

Abstract
Chronic intermittent hypoxia (CIH) is a hallmark manifestation of sleep apnea. A heightened carotid body activity and the resulting chemosensory reflex mediate increased sympathetic nerve activity by CIH. However, the mechanisms underlying heightened carotid body activity by CIH are not known. An elevation of intracellular calcium ion concentration ([Ca2+]i) in glomus cells, the primary oxygen-sensing cells, is an essential step for carotid body activation by hypoxia. In the present study, we examined the effects of CIH on the glomus cell [Ca2+]i response to hypoxia and assessed the underlying mechanisms. Glomus cells were harvested from adult rats or wild-type mice treated with 10 days of either room air (control) or CIH (alternating cycles of 15 s of hypoxia and 5 min of room air; 9 episodes/h; 8 h/day). CIH-treated glomus cells exhibited an enhanced [Ca2+]i response to hypoxia, and this effect was absent in the presence of 2-(4-cyclopropylphenyl)-N-((1R)-1-[5-[(2,2,2-trifluoroethyl)oxo]-pyridin-2-yl]ethyl)acetamide (TTA-A2), a specific inhibitor of T-type Ca2+ channels, and in voltage-gated calcium channel, type 3.2 (CaV3.2), null glomus cells. CaV3.2 knockout mice exhibited an absence of CIH-induced hypersensitivity of the carotid body. CIH increased reactive oxygen species (ROS) levels in glomus cells. A ROS scavenger prevented the exaggerated TTA-A2-sensitive [Ca2+]i response to hypoxia. CIH had no effect on CaV3.2 mRNA levels. CIH augmented Ca2+ currents and increased CaV3.2 protein in plasma membrane fractions of human embryonic kidney-293 cells stably expressing CaV3.2, and either a ROS scavenger or brefeldin-A, an inhibitor of protein trafficking, prevented these effects. These findings suggest that CIH leads to an augmented Ca2+ influx via ROS-dependent facilitation of CaV3.2 protein trafficking to the plasma membrane.
Keywords: sleep apnea, voltage-gated Ca2+ channels, hypertension, protein trafficking, oxidative stress
sleep-disordered breathing with recurrent apnea is a major clinical problem affecting an estimated 9% of adult women and 24% of adult men in the United States (Young et al. 1993). Recurrent apneas are characterized by transient, repetitive cessations of breathing, which can be either due to obstruction of the upper airway (obstructive sleep apnea) or defective generation of respiratory rhythm by the central nervous system (central apnea) (García-Río et al. 2000; Nieto et al. 2000; Ramirez et al. 2013). In severely affected patients, arterial blood oxygen (O2) saturation is reduced to as low as 50% during apneic episodes. Thus chronic intermittent hypoxia (CIH) is a hallmark manifestation of recurrent apnea. Recurrent apnea patients exhibit several comorbidities, including heightened activation of the sympathetic nervous system, and are prone to develop hypertension (Prabhakar et al. 2015). Carotid bodies are the principal sensory organs for detecting changes in arterial blood O2 levels, and the ensuing chemosensory reflex is a potent regulator of sympathetic tone and blood pressure (Fitzgerald and Lahiri 1986; Kumar and Prabhakar 2012). Several lines of evidence from sleep apnea patients and CIH-exposed rodents suggest that heightened carotid body chemosensory reflex contributes to sympathetic activation and hypertension caused by recurrent apnea (Kara et al. 2003; Prabhakar et al. 2015).
An increase in carotid body sensory nerve activity is an essential prerequisite for heightened chemosensory reflex. CIH-exposed rodents exhibit an augmented carotid body sensory nerve response to hypoxia (Del Rio et al. 2010, 2012; Peng and Prabhakar 2003; Peng et al. 2004; Rey et al. 2004). However, the effects of CIH on glomus cells, the primary O2-sensing cells of the carotid body, have received less attention. It is well established that an elevation of intracellular calcium ion concentration ([Ca2+]i) in glomus cells is an essential step for producing sensory nerve excitation by hypoxia (Kumar and Prabhakar 2012). However, the effects of CIH on the glomus cell [Ca2+]i response to hypoxia and the underlying mechanisms have not been examined.
Voltage-gated calcium channels (VGCCs) mediate hypoxia-evoked [Ca2+]i elevation in glomus cells (Kumar and Prabhakar 2012). There are nine pore-forming VGCC Ca2+ channel genes, divided into three families, including the high-threshold VGCC, types 1 and 2 (CaV1 and CaV2), family of Ca2+ channels and the low-threshold CaV3 family of channels. The CaV1 and CaV2 families of VGCCs are heteromeric protein complexes. The T-type Ca2+ channels, members of the CaV3 family, are comprised exclusively of a single pore-forming α1 subunit. T-type Ca2+ channels are products of three different T-type Ca2+ channel genes: CaV3.1, CaV3.2, and CaV3.3 (Catterall 2011; Catterall et al. 2005).
Several studies have documented the role of high-threshold VGCC, especially the L-type (CaV1), in the [Ca2+]i response of glomus cells to hypoxia (Buckler and Vaughan-Jones 1994; e Silva and Lewis 1995; Makarenko et al. 2012; Summers et al. 2000). We recently reported that low-threshold T-type VGCC, especially the CaV3.2, plays an equally important role in mediating hypoxia-induced Ca2+ influx in glomus cells (Makarenko et al. 2015). In the present study, we examined the effects of CIH on the glomus cell [Ca2+]i response to hypoxia and assessed the role of VGCC. Our results demonstrate that CIH augments the [Ca2+]i response to hypoxia in rat and mouse glomus cells, and this effect is mediated primarily by CaV3.2 T-type VGCC. Studies on human embryonic kidney (HEK)-293 cells stably expressing CaV3.2 showed that CIH facilitates trafficking of the CaV3.2 protein to the plasma membrane via reactive oxygen species (ROS) signaling.
METHODS
Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Chicago. All experiments were performed on adult male Sprague-Dawley rats and age- as well as sex-matched CaV3.2 knockout (Cacna1h−/−) and C57BL/6J mice.
Exposure of rats to CIH.
