

Abstract
Glutamate directly activates N-methyl-d-aspartate (NMDA) receptors on presynaptic inhibitory interneurons and enhances GABA release, altering the excitatory-inhibitory balance within a neuronal circuit. However, which class of NMDA receptors is involved in the detection of glutamate spillover is not known. GluN2D subunit-containing NMDA receptors are ideal candidates as they exhibit a high affinity for glutamate. We now show that cerebellar stellate cells express both GluN2B and GluN2D NMDA receptor subunits. Genetic deletion of GluN2D subunits prevented a physiologically relevant, stimulation-induced, lasting increase in GABA release from stellate cells [long-term potentiation of inhibitory transmission (I-LTP)]. NMDA receptors are tetramers composed of two GluN1 subunits associated to either two identical subunits (di-heteromeric receptors) or to two different subunits (tri-heteromeric receptors). To determine whether tri-heteromeric GluN2B/2D NMDA receptors mediate I-LTP, we tested the prediction that deletion of GluN2D converts tri-heteromeric GluN2B/2D to di-heteromeric GluN2B NMDA receptors. We find that prolonged stimulation rescued I-LTP in GluN2D knockout mice, and this was abolished by GluN2B receptor blockers that failed to prevent I-LTP in wild-type mice. Therefore, NMDA receptors that contain both GluN2D and GluN2B mediate the induction of I-LTP. Because these receptors are not present in the soma and dendrites, presynaptic tri-heteromeric GluN2B/2D NMDA receptors in inhibitory interneurons are likely to mediate the cross talk between excitatory and inhibitory transmission.
Keywords: NMDA receptors, cerebellum, inhibitory interneurons, LTP, GABA release
inhibitory synaptic transmission sets the excitability of principal neurons, and, therefore, activity-dependent changes in inhibitory drive are critical for most forms of experience-dependent learning (Chao et al. 2010; Ehrlich et al. 2009; Levelt and Hübener 2012; Marsicano et al. 2002; Nugent et al. 2007; Scelfo et al. 2008; Woodin and Maffei 2011). Growing evidence supports the idea that glutamate is a major regulator of inhibitory transmission (Castillo et al. 2011; McBain and Kauer 2009). Cross talk between excitatory and inhibitory presynaptic terminals offers a powerful means to alter the excitatory/inhibitory balance and so modify the output of neuronal circuitry. Such studies highlight the importance of understanding how glutamate communicates with inhibitory neurons to regulate the release of the inhibitory transmitter GABA.
Glutamate modulates inhibitory transmission through diverse mechanisms (Castillo et al. 2011; Gaiarsa et al. 2002). The best understood presynaptic modification of the strength of inhibitory synapses involves the release of the retrograde signals, endocannabinoids, nitric oxide (NO) and brain-derived neurotrophic factor, which is triggered by the activation of metabotropic glutamate receptors and N-methyl-d-aspartate (NMDA) receptors (NMDARs) in postsynaptic neurons (Castillo et al. 2011). However, glutamate can also directly activate NMDARs on presynaptic GABAergic interneurons to regulate GABA release. This second form of presynaptic plasticity has been commonly observed in several brain regions (Duguid and Smart 2004; Lachamp et al. 2009; Lien et al. 2006; Liu et al. 2007). We have previously shown that glutamate released from parallel fibers (PFs) activates NMDARs in stellate cells (cerebellar inhibitory interneurons) to induce a lasting increase in GABA release probability [long-term potentiation of inhibitory transmission (I-LTP)] (Liu and Lachamp 2006). The change is triggered by activation of NMDARs in presynaptic stellate neurons, but not by the retrograde signals, NO and endocannabinoids. This modulation enhances both evoked and spontaneous inhibitory transmission and markedly alters the activity of the cerebellar inhibitory network (Lachamp et al. 2009). One critical feature of this form of presynaptic plasticity is the heterosynaptic nature, requiring NMDARs on presynaptic inhibitory interneurons to detect glutamate released from nearby excitatory inputs. However, the molecular identity of the NMDARs responsible for the induction of I-LTP and their subcellular localization is not known.
Although NMDARs are localized in the postsynaptic membrane (Petralia et al. 2009), anatomical evidence also supports the existence of NMDARs in the presynaptic terminals of neurons in the neocortex, hippocampus, spinal cord, amygdala and cerebellum (Rodríguez-Moreno et al. 2010). NMDAR immunolabeling has been found in presynaptic axons of many GABAergic neurons, including in the axon terminals of cerebellar interneurons (Paquet and Smith 2000; Petralia et al. 1994) and activation of these receptors elevates calcium levels in axons (Rossi et al. 2012; Shin and Linden 2005) (but also see Christie and Jahr 2008; Clark and Cull-Candy 2002). If these presynaptic NMDARs are activated by glutamate spillover from PF terminals to regulate GABA release, they would need to be able to detect a low concentration of glutamate. One strong candidate is the GluN2D (or NR2D) subunit of NMDA receptors, which is predominantly expressed in GABAergic interneurons, including cerebellar stellate cells (Akazawa et al. 1994; Monyer et al. 1994; Thompson et al. 2000). NMDARs that contain GluN2D subunits exhibit a higher glutamate affinity, lower Mg2+ blockade, and prolonged decay time constant after a brief application of agonist, relative to GluN2A and GluN2B-containing receptors (Cull-Candy et al. 2001; Misra et al. 2000; Siegler Retchless et al. 2012). These unique features enable the receptors to detect a spillover of glutamate from nearby excitatory terminals and give rise to a sustained glutamatergic activation signal. We, therefore, tested the hypothesis that GluN2D-containing receptors in stellate cell induce I-LTP.
Using genetic and pharmacological approaches, we found that both GluN2B and GluN2D receptors are expressed in the dendrites and somata of stellate cells. Our experiments show that genetic deletion of GluN2D abolished the PF stimulation-induced I-LTP in stellate cells. Unexpectedly, this was rescued by prolonging PF stimulation duration, and thus the presence of GluN2D in NMDARs lowered the threshold for induction of I-LTP. Although GluN2B receptor inhibitors prevented such I-LTP in mutant mice, they failed to attenuate I-LTP in wild-type (WT) mice, indicating that these NMDARs contain both GluN2D and GluN2B subunits. Pharmacological inhibition of tri-heteromeric GluN2B/2D (tri-GluN2B/2D) NMDARs prevented the induction of I-LTP in WT mice. We conclude that presynaptic GluN2B/2D NMDARs are required for the induction of I-LTP in WT mice. Incorporation of GluN2D in an NMDAR, therefore, allows a physiologically relevant activity, such as occurs during sensory stimulation, to modulate GABA release.
EXPERIMENTAL PROCEDURES
Cerebellar slice preparation and electrophysiology.
Cerebellar slices were prepared as previously described (Dubois et al. 2012; Liu and Cull-Candy 2000). Briefly, postnatal day (P) 7 to P10 and P18 to P40 WT (The Jackson Laboratory) and GluN2D knockout (KO) (Ikeda et al. 1995) mice on a C57BL/6J background were decapitated, and the cerebellum was isolated. Horizontal or sagittal slices (400 μm) were cut from the cerebellum using a vibratome (Leica VT1200) in an ice-cold artificial cerebrospinal fluid (aCSF) (containing in mM: 81.2 NaCl, 2.4 KCl, 23.4 NaHCO3, 1.4 NaH2PO4, 6.7 MgCl2, 0.5 CaCl2, 23.3 glucose, 69.9 sucrose, pH 7.4). Slices were then maintained in aCSF (in mM: 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 25 glucose, pH 7.4) saturated with 95% O2/5% CO2 at room temperature for at least 30 min before recording. Experimental procedures were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center and followed Penn State University guidelines.
Whole cell patch-clamp recordings were made from cerebellar stellate cells in an O2/CO2-saturated aCSF. Stellate cells were identified by their location in the outer two-thirds of the molecular layer and by the presence of spontaneous action potentials in the cell-attached mode. Analog signals were filtered at 2 or 10 kHz and digitized at 10 kHz (Multiclamp 700A, Axon Instruments). Series resistance was monitored throughout the recordings, and, if this changed by more than 20%, the recordings were discarded. All synaptic and dendritic currents were recorded at near physiological temperature (33–37°C; except for the data in Fig. 1), while single channel currents in outside-out patches were recorded at room temperature to reduce noise.
Fig. 1.
