

Abstract
Allostery has been revealed as an essential property of all proteins. For enzymes, shifting of the structural equilibrium distribution at one site can have substantial impacts on protein dynamics and selectivity. Promising sites of remotely shifting such a distribution by changing the dynamics would be at flexible loops because relatively large changes may be achieved with minimal modification of the protein. A ligand‐selective change of binding affinity to the active site of cyclophilin is presented involving tuning of the dynamics of a highly flexible loop. Binding affinity is increased upon substitution of double Gly to Ala at the hinge regions of the loop. Quenching of the motional amplitudes of the loop slightly rearranges the active site. In particular, key residues for binding Phe60 and His126 adopt a more fixed orientation in the bound protein. Our system may serve as a model system for studying the effects of various time scales of loop motion on protein function tuned by mutations.
Keywords: binding affinity, enzymes, NMR spectroscopy, protein dynamics, selectivity
Modulation of protein functions by tuning of their dynamics is an important step towards understanding the dynamic‐function relationships inside proteins.1 The most obvious way to modify protein function is by mutating amino acid residues at sites that are directly involved in activity.2 Another increasingly popular approach is to alter sites remote from the protein active site. Such allosteric mechanisms are well recognized in multi‐domain proteins but there is growing evidence that they are a common feature in many, if not all, proteins.3 Cyclophilin is one of the most prominent examples of a single domain protein that makes use of intrinsic motions to carry out function.4 Cyclophilin belongs to the isomerase class of enzymes that catalyze the cis–trans isomerization of X‐proline peptide bonds, where X refers to any amino acid.5 Recently, we determined the structural ensemble of cyclophilin that comprises two states, and showed that the loop position is correlated with two structurally similar but distinct states located within the active site.6 Interestingly, in the open loop conformation, the residues in the active site match closely with those in the X‐ray structure in complex with the HIV capsid protein.7 The loop has two double glycine hinge‐like regions at either end, G64 and G65, and G74 and G75. Double substitution to Ala of either glycine pair is expected to render the hinges more rigid. To shift the population towards the binding competent form, we previously stabilized the open state of the loop with a double mutation at the N‐terminal hinge region of the ligand binding loop (G64A and G65A). Consequently, 15N,1H NMR chemical shifts moved closer to those of the bound wild‐type (WT) form, and binding affinity to ligands doubled.6 While the G64A and G65A mutant is not sufficiently stable to allow for structural studies, the C‐terminal hinge region G74A and G75A mutant turned out to be highly stable. Herein, we report the allosteric effects of hinge rigidification within a G74A and G75A cyclophilin mutant. The motional amplitude of the loop was modified, thereby creating a preference for a cis‐locked ligand in the active site.
The cyclophilin glycine double mutant is pre‐organized for peptide binding. To better understand the mechanism of ligand recognition and the effect of loop dynamics on cyclophilin‐peptide binding, we performed NMR chemical shift mapping analysis by comparing peak positions (15N,1H chemical shifts) of cyclophilin WT and mutant, both in free form and bound to cis‐, trans‐ and WT peptide (Supporting Information, Figure S5). For residues experiencing a chemical shift change (CSC) in the presence of the ligand, we observed a similar magnitude of CSC for WT cyclophilin and the glycine double mutant in complex with the WT and trans‐peptides. However, addition of cis‐peptide to the glycine double mutant led only to a minimal chemical shift changes compared to those observed for the WT and trans‐peptides (Figure 1; Supporting Information, Figure S1). Since all experiments were done using saturated concentrations of peptide ligands (1:11 protein to peptide ratio), it is implied that the residues responsible for binding in the double glycine mutant were already in or close to the bound conformation prior to peptide binding. Plotting the differences between CSCs upon cis‐, trans‐ and WT peptide binding for WT protein and mutant, respectively, revealed three hot spot regions involving residues F60, A101 and H126 (Figure 1 d) (F60 and H126 have been reported to be responsible for ligand binding8).
Figure 1.