Awake rats or mice were exposed to a CIH paradigm consisting of 15 s of hypoxia, followed by 5 min of room air (9 episodes/h, 8 h/day), as described previously (Peng et al. 2009; Peng and Prabhakar 2004). Briefly, animals housed in feeding cages were placed in a chamber for exposure to CIH. The animals were unrestrained, freely mobile, and fed ad libitum. The chamber was flushed with alternating cycles of nitrogen and compressed room air so that inspired O2 levels reached 5% O2 during hypoxia and 21% O2 during room air (normoxia). Ambient O2 levels in the chamber were monitored continuously with an O2 analyzer (model OM-11; Beckman Coulter, Brea, CA). A continuous vacuum was created within the chamber to balance the pressure between the inflow and outflow of gases. Inspired carbon dioxide (CO2) levels were maintained at 0.2–0.5% and were monitored continuously by an infrared analyzer (model LB-2; Beckman Coulter). The duration of the gas flow during each hypoxic and normoxic episode was regulated by timer-controlled solenoid valves. Animals exposed to alternating cycles of room air instead of hypoxia for 10 days served as controls. Animals were subjected to either IH or normoxia between 9 AM and 5 PM for 10 consecutive days. In the experiments involving antioxidant treatment, rats received manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP; Alexis Biochemicals, Enzo Life Sciences, Farmingdale, NY), a membrane-permeable superoxide anion scavenger via an intraperitoneal route each morning (5 mg·kg−1·day−1) before they were subjected to a daily regimen of IH. MnTMPyP was given for 10 days. Within 12 h after terminating the 10-day CIH treatment, rats or mice were anesthetized, carotid bodies were dissected, and glomus cells were isolated as described below.
Carotid body sensory nerve activity.
The protocols for recording sensory nerve activity from ex vivo carotid bodies from mice were essentially the same as described previously (Makarenko et al. 2015). Briefly, the carotid body with the sinus nerve was placed in a recording chamber (250 μl vol) and superfused at a rate of 2.5 ml/min with warm (36°C) physiological saline (in mM: 125 NaCl, 5 KCl, 1.8 CaCl2, 2 MgSO4, 1.2 NaH2PO4, 25 NaHCO3, 10 d-glucose, and 5 sucrose). The medium was bubbled with 21% O2-5% CO2. Partial pressure O2 (Po2), partial pressure CO2, and pH of the medium were determined by a blood-gas analyzer (model ABL 5; Radiometer, Copenhagen, Denmark). To facilitate recording of clearly identifiable action potentials, the sinus nerve was treated with 0.1% collagenase for 5 min. Action potentials (2–4 active units) were recorded from one of the nerve bundles with a suction electrode, amplified (alternating current preamplifier, 100–3,000 Hz bandwidth; model P511K; Grass Technologies, Natus Neurology, Middleton, WI), displayed on an oscilloscope (model 5B12N; Tektronix, Beaverton, OR), and stored in a computer via an analog-to-digital (A/D) translation board (PowerLab/8P; ADInstruments, Colorado Springs, CO). “Single” units were selected on the basis of the height and duration of the individual action potentials using a spike discrimination program (Spike Histogram program, PowerLab; ADInstruments).
Primary cultures of glomus cells.
Preparation of primary cultures of glomus cells is essentially the same as described previously (Makarenko et al. 2012; Peng et al. 2014). Briefly, carotid bodies were harvested from rats and mice anesthetized with urethane (1.2 g/kg ip), and glomus cells were dissociated using a mixture of collagenase P (2 mg/ml; Roche Applied Science, Indianapolis, IN), DNase (15 μg/ml; Sigma-Aldrich, St. Louis, MO), and BSA (3 mg/ml; Sigma-Aldrich) at 37°C for 20 min, followed by a 15-min incubation in medium containing DNase (30 μg/ml). Cells were plated on collagen (type VII; Sigma-Aldrich)-coated coverslips and maintained at 37°C in a 7% CO2 + 20% O2 incubator for 12–18 h. The growth medium consisted of F-12K medium (Invitrogen, Thermo Fisher Scientific, Grand Island, NY), supplemented with 1% FBS, insulin-transferrin-selenium (ITS-X; Invitrogen, Thermo Fisher Scientific), and 1% penicillin-streptomycin-glutamine mixture (Invitrogen, Thermo Fisher Scientific).
Real-time RT-PCR.
Carotid bodies were harvested from anesthetized rats (n = 4 rats). Two carotid bodies were pooled, and RNA was extracted using Trizol and reverse transcribed using SuperScript III RT. Real-time RT-PCR was performed using a MiniOpticon system (Bio-Rad Laboratories, Hercules, CA) with the SYBR GreenER Two-Step qRT-PCR Kit (#11764-100; Invitrogen, Thermo Fisher Scientific). Primer sequences for real-time RT-PCR amplification were as follows: 18S forward (fw), GTAACCCGTTGAACCCCATT, 18S reverse (rev), CCATCCAATCGGTAGTAGCG (size, 151; GenBank Accession Number X_01117); CaV3.1 fw, CTTTGACCTGCTGACACTCTG, CaV3.1 rev, GCCATTACAGTCTTGGTGCTCA (size, 185; GenBank Accession Number AF290212); CaV3.2 fw, ACTTGGCCATCGTCCTCCTA, CaV3.2 rev, ATGGTGGGATTGATGGGCAG (size, 101; GenBank Accession Number AF290213); and CaV3.3 fw, GACCCCAGAGCAGTGAGGAT, CaV3.3 rev, TACTTGCTGTCCACGATGCC (size, 155; GenBank Accession Number AF290214). Relative mRNA level was calculated based on the comparative threshold (Ct), known as the 2−[ΔΔ]Ct method, where [ΔΔ]Ct = ΔCt (IH) − ΔCt (normoxia). The ΔCt values of both normoxia and IH are normalized to an internal standard (18s RNA).
Measurements of [Ca2+]i.
Glomus cells were incubated in HBSS with 2 μM Fura-2-AM and 1 mg/ml albumin for 30 min and then washed in a Fura-2-free solution for 30 min at 37°C. The coverslip was transferred to an experimental chamber for determining the changes in [Ca2+]i. Background fluorescence at 340 and 380 nm wavelength was obtained from an area of the coverslip that was devoid of cells. On each coverslip, 5–12 glomus cells were selected (identified by their characteristic clustering), and individual cells were imaged. Image pairs (one at 340 and the other at 380 nm) were obtained every 2 s by averaging 16 frames at each wavelength. Data were collected continuously throughout the experiment. Background fluorescence was subtracted from the cell data obtained at the individual wavelengths. The image obtained at 340 nm was divided by the 380-nm image to obtain ratiometric image. Ratios were converted to free [Ca2+]i using calibration curves constructed in vitro by adding Fura-2 (50 μM-free acid) to solutions containing known concentrations of Ca2+ (0–2,000 nM). The recording chamber was superfused continually with solution from gravity-fed reservoirs.