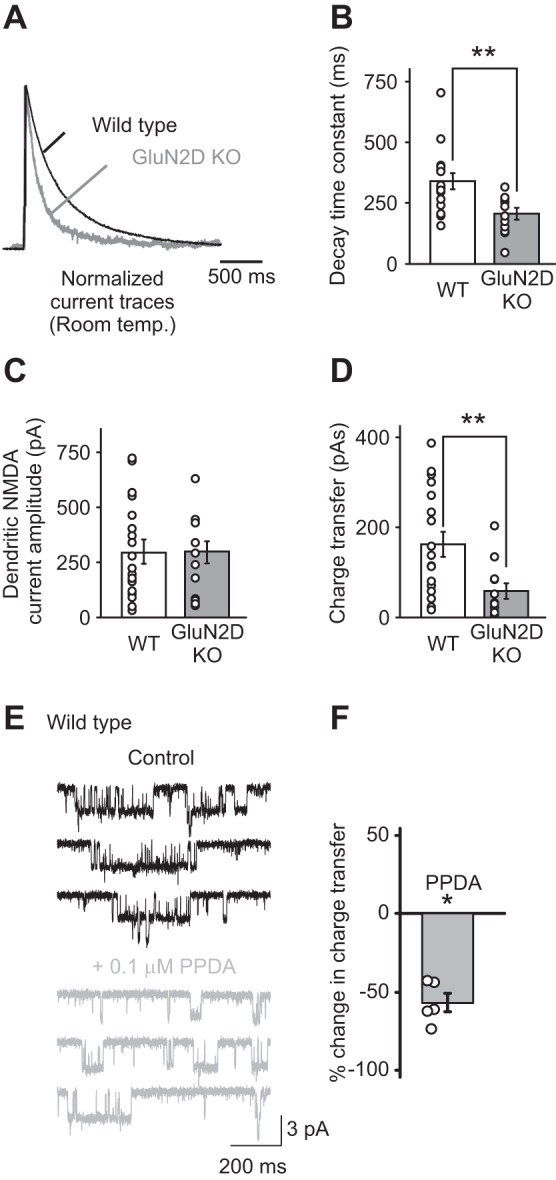
N-methyl-d-aspartate (NMDA) receptors that contain GluN2D subunits are expressed in the soma and dendrites of stellate cells. A: dendritic NMDA receptor currents were evoked by a single-burst stimulation of parallel fibers (PFs) at 100 Hz (4 stimuli) and recorded at +40 mV at room temperature. NMDA receptor currents recorded in stellate cells from GluN2D knockout (KO) mice exhibited a faster decay time compared with wild-type (WT) mice, as shown in the normalized currents (GluN2 KO, gray trace; WT, black trace). B: summary of the decay time constants of NMDA receptor currents recorded in stellate cells from WT and GluN2D KO mice. Deletion of GluN2D accelerated the decay time of evoked dendritic NMDA receptor currents. C: the amplitude of dendritic NMDA receptor-mediated currents. D: the charge transfer of dendritic NMDA receptor-mediated currents, which was calculated as the time integral of NMDA receptor currents, was reduced in GluN2D KO mice. Thus NMDA receptors that contain GluN2D subunits are present in cerebellar stellate cells. Open circles represent data from individual cells. E: outside-out somatic patches were excised from stellate cells from WT mice, and NMDA receptor currents were recorded during the application of 10 μM NMDA and 10 μM glycine. PPDA [(2S*,3R*)-1-(phenanthren-2-carbonyl)piperazine-2,3-dicarboxylic acid; 0.1 μM] was added to the superfusion medium to block GluN2D-containing NMDA receptors. F: PPDA reduced the charge transfer of somatic NMDAR currents. *P < 0.05. **P < 0.01.
I-LTP.
Miniature inhibitory synaptic currents (mIPSCs) were recorded in stellate cells. Cells were voltage clamped at −30 mV in the presence of 0.5 μM tetrodotoxin (TTX) in aCSF, using borosilicate electrodes (6–8 MΩ) filled with a low-chloride pipette solution (in mM: 120 Cs-acetate, 0.4 MgCl2, 0.1 CaCl2, 2.5 MgATP, 0.4 NaGTP, 1.5 NaATP, 10 Cs-EGTA, 5 QX-314 and 10 HEPES, pH 7.3). After obtaining a stable recording for at least 15 min, TTX was washed out for 20 min. PFs were then stimulated with a parallel bipolar electrode (150-μm spacing) that was placed across the molecular layer about 200 μm from the recording electrode. The strength of the stimulation ranged from 5 to 50 V with a duration of 200 μs and was adjusted to evoke NMDAR currents at +40 mV in response to a single burst stimulation (4 stimuli at 100 Hz). I-LTP was then induced using 5 or 15 repeated trains of burst stimulation (repeated every second), while the postsynaptic cell was voltage-clamped at −60 mV. These are physiological relevant stimulation protocols, since sensory stimulation in vivo evokes a burst of action potentials at ∼80 Hz in granule cells (Chadderton et al. 2004; Wilms and Häusser 2015), the axons of which innervate stellate cells. Axonal calcium transients in stellate cells elicited by activation of presynaptic NMDARs reach a peak value at ∼400 ms after glutamate uncaging and remain high at 800 ms (Figs. 2Ad and 5 in Rossi et al. 2012). Thus repeated stimulation of PFs with a 960-ms intertrain interval is expected to lead to a sustained increase in presynaptic calcium levels. Immediately after PF stimulation, TTX was reintroduced into the aCSF, and recordings of mIPSCs were started within 2 min.
Fig. 2.

GluN2B-containing receptors mediated somato-dendritic NMDA receptor currents in stellate cells. A: dendritic NMDA receptor currents were evoked by a train of PF stimulation at 100 Hz (4 stimuli) at 33–37°C. Currents were recorded at +40 mV in stellate cells before (black traces) and during (gray traces) superfusion of an NMDA receptor blocker, ifenprodil (Ifen; 3 μM), a di-heteromeric GluN2B (di-GluN2B) receptor antagonist. Ifenprodil blocked dendritic NMDA receptor currents, suggesting that NMDA receptors that contain GluN2B subunits mediate the dendritic NMDA receptor current. B: summary of the effects of ifenprodil (3 μM) on the amplitude, charge transfer and decay time of NMDA receptor currents at near-physiological temperature (open circles indicate data from individual experiments). C, left: ifenprodil also blocked NMDA receptor currents in outside-out somatic patches excised from stellate cells from WT mice, and thus GluN2B receptors mediate NMDA receptor currents. Right: in GluN2D KO mice, ifenprodil nearly abolished the NMDA-evoked currents from stellate cells. D: the total charge transfer was markedly reduced in WT and abolished in GluN2D KO mice. NMDA receptor currents are mediated by a pure population of di-GluN2B NMDA receptors in mutant mice. Ctrl, control. *P < 0.05. **P < 0.01. ***P < 0.001.
Dendritic NMDAR currents.
Dendritic NMDAR-mediated currents were recorded using a Cs-based pipette solution (in mM: 140 CsCl, 2 NaCl, 10 HEPES, 4 Mg-ATP, 5 QX-314, 5 tetraethylammonium, and 10 Cs-EGTA, pH 7.3). A single train of PF stimulation has been shown to induce a spillover of glutamate and activate extrasynaptic NMDARs (Carter and Regehr 2000; Clark and Cull-Candy 2002; Sun and Liu 2007). Therefore, we evoked NMDAR currents by stimulating PFs using a monopolar electrode (4–8 MΩ, filled with aCSF) with a single train of four depolarizations at 100 Hz. This allowed us to compare our results to other studies that have characterized NMDAR properties using a monopolar stimulating electrode (Carter and Regehr 2000; Clark and Cull-Candy 2002; Sun and Liu 2007). The typical stimulation intensity was 20 V with a duration of 200 μs. Recordings were obtained at +40 mV to relieve the magnesium block of NMDARs, and the PF stimulation protocol was repeated every 30 s. Gabazine (SR-95531, 10 μM) and 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) (5 μM) were included in the aCSF to block GABAA and dl-α-amino-3-hydroxy-5-methylisoxazole-propionic acid (AMPA) receptors, respectively. The evoked currents were recorded for 15 min to obtain a stable baseline before application of NMDAR inhibitors.
Dendritic AMPA currents and paired pulse ratio.
AMPA receptor-mediated excitatory postsynaptic currents (EPSCs) were recorded using the Cs-based internal solution and evoked by stimulating PFs with a monopolar stimulating electrode in the presence of 10 μM 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphanoic acid (R-CPP) (to block NMDAR currents). AMPA currents were recorded at −60 mV following two consecutive PF stimulations (50 Hz) repeated every 3 s. Typical stimulation intensity was 2–5 V with a duration of 200 μs. The paired-pulse ratio of average EPSCs (typically 100–150 events) at the PF-stellate cell synapse was calculated as the ratio of the amplitude of the second EPSC (EPSC2) over the first (EPSC1).
Somatic NMDAR currents in outside-out patches.
Recordings were obtained in a magnesium-free aCSF that contained 0.5 μM TTX, 5 μM NBQX, 10 μM SR-95531 and 0.5 μM strychnine. Somatic outside-out patches were voltage-clamped at −60 mV using borosilicate electrodes (7–15 MΩ) filled with a cesium fluoride-based pipette solution (in mM: 95 CsF, 35 CsCl, 2 NaCl, 1 CaCl2, 4 MgATP, 10 Cs-EGTA, 1 QX-314, 5 tetraethylammonium and 10 HEPES, pH 7.3). If recordings did not contain any spontaneous channel openings, the aCSF was supplemented with 10 μM glycine and 10 μM NMDA. Currents at 0 or −100 mV were also recorded to ensure that the reversal potential was 0 mV. The NMDA-evoked currents were recorded for 10 min to obtain a stable baseline (control period) and then in the presence of a NMDAR inhibitor for at least 10 min. Following washout of each inhibitor, NMDA-evoked currents returned to control levels {%change: R-CPP, 4 ± 10%, n = 16; ifenprodil, −16 ± 12%, n = 5; (2S*,3R*)-1-(phenanthren-2-carbonyl)piperazine-2,3-dicarboxylic acid (PPDA), −5 ± 8%; n = 7, P > 0.05 for all drugs, washout vs. control}. For analysis, current traces were resampled at 10 kHz and filtered at 1 kHz. Events of more than 0.6 pA and lasting more than two filter rise times (332 μs) were then selected over a 1-min period, and charge transfer was calculated.
