7 corresponds to that of the human fetus at 36 weeks postconception, whereas others, using mathematical modeling,37–39 predict that P7 correlates to an even earlier time point in human gestation. In the developing murine retina, with regard to synapse elimination, P7 is thought to correspond to a time point just before birth in humans.40 Thus, the findings presented here may only have direct relevance for the fetal retina and may not necessarily be applicable to the postnatal developing retina. Therefore, in order for these findings to have a clinical impact, future investigation should explore later stages in development more relevant to infants and children.
Furthermore, with regard to developing an imaging tool for anesthesia-induced neurotoxicity, retinal cell apoptosis will carry importance only if there is correlation with the degree of neurodegeneration in the brain. Although we did not directly compare apoptosis in the retina with cell death in the brain of the same animals after isoflurane exposure, we can make some comparisons with a recently studied cohort.22 Compared with the neocortex, hippocampus, and hypothalamus/thalamus, the numbers of TUNEL-positive nuclei and activated caspase-3-positive cells in the retina were relatively increased after an identical 1-hour exposure to isoflurane.22 Although there are limitations with such a comparison, the relative greater degree of neurodegeration seen in the retina may relate to the smaller area of this very thin structure compared with many of the brain regions previously examined but may be because anesthetics are known to affect specific CNS regions differently.22
Despite the limitations, the importance of this work is that we demonstrate for the first time that anesthetics induce neuronal apoptosis in the developing retina. Because the retina provides a window to the CNS and can be imaged noninvasively, our findings create an opportunity to explore anesthesia-induced neuronal degeneration in the developing retina as a potential surrogate for neurotoxicity in the brain. Ultimately, we may be able to develop a noninvasive imaging modality to determine whether anesthesia-induced neuronal apoptosis occurs in infants and children.
Acknowledgments
Funding: NIH/NIGMS R01GM103842-01 (RJL).
Footnotes
The authors declare no conflicts of interest.
Reprints will not be available from the authors.
DISCLOSURES
Name: Ying Cheng, MS.
Contribution: This author helped conduct the study and analyze the data.
Attestation: Ying Cheng has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Linda He.
Contribution: This author helped conduct the study.
Attestation: Linda He has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Vidhya Prasad, MS.
Contribution: This author helped conduct the study.
Attestation: Vidhya Prasad has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Shuang Wang, PhD.
Contribution: This author helped analyze the data and write the manuscript.
Attestation: Shuang Wang has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Richard J. Levy, MD.
Contribution: This author helped design the study, conduct the study, analyze the data, and write the manuscript.
Attestation: Richard J. Levy has seen the original study data, reviewed the analysis of the data, approved the final manuscript, and is the author responsible for archiving the study files.
This manuscript was handled by: Gregory J. Crosby, MD.
References
- 1.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurode-generation in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stefovska VG, Uckermann O, Czuczwar M, Smitka M, Czuczwar P, Kis J, Kaindl AM, Turski L, Turski WA, Ikonomidou C. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann Neurol. 2008;64:434–45. doi: 10.1002/ana.21463. [DOI] [PubMed] [Google Scholar]
- 3.Istaphanous GK, Loepke AW. General anesthetics and the developing brain. Curr Opin Anaesthesiol. 2009;22:368–73. doi: 10.1097/aco.0b013e3283294c9e. [DOI] [PubMed] [Google Scholar]