Measurements of nucleotide autofluorescence.
Autofluorescence images were obtained by using a DM-LFSA Leica microscope (Leica Microsystems GmbH, Wetzlar, Germany) with a 10× objective and ORCA-ER camera (Hamamatsu Photonics K.K., Hamamatsu City, Japan). To detect intrinsic reduced nicotinamide nucleotides, a 4′,6′-diamidino-2-phenylindole (DAPI) filter set (excitation λ of 366 nm/emission λ of 450 nm) was used. Oxidized flavoproteins were identified using an FITC filter set (excitation λ of 460 nm/emission λ of 540 nm). All of the images and fluorescence ratios were processed and analyzed using HCImage software (Hamamatsu Photonics K.K.). During image acquisition, glomus cells were constantly superfused with HBSS solution, pH ∼7.4, fed by gravity. The intensity of background fluorescence was subtracted from the cellular autofluorescence intensity in a standardized fashion. For each glomus cell, the average value of fluorescence from the DAPI signal was divided by the average value of fluorescence from the FITC signal.
Culture of HEK-293 cells stably expressing CaV3.2 channels.
HEK-293 cells, stably expressing CaV3.2 (HEK-CaV3.2 cells; Invitrogen, Thermo Fisher Scientific), were kindly provided by Dr. Dorothy Hanks (The University of Chicago, School of Medicine, Chicago, IL). HEK-CaV3.2 cells were maintained in 100 mm culture dishes (Corning Life Sciences, Lowell, MA) in DMEM, supplemented with 10% FBS, 1% penicillin-streptomycin, 1% l-glutamine, and 50 μg/ml HygroGold (Invitrogen, Thermo Fisher Scientific).
Exposure of HEK-293 cells to IH.
Cell cultures were exposed to IH (1.5% O2 for 30 s, followed by 20% O2 for 5 min at 37°C), as described previously (Yuan et al. 2008). O2 levels were monitored by an electrode (Lazar) placed in the cell culture medium, and ambient O2 levels were monitored by an O2 analyzer (model LB-2; Beckman Coulter). In the experiments involving treatment with drugs, cells were preincubated for 30 min with either drug or vehicle.
Measurements of malondialdehyde.
Cells were homogenized in 10 vol of 20 mM phosphate buffer (pH 7.4) at 4°C, and the resulting homogenate was centrifuged at 500 g for 10 min at 4°C. Malondialdehyde (MDA) levels were analyzed in the supernatant, as described previously (Khan et al. 2011; Ramanathan et al. 2005). Briefly, 100 μl of either sample or the standard was added to 50 μl of 8.1% (wt/vol) SDS, 375 μl of 20% (vol/vol) acetic acid, and 375 μl of 0.8% (wt/vol) thiobarbituric acid. The samples were heated for 60 min in a boiling water bath, followed by incubation on an ice bath for 10 min and centrifuged at 3,000 g for 15 min. The supernatant was removed, and the absorbance of the solution was monitored at 532 nm. MDA was used as a standard, and the levels of MDA were reported in nanomoles/milligram of protein.
Measurements of CaV3.2 T-type Ca2+ current by electrophysiology.
HEK-CaV3.2 cells were voltage clamped in the whole-cell configuration of the patch-clamp technique using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). The currents were filtered at 2 kHz and then digitized at 100 μs per point with an A/D card (National Instruments, Austin, TX). Voltage protocols and data analysis were performed in WinWCP (University of Strathclyde, Scotland, UK). Recordings were made using trypsinized cells (0.25% trypsin-EDTA; Sigma-Aldrich), 2–3 days after plating. All recordings were made at room temperature (20–26°C). CaV3.2 currents were recorded from a holding potential of −90 mV in 10 mV increasing step pulses up to +40 mV. Current-voltage (I–V) curve was generated by analyzing the peak current at each voltage step of 200 ms duration from −90 to +40 mV. The electrode potential was adjusted to give zero current between the pipette solution and bath solution before the cells were attached. No correction for liquid junction potential was applied. The values for the cell capacitance varied between 10 and 25 pF. Current density of the peak current at −30 mV was measured by dividing the peak current with the corresponding cell capacitance. Recordings were made using patch pipettes that were made with a micropipette puller (Narishige Scientific Instrument, Tokyo, Japan) and borosilicate glass (World Precision Instruments, Sarasota, FL). After fire polishing, final electrode resistance, when filled with the cesium chloride-based patch-pipette solution, was 2 MΩ. Electrodes were filled with the following solution: 100 mM CsCl, 5 mM MgCl2, 40 mM HEPES, 10 mM EGTA, and 2 mM ATP, pH 7.3 (adjusted by cesium hydroxide, 0.295 osmol/kgH2O). Recordings were made in a tetraethylammonium (TEA)-based extracellular solution containing the following solution: 140 mM TEA-Cl2, 10 mM glucose, 10 mM HEPES, and 10 mM CaCl2, pH 7.3 (adjusted by TEA-hydroxide). Nickel(II) chloride was prepared as a 100-mM stock solution in distilled water and then added directly to the TEA recording solution to a final concentration of 100 μM.
Isolation of membrane fractions.
HEK-293 cells, exposed to either normoxia or IH, were homogenized in 25 mM Tris-HCl buffer, pH 7.5, containing 250 mM sucrose, 20 mM MgCl2, 0.1 mM EGTA, 0.2 mM PMSF, 1 mM 2-aminoethylisothiouronium bromide, protease (Roche Applied Science), and phosphatase (EMD Biosciences, San Diego, CA) inhibitor cocktail. The extract was centrifuged at 5,500 g for 15 min to remove the nuclear and mitochondrial fractions, and the resulting supernatant was centrifuged further at 40,000 g for 30 min. Both the supernatant and the pellet containing the soluble and the membrane fraction, respectively, were stored at −80°C until further analysis by Western blot assay.
Western blot analysis.
Western blotting was performed as described previously (Yuan et al. 2005). Briefly, cell extracts (20 μg) were fractionated by 8% polyacrylamide-SDS gel electrophoresis and transferred to a polyvinyl pyrrolidone difluoride membrane (Immobilon-P; EMD Millipore, Bedford, MA). The membrane was blocked with Tris-buffered saline, containing 5% nonfat milk, and incubated with an anti-CaV3.2 polyclonal antibody (1:500 dilution; Alamone Labs, Jerusalem, Israel) in blocking buffer. Membranes were treated with goat anti-rabbit secondary antibody conjugated with horseradish peroxidase (1:2,000 dilution; EMD Millipore), and immune complexes were visualized using an enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ). The blots were scanned and quantified using Scion Image software (National Institutes of Health, Bethesda, MD). Similar procedures were used for immunoblot analysis of cadherin (1:3,000 dilution; Sigma-Aldrich).
Drugs and chemicals.
All stock solutions were made fresh before the experiments. 2-(4-Cyclopropylphenyl)-N-((1R)-1-[5-[(2,2,2-trifluoroethyl)oxo]-pyridin-2-yl]ethyl)acetamide (TTA-A2; Alamone Labs), nifedipine (Sigma-Aldrich), and MnTMPyP (Enzo Life Sciences) were obtained from commercial sources. Nifedipine stock solution was made in HBSS, TTA-A2 stock solution was made in DMSO (Sigma-Aldrich), and pH of final solutions was adjusted to ∼7.4. Desired concentrations of drugs were added to glomus cell culture plates.
Analysis of data.
Average data are presented as means ± SE. Statistical significance was assessed by Student's t-test for statistical comparisons between two groups and one-way ANOVA, followed by Tukey's test for multiple group comparisons. P < 0.05 was considered significant.
RESULTS
CIH augments Ca2+ influx by hypoxia in glomus cells via CaV3.2 T-type VGCC.
Glomus cell [Ca2+]i responses to hypoxia (medium Po2 34 ± 3 mmHg) were determined in control and CIH-exposed rats. Hypoxia increased [Ca2+]i levels in control glomus cells, and this response increased approximately twofold in CIH-treated cells (Fig. 1, A and B). The [Ca2+]i response to hypoxia was examined in the presence of TTA-A2, a selective blocker of T-type VGCC (Martin et al. 2000; Randall and Tsien 1997; Shipe et al. 2008). TTA-A2 prevented the augmented Ca2+ influx by CIH, and the magnitude of the remaining [Ca2+]i response to hypoxia was the same as in control glomus cells, which were blocked by nifedipine, an L-type VGCC inhibitor (Fig. 1, A and B). Nifedipine, by itself, inhibited the [Ca2+]i response in CIH-treated glomus cells by 40 ± 4%, as opposed to 60 ± 8.5% inhibition in control glomus cells (P < 0.05), suggesting that nifedipine was less effective in blocking the augmented [Ca2+]i response by CIH compared with TTA-A2.
Fig. 1.

Effect of chronic intermittent hypoxia (CIH) on hypoxia-evoked calcium ion (Ca2+) influx in glomus cells. A: examples of hypoxia [Hx; partial pressure of oxygen (Po2) 34 ± 3 mmHg]-induced intracellular Ca2+ concentration ([Ca2+]i) responses of glomus cells from rats treated with either normoxia (N; n = 35 cells) or CIH (n = 27 cells) for 10 days. [Ca2+]i responses to hypoxia in CIH-treated cells were monitored with and without 25 μM 2-(4-cyclopropylphenyl)-N-((1R)-1-[5-[(2,2,2-trifluoroethyl)oxo]-pyridin-2-yl]ethyl)acetamide [TTA-A2 (TTA); n = 31 cells] and 25 μM TTA-A2 + 10 μM nifedipine (TTA + Nif; n = 29 cells). Horizontal bars represent the duration of the hypoxic challenge and TTA-A2 and TTA-A2 + nifedipine applications. B: average data (means ± SE) of hypoxia-induced [Ca2+]i responses were presented as stimulus-evoked response minus baseline levels {Δ [Ca2+]i (nM)}. **P < 0.01; n.s., not significant (P > 0.05).
CaV3.2 is the primary T-type VGCC expressed in glomus cells (Makarenko et al. 2015). To assess the selective contribution of CaV3.2, studies were performed on glomus cells from wild-type and Cacna1h−/− mice. CIH-treated wild-type glomus cells exhibited robust facilitation of Ca2+ influx by hypoxia compared with control cells (Fig. 2, A and B). CaV3.2 null glomus cells, before CIH, exhibited a reduced [Ca2+]i response to hypoxia compared with wild-type cells (Fig. 2, A and B). CIH was ineffective in augmenting Ca2+ influx by hypoxia in glomus cells from CaV3.2 null mice (Fig. 2, A and B). TTA-A2 had no effect on the Ca2+ response to hypoxia in CIH-treated CaV3.2 null glomus cells, and the remaining Ca2+ influx by hypoxia was blocked by nifedipine (Fig. 2, A and B).
Fig. 2.

Effect of CIH on the [Ca2+]i response to hypoxia in glomus cells from wild-type (WT) and voltage-gated calcium channel (VGCC), type 3.2 (CaV3.2), knockout (Cacna1h−/−) mice. A: examples of the [Ca2+]i response to hypoxia (Po2 34 ± 3 mmHg) in glomus cells from WT and Cacna1h−/−, treated with either normoxia (WT, n = 22 cells; Cacna1h−/−, n = 18 cells) or CIH (WT, n = 26 cells; Cacna1h−/−, n = 19 cells) for 10 days. The effects of CIH on the [Ca2+]i response to hypoxia were examined in Cacna1h−/− glomus cells in the presence and absence of 25 μM TTA-A2 (n = 17 cells) or 10 μM nifedipine (n = 15 cells). Horizontal bars represent the duration of hypoxia, TTA-A2, and nifedipine applications. B: average data (means ± SE) of the effects of CIH on the [Ca2+]i responses to hypoxia in glomus cells. The data presented are stimulus-evoked response minus baseline levels. **P < 0.01; n.s., not significant (P > 0.05).
To establish the functional significance of CaV3.2 VGCC, the carotid body sensory nerve response to hypoxia was recorded in control and CIH-treated wild-type and CaV3.2 null mice. Before CIH, CaV3.2 null mice exhibited a reduced carotid body sensory nerve response to hypoxia compared with wild-type mice (Fig. 3, A and B). Following CIH, wild-type carotid bodies responded with an augmented sensory nerve response to hypoxia, and this effect was nearly absent in CaV3.2 null carotid bodies (Fig. 3, A and B).
Fig. 3.

Effect of CIH on sensory nerve response of the carotid body to hypoxia in WT and Cacna1h−/− mice. A: representative examples of carotid body (CB) sensory nerve responses [impulses per second (imp/s)] to hypoxia (Po2 ∼35 mmHg) in WT and Cacna1h−/− mice exposed to either normoxia or CIH. Horizontal bars represent the duration of the hypoxic challenge. Inset: superimposed action potentials of a “single” fiber from which data were derived. B: average data (means ± SE) of carotid body sensory nerve response to hypoxia presented as stimulus-evoked activity minus baseline activity (Δimp/s). Each bar represents the data from 10–16 carotid bodies. **P < 0.01; n.s., not significant (P > 0.05).
ROS mediate the effects of CIH.
Cellular and systemic responses to CIH are mediated by ROS signaling (Prabhakar et al. 2015). To determine whether ROS contribute to the enhanced Ca2+ influx by CIH, ROS levels were determined in glomus cells by two independent approaches: one by monitoring the ratio of reduced/oxidized nucleotides by autofluorescence (del V Cano et al. 2008) and the other by measuring MDA levels, which represent oxidized lipids (Janero 1990). In CIH-exposed cells, the ratio of reduced/oxidized nucleotides decreased, and MDA levels increased compared with controls (normoxia), and these effects were absent following treatment with MnTMPyP, a membrane-permeable ROS scavenger (Fig. 4, A–C).
Fig. 4.

Effect of CIH on reactive oxygen species (ROS) levels in rat glomus cells. A: autofluorescence of reduced [excitation (Ex) λ366 nm/emission (Em) λ450 nm] and oxidized nucleotides (excitation λ460 nm/emission λ540 nm) was monitored as an index of ROS in rat glomus cells. Fluorescent and differential contrasting (DIC) photomicrographs of glomus cells treated with normoxia (top), CIH (middle), or CIH + manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP; bottom). Squares in each photomicrograph represent magnified (2×) images of the region denoted by dotted lines. B: quantitative analysis of the ratios of reduced/oxidized nucleotide autofluorescence intensity (means ± SE) in glomus cells treated with normoxia (n = 42 cells), CIH (n = 58 cells), or CIH and MnTMPyP (n = 49 cells). C: malondialdehyde (MDA) levels, representing the oxidized lipids, were analyzed as an index of ROS in carotid bodies from rats treated with normoxia, CIH, or CIH and MnTMPyP. Rats were treated every day for 10 days with MnTMPyP (5 mg/kg ip) before a daily regimen of CIH treatment. Data represent means ± SE from 3 independent experiments (4 carotid bodies/experiment). **P < 0.01; n.s., not significant (P > 0.05).
The augmented [Ca2+]i response to hypoxia in CIH-treated cells was prevented by MnTMPyP (Fig. 5, A and B). TTA-A2 was ineffective in reducing the [Ca2+]i response to hypoxia in CIH cells treated with MnTMPyP (Fig. 5, A and B).
Fig. 5.

Role of ROS in CIH-induced Ca2+ responses to hypoxia in rat glomus cells. A: examples of hypoxia (Po2 34 ± 3 mmHg)-induced [Ca2+]i responses of glomus cells from rats treated with normoxia (n = 49 cells), CIH (n = 57 cells), or CIH and MnTMPyP (n = 38 cells), with and without 25 μM TTA-A2 (n = 44 cells). Horizontal bars represent the duration of the hypoxia and TTA-A2 challenges. B: average data (means ± SE) of hypoxia-induced [Ca2+]i responses of glomus cells presented as stimulus-evoked response minus baseline levels. **P < 0.01; n.s., not significant (P > 0.05).
Effect of CIH on mRNAs encoding T-type VGCC subtypes.
Real-time RT-PCR analysis was performed on carotid bodies to determine whether CIH affects the expression of mRNAs encoding T-type VGCC subtypes. However, the mRNA levels of α1G, α1H, and α1I, which encode the pore-forming subunits of CaV3.1, -3.2, and -3.3, respectively, were comparable between CIH-treated and control carotid bodies (Fig. 6).
Fig. 6.
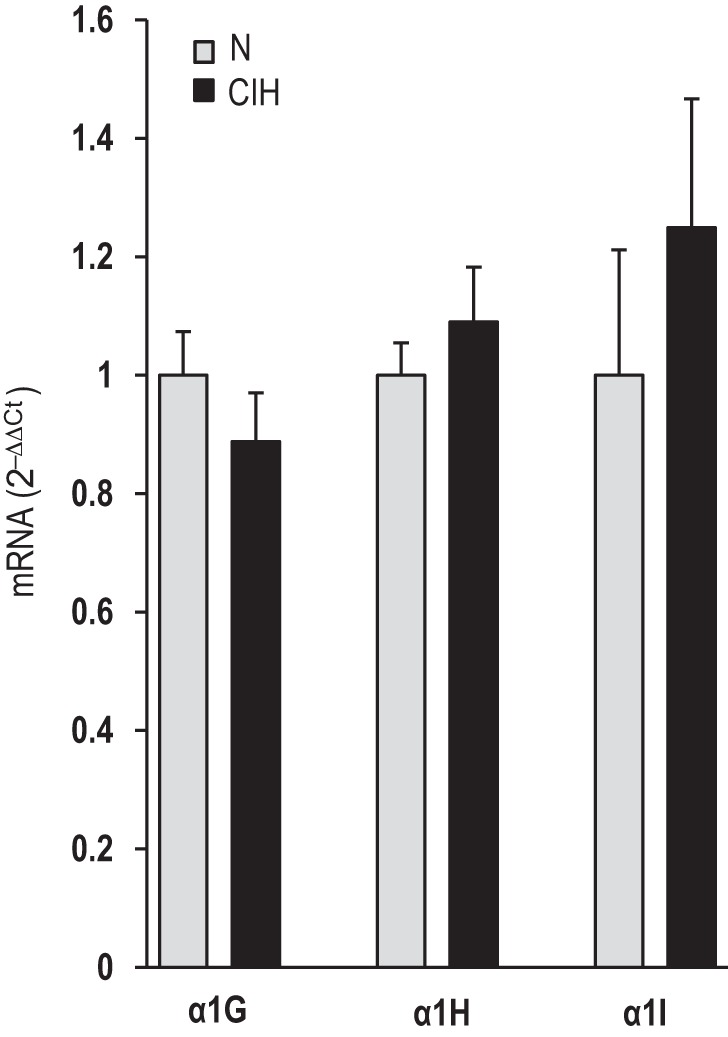
Effects of CIH on mRNAs encoding T-type VGCC subtypes in the rat carotid body. Real-time RT-PCR analysis of mRNAs encoding α1G, α1H, and α1I subunits of T-type Ca2+ channels in rat carotid bodies. The values of mRNAs are normalized to 18S mRNA and expressed as comparative threshold (Ct; 2−[ΔΔ]Ct). Data presented are means ± SE from 4 individual experiments performed in triplicate.
CIH facilitates CaV3.2 protein trafficking through ROS signaling.
The mechanisms underlying activation of CaV3.2 by CIH-induced ROS were examined. We hypothesized that altered kinetics of the channel activation and/or increased trafficking of the CaV3.2 protein to the plasma membrane contribute to CaV3.2 activation by ROS. However, the testing of these possibilities in glomus cells was found to be technically challenging because of the small size of the carotid body, which weighs only ∼60–80 μg in rats. To circumvent this limitation, studies were performed on HEK-293 cells stably expressing CaV3.2 protein.
Patch-clamp analysis showed an absence of Ca2+ currents in wild-type HEK-293 cells, whereas CaV3.2 HEK cells showed a robust T-type Ca2+ current, which was blocked by TTA-A2 (Fig. 7, A and B). CaV3.2-expressing HEK cells were treated with either normoxia or 60 cycles of IH (IH60), wherein each cycle consisted of 30 s of hypoxia (1.5% O2) and 5 min of normoxia (20% O2). In IH60-treated cells, Ca2+ currents were augmented, but neither the current kinetics nor the I–V relation was affected (Fig. 8, A and B). Ca2+ currents normalized to cell capacitance showed a significant increase in the current density in IH60-treated cells compared with control normoxic cells (Fig. 8C).
Fig. 7.

Characterization of Ca2+ currents in human embryonic kidney (HEK) cells stably expressing CaV3.2. A: representative examples of Ca2+ currents recorded from WT and CaV3.2-expressing HEK cells. Currents were recorded from a holding potential of −90 up to −30 mV. Ca2+ current in CaV3.2-HEK cells was recorded with and without 25 μM TTA-A2. B: average data (means ± SE) of peak current density in WT HEK (WT; n = 5 cells), CaV3.2-HEK cells (n = 10 cells), and CaV3.2-HEK cells in the presence of TTA-A2 (black bar; n = 5 cells). *P < 0.05.
Fig. 8.

Effect of IH on Ca2+ current in CaV3.2-HEK cells. A: representative Ca2+ currents from CaV3.2-HEK cells treated with either normoxia or 60 cycles of IH (IH60). Currents were recorded from a holding potential of −90 to +40 mV in 10 mV steps every 10 s. B: normalized current-voltage relation of CaV3.2 currents in cells treated with normoxia and IH60. C: normalized peak current density obtained with a step to −30 mV for normoxia- and IH60-treated CaV3.2-HEK cells. Normoxia, n = 32 cells; IH60, n = 28 cells (B and C). Data presented are means ± SE. **P < 0.01.
MDA levels were increased in CaV3.2 HEK cells treated with IH60 (Fig. 9A). MnTMPyP prevented the elevated MDA levels, the augmented Ca2+ current, and the increased current density in IH60-treated cells (Fig. 9, A–C).
Fig. 9.

ROS mediates the augmented Ca2+ current in IH-treated CaV3.2-HEK cells. A: MDA levels were determined as an index of ROS generation. Average data (means ± SE) of MDA in CaV3.2-HEK cells treated with normoxia or IH60 or IH60 in the presence of 50 μM MnTMPyP (n = 3 independent experiments each). B: representative Ca2+ currents from CaV3.2-HEK cells treated with normoxia or IH60, with and without 50 μM MnTMPyP. The recording protocols were same as in Fig. 8. C: average (means ± SE) normalized peak current density obtained with a step to −30 mV for normoxia (n = 9 cells)-, IH60 (n = 7 cells)-, and IH60 + 50 μM MnTMPyP (n = 9 cells)-treated CaV3.2-HEK cells. **P < 0.01; n.s., not significant (P > 0.05).
To determine whether IH facilitates translocation of CaV3.2 protein to the plasma membrane, Western blot analysis was performed on total and membrane fractions of CaV3.2 HEK-293 cells. Pan-cadherin protein expression was monitored as a marker of membrane fractions. In cells exposed to IH60, CaV3.2 protein expression in the membrane fraction, as well as the ratio of plasma membrane/total CaV3.2 protein expression, was significantly higher compared with control cells (Fig. 10, A and B). Brefeldin-A (BFA), a blocker of protein trafficking, prevented the increased membrane expression, the ratio of plasma membrane/total CaV3.2 protein expression, and the increased CaV3.2 current density in IH60-treated cells (Fig. 10, A–C).
Fig. 10.

IH facilitates CaV3.2 protein trafficking. A: representative Western blot (top) and densitometric analysis (bottom) of plasma membrane fractions of CaV3.2 and pan-cadherin (marker of membrane fraction) proteins of normoxia- and IH60-treated CaV3.2-HEK cells in the presence and absence of 30 ng/ml brefeldin A (BFA), a blocker of protein trafficking. B: densitometric analysis of Western blots of plasma membrane fractions of CaV3.2 expressed as a percentage of total cell CaV3.2 protein of normoxia- and IH60-treated CaV3.2-HEK cells in the presence and absence of 30 ng/ml BFA. C: average (means ± SE) normalized peak current density obtained with a step to −30 mV of CaV3.2-HEK cells treated with normoxia (n = 17 cells), IH60 (n = 18 cells), and IH60 + 30 nM BFA (n = 19 cells). **P < 0.01; n.s., not significant (P > 0.05).
DISCUSSION
The present study demonstrates that CIH augments hypoxia-induced Ca2+ in glomus cells of rat and mouse carotid bodies. The following findings suggest that T-type VGCC, especially the CaV3.2, mediates the augmented Ca2+ influx by CIH: 1) TTA-A2, a selective blocker of T-type VGCC, prevented the exaggerated [Ca2+]i response to hypoxia in CIH-treated glomus cells; 2) the augmented Ca2+ response by CIH was absent in CaV3.2 null glomus cells; and 3) nifedipine, a blocker of L-type VGCC, was ineffective in preventing the augmented Ca2+ response of glomus cells by CIH. TTA-A2 is a pan-T-type VGCC channel blocker that inhibits CaV3.1, -3.2, and -3.3 (Martin et al. 2000; Randall and Tsien 1997; Shipe et al. 2008). However, TTA-A2 had no effect on the glomus cell response to CIH in CaV3.2 null mice, suggesting that T-type VGCC, other than CaV3.2, plays little or no role in mediating the augmented glomus cell [Ca2+]i response to hypoxia by CIH. The absence of a heightened carotid body sensory nerve response to hypoxia in CIH-treated Cacna1h−/− mice suggests a critical role for CaV3.2 in mediating hypersensitivity of the carotid body to hypoxia by CIH.
Previous studies have shown that L-type VGCC plays an important role in hypoxia-induced Ca2+ influx in glomus cells (Buckler and Vaughan-Jones 1994; Makarenko et al. 2012). Consistent with these studies, nifedipine, an L-type VGCC blocker, abolished the normal [Ca2+]i response to hypoxia in the presence of TTA-A2 in CIH-treated glomus cells. It follows that the augmented Ca2+ influx by hypoxia is due to selective recruitment of T-type VGCC, primarily the CaV3.2, by CIH. Ortiz et al. (2013) reported that whereas CIH had no effect on the resting membrane potential of glomus cells, it modestly augments hypoxia-induced depolarization of rat glomus cells. Given that CaV3.2 is a low-threshold, activated T-type VGCC, it is ideally suited for transducing CIH-induced, modest depolarization of glomus cells into a robust [Ca2+]i elevation by hypoxia.
Our study further identified ROS signaling as an important cellular mechanism mediating the effects of CIH on T-type VGCC. Evidence includes the following: 1) ROS levels were increased in CIH-treated glomus, as well as in CaV3.2-HEK cells, and 2) MnTMPyP, a membrane-permeable ROS scavenger, not only prevented the CIH-induced ROS generation but also the augmented Ca2+ influx by hypoxia in glomus cells, as well as the increased Ca2+ current density in CaV3.2-HEK cells. The finding that TTA-A2 had no further effect on the hypoxia-induced Ca2+ influx in MnTMPyP-treated CIH glomus cells further suggests that T-type VGCC is the target of ROS signaling activated by CIH. Previous studies showed that CIH-induced ROS generation in the carotid body is complex and involves multiple oxidases, including the NADPH oxidase 2 (Peng et al. 2009) and xanthine oxidase (Nanduri et al. 2013), as well as inhibition of complex I of the mitochondrial electron transport chain (Khan et al. 2011) and decreased antioxidant enzyme activity (Nanduri et al. 2009). It remains to be established which of these sources of ROS contributes to the recruitment of CaV3.2 by CIH.
A previous study showed that CIH increases the CaV3.2 mRNA expression in neonatal rat adrenal medullary chromaffin cells (Souvannakitti et al. 2010). However, we found that CaV3.2 mRNA levels were unaltered in CIH-treated adult rat carotid bodies. Thus the transcriptional activation is unlikely to account for the enhanced CaV3.2 function in adult glomus cells by CIH. Studies on CaV3.2-HEK cells provided important mechanistic insights into how IH-induced ROS facilitate the CaV3.2 function. Although IH increased the CaV3.2 currents, it had no effect on either the current kinetics or the I–V relation, indicating that altered kinetics of channel activation is unlikely to mediate the enhanced CaV3.2 function. The following findings suggest that increased trafficking of the CaV3.2 protein to the plasma membrane accounts for the increased CaV3.2 current density by IH: 1) CaV3.2 protein expression increased in the membrane fractions of IH-treated CaV3.2-HEK cells, and this effect was prevented by BFA, a potent blocker of protein trafficking, and 2) BFA treatment blocked the increased CaV3.2 current density by IH. Although we were unable to demonstrate a similar phenomenon in glomus cells, due to the small size of the carotid body, the findings from CaV3.2-expressing HEK-293 cells suggest that post-translational mechanisms involving increased trafficking of channel protein to the membrane as a potential mechanism underlying increased CaV3.2 function by CIH.
In summary, the present study shows that CIH augments the [Ca2+]i response to acute hypoxia in rat and mouse glomus cells, and this effect is mediated by ROS-dependent activation of T-type VGCC, especially the CaV3.2 subtype. Studies on Cacna1h−/− mice showed that CaV3.2-mediated, enhanced Ca2+ influx is a major cellular mechanism contributing to the augmented carotid body sensory nerve response to hypoxia in CIH-treated rodents. In addition to sensitizing the hypoxic sensory response, CIH leads to plasticity of the carotid body manifested as sensory long-term facilitation (LTF), which is characterized by a long-lasting increase in baseline carotid body sensory nerve activity following repetitive hypoxia (Peng et al. 2003). The role of T-type VGCC in the induction of sensory LTF by CIH, however, remains to be elucidated.
GRANTS
Support for this research was provided by the National Heart, Lung, and Blood Institute (Grant PO1 HL-090554).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: V.V.M. and N.R.P. conception and design of research; V.V.M., G.U.A., Y.-J.P., S.A.K., and J.N. performed experiments; V.V.M., G.U.A., Y.-J.P., S.A.K., and J.N. analyzed data; V.V.M. prepared figures; V.V.M. and N.R.P. drafted manuscript; G.K.K., A.P.F., and N.R.P. edited and revised manuscript; N.R.P. interpreted results of experiments; N.R.P. approved final version of manuscript.
REFERENCES
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol 476: 423–428, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3: a003947, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 57: 411–425, 2005. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Iturriaga R. Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. Eur Respir J 36: 143–150, 2010. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Parga MJ, Madrid C, Iturriaga R. Carotid body inflammation and cardiorespiratory alterations in intermittent hypoxia. Eur Respir J 39: 1492–1500, 2012. [DOI] [PubMed] [Google Scholar]
- del V Cano M, Reyes JM, Park CY, Gao X, Mori K, Chuck RS, Gehlbach PL. Demonstration by redox fluorometry that sulforaphane protects retinal pigment epithelial cells against oxidative stress. Invest Ophthalmol Vis Sci 49: 2606–2612, 2008. [DOI] [PubMed] [Google Scholar]
- e Silva MJ, Lewis DL. L- and N-type Ca2+ channels in adult rat carotid body chemoreceptor type I cells. J Physiol 489: 689–699, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald R, Lahiri S. Reflex responses to carotid/aortic body stimulation. In: Handbook of Physiology: Respiratory System: Control of Breathing. Bethesda, MD: American Physiological Society, 1986, p. 313–362. [Google Scholar]
- García-Río F, Racionero MA, Pino JM, Martínez I, Ortuño F, Villasante C, Villamor J. Sleep apnea and hypertension. Chest 117: 1417–1425, 2000. [DOI] [PubMed] [Google Scholar]
- Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med 9: 515–540, 1990. [DOI] [PubMed] [Google Scholar]
- Kara T, Narkiewicz K, Somers VK. Chemoreflexes—physiology and clinical implications. Acta Physiol Scand 177: 377–384, 2003. [DOI] [PubMed] [Google Scholar]
- Khan SA, Nanduri J, Yuan G, Kinsman B, Kumar GK, Joseph J, Kalyanaraman B, Prabhakar NR. NADPH oxidase 2 mediates intermittent hypoxia-induced mitochondrial complex I inhibition: relevance to blood pressure changes in rats. Antioxid Redox Signal 14: 533–542, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol 2: 141–219, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenko VV, Nanduri J, Raghuraman G, Fox AP, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am J Physiol Cell Physiol 303: C916–C923, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenko VV, Peng YJ, Yuan G, Fox AP, Kumar GK, Nanduri J, Prabhakar NR. CaV3.2 T-type Ca2 channels in H2S-mediated hypoxic response of the carotid body. Am J Physiol Cell Physiol 308: C146–C154, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RL, Lee JH, Cribbs LL, Perez-Reyes E, Hanck DA. Mibefradil block of cloned T-type calcium channels. J Pharmacol Exp Ther 295: 302–308, 2000. [PubMed] [Google Scholar]
- Nanduri J, Vaddi DR, Khan SA, Wang N, Makerenko V, Prabhakar NR. Xanthine oxidase mediates hypoxia-inducible factor-2α degradation by intermittent hypoxia. PLoS One 8: e75838, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA 106: 1199–1204, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 283: 1829–1836, 2000. [DOI] [PubMed] [Google Scholar]
- Ortiz FC, Del Rio R, Ebensperger G, Reyes VR, Alcayaga J, Varas R, Iturriaga R. Inhibition of rat carotid body glomus cells TASK-like channels by acute hypoxia is enhanced by chronic intermittent hypoxia. Respir Physiol Neurobiol 185: 600–607, 2013. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Makarenko VV, Nanduri J, Vasavda C, Raghuraman G, Yuan G, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Inherent variations in CO-H2S-mediated carotid body O2 sensing mediate hypertension and pulmonary edema. Proc Natl Acad Sci USA 111: 1174–1179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci 29: 4903–4910, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA 100: 10073–10078, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol (1985) 96: 1236–1242, 2004. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol (1985) 94: 2342–2349, 2003. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Rennison J, Prabhakar NR. Intermittent hypoxia augments carotid body and ventilatory response to hypoxia in neonatal rat pups. J Appl Physiol (1985) 97: 2020–2025, 2004. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Kumar GK, Nanduri J. Peripheral chemoreception and arterial pressure responses to intermittent hypoxia. Compr Physiol 5: 561–577, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan L, Gozal D, Siegel JM. Antioxidant responses to chronic hypoxia in the rat cerebellum and pons. J Neurochem 93: 47–52, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez JM, Garcia AJ, Anderson TM, Koschnitzky JE, Peng YJ, Kumar GK, Prabhakar NR. Central and peripheral factors contributing to obstructive sleep apneas. Respir Physiol Neurobiol 189: 344–353, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall AD, Tsien RW. Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology 36: 879–893, 1997. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol 560: 577–586, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipe WD, Barrow JC, Yang ZQ, Lindsley CW, Yang FV, Schlegel KA, Shu Y, Rittle KE, Bock MG, Hartman GD, Tang C, Ballard JE, Kuo Y, Adarayan ED, Prueksaritanont T, Zrada MM, Uebele VN, Nuss CE, Connolly TM, Doran SM, Fox SV, Kraus RL, Marino MJ, Graufelds VK, Vargas HM, Bunting PB, Hasbun-Manning M, Evans RM, Koblan KS, Renger JJ. Design, synthesis, and evaluation of a novel 4-aminomethyl-4-fluoropiperidine as a T-type Ca2+ channel antagonist. J Med Chem 51: 3692–3695, 2008. [DOI] [PubMed] [Google Scholar]
- Souvannakitti D, Nanduri J, Yuan G, Kumar GK, Fox AP, Prabhakar NR. NADPH oxidase-dependent regulation of T-type Ca2+ channels and ryanodine receptors mediate the augmented exocytosis of catecholamines from intermittent hypoxia-treated neonatal rat chromaffin cells. J Neurosci 30: 10763–10772, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers BA, Overholt JL, Prabhakar NR. Augmentation of L-type calcium current by hypoxia in rabbit carotid body glomus cells: evidence for a PKC-sensitive pathway. J Neurophysiol 84: 1636–1644, 2000. [DOI] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328: 1230–1235, 1993. [DOI] [PubMed] [Google Scholar]
- Yuan G, Nanduri J, Bhasker CR, Semenza GL, Prabhakar NR. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J Biol Chem 280: 4321–4328, 2005. [DOI] [PubMed] [Google Scholar]
- Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol 217: 674–685, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]