Data analysis.
Clampfit 9.0 (Axon Instruments) was used for the analysis of dendritic NMDAR currents and mIPSCs. The average dendritic NMDAR-mediated current was obtained by aligning all of the traces to the stimulus artifact (typically 10 events). The inhibition of NMDAR currents by antagonists was calculated from the amplitude and integral of EPSCs (charge transfer) before and after the addition of an inhibitor. The decay time constant was obtained by fitting the decay phase of the EPSCs with a two-exponential function. mIPSCs were selected using an event detection template. The average frequency and amplitude of mIPSCs were calculated over periods of 5 min. The group data in the I-LTP experiment in WT mice induced by 5 and 15 trains of PF stimulation include several cells from our laboratory's previous published results (Lachamp et al. 2009), since no difference was observed between the two data sets. During the induction of I-LTP, the PF stimulation-evoked currents were recorded at −60 mV in postsynaptic stellate cells. This current consisted of a fast component mediated via AMPA receptors and a slow component. The latter is mediated by NMDARs because it was blocked by 10 μM R-CPP, as we have shown previously (Fig. 6C in Liu and Lachamp 2006). Since the decay time constant of the fast component of the current was 8 ms (7.5 ± 0.9 ms, n = 7, not shown), we determined the charge transfer of the slow component of the current 8–500 ms after the last of the four stimuli. Because the amplitude of dendritic NMDAR-mediated currents evoked by local glutamate uncaging is fivefold greater than axonal NMDAR currents (Fig. 3C in Rossi et al. 2012), the currents recorded during PF stimulation were largely mediated by somato-dendritic receptors.
Fig. 6.

Activation of tri-heteromeric GluN2B/2D (tri-GluN2B/2D) NMDA receptors, but not di-GluN2B receptors, induced I-LTP in WT mice. A 15-train PF stimulation protocol was used to induce an increase in mIPSC frequency (I-LTP) in WT and GluN2D KO mice. A and B, left: representative traces of mIPSCs (outward currents) before (top) and after (bottom) PF stimulation in WT (A) and KO (B) mice. Center: time course of mIPSC frequency (top) and amplitude (bottom). Right: group data (means ± SE) showing the time course of mIPSCs frequency (circles) and amplitude (triangles). A, left and center: ifenprodil (3 μM), a di-GluN2B antagonist, was applied during a 15-train PF stimulation protocol and failed to prevent I-LTP in WT mice. Right: summary of the effect of ifenprodil on the long-lasting increase in mIPSC frequency induced by 15 trains of PF stimulation (n = 6). Light gray area shows the means ± SE of mIPSC frequency after PF stimulation without GluN2B antagonist in WT mice (from Fig 5A) for comparison. B: in GluN2D KO animals, I-LTP was blocked by GluN2B antagonists (3 μM ifenprodil or 5 μM RO 04–5595, n = 8). For comparison, the light gray area shows the means ± SE of mIPSC frequency after PF stimulation without GluN2B antagonist in GluN2D KO mice (from Fig 5B). C and D: mean mIPSC frequency (C) and amplitude (D) in individual cells following 15 trains of PF stimulation in WT and mutant mice. E: summary of the effects of GluN2B antagonists on the long-lasting increase in mIPSC frequency induced by 15 trains of PF stimulation. GluN2B antagonists effectively blocked I-LTP only in GluN2D KO mice, suggesting that activation of di-GluN2B receptors induced a lasting increase in GABA release in mutant mice. Ifenprodil failed to prevent I-LTP in WT mice, and thus NMDA receptors that contain both GluN2D and GluN2B subunits are responsible for the induction of I-LTP in WT mice. *P < 0.05.
Fig. 3.

Genetic deletion of GluN2D subunits abolished long-term potentiation of inhibitory transmission (I-LTP) induced by threshold PF stimulation. A and B: miniature inhibitory synaptic currents (mIPSCs) were recorded in stellate cells in the presence of tetrodotoxin (TTX), and PFs were stimulated with 5 trains of 4 pulses at 100 Hz (5×) after washing out TTX for 20 min, in WT (A) and KO (B) mice. Left: representative traces of mIPSCs recorded at −30 mV (outward currents) before (top) and after (bottom) PF stimulation. Center: corresponding time course of mIPSC frequency (top) and amplitude (bottom). Right: group data shown as means ± SE showing the time course of mIPSC frequency (top) and amplitude (bottom) following PF stimulation in WT (open symbols; A) and in GluN2D KO mice (gray symbols; B; for comparison the light gray area shows the mean ± SE of mIPSC frequency after PF stimulation in WT mice). Insets: representative recordings at −60 mV during PF stimulation in stellate cells. Five trains of PF stimulation were sufficient to trigger I-LTP in stellate cells from WT mice (n = 8), but failed to induce a change in mIPSCs in GluN2D KO mice (n = 6). Therefore, NMDA receptors that contain GluN2D subunits mediate the induction of I-LTP. C and D: group data showing mIPSC frequency (C) and amplitude (D) of individual cells before (15 min average before TTX washout) and after (average 15–30 min after stimulation) PF stimulation in WT (left) and GluN2D KO (right) mice. Only mIPSC frequency from WT animals increased following 5 trains of PF stimulation. E: group data showing that 5 trains of PF stimulation increased mIPSC frequency in individual cells from WT, but not GluN2D KO, mice. *P < 0.05. **P < 0.01. NS, nonsignificant.
No statistical method was used to predetermine sample sizes, but they are similar to previous studies (Lachamp et al. 2009; Liu and Cull-Candy 2000; Sun and Liu 2007). Data sets were obtained from at least three different litters, and animals from either sex were assigned randomly to the different experimental conditions. All values are means ± SE, and a P value < 0.05 was considered as significant. All tests were performed on primary data (not normalized). Normality and equality of the variances were assessed, and statistical tests were chosen accordingly. These mostly included one-way or two-way ANOVA with repeated measurements (except for Fig. 11), and Tukey post hoc procedures were applied when needed. For detailed statistical analysis, see Table 1.
Fig. 11.

The induction of I-LTP is independent of dendritic currents evoked by PF stimulation and of the initial mIPSC frequency. A: during the induction of I-LTP, postsynaptic currents evoked by PF stimulation were recorded at −60 mV. The total charge transfer of the slow component of the inward current was measured starting at 8 ms after the last stimulus during each train (see experimental procedures). This is shown as the shaded area in the representative example (truncated at 135 ms). Individual values are represented by open circles, and the median value is shown as a horizontal bar. There was no significant difference between groups (see Table 1). B: plot of the change in mIPSC frequency after PF stimulation in all individual cells as a function of the total charge transfer of postsynaptic currents evoked by PF stimulation. Regression line is gray dotted line. Together these results suggest that the induction of I-LTP was not correlated with the magnitude of the charge transfer of the postsynaptic currents recorded during PF stimulation. C: plot of the change in mIPSC frequency (%) after PF stimulation (I-LTP) in all cells as a function of initial mIPSC frequency (n = 59). There was no correlation between the magnitude of I-LTP and the initial mIPSC frequency. D: summary of individual mIPSC frequency for each experimental condition.
Table 1.
Statistics
| Figure | Conditions | n/Group | Analysis | Factor Analyzed | F Value | P Value |
|---|---|---|---|---|---|---|
| 1B | Genotype | 18–12 | Unpaired t-test, equal variance | NMDA current decay time | 0.003 | |
| 1C | Genotype | 18–12 | Unpaired t-test, equal variance | NMDA current amplitude | 0.210 | |
| 1D | Genotype | 18–12 | Unpaired t-test, equal variance | NMDA current charge transfer | 0.009 | |
| 1F | PPDA in stellate cell WT | 5 | Paired t-test | Charge transfer | 0.019 | |
| 2B | Ifen in WT (physiol. temperature) | 7 | Paired t-test | NMDA current amplitude | <0.001 | |
| Ifen in WT (physiol. temperature) | 7 | Paired t-test | NMDA current charge transfer | <0.001 | ||
| Ifen in WT (physiol. temperature) | 7 | Paired t-test | NMDA current decay time | 0.016 | ||
| 2D | Ifen in stellate cell WT | 5 | Paired t-test | Charge transfer | 0.007 | |
| Ifen in SC GluN2D KO | 4 | Paired t-test | Charge transfer | 0.001 | ||
| 3C | Interaction genotype/stimulation | 6–8 | 2-way RM ANOVA | mIPSC frequency | F(1,27) = 14.430 | 0.003 |
| Stimulation in WT | 8 | Tukey | mIPSC frequency | 0.020 | ||
| Stimulation in GluN2D KO | 6 | Tukey | mIPSC frequency | 0.167 | ||
| 3D | Interaction genotype/stimulation | 6–8 | 2-way RM ANOVA | mIPSC amplitude | F(1,27) = 0.662 | 0.432 |
| Stimulation in WT | 8 | Tukey | mIPSC amplitude | 0.089 | ||
| Stimulation in GluN2D KO | 6 | Tukey | mIPSC amplitude | 0.608 | ||
| 4A | Genotype | 6–7 | Unpaired t-test, equal variance | Paired pulse ratio | 0.761 | |
| 4B | Genotype | 36–58 | Kolmogorov-Smirnov test | mIPSC frequency | Ks stat 0.155172 | 0.659 |
| Genotype | 33–36 | Kolmogorov-Smirnov test | mIPSC amplitude | Ks stat 0.143939 | 0.868 | |
| 4D | PPDA in WT | 5 | Paired t-test | mIPSC frequency | 0.502 | |
| Ifen in WT | 5 | Paired t-test | mIPSC frequency | 0.129 | ||
| R-CPP in WT | 5 | Paired t-test | mIPSC frequency | 0.095 | ||
| 5C | Stimulation in GluN2D KO | 9 | 1-way RM ANOVA | mIPSC frequency | F(1,17) = 10.522 | 0.012 |
| 5D | Stimulation in GluN2D KO | 9 | 1-way RM ANOVA | mIPSC amplitude | F(1,17) = 0.006 | 0.939 |
| 6C | Interaction genotype/stimulation | 6–8 | 2-way RM ANOVA | mIPSC frequency | F(1,27) = 8.917 | 0.011 |
| Stimulation in WT | 6 | Tukey | mIPSC frequency | 0.022 | ||
| Stimulation in GluN2D KO | 8 | Tukey | mIPSC frequency | 0.153 | ||
| 6D | Interaction genotype/stimulation | 6–8 | 2-way RM ANOVA | mIPSC amplitude | F(1,27) = 0.621 | 0.446 |
| Stimulation in WT | 6 | Tukey | mIPSC amplitude | 0.579 | ||
| Stimulation in GluN2D KO | 8 | Tukey | mIPSC amplitude | 0.625 | ||
| 7D | Interaction genotype/stimulation | 5–6 | 2-way RM ANOVA | Charge transfer | F(2,31) = 4.214 | 0.039 |
| PPDA in GluN2D KO | 6 | Tukey | Charge transfer | 0.906 | ||
| PPDA in Golgi cells | 5 | Tukey | Charge transfer | 0.004 | ||
| PPDA in Purkinje cells | 5 | Tukey | Charge transfer | 0.015 | ||
| 8D | Interaction genotype/stimulation | 6–7 | 2-way RM ANOVA | Charge transfer | F(2,37) = 4.258 | 0.033 |
| R-CPP in GluN2D KO | 6 | Tukey | Charge transfer | 0.997 | ||
| R-CPP in Golgi cells | 7 | Tukey | Charge transfer | 0.002 | ||
| R-CPP in Purkinje cells | 6 | Tukey | Charge transfer | 0.938 | ||
| 9B | R-CPP in WT | 8 | 1-way RM ANOVA | mIPSC frequency | F(1,15) = 3.227 | 0.115 |
| 10B | R-CPP in Stellate cell WT | 5 | Paired t-test | Charge transfer | 0.247 | |
| 10D | R-CPP in WT | 5 | Paired t-test | NMDA current amplitude | 0.053 | |
| 10E | R-CPP in WT | 5 | Paired t-test | NMDA current charge transfer | 0.598 | |
| 10F | R-CPP in WT | 5 | Paired t-test | NMDA current decay time | 0.207 | |
| 11A | Comparison between groups | 6–9 | 1-way ANOVA | Charge transfer | F(5,42) = 1.266 | 0.299 |
| 11B | Interaction %change in mIPSC frequency/charge transfer | 57 | Pearson product moment correlation | Charge transfer | R2 = 0.008 | 0.518 |
| 11C | Interaction %change in mIPSC frequency/initial mIPSC frequency | 59 | Pearson product moment correlation | mIPSC frequency | R2 < 0.001 | 0.976 |
| 11A legend | Interaction %change in mIPSC frequency/charge transfer | 8 | Pearson product moment correlation | Charge transfer | R2 = 0.175 | 0.302 |
| Methods | Washout R-CPP | 16 | Paired t-test | NMDA current charge transfer | 0.114 | |
| Methods | Washout Ifen | 5 | Paired t-test | NMDA current charge transfer | 0.367 | |
| Methods | Washout PPDA | 7 | Paired t-test | NMDA current charge transfer | 0.246 |
n, No. of mice. PPDA, (2S*,3R
)-1-(phenanthren-2-carbonyl)piperazine-2,3-dicarboxylic acid;
WT, wild type; Ifen, ifenprodil; SC, stellate cells; KO, knockout; R-CPP, 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphanoic acid; NMDA, N-methyl-d-aspartate; mIPSC, miniature inhibitory synaptic currents; RM, repeated measures. Bolded P values indicate a significant effect (P < 0.05).
RESULTS
Stellate cells express functional GluN2D and GluN2B receptors.
We first determined whether functional GluN2D-containing receptors are present in the dendrites of stellate cells. While NMDARs are not present at the synapse, high-frequency stimulation of PF triggers a glutamate spillover, which can evoke currents mediated by NMDARs. These receptors are presumably located on dendrites but at extrasynaptic sites (Carter and Regehr 2000; Clark and Cull-Candy 2002; Sun and Liu 2007). Because both recombinant and native di-heteromeric GluN2D (di-GluN2D) receptors display a slow decay time when characterized at room temperature (Misra et al. 2000), we measured dendritic NMDAR currents in response to a train of PF stimulation at 100 Hz (4 stimuli) in WT and GluN2D KO mice (Ikeda et al. 1995). We found that deletion of GluN2D subunits accelerated the decay kinetics of dendritic NMDAR currents (Fig. 1A). The decay time constant of NMDAR currents decreased from 338 ± 30 ms (n = 18) in WT to 203 ± 23 ms (n = 12; P < 0.002; Fig. 1B) in GluN2D KO mice. This is in agreement with the presence of NMDARs that contain GluN2D subunits (Cull-Candy et al. 2001). The total charge transfer of NMDA currents decreased by ∼60% in mutant mice (Fig. 1D). Furthermore, 0.1 μM PPDA, which blocks GluN2D-containing NMDARs, reduced the NMDA-evoked currents in outside-out patches from WT stellate cells by 57 ± 6% (n = 5; P < 0.01; Fig. 1, E and F). These results indicate that GluN2D-containing receptors mediate the dendritic and somatic NMDAR currents in stellate cells.
We next investigated the contribution of GluN2B-containing NMDARs to dendritic NMDAR currents in WT mice. At 33–37°C, 3 μM ifenprodil inhibited the amplitude of dendritic NMDAR currents by 32 ± 6% (before, 427 ± 103; ifenprodil, 291 ± 89 pA; n = 7; P < 0.001) and reduced the charge transfer by 20 ± 7% (P < 0.001). Thus GluN2B receptors are present in the dendrites of stellate cells, consistent with a report that GluN2B, but not GluN2A-containing NMDARs, mediate dendritic NMDAR currents in rat stellate/basket cells (Bidoret et al. 2015). Ifenprodil also prolonged the decay time constant in WT mice from 163 ± 10 ms to 201 ± 15 ms (Fig. 2, A and B; P < 0.05), consistent with the presence of GluN2D-containing NMDARs. Furthermore, we found that 3 μM ifenprodil inhibited NMDA-evoked currents in outside-out patches from WT stellate cells by 47 ± 9% (n = 5; P < 0.01; Fig. 2, C and D). A marked increase in the blockade of NMDAR currents by ifenprodil in GluN2D KO mice to ∼90% inhibition, compared with WT mice, indicates that somatic NMDAR currents are largely mediated by GluN2B and GluN2D receptors.
Deletion of GluN2D subunits prevents I-LTP.
We next investigated the role of the GluN2D subunit in the induction of I-LTP. We recorded mIPSCs from stellate cells to monitor spontaneous GABA release and then stimulated PFs to evoke glutamate release. In vivo studies show that sensory stimulation evokes a burst of three to four action potentials at ∼80 Hz in rats (Chadderton et al. 2004). We therefore stimulated PFs using five trains of four depolarizations at 100 Hz and found that this induced a lasting increase in mIPSC frequency of 55 ± 23% (prestimulation 1.3 ± 0.4 Hz; poststimulation 2.0 ± 0.6 Hz; n = 8; P < 0.001) in most neurons tested (7 of 8 cells), consistent with our laboratory's previous observation (Lachamp et al. 2009). The mIPSC amplitude was not altered (prestimulation 22.7 ± 0.8 pA; poststimulation 20.2 ± 1.8 pA), indicating an increase in spontaneous GABA release from stellate cells in WT mice (Fig. 3, A, C–E). However, we found that this threshold stimulation protocol failed to produce a sustained increase in mIPSC frequency in all six stellate cells tested from GluN2D KO mice (prestimulation 1.5 ± 0.5 Hz; poststimulation 1.3 ± 0.4 Hz) and the amplitude remained unaltered (prestimulation 32 ± 4 pA; poststimulation 32 ± 3 pA; Fig. 3, B–E).
To test whether this change was due to reduced glutamate release from PFs in mutant mice, we stimulated PFs with two consecutive stimuli, and EPSCs were recorded at −60 mV in the presence of R-CPP (10 μM) and picrotoxin (100 μM) to block NMDA and GABA receptors, respectively. The paired pulse ratio of evoked EPSCs did not change in the GluN2D KO mice (WT 1.9 ± 0.2, n = 7; GluN2D KO 2.0 ± 0.2, n = 6; Fig. 4A), indicating that the probability of glutamate release from PFs in mutant mice was not altered. Furthermore, mIPSC frequency was not altered in GluN2D KO mice (WT 2.2 ± 0.3 Hz, n = 49; GluN2D KO 2.1 ± 0.3 Hz, n = 36; Fig. 4B), suggesting that basal spontaneous GABA release was also unaffected. Thus deletion of GluN2D subunits prevents the NMDAR-dependent, lasting increase in GABA release induced using a threshold stimulation protocol.
Fig. 4.

NMDA receptors do not regulate basal spontaneous GABA release in cerebellar stellate cells. A, top: representative recordings of excitatory postsynaptic currents (EPSCs) evoked by PF stimulation with two consecutive stimuli in WT (left) and GluN2D KO mice (right). Bottom: the paired-pulse ratio of EPSCs at the PF-stellate cell synapse (the ratio of amplitude of EPSC2/EPSC1) was not modified in GluN2D KO mice (WT n = 7, GluN2D KO n = 6), indicating that the release probability of glutamate from PFs was not altered. B: cumulative distribution of mIPSC frequency (left) and amplitude (right) in WT (solid line) and GluN2D KO mice (dashed line). mIPSC frequency was not altered in GluN2D KO mice (n = 36) compared with WT mice (n = 49). Thus deletion of GluN2D did not change basal spontaneous GABA release from stellate cells. C: representative traces of mIPSCs (outward currents) in WT (left) and GluN2D KO (right) mice. The bottom traces are enlargements of regions designated by the dashed lines. D: NMDA receptor blockers did not alter basal mIPSC frequency recorded at −60 mV using a CsCl-based pipette solution in WT stellate cells. After a stable baseline was obtained in the presence of 0.5 μM TTX and 5 μM 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione to isolate mIPSCs, PPDA (left, 0.1 μM, n = 5), ifenprodil (center, 3 μM, n = 5) or 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phosphanoic acid (R-CPP; right, 0.2 μM, n = 5) were added to the artificial cerebrospinal fluid for at least 20 min.
GluN2D sets a low threshold for induction of I-LTP.
Tri-heteromeric receptors are composed of two GluN1 and two distinct GluN2 subunits and are thought to be the majority of the native NMDARs in the hippocampus (Rauner and Köhr 2011). NMDARs that contain both GluN2B and GluN2D subunits have been described in cerebellar Golgi cells and substantia nigra dopaminergic neurons (Brickley et al. 2003; Brothwell et al. 2008; Jones and Gibb 2005). While NMDARs containing two GluN2D subunits have low channel conductance and opening probability, tri-GluN2B/2D receptors exhibit distinct biophysical characteristics, with additional large 50-pS conductance channel openings and low sensitivity to Mg2+ blockade (Huang and Gibb 2014; Misra et al. 2000). Thus these receptors can be activated at more hyperpolarized potentials than di-GluN2B receptors and generate a larger current than di-GluN2D receptors. Prominent GluN2A/B staining was observed in the axonal terminals of cerebellar basket cells (Petralia et al. 1994). This raises the possibility that tri-GluN2B/2D NMDARs could be present in axons and induce I-LTP in WT mice.
Deletion of GluN2D would convert a tri-GluN2B/2D NMDAR to di-GluN2B receptor. Because GluN2D receptors have a higher affinity for glutamate than GluN2B receptors, we predicted that prolonging PF stimulation would increase the likelihood of activation of low-affinity NMDARs by spillover glutamate and thus rescue I-LTP in GluN2D KO mice. To test this possibility, we increased the PF stimulation to 15 trains.
As we have shown previously, 15 trains of PF stimulation induced a sustained increase in the frequency of mIPSCs (74 ± 14%; prestimulation 4.2 ± 1.0 Hz; poststimulation 7.8 ± 2.4 Hz; n = 8; P < 0.001) with little effect on the amplitude (prestimulation 39 ± 6 pA, poststimulation 35 ± 5 pA; Fig. 5A) in WT mice (Lachamp et al. 2009). In contrast to the five-train stimulation, this protocol successfully induced a sustained increase in the frequency of mIPSCs in GluN2D KO mice (Fig. 5, B, C, and E). The frequency of mIPSCs increased by 91 ± 25% (prestimulation 1.3 ± 0.4 Hz; poststimulation 2.2 ± 0.6 Hz; n = 9; P < 0.05; Fig. 5, B and C) over a period of 30 min relative to control without changing the mIPSC amplitude (prestimulation 24.2 ± 4.4 pA; poststimulation 24.1 ± 4.4 pA, Fig. 5, B and D). Thus the presence of GluN2D in NMDARs lowered the threshold for induction of I-LTP.
Fig. 5.

Genetic deletion of GluN2D subunits increased the threshold of induction of I-LTP. A prolonged stimulation protocol, 15 trains of PF stimulation, was used to induce an increase in mIPSC frequency (I-LTP) in both WT and GluN2D KO mice. A and B, left: representative traces of mIPSCs (outward currents) before (top) and after (bottom) PF stimulation in WT (A) and KO (B) mice. Center: time course of mIPSC frequency (top) and amplitude (bottom). Right: group data (means ± SE) showing the time course of mIPSC frequency (top) and amplitude (bottom). Insets: representative traces during PF stimulation. C and D: in WT mice, 15 trains of PF stimulation induced a lasting increase in mIPSC frequency (C) without any change in the amplitude (n = 8; D), suggesting an increase in spontaneous GABA release from stellate cells (I-LTP). In GluN2D KO mice, this stimulation protocol also induced a sustained increase in mIPSCs frequency without a change in the amplitude (n = 9). E: group data showing that 15 trains of PF stimulation were able to rescue I-LTP in GluN2D KO mice. Therefore, incorporation of GluN2D subunits in NMDA receptors in WT mice lowers the threshold for the induction of I-LTP. *P < 0.05.
We next investigated the possibility that tri-GluN2B/2D NMDARs induce I-LTP in stellate cells from WT mice. Deletion of GluN2D is predicted to alter the tri-GluN2B/2D receptors to di-GluN2B receptors, which contain two GluN2B subunits. Ifenprodil at 3 μM has been shown to inhibit di-GluN2B receptors, but not tri-GluN2B/2D NMDARs (Brickley et al. 2003). Application of ifenprodil (3 μM) did not block the 15-train PF stimulation-induced increase in mIPSC frequency (95 ± 37%; prestimulation 1.2 ± 0.3 Hz; poststimulation 2.4 ± 0.8 Hz; n = 6; P < 0.05; Fig. 6, A, C, and E) and did not alter the basal mIPSC frequency (Fig. 4D) in WT mice. Thus di-GluN2B receptors are not necessary for the induction of I-LTP. We, therefore, determined whether activation of di-GluN2B receptors triggered I-LTP in GluN2D KO mice. In contrast to WT mice, application of the selective antagonists ifenprodil (3 μM) and RO 04–5595 (5 μM) during a 15-train PF stimulation protocol completely prevented the induction of I-LTP (prestimulation 2.2 ± 0.8 Hz; poststimulation 1.6 ± 0.6 Hz; Fig. 6, B–E) in mutant mice. These results indicate that activation of di-GluN2B receptors induces a lasting increase in GABA release only in mutant mice but not in WT mice (Fig. 6E). Therefore, tri-GluN2B/2D receptors in WT mice are responsible for the induction of I-LTP.
Pharmacological blockade of the PF stimulation-induced lasting increase in GABA release.
Our results so far suggest that NMDARs that are responsible for I-LTP contain both GluN2B and GluN2D subunits. NMDARs are located in both the dendrites and axon terminals of stellate cells. Our laboratory has previously shown that neither the release of NO or endocannabinoids, nor a calcium rise in postsynaptic stellate cells, is required for I-LTP (Lachamp et al. 2009). Thus postsynaptic NMDAR activation is unlikely to trigger I-LTP. Axonal NMDARs can be activated by glutamate (Duguid et al. 2007; Rossi et al. 2012) and, therefore, are strong candidates for induction of I-LTP. It has also been shown that activation of somato-dendritic NMDARs can lead to depolarization of axon terminals of stellate cells and enhances GABA release (Christie and Jahr 2008). Thus I-LTP can potentially be induced either by direct activation of axonal receptors, or by activation of somato-dendritic receptors. The latter model makes two predictions. First inhibition of somato-dendritic receptors should prevent I-LTP. Our results, however, show that GluN2B blockers inhibited dendritic NMDARs, but failed to block I-LTP (Figs. 2E and 6A). Second inhibitors that block I-LTP should also inhibit NMDAR currents in the soma or dendrites. To address this issue, we tested the ability of two inhibitors to block tri-GluN2B/2D receptors and determined whether these inhibitors prevented the induction of I-LTP.
First, PPDA exhibits a moderate preference for GluN2C and GluN2D over GluN2A and GluN2B recombinant receptors. Thus PPDA at 0.1 μM is reported to preferentially block GluN2C/D-containing NMDARs (Feng et al. 2004). To determine whether 0.1 μM PPDA inhibits native GluN2D-containing receptors, we took advantage of the well-characterized expression of tri-GluN2B/2D (and di-GluN2B) receptors in P7-10 Golgi cells, and di-GluN2D receptors in Purkinje cells (Brickley et al. 2003; Misra et al. 2000). We excised outside-out patches from somata of these neurons and evoked NMDAR currents by application of NMDA and glycine. Renzi et al. (2007) have shown that ∼25% of Purkinje cell patches also have large-conductance NMDAR currents due to GluN2A and 2B receptors. However, such currents were not detected in our patches, and, therefore, NMDAR currents in these patches were mainly mediated by di-GluN2D receptors. We found that 0.1 μM PPDA inhibited NMDA-evoked currents in outside-out patches excised from Golgi cells (−45 ± 13%; n = 5; P < 0.01; Fig. 7, B and D). PPDA also reduced the somatic single-channel currents mediated via di-GluN2D NMDARs in Purkinje cells (−59 ± 13%; n = 5; P < 0.05; Fig. 7, C and D). In contrast, PPDA at 0.1 μM did not inhibit somatic currents mediated by di-GluN2B receptors in stellate cells from P18 GluN2D KO mice (3 ± 9%; n = 6; P > 0.05; Fig. 7, A and D; the predicted inhibition of recombinant di-GluN2B receptors is ∼15–20%). Thus 0.1 μM PPDA inhibits both tri-GluN2B/2D and di-GluN2D receptors, but not di-GluN2B receptors (Fig. 7D) in cerebellar neurons. When 0.1 μM PPDA was applied during a 15-train PF stimulation protocol, PF stimulation failed to enhance GABA release (mIPSC frequency: prestimulation 4.2 ± 1.1 Hz; poststimulation 4.0 ± 1.0 Hz; n = 6; Fig. 7, E and F). Application of PPDA alone did not alter the frequency of mIPSCs (Fig. 4D). This result is consistent with the idea that GluN2D receptors are critically involved in the induction of I-LTP in stellate cells.
Fig. 7.

A low concentration of PPDA inhibited GluN2D-containing NMDA receptors and blocked I-LTP. A–D: assessment of the selectivity of 0.1 μM PPDA on native NMDAR subtypes in somatic patches from stellate cells (GluN2D KO; A), Golgi cells (WT; B), and Purkinje cells (WT; C). In WT mice postnatal days 8–10, Golgi cells are known to have a mixed population of di-GluN2B and tri-GluN2B/2D receptors (Brickley et al. 2003), whereas mainly di-GluN2D receptors are present in Purkinje cells (Renzi et al. 2007). D: PPDA (0.1 μM) reduced the charge transfer of NMDA receptor currents in both Purkinje cells (n = 5) and Golgi cells (n = 5), but not in stellate cells from GluN2D KO mice (n = 6). Thus 0.1 μM PPDA inhibits GluN2D-containing NMDA receptors but not di-GluN2B NMDA receptors. E: application of 0.1 μM PPDA during TTX washout and during the PF stimulation in WT animals. Left: representative traces of mIPSCs before (top) and after (bottom) PF stimulation. Center: time course of mIPSC frequency (top) and amplitude (bottom) from the corresponding recordings on the left. Right: group data (means ± SE) showing the time course of mIPSC frequency (circles) and amplitude (triangles). Light gray area shows the means ± SE of mIPSC frequency after PF stimulation without antagonist for comparison (Fig 5A). F: application of 0.1 μM PPDA during PF stimulation abolished I-LTP (n = 7). For drug effects: *P < 0.05, **P < 0.01. For the interaction between drugs and cell types: #P < 0.05.
Second, a low concentration of CPPene has been shown to inhibit recombinant tri-GluN2B/2D receptors (IC50 = 0.06 μM), di-GluN2B receptors (IC50 = 0.14 μM), but not di-GluN2D receptors (IC50 = 1.8 μM) (Buller and Monaghan 1997). We next tested whether 0.2 μM R-CPP, the parent compound of d-CPPene, blocked tri-GluN2B/2D, but not di-GluN2D, and determined its effects on di-GluN2B receptors in neurons. R-CPP at 0.2 μM did not inhibit somatic GluN2B NMDAR currents in stellate cells from GluN2D KO mice (4 ± 6%; Fig. 8, A and D), which could be due to a reduced inhibitory potency of R-CPP relative to CPPene (Lowe et al. 1990). Application of 0.2 μM R-CPP reversibly inhibited NMDA-evoked currents mediated via tri-GluN2B/2D NMDARs in outside-out patches from the somata of Golgi cells (−54 ± 8%; n = 7; P < 0.01; washout: −6 ± 12%; n = 5; Fig. 8, B and D), but did not block NMDAR currents in patches from Purkinje cells (7 ± 9%; n = 6; P > 0.05; Fig. 8, C and D). Thus in cerebellar neurons, R-CPP at 0.2 μM blocks tri-GluN2B/2D NMDARs rather than di-GluN2D or di-GluN2B receptors.
Fig. 8.

A: low concentration of R-CPP inhibited tri-GluN2B/2D receptors, but not di-GluN2B or di-GluN2D receptors. A–C: the effects of 0.2 μM R-CPP on native NMDA receptors containing the GluN2B and/or GluN2D subunits was assessed in outside-out patches excised from the somata of 3 types of neurons. NMDA receptor currents in somatic patches from stellate cells (postnatal days 18–21 GluN2D KO; A), Golgi cells (B), and Purkinje cells (C) were evoked by bath application of 10 μM NMDA and 10 μM glycine. R-CPP 0.2 μM blocked NMDA receptor currents in outside-out patches from Golgi cells. D: group data show that 0.2 μM R-CPP reduced the charge transfer of NMDA receptor currents in patches from Golgi cells (GC; n = 7) but not from Purkinje cells (PC; n = 6) or stellate cells (SC) from GluN2D KO mice (n = 6). These results demonstrate that a low concentration of R-CPP blocked tri-GluN2B/2D receptors but not di-GluN2B or di-GluN2D receptors. For drug effects: **P < 0.01. For the interaction between drugs and cell types: #P < 0.05.
If tri-GluN2B/2D NMDARs are involved in I-LTP induction, a low concentration of R-CPP should block I-LTP. Indeed we found that application of 0.2 μM R-CPP during PF stimulation prevented the induction of I-LTP in WT mice (prestimulation 1.1 ± 0.4 Hz; poststimulation 0.8 ± 0.3 Hz; n = 8; P > 0.05; Fig. 9), but did not modify basal GABA release (Fig. 4D). This is consistent with the idea that tri-GluN2B/2D receptors are required to induce I-LTP in stellate cells.
Fig. 9.

A low concentration of R-CPP prevented I-LTP in WT mice. To test the effects of 0.2 μM R-CPP on the induction of I-LTP, 0.2 μM R-CPP was applied during TTX washout and during the 15-train PF stimulation in WT mice (see Fig. 4). A, left and center: representative mIPSC traces and time course of mIPSC frequency and amplitude from a typical recording. Right: group data (means ± SE) showing that R-CPP at 0.2 μM prevented the PF stimulation-induced increase in GABA release (n = 8). For comparison, the light gray area shows the means ± SE of mIPSC frequency after PF stimulation without antagonist found in Fig 5A. B: the frequency of mIPSCs was not altered following PF stimulation (15 min before TTX washout and 15–30 min after PF stimulation). This supports the idea that activation of tri-GluN2B/2D NMDA receptors is required for the induction of I-LTP.
Our results show a low concentration of R-CPP and PPDA can block tri-GluN2B/2D receptors, but they are likely to also inhibit recombinant GluN2A and GluN2C receptors, respectively (Buller and Monaghan 1997; Feng et al. 2004). Because ifenprodil blocked I-LTP in GluN2D KO mice, GluN2A and GluN2C receptors are unlikely to be involved in the induction of I-LTP. The effect of a low concentration of R-CPP on other NMDAR subtypes remains to be determined.
Do I-LTP inhibitors also block somato-dendritic NMDAR currents?
The dendritic model in which activation of dendritic NMDARs is necessary for the induction of presynaptic I-LTP predicts that inhibitors that block I-LTP should also inhibit NMDAR currents in the soma or dendrites. We, therefore, determined the effect of a low concentration of R-CPP on somato-dendritic NMDAR currents in WT mice.
Application of R-CPP at 0.2 μM did not block the NMDAR current in somatic patches from stellate cells (1 ± 4%; n = 5; Fig. 10, A and B). Because a low concentration of R-CPP inhibits NMDA-evoked currents in outside-out patches from cerebellar Golgi cells that express tri-GluN2B/2D receptors, these receptors are unlikely to be present in the somata of stellate cells.
Fig. 10.

A low concentration of R-CPP did not inhibit somato-dendritic NMDA receptor currents in stellate cells. A and B: outside-out somatic patches were excised from stellate cells from WT mice (A), and NMDA receptor currents were recorded during the application of 10 μM NMDA and 10 μM glycine (B). After establishment of a stable baseline, R-CPP (0.2 μM) that blocks tri-GluN2B/2D was added to the superfusion medium. B: group data showing that R-CPP had no effect, suggesting that tri-GluN2B/2D NMDA receptors are not expressed in the somata of stellate cells. C: dendritic NMDA receptor currents were evoked by a train of PF stimulation at 100 Hz (4 stimuli). Currents were recorded at +40 mV in stellate cells before (black traces) and during (gray traces) superfusion of an NMDA receptor blocker. R-CPP (0.2 μM) did not reduce the PF-evoked NMDA receptor current. D–F: R-CPP (0.2 μM) did not alter the amplitude (D), charge transfer (E), and decay time (F) of dendritic NMDA receptor currents (open circles indicate data from individual experiments). In WT mice, NMDARs that are sensitive to a low concentration of R-CPP, including tri-GluN2B/2D receptors, are not present on the soma of stellate cells. R-CPP prevented I-LTP but did not inhibit dendritic currents; therefore, I-LTP, which requires tri-GluN2B/2D receptors (Figs. 5 and 6), is not triggered by a somato-dendritic depolarization.
We then stimulated PFs at 100 Hz (4 stimuli) to activate dendritic NMDARs at 33–37°C and found that 0.2 μM R-CPP had no effect on the amplitude of dendritic NMDAR currents (prestimulation 463 ± 85 pA; R-CPP 412 ± 79 pA; n = 5) or total charge transfer (3 ± 3%). The decay time also remained unaltered (prestimulation 219 ± 17 ms; R-CPP 234 ± 22 ms; Fig. 10, C–F). Thus NMDARs that are sensitive to a low concentration of R-CPP, including tri-GluN2B/2D receptors, are absent from the somata and dendrites of stellate cells. The IC50 of R-CPP for NMDAR subtypes may differ between somatic and dendritic receptors, which were activated by application of NMDA and spillover glutamate, respectively. We, therefore, compared the effects of 0.2 μM R-CPP on I-LTP with that on dendritic NMDARs, since in the dendritic model, activation of dendritic NMDARs is responsible for the induction of I-LTP. Because 0.2 μM R-CPP completely prevented the induction of I-LTP, but failed to block dendritic currents (Fig. 9), the tri-GluN2B/2D receptors that are responsible for the induction of I-LTP are unlikely to be located in the somata and dendrites of stellate cells. Consistent with this idea, we found no correlation between the degree of I-LTP and the total charge transfer of the slow component of the dendritic currents recorded at −60 mV during PF stimulation (Fig. 11). The latter is presumably mediated in large part by postsynaptic GluN2D-containing receptors due to their lower Mg2+ blockade compared with GluN2B receptors (Lachamp et al. 2009). These results are not in agreement with the prediction of a model in which activation of dendritic NMDARs induces I-LTP. Therefore, NMDARs located in axons rather than in dendrites are most likely to induce I-LTP.
DISCUSSION
Our results address a fundamental issue concerning synaptic cross talk: what types of presynaptic NMDARs in inhibitory interneurons sense glutamate release and modulate GABA release? We found that tri-GluN2B/2D receptors are responsible for the induction of I-LTP, and these receptors are absent in dendrites. This result indicates that GluN2D-containing NMDARs at the presynaptic terminals sense the low concentration of glutamate that spills over from neighboring excitatory inputs. These receptors thus mediate the cross talk between excitatory and inhibitory transmission.
Which type of NMDARs induce I-LTP?
The subunit composition of NMDARs controls a number of biophysical properties of the channel, as well as their subcellular localization (Cull-Candy et al. 2001; Petralia et al. 2009; Siegler Retchless et al. 2012; Yuan et al. 2009) and, therefore, endows them with different roles in regulating synaptic transmission. Postsynaptic NMDARs that contain GluN2A and GluN2B subunits are important for the induction of long-term plasticity at excitatory synapses (Wyllie et al. 2013). Many GABAergic interneurons express high levels of GluN2D mRNA (Akazawa et al. 1994; Monyer et al. 1994; Thompson et al. 2000). However, the role of GluN2D receptors in inhibitory neurons is not known. GluN2D has a high glutamate binding affinity and, therefore, may detect a low concentration of glumate that has spilled over from nearby excitatory inputs. In vivo studies show that somatosensory stimulation evokes a short burst of action potentials in PF inputs to stellate cells (Chadderton et al. 2004). Our finding that deletion of GluN2D abolished the I-LTP induced by a few trains of PF stimulation reveals that GluN2D receptors are critical for triggering a lasting increase in GABA release from inhibitory interneurons. Using a combined pharmacological and genetic approach, we identify these receptors as tri-GluN2B/2D receptors, and the presence of GluN2D lowers the threshold of induction of I-LTP. This result is in agreement with previous observations that a NMDA-evoked enhancement in mIPSC frequency displayed a low sensitivity to ifenprodil and Mg2+ blockade, a characteristic of tri-GluN2B/2D receptors (Glitsch and Marty 1999; Huang and Gibb 2014; Rossi et al. 2012). Such tri-GluN2B/2D receptors are also present in substantia nigra dopaminergic neurons (Brothwell et al. 2008; Jones and Gibb 2005) and may be involved in the regulation of neurotransmitter release from these cells. Indeed deletion of GluN2D subunits reduces dopamine levels in the prefrontal cortex and hippocampus (Miyamoto et al. 2002) and alters locomotor activity (Hagino et al. 2010). Therefore GluN2D receptors are likely to play a major role in the glutamate-dependent modulation of neurotransmitter release.
Where are the tri-GluN2B/2D receptors located?
We found that a low concentration of R-CPP completely abolished I-LTP, but had no effect on somato-dendritic NMDAR currents. In contrast, ifenprodil partially blocked somato-dendritic NMDARs, but failed to prevent I-LTP. Therefore, activation of dendritic NMDARs is not required for the induction of I-LTP, and thus NMDARs responsible for I-LTP are most likely present in axons. Burst stimulation of PFs can also activate NMDARs in the dendrites (Carter and Regehr 2000; Clark and Cull-Candy 2002; Sun and Liu 2007), producing a sustained depolarization (Carter and Regehr 2000), which activates calcium channels in axons and enhances GABA release from stellate cells (Christie and Jahr 2008; Pugh and Jahr 2011). However, our results do not support the idea that an NMDAR-dependent somato-dendritic depolarization electrotonically spreads to the axon to induce I-LTP. Although NMDARs can act as both metabotropic and ionotropic receptors (Nabavi et al. 2013), we have previously shown that calcium entry via NMDARs is required for the induction of I-LTP (Liu and Lachamp 2006); thus a metabotropic action of somato-dendritic NMDARs is unlikely to trigger I-LTP. Our results suggest that spillover glutamate acts on NMDARs at the presynaptic terminals of GABAergic interneurons to induce I-LTP, which is supported by a variety of other reports. First, glutamate uncaging at axonal locations in stellate cells evoked a calcium transient in the axon terminals via NMDAR activation (Rossi et al. 2012). Second, glutamate release from isolated Purkinje cells induces an NMDAR-dependent increase in GABA release from presynaptic terminals (Duguid et al. 2007). Third, GluN2A/B immunoreactivity is present in basket cell terminals (Petralia et al. 1994) in the cerebellum. Fourth, functional NMDA channel currents with high and low conductance, a characteristic of tri-GluN2B/2D receptors (Brickley et al. 2003; Jones and Gibb 2005), have been detected in the enlarged varicosities of cultured stellate/basket cell axon terminals following NMDA treatment (Fiszman et al. 2005). Thus GluN2D-containing NMDARs at presynaptic sites may modulate GABA release in other neurons, since GluN1 subunits have been found in the axon terminals of other GABAergic interneurons and GluN2D mRNA is present in many inhibitory interneurons (Akazawa et al. 1994; Monyer et al. 1994; Paquet and Smith 2000; Thompson et al. 2000).
Glutamate acts on NMDARs in presynaptic and postsynaptic neurons to regulate GABA release via distinct mechanisms. It is known that glutamate receptors in postsynaptic neurons can trigger the release of retrograde signals that modulate GABA release (Castillo et al. 2011). While a metabotropic glutamate receptor-evoked release of endocannabinoids suppresses GABA release, NMDAR activation promotes an NO-dependent enhancement of GABA secretion (Adermark and Lovinger 2009; Chevaleyre et al. 2007; Chevaleyre and Castillo 2003; Jiang et al. 2010; Marsicano et al. 2002; Nugent et al. 2007). However, I-LTP induction in stellate cells is not induced by retrograde signals, because our laboratory has previously shown that loading postsynaptic stellate cells with BAPTA or the use of NO synthase inhibitors failed to prevent I-LTP (Lachamp et al. 2009). Here we found that tri-GluN2B/2D NMDARs in presynaptic inhibitory interneurons detect the spillover of glutamate and lead to an enhancement of GABA release. The ability to modulate GABA release in the cerebellum is critical for the physiological functioning of this brain region, because an increase in GABA release from stellate/basket cells alters motor coordination and has also been implicated in neurological disorders, such as episodic ataxia (Herson et al. 2003). Our finding that activation of presynaptic tri-GluN2D/2B receptors induces I-LTP opens the possibility for selectively modulating GABA release without affecting plasticity at those excitatory synapses that require postsynaptic NMDARs that contain GluN2A and GluN2B subunits (Wyllie et al. 2013).
GRANTS
This work was supported by National Science Foundation Grant IBN-0344559 and National Institutes of Health Grants NS-58867 and MH-095948 (S. J. Liu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.J.D., P.M.L., L.S., and S.J.L. conception and design of research; C.J.D., P.M.L., and L.S. performed experiments; C.J.D., P.M.L., and L.S. analyzed data; C.J.D., P.M.L., L.S., and S.J.L. interpreted results of experiments; C.J.D., P.M.L., and L.S. prepared figures; C.J.D. and S.J.L. drafted manuscript; C.J.D. and S.J.L. edited and revised manuscript; C.J.D., P.M.L., L.S., M.M., and S.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Daniel Monaghan, Matthieu Maroteaux, Iaroslav Savtchouk, Yu Liu, and Matthew Whim for experimental advice and helpful discussions.