The cyclophilin glycine double mutant is pre‐organized for peptide binding. a)–c) Chemical shift changes (CSC) and details of [15N,1H]‐Transverse relaxation optimized spectroscopy (TROSY) spectra are shown for protein bound to: a) cis‐peptide; b) trans‐peptide; and c) WT peptide (WT). d) The difference in CSC for WT cyclophilin and G74A and G75A bound to the respective ligands (CSCwild‐type/peptide−CSCG74A/G75A/peptide) is plotted as a function of amino acid residues. Arrows indicate the residues responsible for ligand binding. The cis‐peptide shows a clear trend not seen for the WT and trans‐ligand. e)–h) Spectral expansions are shown for the active site residues Cys52, Phe60, Ala101 and His126, for free and bound WT and mutant G74A and G75A. The top and bottom panel of each pair represent the WT protein and the G74A and G75A mutant, respectively, in free protein form (red), bound to cis‐peptide (cyan), trans‐peptide (green), and WT peptide (blue). All of the experiments were done with an 11‐fold excess of the peptide ligand (0.3 mm protein and 3.4 mm ligand). Residues in the vicinity of the active site residues are also labeled. Arrows indicate the chemical shift changes induced upon addition of ligand.
Loop residues are less mobile in the glycine double mutant. To verify that the glycine‐to‐alanine double mutation indeed rigidifies the loop, we performed nuclear Overhauser effect spectroscopy (NOESY) measurements on mutant and WT cyclophilin. Figure 2 shows slices through the 1H–1H plane for residues G65, D66, T68, and H70 located on the loop, which exhibit a considerable number of cross peaks to the rest of the protein. To further investigate the implications of loop dynamics upon mutation, we measured and compared the transverse relaxation rates R 2 of the WT and mutant using a Hahn–echo‐type experiment, which is sensitive to exchange contribution on the micro‐ to millisecond time scale.9 Figure 2 shows R 2 rates plotted as a function of the amino acid sequence. In WT cyclophilin, loop residues as well as residues 135 and 149 undergo detectable exchange, as revealed by increased R 2 rates (Figure 2, Figure 3; Supporting Information, Figure S2). Upon introduction of the double alanine at position 74 and 75, the pattern of motion at and around the loop changed dramatically. The R 2 rates of the loop residues decreased to values typical of the rest of the protein, while a substantial rise in R 2 was noticed for residues 72, 77, 78, 82, 83, and 86 (Figure 2 b). Residues 72, 77, and 78 are close to the mutation site; therefore, their R 2 increases could be directly related to the amino acid change at 74 and 75. Residues 82 and 83 form part of a hub known to induce a conformational switch from a minor to a major conformational state.4 This suggests that the double glycine mutation can induce exchange in this region through long‐range effects. The origin of an increased R 2 rate for residue 86 is not clear, but its peak completely broadens out in the free WT protein, indicating that it undergoes even more exchange in the free protein. We performed relaxation measurements in the presence of different peptide ligands to see how the R 2 rates change in bound WT and mutant cyclophilin (Figure 2 a and b). Interestingly, the R 2 rates for WT and mutant cyclophilin were very similar when bound to the WT peptide and or peptide in trans‐conformation, except in the case of residues 135 and 149 (Figure 2). However, when bound to the peptide in the cis‐conformation a general increase in R 2 was observed around residues 93–126 for the double glycine mutant (Figure 2 b).
Figure 2.
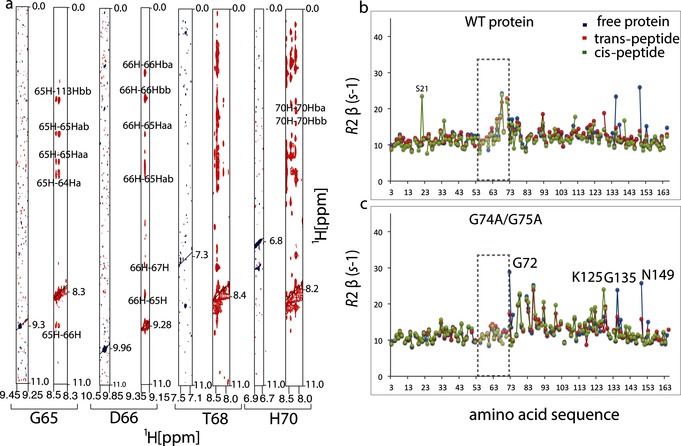
Loop residues are less mobile in the glycine double mutant. a) Cross‐sections of [1H,1H]‐NOESY spectra showing cross peaks for G65, D66, T68, and H70 for WT cyclophilin (blue) and G74A and G75A mutant (red). Increased intensities of cross peaks indicate that loop residues make substantial contact with the rest of the protein and that they are less mobile in the mutant than in the WT protein. b), c) Transverse R 2 NMR relaxation rates plotted as functions of the amino acid residues for the free WT cyclophilin and mutant (blue), and bound to the trans‐peptide (red) and cis‐peptide (green). In the double glycine mutant, the increased R 2 rates of loop residues reduce to R 2 values typical of the rest of the protein (gray rectangle), except around the residues near the mutation site.