- 4.Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, Dissen GA, Creeley CE, Olney JW. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–41. doi: 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Istaphanous GK, Howard J, Nan X, Hughes EA, McCann JC, McAuliffe JJ, Danzer SC, Loepke AW. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology. 2011;114:578–87. doi: 10.1097/ALN.0b013e3182084a70. [DOI] [PubMed] [Google Scholar]
- 6.Olney JW, Young C, Wozniak DF, Ikonomidou C, Jevtovic-Todorovic V. Anesthesia-induced developmental neuroapoptosis. Does it happen in humans? Anesthesiology. 2004;101:273–5. doi: 10.1097/00000542-200408000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Rizzi S, Ori C, Jevtovic-Todorovic V. Timing versus duration: determinants of anesthesia-induced developmental apoptosis in the young mammalian brain. Ann N Y Acad Sci. 2010;1199:43–51. doi: 10.1111/j.1749-6632.2009.05173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR, Weaver AL, Warner DO. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiMaggio C, Sun LS, Kakavouli A, Byrne MW, Li G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J Neurosurg Anesthesiol. 2009;21:286–91. doi: 10.1097/ANA.0b013e3181a71f11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flick RP, Katusic SK, Colligan RC, Wilder RT, Voigt RG, Olson MD, Sprung J, Weaver AL, Schroeder DR, Warner DO. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011;128:e1053–61. doi: 10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalkman CJ, Peelen L, Moons KG, Veenhuizen M, Bruens M, Sinnema G, de Jong TP. Behavior and development in children and age at the time of first anesthetic exposure. Anesthesiology. 2009;110:805–12. doi: 10.1097/ALN.0b013e31819c7124. [DOI] [PubMed] [Google Scholar]
- 12.Sprung J, Flick RP, Katusic SK, Colligan RC, Barbaresi WJ, Bojanić K, Welch TL, Olson MD, Hanson AC, Schroeder DR, Wilder RT, Warner DO. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin Proc. 2012;87:120–9. doi: 10.1016/j.mayocp.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ing C, DiMaggio C, Whitehouse A, Hegarty MK, Brady J, von Ungern-Sternberg BS, Davidson A, Wood AJ, Li G, Sun LS. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics. 2012;130:e476–85. doi: 10.1542/peds.2011-3822. [DOI] [PubMed] [Google Scholar]
- 14.Chan WY, Lorke DE, Tiu SC, Yew DT. Proliferation and apoptosis in the developing human neocortex. Anat Rec. 2002;267:261–76. doi: 10.1002/ar.10100. [DOI] [PubMed] [Google Scholar]
- 15.Blomgren K, Leist M, Groc L. Pathological apoptosis in the developing brain. Apoptosis. 2007;12:993–1010. doi: 10.1007/s10495-007-0754-4. [DOI] [PubMed] [Google Scholar]
- 16.Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–33. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dabrowski A, Umemori H. Orchestrating the synaptic network by tyrosine phosphorylation signalling. J Biochem. 2011;149:641–53. doi: 10.1093/jb/mvr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanno H, Shen X, Kuru N, Bormuth I, Bobsin K, Gardner HA, Komljenovic D, Tarabykin V, Erzurumlu RS, Tucker KL. Control of postnatal apoptosis in the neocortex by RhoA-subfamily GTPases determines neuronal density. J Neurosci. 2010;30:4221–31. doi: 10.1523/JNEUROSCI.3318-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vecino E, Hernández M, García M. Cell death in the developing vertebrate retina. Int J Dev Biol. 2004;48:965–74. doi: 10.1387/ijdb.041891ev. [DOI] [PubMed] [Google Scholar]
- 20.Cordeiro MF, Migdal C, Bloom P, Fitzke FW, Moss SE. Imaging apoptosis in the eye. Eye (Lond) 2011;25:545–53. doi: 10.1038/eye.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson SA, Young C, Olney JW. Isoflurane-induced neuroapoptosis in the developing brain of nonhypoglycemic mice. J Neurosurg Anesthesiol. 2008;20:21–8. doi: 10.1097/ANA.0b013e3181271850. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Y, Levy RJ. Subclinical carbon monoxide limits apoptosis in the developing brain after isoflurane exposure. Anesth Analg. 2014;118:1284–92. doi: 10.1213/ANE.0000000000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordeiro MF, Guo L, Luong V, Harding G, Wang W, Jones HE, Moss SE, Sillito AM, Fitzke FW. Real-time imaging of single nerve cell apoptosis in retinal neurodegeneration. Proc Natl Acad Sci U S A. 2004;101:13352–6. doi: 10.1073/pnas.0405479101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Y, Corbin JG, Levy RJ. Programmed cell death is impaired in the developing brain of FMR1 mutants. Dev Neurosci. 2013;35:347–58. doi: 10.1159/000353248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–27. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 26.Singh BK, Tripathi M, Chaudhari BP, Pandey PK, Kakkar P. Natural terpenes prevent mitochondrial dysfunction, oxidative stress and release of apoptotic proteins during nimesulide-hepatotoxicity in rats. PLoS One. 2012;7:e34200. doi: 10.1371/journal.pone.0034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012;31:152–81. doi: 10.1016/j.preteyeres.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Bouchier-Hayes L, Muñoz-Pinedo C, Connell S, Green DR. Measuring apoptosis at the single cell level. Methods. 2008;44:222–8. doi: 10.1016/j.ymeth.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sasaki C, Kitagawa H, Zhang WR, Warita H, Sakai K, Abe K. Temporal profile of cytochrome c and caspase-3 immunoreactivities and TUNEL staining after permanent middle cerebral artery occlusion in rats. Neurol Res. 2000;22:223–8. doi: 10.1080/01616412.2000.11741065. [DOI] [PubMed] [Google Scholar]
- 30.Weishaupt JH, Diem R, Kermer P, Krajewski S, Reed JC, Bähr M. Contribution of caspase-8 to apoptosis of axotomized rat retinal ganglion cells in vivo. Neurobiol Dis. 2003;13:124–35. doi: 10.1016/s0969-9961(03)00032-9. [DOI] [PubMed] [Google Scholar]
- 31.Nickells RW. The cell and molecular biology of glaucoma: mechanisms of retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2012;53:2476–81. doi: 10.1167/iovs.12-9483h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X, Lu Y, Dong Y, Zhang G, Zhang Y, Xu Z, Culley DJ, Crosby G, Marcantonio ER, Tanzi RE, Xie Z. The inhalation anesthetic isoflurane increases levels of proinflammatory TNF-α, IL-6, and IL-1β. Neurobiol Aging. 2012;33:1364–78. doi: 10.1016/j.neurobiolaging.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Istaphanous GK, Ward CG, Nan X, Hughes EA, McCann JC, McAuliffe JJ, Danzer SC, Loepke AW. Characterization and quantification of isoflurane-induced developmental apoptotic cell death in mouse cerebral cortex. Anesth Analg. 2013;116:845–54. doi: 10.1213/ANE.0b013e318281e988. [DOI] [PubMed] [Google Scholar]
- 34.Cusato K, Bosco A, Linden R, Reese BE. Cell death in the inner nuclear layer of the retina is modulated by BDNF. Brain Res Dev Brain Res. 2002;139:325–30. doi: 10.1016/s0165-3806(02)00570-9. [DOI] [PubMed] [Google Scholar]
- 35.Young RW. Cell death during differentiation of the retina in the mouse. J Comp Neurol. 1984;229:362–73. doi: 10.1002/cne.902290307. [DOI] [PubMed] [Google Scholar]
- 36.Péquignot MO, Provost AC, Sallé S, Taupin P, Sainton KM, Marchant D, Martinou JC, Ameisen JC, Jais JP, Abitbol M. Major role of BAX in apoptosis during retinal development and in establishment of a functional postnatal retina. Dev Dyn. 2003;228:231–8. doi: 10.1002/dvdy.10376. [DOI] [PubMed] [Google Scholar]
- 37.Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- 38.Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev. 2002;8:30–8. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- 39.Loepke AW, McCann JC, Kurth CD, McAuliffe JJ. The physiologic effects of isoflurane anesthesia in neonatal mice. Anesth Analg. 2006;102:75–80. doi: 10.1213/01.ANE.0000181102.92729.B8. [DOI] [PubMed] [Google Scholar]
- 40.Translating time. Available at: www.translatingtime.net. Accessed January 21, 2014.