REFERENCES
- Adermark L, Lovinger DM. Frequency-dependent inversion of net striatal output by endocannabinoid-dependent plasticity at different synaptic inputs. J Neurosci 29: 1375–1380, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-d-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol 347: 150–160, 1994. [DOI] [PubMed] [Google Scholar]
- Bidoret C, Bouvier G, Ayon A, Szapiro G, Casado M. Properties and molecular identity of NMDA receptors at synaptic and non-synaptic inputs in cerebellar molecular layer interneurons. Front Synaptic Neurosci 7: 1, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Misra C, Mok M, Mishina M, Cull-Candy SG. NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDA receptor subtype restricted to extrasynaptic sites. J Neurosci 23: 4958–4966, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothwell SLC, Barber JL, Monaghan DT, Jane DE, Gibb AJ, Jones S. NR2B- and NR2D-containing synaptic NMDA receptors in developing rat substantia nigra pars compacta dopaminergic neurones. J Physiol 586: 739–750, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller AL, Monaghan DT. Pharmacological heterogeneity of NMDA receptors: characterization of NR1a/NR2D heteromers expressed in Xenopus oocytes. Eur J Pharmacol 320: 87–94, 1997. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci 20: 4423–4434, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Chiu CQ, Carroll RC. Long-term plasticity at inhibitory synapses. Curr Opin Neurobiol 21: 328–338, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature 428: 856–860, 2004. [DOI] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JLR, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468: 263–269, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38: 461–472, 2003. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron 54: 801–812, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Dendritic NMDA receptors activate axonal calcium channels. Neuron 60: 298–307, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci 22: 4428–4436, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327–335, 2001. [DOI] [PubMed] [Google Scholar]
- Dubois C, Ramamoorthy P, Whim M, Liu S. Activation of NPY type 5 receptors induces a long-lasting increase in spontaneous GABA release from cerebellar inhibitory interneurons. J Neurophysiol 107: 1655–1665, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Pankratov Y, Moss GWJ, Smart TG. Somatodendritic release of glutamate regulates synaptic inhibition in cerebellar Purkinje cells via autocrine mGluR1 activation. J Neurosci 27: 12464–12474, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci 7: 525–533, 2004. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron 62: 757–771, 2009. [DOI] [PubMed] [Google Scholar]
- Feng B, Tse HW, Skifter DA, Morley R, Jane DE, Monaghan DT. Structure-activity analysis of a novel NR2C/NR2D-preferring NMDA receptor antagonist: 1-(phenanthrene-2-carbonyl) piperazine-2,3-dicarboxylic acid. Br J Pharmacol 141: 508–516, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszman ML, Barberis A, Lu C, Fu Z, Erdélyi F, Szabó G, Vicini S. NMDA receptors increase the size of GABAergic terminals and enhance GABA release. J Neurosci 25: 2024–2031, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiarsa JL, Caillard O, Ben-Ari Y. Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci 25: 564–570, 2002. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci 19: 511–519, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagino Y, Kasai S, Han W, Yamamoto H, Nabeshima T, Mishina M, Ikeda K. Essential role of NMDA receptor channel ε4 subunit (GluN2D) in the effects of phencyclidine, but not methamphetamine. PloS One 5: e13722, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herson PS, Virk M, Rustay NR, Bond CT, Crabbe JC, Adelman JP, Maylie J. A mouse model of episodic ataxia type-1. Nat Neurosci 6: 378–383, 2003. [DOI] [PubMed] [Google Scholar]
- Huang Z, Gibb AJ. Mg2+ block properties of triheteromeric GluN1-GluN2B-GluN2D NMDA receptors on neonatal ratsubstantia nigra pars compacta dopaminergic neurones. J Physiol 592: 2059–2078, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Araki K, Takayama C, Inoue Y, Yagi T, Aizawa S, Mishina M. Reduced spontaneous activity of mice defective in the epsilon 4 subunit of the NMDA receptor channel. Brain Res Mol Brain Res 33: 61–71, 1995. [DOI] [PubMed] [Google Scholar]
- Jiang B, Huang S, de Pasquale R, Millman D, Song L, Lee HK, Tsumoto T, Kirkwood A. The maturation of GABAergic transmission in visual cortex requires endocannabinoid-mediated LTD of inhibitory inputs during a critical period. Neuron 66: 248–259, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Gibb AJ. Functional NR2B- and NR2D-containing NMDA receptor channels in rat substantia nigra dopaminergic neurones. J Physiol 569: 209–221, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachamp PM, Liu Y, Liu SJ. Glutamatergic modulation of cerebellar interneuron activity is mediated by an enhancement of GABA release and requires protein kinase A/RIM1alpha signaling. J Neurosci 29: 381–392, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levelt CN, Hübener M. Critical-period plasticity in the visual cortex. Annu Rev Neurosci 35: 309–330, 2012. [DOI] [PubMed] [Google Scholar]
- Lien CC, Mu Y, Vargas-Caballero M, Poo M. Visual stimuli-induced LTD of GABAergic synapses mediated by presynaptic NMDA receptors. Nat Neurosci 9: 372–380, 2006. [DOI] [PubMed] [Google Scholar]
- Liu S, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405: 454–458, 2000. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Lachamp P. The activation of excitatory glutamate receptors evokes a long-lasting increase in the release of GABA from cerebellar stellate cells. J Neurosci 26: 9332–9339, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang LI, Tao HW. Heterosynaptic scaling of developing GABAergic synapses: dependence on glutamatergic input and developmental stage. J Neurosci 27: 5301–5312, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DA, Neijt HC, Aebischer B. d-CPP-ene (SDZ EAA 494), a potent and competitive N-methyl-d-aspartate (NMDA) antagonist: effect on spontaneous activity and NMDA-induced depolarizations in the rat neocortical slice preparation, compared with other CPP derivatives and MK-801. Neurosci Lett 113: 315–321, 1990. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530–534, 2002. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Kauer JA. Presynaptic plasticity: targeted control of inhibitory networks. Curr Opin Neurobiol 19: 254–262, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra C, Brickley S, Wyllie D, Cull-Candy SG. Slow deactivation kinetics of NMDA receptors containing NR1 and NR2D subunits in rat cerebellar Purkinje cells. J Physiol 525: 299–305, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Yamada K, Noda Y, Mori H, Mishina M, Nabeshima T. Lower sensitivity to stress and altered monoaminergic neuronal function in mice lacking the NMDA receptor epsilon 4 subunit. J Neurosci 22: 2335–2342, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529–540, 1994. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A 110: 4027–4032, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature 446: 1086–1090, 2007. [DOI] [PubMed] [Google Scholar]
- Paquet M, Smith Y. Presynaptic NMDA receptor subunit immunoreactivity in GABAergic terminals in rat brain. J Comp Neurol 423: 330–347, 2000. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Al-Hallaq RA, Wenthold RJ. Trafficking and targeting of NMDA receptors. In: Biology of the NMDA Receptor. Frontiers in Neuroscience, edited by Van Dongen AM. Boca Raton, FL: CRC, 2009, chapt. 8. [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci 14: 6102–6120, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh JR, Jahr CE. NMDA receptor agonists fail to alter release from cerebellar basket cells. J Neurosci 31: 16550–16555, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauner C, Köhr G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-Methyl-d-aspartate receptor population in adult hippocampal synapses. J Biol Chem 286: 7558–7566, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzi M, Farrant M, Cull-Candy SG. Climbing-fibre activation of NMDA receptors in Purkinje cells of adult mice. J Physiol 585: 91–101, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Banerjee A, Paulsen O. Presynaptic NMDA receptors and spike timing-dependent depression at cortical synapses. Front Synaptic Neurosci 2: 18, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi B, Ogden D, Llano I, Tan YP, Marty A, Collin T. Current and calcium responses to local activation of axonal NMDA receptors in developing cerebellar molecular layer interneurons. PloS One 7: e39983, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scelfo B, Sacchetti B, Strata P. Learning-related long-term potentiation of inhibitory synapses in the cerebellar cortex. Proc Natl Acad Sci U S A 105: 769–774, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Linden DJ. An NMDA receptor/nitric oxide cascade is involved in cerebellar LTD but is not localized to the parallel fiber terminal. J Neurophysiol 94: 4281–4289, 2005. [DOI] [PubMed] [Google Scholar]
- Siegler Retchless B, Gao W, Johnson JW. A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat Neurosci 15: 406–413, S1–S2, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Liu SJ. Activation of extrasynaptic NMDA receptors induces a PKC-dependent switch in AMPA receptor subtypes in mouse cerebellar stellate cells. J Physiol 583: 537–553, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Drewery DL, Atkins HD, Stephenson FA, Chazot PL. Immunohistochemical localization of N-methyl-d-aspartate receptor NR1, NR2A, NR2B and NR2C/D subunits in the adult mammalian cerebellum. Neurosci Lett 283: 85–88, 2000. [DOI] [PubMed] [Google Scholar]
- Wilms CD, Häusser M. Reading out a spatiotemporal population code by imaging neighbouring parallel fibre axons in vivo. Nat Commun 6: 6464, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodin MA, Maffei A. (editors) Inhibitory Synaptic Plasticity. New York: Springer, 2011. [Google Scholar]
- Wyllie DJA, Livesey MR, Hardingham GE. Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74: 4–17, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci 29: 12045–12058, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]