Figure 3.
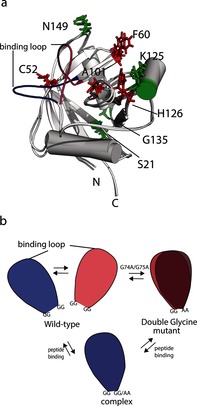
Structural representation of binding and dynamic residues. Indicated on the previously determined cyclophilin two‐state ensemble are the residues that experience: a) Large CSC upon binding to the cis‐peptide (red) and R 2 change upon peptide binding (green); b) Mechanistic representation for loop opening and closing of the WT, G74A and G75A mutant and the complex. The binding loop in the open (blue) and closed state (magenta) are also shown.
The G74A and G75A double mutant has a higher affinity for cis‐peptide. If the double glycine mutant possesses a pre‐organized binding site (as suggested by CSC, NOESY, and relaxation data) then the affinity of G74A and G75A/cis‐peptide interaction should be higher than that of the WT/cis‐peptide interaction. Indeed, the K d for the G74A and G75A/cis‐peptide was 50±30 μm, while that for the WT cyclophilin was 170±70 μm at 4 °C using fluorescence resonance energy transfer (FRET)10 based assays (see Figure 4). (A similar K d trend was observed with chemical‐shift analysis of individual binding residues at 26 °C; Supporting Information, Figure S4.) K d measurements were further corroborated with WT cyclophilin/WT peptide (164±32 μm) and G74A and G75A/WT peptide (107±30 μm) interactions, indicating that the double glycine mutant had an improved affinity for the WT peptide while the affinities for trans‐peptide remained virtually unchanged (94±20 μm compared to 101±38 μm; Supporting Information, Figure S4). The fact that the WT peptide alters K d upon mutation, whereas trans‐ does not, implies that the cis state causes the change. The enhanced affinity observed for the double glycine mutant is a result of its ability to efficiently accommodate the cis‐peptide because of its pre‐organization at the active site, as corroborated by CSC mapping analysis, relaxation measurements and NOESY data.
Figure 4.
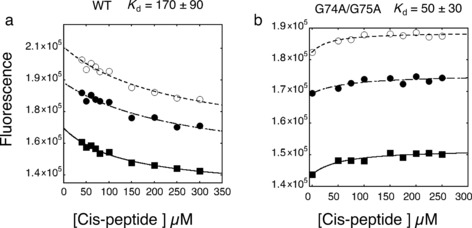
Equilibrium binding constants for cis‐peptide binding were determined from a plot of the fluorescence change (FRET) versus peptide concentration. The K d of the G74A/G75A mutant is 50±30 μm, while that for the WT is 170±90 μm. K d was determined by fitting of the data to a standard equation describing the equilibrium binding of two molecules.10 Fits from three different wavelengths are shown. Measurements were performed at 4 °C. K d values are the averages of three different wavelengths and propagated errors are reported.
In summary, we set out to investigate how enzyme‐substrate selectivity can be fine‐tuned by dynamics. We chose the enzyme cyclophilin for two reasons; it contains a dynamic loop (residues 65–72) that samples two conformational sub‐states and it binds both a cis‐ and trans‐oriented peptide. Binding is coupled to loop motions. With that in mind, we introduced a double glycine‐to‐alanine mutation at the C‐terminal hinge region of the loop (Figure 3 b). This mutation increased binding competence to cis‐peptide relative to the WT cyclophilin. Using NOESY analysis and relaxation measurements, we demonstrated that the mutation does indeed narrow the space sampled by the loop. Subsequently, we investigated the molecular basis of the tuning effect on the active site by NMR CSC mapping analysis. CSC analysis showed that the active site, including residues F60, A101, H126 and C52, are slightly rearranged in the mutant compared to the WT with substantial reduction in exchange contributions to the transverse relaxation R 2 rates for the loop residues. We conclude that CSCs are caused by both structural and dynamical re‐organization and that cis‐ligand binding in the mutant experiences less rearrangement compared to WT cyclophilin. Additionally, increased R 2 rates were observed for S21, K125, G135 and N149, which correlated with the ability of the mutant to bind preferentially to the cis‐peptide. Together, these findings indicate that by altering the dynamics of the loop movement distal to an enzyme active site, enzyme function may be influenced substantially. Double glycine substitution has a preference for the cis‐ligand. Our previously reported double mutation at the N‐terminal hinge of the same loop doubled the affinity of both the trans‐ and WT ligands.6 On the other hand, the mutation investigated here maintained affinity to the trans‐peptide, and increased affinity to the WT ligand. This observation can be explained by increased affinity for cis‐peptide binding, and unaltered trans‐peptide binding. In another example of cis‐trans isomerase, human pin1 binding to a cis‐ and or trans‐locked peptide resulted in an altered dynamic behavior of the protein.11 Interestingly, the trans‐locked peptide produced a weaker response, which we also observed for the R 2 rates of the cyclophilin mutant, but not for the WT. It has been pointed out that the time‐scales of loop motion and confomational plasticity may play an important role in activity, selectivity and the rate of enzyme catalysis.4, 12 Mutation of histidine 70 in cyclophilin to alanine within the loop caused a minimal change in dynamics as probed by CPMG NMR measurements, and therefore did not alter affinity.4 More recently, it has been shown that altered loop dynamics of two escape‐mutants of HIV‐capsid protein are also relevant to interaction with cyclophilin.13 Direct evidence of the influence of dynamics in single domain protein binding and selectivity has been found in a few other cases, both experimetally12b and computationally.14 However, the concept of conformational selection and its effect on protein function is becoming increasingly popular.3d–3g Our system opens up possibilities to introduce specific time scales of loop motion by further mutations. Linking these time scales to catalytic activities would help to elucidate the role of time‐scale dependent dynamic allostery.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by a Wenner‐Gren Stiftelsern WG‐13 Postdoctoral fellowship held by C. N. Chi, the Swiss National Science Foundation with Grant 140214 and an ETH Research Grant ETH‐04 13‐1 awarded to B. Vögeli. Special thanks to Prof. Dr. Roland Riek for his critical comments on the manuscript.
B. Vögeli, S. Bibow, C. N. Chi, Angew. Chem. Int. Ed. 2016, 55, 3096.
References
- 1.
- 1a. Bahar I., Lezon T. R., Yang L. W., Eyal E., Annu. Rev. Biophys. 2010, 39, 23–42; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Gibbs A. C., J. Med. Chem. 2014, 57, 7819–7837; [DOI] [PubMed] [Google Scholar]
- 1c. van den Bedem H., Fraser J. S., Nat. Methods 2015, 12, 307–318; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Fenwick R. B., van den Bedem H., Fraser J. S., Wright P. E., Proc. Natl. Acad. Sci. USA 2014, 111, E445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fersht A., Structure and Mechanism in Protein Science; A Guide to Enzyme Catalysis and Protein Folding, W. H. Freeman and Company, New York, 1998. [Google Scholar]
- 3.
- 3a. Monod J., Wyman J., Changeux J. P., J. Mol. Biol. 1965, 12, 88–118; [DOI] [PubMed] [Google Scholar]
- 3b. D. E. Koshland, Jr. , Nemethy G., Filmer D., Biochemistry 1966, 5, 365–385; [DOI] [PubMed] [Google Scholar]
- 3c. Kamerlin S. C. L., Warshel A., Proteins Struct. Funct. Bioinf. 2010, 78, 1339–1375; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Gunasekaran K., Ma B., Nussinov R., Proteins Struct. Funct. Bioinf. 2004, 57, 433–443; [DOI] [PubMed] [Google Scholar]
- 3e. Ma B., Nussinov R., Curr. Opin. Chem. Biol. 2010, 14, 652–659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3f. Nussinov R., Tsai C.-J., Curr. Opin. Struct. Biol. 2015, 30, 17–24; [DOI] [PubMed] [Google Scholar]
- 3g. Tsai C. J., del Sol A., Nussinov R., J. Mol. Biol. 2008, 378, 1–11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3h. Chi C. N., Bach A., Engstrom A., Wang H., Stromgaard K., Gianni S., Jemth P., Biochemistry 2009, 48, 7089–7097. [DOI] [PubMed] [Google Scholar]
- 4. Eisenmesser E. Z., Millet O., Labeikovsky W., Korzhnev D. M., Wolf-Watz M., Bosco D. A., Skalicky J. J., Kay L. E., Kern D., Nature 2005, 438, 117–121. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi N., Hayano T., Suzuki M., Nature 1989, 337, 473–475. [DOI] [PubMed] [Google Scholar]
- 6.C. N. Chi, B. Vogeli, S. Bibow, D. Strotz, J. Orts, P. Guntert, R. Riek, Angew. Chem. Int. Ed 2015, 54, 11657–11661. [DOI] [PubMed]
- 7. Gamble T. R., Vajdos F. F., Yoo S., Worthylake D. K., Houseweart M., Sundquist W. I., Hill C. P., Cell 1996, 87, 1285–1294. [DOI] [PubMed] [Google Scholar]
- 8. Zydowsky L. D., Etzkorn F. A., Chang H. Y., Ferguson S. B., Stolz L. A., Ho S. I., Walsh C. T., Protein Sci. 1992, 1, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lakomek N.-A., Kaufman J. D., Stahl S. J., Louis J. M., Grishaev A., Wingfield P. T., Bax A., Angew. Chem. Int. Ed. 2013, 52, 3911–3915; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4003–4007. [Google Scholar]
- 10. Chi C. N., Elfstrom L., Shi Y., Snall T., Engstrom A., Jemth P., Proc. Natl. Acad. Sci. USA 2008, 105, 4679–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Namanja A. T., Wang X. J., Xu B., Mercedes-Camacho A. Y., Wilson B. D., Wilson K. A., Etzkorn F. A., Peng J. W., J. Am. Chem. Soc. 2010, 132, 5607–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Camilloni C., Sahakyan A. B., Holliday M. J., Isern N. G., Zhang F., Eisenmesser E. Z., Vendruscolo M., Proc. Natl. Acad. Sci. USA 2014, 111, 10203–10208; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Michielssens S., Peters J. H., Ban D., Pratihar S., Seeliger D., Sharma M., Giller K., Sabo T. M., Becker S., Lee D., Griesinger C., de Groot B. L., Angew. Chem. Int. Ed. 2014, 53, 10367–10371; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10535–10539; [Google Scholar]
- 12c. Boehr D. D., McElheny D., Dyson H. J., Wright P. E., Science 2006, 313, 1638–1642; [DOI] [PubMed] [Google Scholar]
- 12d. Nashine V. C., Hammes-Schiffer S., Benkovic S. J., Curr. Opin. Chem. Biol. 2010, 14, 644–651; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12e. Whittier S. K., Hengge A. C., Loria J. P., Science 2013, 341, 899–903; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12f. Ådén J., Verma A., Schug A., Wolf-Watz M., J. Am. Chem. Soc. 2012, 134, 16562–16570. [DOI] [PubMed] [Google Scholar]
- 13. Lu M., Hou G., Zhang H., Suiter C. L., Ahn J., Byeon I.-J. L., Perilla J. R., Langmead C. J., Hung I., Gor'kov P. L., Gan Z., Brey W., Aiken C., Zhang P., Schulten K., Gronenborn A. M., Polenova T., Proc. Natl. Acad. Sci. USA 2015, 112, 14617–14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fuchs J. E., Huber R. G., Waldner B. J., Kahler U., von Grafenstein S., Kramer C., Liedl K. R., PLoS ONE 2015, 10, e0140713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary