

Abstract
The intracellular concentration of Cl− ([Cl−]i) in neurons is a highly regulated variable that is established and modulated by the finely tuned activity of the KCC2 cotransporter. Despite the importance of KCC2 for neurophysiology and its role in multiple neuropsychiatric diseases, our knowledge of the transporter's regulatory mechanisms is incomplete. Recent studies suggest that the phosphorylation state of KCC2 at specific residues in its cytoplasmic COOH terminus, such as Ser940 and Thr906/Thr1007, encodes discrete levels of transporter activity that elicit graded changes in neuronal Cl− extrusion to modulate the strength of synaptic inhibition via Cl−-permeable GABAA receptors. In this review, we propose that the functional and physical coupling of KCC2 to Cl−-sensitive kinase(s), such as the WNK1-SPAK kinase complex, constitutes a molecular “rheostat” that regulates [Cl−]i and thereby influences the functional plasticity of GABA. The rapid reversibility of (de)phosphorylation facilitates regulatory precision, and multisite phosphorylation allows for the control of KCC2 activity by different inputs via distinct or partially overlapping upstream signaling cascades that may become more or less important depending on the physiological context. While this adaptation mechanism is highly suited to maintaining homeostasis, its adjustable set points may render it vulnerable to perturbation and dysregulation. Finally, we suggest that pharmacological modulation of this kinase-KCC2 rheostat might be a particularly efficacious strategy to enhance Cl− extrusion and therapeutically restore GABA inhibition.
Keywords: epilepsy, hyperexcitability, inhibitory synaptic transmission, K-Cl cotransport, WNK kinases
the strength and efficacy of hyperpolarizing synaptic inhibition via type A γ-aminobutyric acid receptor (GABAAR) and glycine receptor (GlyR), Cl−-permeable ion channels, is modulated by the intracellular concentration of Cl− ([Cl−]i) in postsynaptic neurons. Genetics has firmly established the necessity of the Cl−-extruding cation-chloride cotransporter (CCC) KCC2 (SLC12A5) for establishing and maintaining the low [Cl−]i of postsynaptic neurons that is required for inhibition (Gagnon and Delpire 2013). Within the past few years, several reviews have highlighted the role of KCC2 and its regulation in neurophysiology and its potential as a pharmacotherapeutic target for a number of neuropsychiatric disorders, like epilepsy, spasticity, neuropathic pain, and autism (Arroyo et al. 2013; Benarroch 2013; Deidda et al. 2014; Gagnon and Delpire 2013; Hartmann and Nothwang 2015; Kahle et al. 2015; Medina et al. 2014; Puskarjov et al. 2014a). However, our knowledge of the mechanisms that regulate KCC2 activity is still rudimentary. How is KCC2 activity regulated to maintain [Cl−]i within a precise range to ensure an appropriate strength of inhibition in particular contexts (e.g., in developing neurons or during intense neuronal activity)? Do these mechanisms become dysregulated in neurological disease? Could a more detailed biochemical knowledge of these mechanisms be exploited to treat neurological diseases featuring impaired GABA inhibition? These are important questions with potential clinical ramifications.
Here we provide an update and review of recently discovered kinase-mediated phosphoregulatory mechanisms of KCC2 that have been shown to provide an additional mode of ionic plasticity to fine-tune the inhibitory drive of neuronal circuits. Evidence has shown that the experimental modulation of KCC2 (de)phosphorylation can considerably alter the magnitude of KCC2 activity depending on the sites that are manipulated, and the recent finding that Cl−-sensitive kinases associate with and regulate KCC2 activity (Alessi et al. 2014; Friedel et al. 2015) suggests that KCC2 phosphorylation is a dynamic mechanism that changes in response to inputs from intracellular signaling cascades. For example, the dephosphorylation of KCC2 at Thr906/Th1007 stimulates KCC2 >25-fold over baseline activity levels by preventing WNK1-SPAK kinase-dependent inhibitory phosphorylation at these sites (de Los Heros et al. 2014; Friedel et al. 2015; Rinehart et al. 2009; Titz et al. 2015). This is the most potent known mechanism to date of KCC2 stimulation and is sufficient to trigger hyperpolarizing GABAA responses in even high extracellular K+ concentrations and in neurons with extremely negative resting membrane potentials (Titz et al. 2015). Functionally, KCC2 Thr906/Thr1007 phosphorylation inhibits KCC2 in immature neurons and is necessary to maintain the depolarizing action of GABA (Friedel et al. 2015), which has long been known to be critical for multiple aspects of brain development. Conversely, phosphorylation at Ser940 stimulates KCC2 activity by increasing its expression at the plasma membrane, decreases in Ser940 phosphorylation have been noted in rodent models of seizures (Silayeva et al. 2015), and human mutations in KCC2 resulting in the downregulation of Ser940 phosphorylation have been associated with idiopathic generalized epilepsy (Kahle et al. 2014b) and familial febrile seizures (Puskarjov et al. 2014b).
Setting these recent exciting findings within a framework of systems dynamics nomenclature that has not previously been applied to cellular Cl− homeostasis, we propose a novel model in which the coupling of Cl−-sensitive regulatory kinases with KCC2 comprises a molecular “rheostat” that senses changes in [Cl−]i, transduces this signal to KCC2, and by multisite phosphorylation appropriately tunes KCC2 activity to elicit graded changes in neuronal [Cl−]i to modulate GABA neurotransmission. We suggest that the versatility of this adaptation mechanism, while important for homeostasis, might create susceptibility to dysregulation in disease. Finally, pharmacological modulation of this kinase-KCC2 rheostat might be a strategy to enhance Cl− extrusion and therapeutically restore GABA inhibition.
Dependence of GABA and Glycine Neurotransmission upon [Cl−]i Homeostasis
Glutamate excitatory neurotransmission is mediated by the transmembrane movement of cations (e.g., Na+), whereas GABAAR- and GlyR-mediated synaptic neurotransmission is influenced by the electrochemical driving force for Cl− (and to a lesser extent, HCO3−) across the neuronal membrane. Mature CNS neurons are hyperpolarized by the opening of a Cl− conductance associated with the activation of GABAARs and GlyRs, moving the membrane potential away from the threshold of action potential firing. As GABA is released from interneurons and hyperpolarizes the postsynaptic membrane of larger projection neurons, this phenomenon is called postsynaptic inhibition (Fig. 1A). As discussed below, this is made possible by the postsynaptic neuron's [Cl−]i, which sets the electrochemical potential equilibrium of Cl− (ECl) below resting membrane potential. In contrast, in the peripheral nervous system (PNS), interneurons release GABA onto terminals that are loaded with, instead of depleted of, Cl−. As GABAAR-mediated depolarization of the terminal inhibits propagation of an incoming signal from the periphery and the GABA effect on the presynaptic side, this phenomenon is called presynaptic inhibition (Fig. 1B). In these two situations, intracellular Cl− is not at equilibrium and the opening of GABAARs or GlyRs hyperpolarizes or depolarizes the neuronal membrane. If Cl− was instead at equilibrium, opening of an anion conductance would fail to produce a net movement of Cl− and a change in membrane potential but would still result in an increase in membrane conductance (and decrease in resistance) leading to changes in the excitability properties of the membrane. This shunting phenomenon diminishes the efficacy of glutamatergic (excitatory) currents and is therefore “inhibitory.” While these features of GABAergic and glycinergic signaling endow neurons with a remarkable functional plasticity, they also render neurons susceptible to dysfunction when [Cl−]i, is perturbed. Therefore, molecular mechanisms have evolved to maintain precise homeostasis of [Cl−]i in neurons.
Fig. 1.
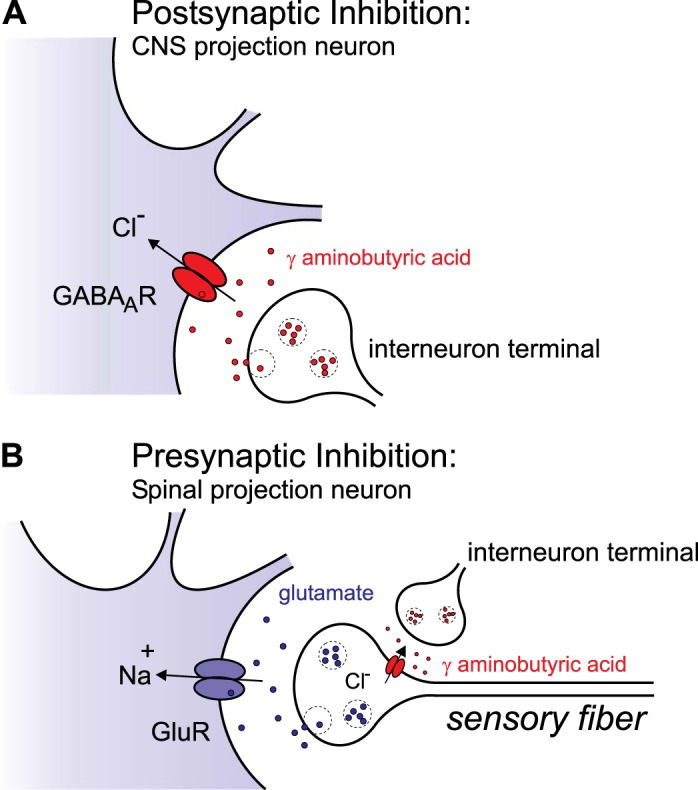
Postsynaptic and presynaptic inhibition. A: the terminal of an inhibitory interneuron synapses onto a CNS projection neuron. Upon depolarization of the interneuron terminal, voltage-gated Ca2+ channels are activated, leading to Ca2+ entry, vesicular fusion, and release of GABA. On the postsynaptic membrane, GABA binds to receptors, triggering the inward movement of Cl− and membrane hyperpolarization. B: the terminal of an inhibitory interneuron synapses onto the terminal of a sensory fiber. As this terminal is loaded with Cl−, activation of GABA receptor leads to Cl− efflux and membrane depolarization. This depolarization weakens the synapse and prevents the release of glutamate and the transfer of signal from the periphery to the spinal projection neuron.
Molecular Determinants of [Cl−]i in Neurons
It was believed prior to the 1970s that Cl− was passively distributed across the plasma membrane while Na+ and K+ were actively transported (reviewed in Alvarez-Leefmans and Delpire 2009b). According to this view, [Cl−]i could change only if the external concentration of Cl− ([Cl−]o) or the neuronal membrane potential changed. For GABAergic and glycinergic neurons, this constraint had profound consequences, since when Cl− is at electrical equilibrium opening of an anion conductance would fail to produce a net movement of Cl− and membrane hyperpolarization or depolarization.
What are the factors that determine the electrical equilibrium of Cl− and/or [Cl−]i? There are likely four major factors: 1) [Cl−]o, 2) the membrane potential, 3) the distribution of Donnan charges inside and outside the cell, and 4) the transport pathways that allow the membrane permeation of Cl− (Fig. 2A). The external Cl− (110 mM) and the membrane potential (−65 to −70 mV) are the two parameters that are mostly cited. Based on the Nernst equation that considers the concentration gradients and the membrane potential, [Cl−]i is at equilibrium at extremely low values (8–10 mM) compared with [Cl−]o. However, the cell is a much more complex system with, for instance, large impermeant macromolecules that carry numerous negative charges repelling diffusible anions (Donnan 1911; Glykys et al. 2014). Additionally, the transport of Cl− across the plasma membrane does not occur in isolation but is often coupled to the movement of other ionic species and sometimes even water. All these “complicating” factors have been discussed in a recent review (Delpire and Staley 2014).
Fig. 2.

A: factors influencing the intracellular Cl− concentration. Bottom left: K+ permeability generates an electropositive external environment, driving Cl− out of the cell. Top right: inward Cl− gradient due to high extracellular Cl− concentration ([Cl−]o). Bottom right: the presence of negative charges on nondiffusible macromolecules decreases the need for diffusible anions in the cytosol. Top left: major secondary active Cl− transport mechanisms create a pathway for Cl− movement across the plasma membrane. B: family of SLC12A transporters showing 2 major branches: the Na+-dependent (K+)-Cl− cotransporters (in black) and the Na+-independent K-Cl cotransporters (in blue). Two orphan members, CCC8 and CCC9, are also displayed. Length of tree branches can be compared to the reference bar, which represents 0.1 amino acid substitutions per site.
Hyperpolarizing inhibitory postsynaptic potentials were first observed in the early 1970s in cat spinal motor neurons (Llinas et al. 1974; Lux 1971) and consistently observed thereafter (for review see Alvarez-Leefmans and Delpire 2009a). In fact, the concept that GABA elicits membrane hyperpolarization is now textbook material and unequivocally indicates that [Cl−]i is lower than its thermodynamic potential equilibrium concentration. As energy is required, the key question is, What type of energy is involved? The most direct source of energy and transport mechanism would be ATP hydrolysis by a Cl− pump. While some biochemical evidence for a “Cl−-ATPase” has been presented (Hattori et al. 1998), the fact that this ATPase was sensitive to ethacrynic acid suspiciously indicated a functional coupling between Na+-K+-2Cl− cotransporter [which is inhibited by ethacrynic acid] and the Na+-K+-ATPase. This coupling introduces the concept of secondary active transport mechanisms that use the stored energy of the ion gradients generated by the primary active Na+-K+-ATPase. Among them are the SLC12A family transporters, also known as the cation-chloride cotransporters (CCCs). As indicated in Fig. 2B, this transport family is comprised of two subfamilies: the inward (Na+ dependent) transporters (such as the Na+-K+-2Cl− cotransporters) and the outward (Na+ independent) transporters (such as the K+-Cl− cotransporters).
The Nervous System-Specific KCC2 Cotransporter
K+-Cl− cotransport is mediated by four transporters (KCC1–KCC4) encoded by SLC12A4–SLC12A7 genes. Na+-K+-2Cl− cotransport is mediated by NKCC1 and NKCC2, encoded by SLC12A2 and SLC12A1 genes, respectively. While all K+-Cl− cotransporters are expressed in the nervous system, KCC2 is the most abundant and relevant isoform in neurons (Lu et al. 1999; Payne 1997; Payne et al. 1996). KCC2 is transcribed from two distinct promoters, giving rise to KCC2a and KCC2b (Uvarov et al. 2007). The “b” isoform is the most abundant form of the transporter and is upregulated during postnatal brain development (Clayton et al. 1998; Lu et al. 1999). While most of the physiology of K+-Cl− cotransport was originally worked out in red blood cells (RBCs) (Lauf et al. 1992), the functional identification of the transporter (Misgeld et al. 1986; Thompson et al. 1988) was made in neurons well before its cloning (Payne et al. 1996). Tight coupling between K+ and Cl− transport in a strict 1:1 stoichiometry was demonstrated in rabbit RBCs (Jennings and Adame 2001). By using the large K+ gradient that exists across the neuronal membrane, the cotransporter is able to drive Cl− against its electrochemical potential equilibrium, and this in a completely electroneutral fashion. Thus KCC2 is often regarded as the mechanism that behaves like a Cl− pump, creating the driving force for Cl− entry into the cell and membrane hyperpolarization upon GABA release (for reviews see Blaesse et al. 2009; Delpire 2000; Kahle et al. 2008b).
Is KCC2 responsible for setting [Cl−]i or does it instead provide an efficient pathway for the movement of Cl− across the neuronal membrane? These two functions are interconnected and not easily distinguishable. The fact that KCC2 provides a rapid and efficient pathway for Cl− recovery after loading has been demonstrated experimentally (Rivera et al. 1999; Zhu et al. 2005). Opening of GABA-mediated Cl− conductance results in Cl− entry in the neuron, which is enhanced by frequent depolarization of the membrane upon sustained excitatory activity. Thus, as timing is a key factor, the ability of the neuron to promptly restore its Cl− concentration after loading is critical. In a mature neuron this function is fulfilled by KCC2, and a brisk postnatal upregulation of KCC2 activity is indeed required for the excitatory-inhibitory GABA sequence during neurodevelopment. Molecular genetics has shed particular light on the necessity of KCC2 for neuronal Cl− homeostasis and GABA/glycine function: genetic knockout of KCC2 in mice (Hubner et al. 2001; Khalilov et al. 2011; Zhu et al. 2005, 2008), flies (Hekmat-Scafe et al. 2006), and worms (Tanis et al. 2009) elevates neuronal [Cl−]i, depolarizes the equilibrium potential of GABA (EGABA), impairs inhibition, and causes neuronal hyperexcitability. A similar electrophysiological phenotype is seen in multiple models of neurological and psychiatric disease associated with decreased functional expression of KCC2, including autism (Cellot and Cherubini 2014), schizophrenia (Hyde et al. 2011; Kalkman 2011), epilepsy (Kahle et al. 2014b; Silayeva et al. 2015), spasticity (Boulenguez et al. 2010), and neuropathic pain (Coull et al. 2003). These data collectively reveal the necessity of KCC2 for the homeostasis of neuronal [Cl−]i and appropriate function of GABA. However, homeostasis implies the existence of mechanisms that sense perturbations and initiate dynamic, adaptive responses to restore equilibrium. Have we been thinking about KCC2 activity in too static a manner?
Neuronal [Cl−]i: a Regulated Variable (Stock) Controlled by KCC2-Mediated Cl− Efflux (Flow)?
Despite variations in the external environment, the internal milieu is kept relatively constant, allowing biological processes to proceed (Bernard 1878). “Homeostasis” describes how key variables in the internal milieu are maintained within an acceptable range by feedback mechanisms that compare the actual value of the so-called “regulated variable” (e.g., blood glucose or blood pressure) to a desired value or set point (Cannon 1929; Hardy 1965). In contrast, “controlled variables” are activities, or rates, of the processes that contribute to the stability of the regulated variables (e.g., the rate of insulin secretion regulates blood glucose concentration or the rate of renal NaCl reabsorption regulates intravascular blood volume) (Cabanac 2006). In the nomenclature of systems dynamics, regulated variables are the “Stocks” and controlled variables the “Flows” of a system. Flows increase (in-flows) or decrease (out-flows) the value of the regulated variable depending on physiological need, or in response to pathophysiological perturbation.
Given the tight control with which neuronal [Cl−]i is regulated, and the important role played by KCC2 in this process, [Cl−]i might be considered a regulated variable (or Stock) and the rate of KCC2 transport a controlled variable (or Flow) (Fig. 3). KCC2 itself could be considered a “Plant” of the system, that is, an effector component of the homeostatic circuit regulated by a “Controller” (that senses or monitors the value of the regulated variable and generates an appropriate Signal that regulates Plant activity) to effectively adjust the value of the regulated variable. Since KCC2 mediates Cl− efflux in most physiological conditions in the brain and spinal cord, KCC2 activity could be more specifically considered an out-flow of the system, where increased rates of KCC2 transport extrude Cl− and decrease neuronal [Cl−]i and decreased rates of KCC2 transport raise [Cl−]i. This type of model could also incorporate the activity level of the related Cl−-importing NKCC1 cotransporter as an in-flow of the system, although evidence showing clear coexpression of NKCC1 and KCC2 in neurons and the importance of NKCC1 in regulating [Cl−]i in immature CNS neurons is unclear (reviewed in Delpire and Staley 2014).
Fig. 3.

System dynamics concept of “Stock” [intracellular concentration of Cl− ([Cl−]i)] and “Flow” (KCC2 activity). The master signal is the WNK-SPAK kinases. The kinases phosphorylate and activate the “in-flow” NKCC1 and inactivate the “out-flow” KCC2, resulting in an intracellular Cl− concentration that tips the balance from GABA hyperpolarization (in purple) to GABA shunting (in yellow) and GABA depolarization (in orange). Small graded changes in the phosphorylation status of KCC2 (like a rheostat) will introduce graded changes in intracellular Cl− and GABA effect.
Given the above considerations, several key questions emerge: 1) What are the biochemical mechanisms that encode different rates of KCC2 transport activity? 2) What is the molecular identity of the intracellular Cl− sensor (Controller) that detects changes in [Cl−]i? 3) What are the associated molecules that transduce Controller-triggered Signals to KCC2 at the plasma membrane?
Regulated Phosphorylation of KCC2: Rheostat of Neuronal [Cl−]i and GABA Function?
Tuning the inhibitory tone of GABA and glycine likely involves graded changes in [Cl−]i (Titz et al. 2015). This first starts at the transcriptional level, where expression of KCC2 (and possibly NKCC1) is regulated in both temporal and spatial fashions (Clayton et al. 1998; Dzhala et al. 2005; Lu et al. 1999; Plotkin et al. 1997; Rivera et al. 1999). Expression of NKCC1 and KCC2 is also influenced by epigenetic factors (Lee et al. 2010a; Yeo et al. 2012). A second step of regulation involves the insertion and retrieval of KCC2 to and from the plasma membrane, respectively—a process that is in part modulated by phosphorylation at residue Ser940. Indeed, protein kinase C (PKC) directly phosphorylates KCC2 at Ser940 (Lee et al. 2007), thereby stimulating cell surface expression and intrinsic transport rate of KCC2, leading to an overall increase in transporter activity. Reversibly, dephosphorylation of Ser940 is thought to mediate an activity-dependent reduction of KCC2 function through a decrease in transporter stability at the plasma membrane (Lee et al. 2010b) and/or enhanced endocytosis (Chamma et al. 2013). Note that cell surface KCC2 expression occurs predominantly at synaptic densities (Báldi et al. 2010; Chamma et al. 2013). Interestingly, this effect is in opposite direction to the effect of PKC on NKCC1 cell surface expression (Mykoniatis et al. 2010). Finally, fine-tuning of KCC2 activity occurs by kinases and phosphatases affecting the intrinsic transport properties of the transporter. Fine-tuning could also be activity dependent and thus part of plasticity of the inhibitory system, as indicated by two independent mechanisms. The first mechanism involves intracellular Ca2+ and control of gene expression, with depolarizing responses in immature neurons raising intracellular Ca2+ levels that result in increased KCC2 expression (Ganguly et al. 2001). A second mechanism likely involves changes in [Cl−]i and control of KCC2 transport activity by the SPAK/OSR1 (Gagnon et al. 2006a) and WNK kinases (Piala et al. 2014), the activities of which are sensitive to the surrounding Cl− concentration (see below).
Work in the late 1980s and early 1990s established that K+-Cl− cotransport activity is modulated by phosphorylation-dephosphorylation mechanisms (Jennings and al-Rohil 1990; Jennings and Schultz 1991; Sachs 1988). The transporter is stimulated by okadaic acid and calyculin A, indicating that protein phosphatase 1 and 2A (PP1 and PP2A)-dependent dephosphorylation is important for KCC activation in certain cellular contexts, such as in response to cell swelling. This property was later demonstrated with human KCC2 (Song et al. 2001). Note that this is opposite to the regulation of the Na+-dependent CCCs like NKCC1, which are stimulated by phosphorylation instead of dephosphorylation (Lytle and Forbush 1992). This regulation is reminiscent of the opposite effects of osmolarity on both types of cotransporters, with KCCs activated by hypotonicity and the NKCCs activated by hypertonicity (Jennings and Schultz 1991; Lytle and Forbush 1992). This “yin-and-yang” of NKCC1 vs. KCC2 regulation suggested that a common regulatory mechanism is at play for the two subfamilies of transporters, at least in certain cellular contexts. Over the last decade, yeast two-hybrid screens and the genetic investigation of rare Mendelian disorders have identified key regulatory kinases of the CCCs (Gagnon and Delpire 2012; McCormick and Ellison 2011). A large-scale yeast two-hybrid screen with the amino-terminal tail of KCC3 first identified the Ste20p-related kinase SPAK as a binding partner of KCC3 and all CCCs (Piechotta et al. 2002), whereas a follow-up screen with the carboxy-terminal tail of SPAK identified WNK2 and WNK4 as interactors of SPAK (Piechotta et al. 2003). In parallel, mutations in human WNK1 and WNK4 revealed their role in modulating thiazide-sensitive Na+-Cl− cotransporter (NCC) activity via phosphorylation in the distal convoluted tubule (DCT) of the nephron (McCormick and Ellison 2011; Wilson et al. 2001).
The current model of WNK-SPAK/OSR1 kinase-mediated regulation of the CCCs (Fig. 4) based on heterologous expression of WT and mutant transporters and kinases proposes that WNK kinases lie upstream of the SPAK/OSR1 (Gagnon et al. 2006a, 2007a; Vitari et al. 2005, 2006). Through protein-protein interaction via an RFxV (Arg-Phe-Xaa-Val) motif in the WNK kinases and the CCCs, and a so-called 92-residue CCT (conserved COOH terminal) domain in the SPAK/OSR1 kinases, WNKs phosphorylate and activate SPAK/OSR1, which in turn bind to the CCCs at their amino terminus and phosphorylate Thr or Ser residues at the amino terminus of the NKCCs or the carboxy terminus of the KCCs. Phosphorylation occurs at Thr207, Thr212, and Thr217 for human NKCC1 (Darman and Forbush 2002; Vitari et al. 2006) and Thr906 and Thr1007 for human KCC2 (Rinehart et al. 2009). A recent study has shown that SPAK/OSR1 directly phosphorylates KCC2 on Thr1007 (and KCC3 at the homologous residue, Thr991) (de Los Heros et al. 2014).
Fig. 4.

Regulation of KCC2 by phosphorylation. A: opposite to NKCC1, it is dephosphorylation that activates KCC2. The kinases that stimulate NKCC1 and inhibit KCC2 are SPAK or OSR1 (mammalian Ste20p-like kinases). Their activity is also dependent on phosphorylation by upstream WNK kinases. B: equilibrium between silent and active transporter in the membrane involves the phosphorylation of 2 carboxy-terminal tail threonine residues (Thr906 and Thr1007). Stability of the transporter at the membrane depends on the phosphorylation status of neighboring residue Ser940. The activity and phosphorylation status of KCC2 at Thr906 and Thr1007 of retrieved transporters is unknown.
Through systematic alanine mutagenesis of known Ser/Thr/Tyr phosphosites (genetically preventing phosphorylation and mimicking dephosphorylation), Titz and coworkers recently demonstrated that modulation of the phosphorylation state of KCC2 at specific residues in the carboxy terminus can significantly enhance or diminish KCC2 activity (Titz et al. 2015). Mutagenesis increased KCC2 activity in the following order: T1087A (smallest increase), T6A, KCC2-WT, S25A + S26A, S937A, T1007A, T906A + T1007A (largest increase). These data corroborated and extended earlier work in HEK293 cells (de Los Heros et al. 2014; Rinehart et al. 2009) that showed that dual dephosphorylation of KCC2 at Thr906 and Thr1007 potently activates KCC2 and further demonstrated that this enhanced activity in neurons reaches sufficiently high values (approaching thermodynamic equilibrium) such that hyperpolarizing GABAA responses may be obtained in neurons even with extremely negative resting membrane potentials or bathed in high extracellular K+ concentrations ([K+]o). This latter property is interesting and suggests that mechanisms such as KCC2 Thr906/Thr1007 dephosphorylation might allow neurons to adjust Cl−-dependent inhibitory responses in the presence of elevated [K+]o (e.g., that might favor epileptic discharge) (Thompson and Gahwiler 1989), thereby counteracting increased Cl− influx through conductive pathways and maintaining an adaptive, strong hyperpolarizing inhibition. KCC2 Thr906/Thr1007 dephosphorylation might similarly be exploited during acute shifts in the membrane potential to more negative values (e.g., through activation of a K+ conductance), allowing the maintenance of hyperpolarizing GABA inhibition.
The activities of the NKCCs and the KCCs are reciprocally responsive to changes in [Cl−]i that parallel their phosphorylation state (Alessi et al. 2014; Gagnon et al. 2006b; Kahle et al. 2010). Cl− directly binds to the catalytic site of WNK1 and stabilizes the inactive conformation of the kinase to prevent autophosphorylation and activation (Piala et al. 2014). Cl− has also been shown to directly inhibit SPAK/OSR1 kinase activity (Gagnon et al. 2006a). Thus one simple hypothesis is that these kinases may act as the primary sensors of [Cl−]i, transducing this information to KCC2 (and NKCC1) at the plasma membrane via changes in phosphorylation. In this model, as [Cl−]i decreases WNK-SPAK/OSR1 kinase activities increase, thereby enhancing NKCC1/KCC2 phosphorylation, with a net effect of increasing net Cl− influx (Fig. 3). As [Cl−]i increases, kinase activity is silenced and cellular phosphatases (such as PP1) (Bize et al. 1999; Gagnon and Delpire 2010) decrease NKCC1/KCC2 phosphorylation, leading to a reduction in [Cl−]i. In this paradigm, the coupling of Cl−-sensitive kinase with CCCs would comprise a molecular rheostat that adaptively tunes [Cl−]i and the response to GABA/glycine in response to changes in [Cl−]i, as occurs during normal or perturbed neuronal activity. In the system dynamics model of neuronal Cl− homeostasis (Fig. 3), the WNK-SPAK/OSR1 kinases could be considered Controllers whose level of autophosphorylation would encode specific values of [Cl−]i, and this change in catalytic activity would then get transduced as a signal to elicit changes in KCC2 (and possibly NKCC1) phosphorylation and activity. As such, a WNK1 and possibly SPAK/OSR1 Cl− sensor would perform similarly to other Controllers in homeostatic circuits, such as TRP channels, ASICs, and GPCRs, which monitor temperature, pH, and fatty acids, respectively (Briscoe et al. 2003; Oh et al. 2010).
Developmental Cl− Rheostasis and the GABA Excitatory-Inhibitory Sequence
Could a WNK1-SPAK kinase-KCC2 coupled rheostat be effective in the developmental excitatory-inhibitory GABA sequence? Depolarizing and even excitatory action of GABA due to elevated [Cl−]i is an evolutionarily conserved hallmark of immature neurons (Ben-Ari et al. 1989; Owens et al. 1996) that is required for neuronal proliferation, migration, and synaptogenesis (Aguado et al. 2003; Bortone and Polleux 2009; Fiumelli et al. 2013) and is related to the delayed postnatal increase in Cl−-extruding KCC2 cotransporter activity (Rivera et al. 1999). However, the mechanisms underlying the developmental switch in KCC2 activity are not well understood, and it is unclear whether changes in the amount of KCC2 protein alone, or changes in transporter regulation, might also be involved in the net increase of KCC2 activity. Previous work has shown a discrepancy between the protein expression of KCC2 and the KCC2-dependent Cl− extrusion capacity of immature neurons, suggesting other factors (Blaesse et al. 2006). In addition, other work has shown that the overall expression level of KCC2 is high enough in neonatal hippocampus that rapid, activity-dependent stimulation of KCC2 can elicit a negative shift in EGABA close to the adult level (Khirug et al. 2010). We recently showed that KCC2 is more highly phosphorylated at Thr906/Thr1007 in immature neurons compared with mature neurons and that the cotransporter forms a physical complex with WNK1 and SPAK in the developing brain (Friedel et al. 2015). Dominant-negative mutation, genetic knockdown, or chemical inhibition of WNK1 in immature neurons (but not mature neurons) is sufficient to trigger a hyperpolarizing shift in GABA activity by enhancing KCC2-mediated Cl− extrusion secondary to a reduction of Thr906/Thr1007 inhibitory phosphorylation (Friedel et al. 2015). These results extended previous work by Rinehart et al. (2009), who showed that KCC2 Thr906 phosphorylation inversely correlates with KCC2 activity in the developing mouse brain, and Inoue et al. (2012), who showed a phosphorylation-dependent inhibitory effect of taurine on KCC2 activity in immature neurons that was recapitulated by WNK1 overexpression in the absence of taurine. Together, these compelling data suggest that a postnatal decrease in WNK1-regulated inhibitory phosphorylation of KCC2 also contributes to increased KCC2 function (Fig. 5), and thus to the excitatory-to-inhibitory GABA shift that occurs during development. This also raises the possibility that dysfunctional phosphoregulation of these sites could be important in certain neurodevelopmental pathologies, like autism or neonatal seizures. An important issue of future investigation will be to determine how the increased levels of Cl− in immature neurons affect WNK1 kinase activity. Could taurine, a factor known to activate WNK1 in immature neurons, achieve this by decreasing the sensitivity of WNK1 to Cl−?
Fig. 5.
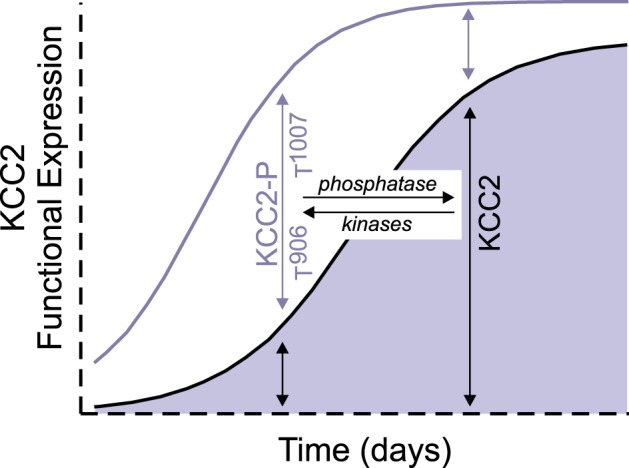
Developmental regulation of KCC2 function. During development, expression of KCC2 increases. We hypothesize that during this increased expression the cotransporter is mostly phosphorylated at residues Thr906 and Thr1007 and therefore functionally silent. Upon neuronal maturation, the level of phosphorylation decreases and the number of active transporters in the membrane (area under blue curve) increases. Thus the KCC2 activity, which increases during development, is a combination of an increased number of transporters and increased activity of these transporters by dephosphorylation.
Multisite Phosphorylation: Combinatorial Control of KCC2 by Different Signaling Inputs
KCC2 contains multiple phosphorylation sites that are targets of different kinases, increasing the combinatorial number of transporter states. Indeed, residue Ser940, identified by the Moss lab, is probably the functionally best-characterized KCC2 phosphorylation site to date (reviewed in Kahle et al. 2013) and already found to have clinical relevance in humans (Kahle et al. 2014b). Similarly, the dephosphorylation of these residues is mediated by different phosphatases, which are themselves are highly regulated molecules. In neurons, glutamate-induced NMDA receptor activation and the associated influx of Ca2+ promote PP1-dependent KCC2 Ser940 dephosphorylation that coincides with a deficit in hyperpolarizing GABAergic inhibition resulting from the loss of KCC2 activity (Lee et al. 2011). This is interesting but raises the question of how PP1 might be utilized to activate KCC2 via Thr906/Thr1007 dephosphorylation but also inhibit KCC2 via Ser940 dephosphorylation. Such an effect could be achieved by tethering PP1 activity to different regulatory subunits. Or, perhaps like NKCC1, KCC2 could itself serve as the PP1 regulatory subunit or scaffold affecting not only the phosphorylation state of the cotransporter but also the phosphorylation status of kinases that are bound to it (Darman et al. 2001; Gagnon et al. 2007b). Interestingly, Silayeva et al. recently found that status epilepticus (SE), as modeled by kainate injection in mice, results in the glutamate-dependent dephosphorylation (and subsequent internalization of KCC2) of Ser940 and that kainate injection in KCC2 Ser940Ala knockin mice induced lethality and subsequent entrance into SE within 30 min (Silayeva et al. 2015). Impaired KCC2 activity related to a maladaptive decrease in Ser940 phosphorylation has also proven relevant for the adaptive and pathological endocrine response to stress (MacKenzie and Maguire 2015; Sarkar et al. 2011) and spasticity associated with brain and spinal cord injury (Toda et al. 2014). Recent work by the Nothwang laboratory has identified other potential sites of KCC2 phosphorylation, including Ser937 (Weber et al. 2014). A genetic mutant KCC2 (S937D), mimicking phosphorylation, results in a significant upregulation of transport activity. The physiological relevance of this site has yet to be determined.
Kinase-KCC2 Coupling: Versatility and Vulnerability
The versatility and flexibility of kinase-controlled KCC2 regulation may enable efficient adaptation to physiological stress but may also expose the systems in which it operates to dysregulation and the development of disease. In contrast to homeostatic systems with fixed set points, those with adjustable set points are more capable of responding to perturbations but are also more vulnerable to dysregulation (Kotas and Medzhitov 2015). Diseases, acute or chronic, can result when homeostatic systems become locked in maladaptive alternative, “stable” states (Bateson et al. 2004).
A great genetic example of this is pseudohypoaldosteronism type II (OMIM 145260; PHAII), a human autosomal dominant disease featuring NaCl-sensitive hypertension due to constitutive activity of the thiazide-sensitive NCC, a close renal-specific cousin of KCC2. PHAII results from dysregulated WNK1-mediated NCC phosphorylation via SPAK kinase, and WNK1 mutations result in constitutively phosphorylated and active NCC. In the kidney DCT, low [Cl−]i stimulates WNK kinases to phosphorylate and activate NCC (McCormick et al. 2011). Could injury-induced (e.g., inflammation or ischemia) or genetically encoded derangements in WNK kinase-mediated Cl− sensing inappropriately change the Cl− set point and thereby, in analogous fashion to its renal phenotype, cause dysregulated signaling output to KCC2, locking KCC2 activity in “inappropriate” state? Relevant for this hypothesis is the finding that mutations in the nervous system-specific isoform of WNK1, WNK1/HSN2, cause a separate disease named hereditary sensory and autonomic neuropathy type II (HSANII), featuring congenital pain insensitivity (OMIM 201300) (Shekarabi et al. 2008). WNK1/HSN2 is highly expressed in the dorsal horn of the spinal cord, an important site of pain processing and KCC2 function. Could mutation in WNK1/HSN2 alter the KCC2-dependent Cl− handling of dorsal horn postsynaptic neurons and thereby dysregulate GABAAR-mediated signaling in HSANII patients?
Decreased KCC2 activity, higher neuronal [Cl−]i, and disinhibited GABA signaling accompany multiple diseases associated with ischemia and inflammation, including neuropathic pain resulting from peripheral nerve injury (Coull et al. 2005), motor spasticity due to spinal cord injury (Boulenguez et al. 2010), or ischemic stroke (Jaenisch et al. 2010; Toda et al. 2014). Inflammation is a usually protective response that is triggered to restore function when homeostatic mechanisms fail. However, to do this, inflammatory signals target the same nodes that normally execute homeostatic functions, altering the flow of Plants or the sensitivity of Controllers to elicit set-point changes that, with chronicity, can turn maladaptive and result in chronic disease states (Chovatiya and Medzhitov 2014; Kotas and Medzhitov 2015). The normal kinase signaling pathways that dynamically regulate KCC2, which, like other cellular signaling cascades, have a multiplicity of other functions, may become inappropriately activated by inflammation or associated forms of cell stress (e.g., osmotic) and trigger maladaptive set-point changes in [Cl−]i. For example, it is interesting that KCC2, which mediates isotonic Cl− transport for the majority of its functions in the CNS, has evolutionarily retained the swelling-regulated dephosphorylation mechanism (Thr906/Thr1007) operative in the other KCCs for cell volume homeostasis in other cell types, like RBCs. This precise and powerful mechanism of KCC regulation has likely been co-opted and retained in the evolutionary toolbox for nonswelling purposes for KCC2 (e.g., in the developmental switch in GABA function, see above). However, extracellular osmotic stress, as can occur in the context of inflammation and tissue injury due to blood-brain barrier breakdown and cell lysis, could trigger the inappropriate activation of this phosphorylation mechanism not usually exposed to changes in osmotic stimuli in the healthy CNS.
Targeting Kinase-KCC2 Coupling: a Method of Therapeutically Fine-Tuning GABA Activity?
Paradoxically, the adjustability of a kinase-KCC2 rheostat might represent a particularly effective mechanism that could be pharmacologically exploited to restore ionotropic inhibition. Given the limitations of targeting the GABA system directly (e.g., with benzodiazepines or barbiturates), an alternative approach for psychotropic and analgesic drug development has been the concept of facilitating neuronal Cl− extrusion to restore GABAergic inhibition by activating KCC2 (Doyon et al. 2013; Kahle et al. 2014a). Recently, a few groups have developed innovative high-throughput assays to screen for compounds that modulate KCC2 activity (Delpire et al. 2009, 2012; Gagnon et al. 2013), and one drug shows promise as a KCC2-dependent Cl− extrusion enhancer with therapeutic effect in a model of neuropathic pain (Gagnon et al. 2013). These early but encouraging results require validation, but they establish the validity in vivo of the concept of GABA modulation via the pharmacological targeting of CCC-dependent Cl− transport (Gagnon et al. 2013; Kahle et al. 2014a; Kaila et al. 2014). Could CCC phosphoregulatory mechanisms, normally employed to modulate transporter activity in response to perturbation or biological need, be harnessed to stimulate the KCCs (or inhibit NKCC1) for therapeutic benefit in disease states featuring an accumulation of intracellular Cl−?
Coincident inhibition of NKCC1-mediated Cl− loading and activation of KCC2-mediated Cl− extrusion via WNK-SPAK kinase inhibition could be one such approach (Kahle et al. 2014a). Targeting NKCC1/KCC2 activity would not affect neuronal excitability directly but would modulate the efficacy of endogenous GABAergic inhibition, potentially yielding increased specificity and wider therapeutic windows. All other efforts at developing CCC inhibitor drugs to date have focused on targeting NKCC1 and KCC2 individually. Because CCCs also work in concert with one another and other Cl− channels to achieve homeostasis, it is unknown whether targeting one CCC (e.g., NKCC1) could be compensated by other CCCs. Since WNK-SPAK/OSR1 kinase cascade regulates both NKCC1 and KCC2, but do so in reciprocal fashion, kinase inhibition would be expected to promote combined NKCC1 inhibition and KCC2 stimulation (Alessi et al. 2014; de Los Heros et al. 2014; Gagnon and Delpire 2012; Kahle et al. 2008a; Rinehart et al. 2009) (Fig. 3), yielding a net facilitation of Cl− extrusion. Therefore, WNK-SPAK inhibition may be more efficacious than poorly CNS-penetrant loop diuretics (bumetanide; see Tyzio et al. 2014) or KCC2 activators (Gagnon et al. 2013).
Moreover, since the WNK kinases might also be the Cl− sensors that detect changes in intracellular Cl− (Piala et al. 2014), inhibiting these molecules might prevent feedback mechanisms that would counter the effects of targeting NKCC1 or KCC2 alone. The approach might be a tenable method of restoring ionotropic inhibition for a wide range of disorders, including both neurodevelopmental diseases such as neonatal seizures, autism, and schizophrenia and adult disorders such as neuropathic pain, temporal lobe epilepsy, and spasticity.
Another alluring possible therapeutic strategy is preventing maladaptive increases in KCC2 Ser940 dephosphorylation. Since seizures affect GABA function of corticotropin-releasing hormone, which acts on the hypothalamic-pituitary-adrenal (HPA) axis (O'Toole et al. 2014), could antagonizing KCC2 Ser940 dephosphorylation counter the hyperexcitability of the HPA axis in disorders like anxiety, major depression, and posttraumatic stress disorder? These are fascinating questions worthy of future investigation. These findings may be relevant for human epileptigenesis; indeed, mutations in human KCC2 associated with idiopathic generalized epilepsy at Arg952 and/or Arg1049 have been shown to disrupt the phosphorylation of KCC2 at Ser940 (Kahle et al. 2014b).
Conclusions
Regulated phosphorylation of KCC2 likely represents an effective mechanism to fine-tune [Cl−]i, and by doing so, modulate GABA/glycineric inhibition and neuronal plasticity. This is likely achieved by multiple signaling pathways phosphorylating different residues, such as PKC modulation of Ser940, which are operative in specific physiological contexts. Recent work has shown that the experimental modification of KCC2 phosphorylation can considerably affect KCC2 activity, with transport rates approaching thermodynamic equilibrium when, for example, Thr906/Th1007 phosphorylation is prevented (Titz et al. 2015). This suggests that inhibition of KCC2 Thr906/Thr1007 phosphorylation directly (via drug or antibody binding) or indirectly (e.g., via kinase inhibitors that target WNK-SPAK kinase signaling) might be a novel way of facilitating neuronal Cl− extrusion for therapeutic effect. The regulated phosphorylation of KCC2 Ser940 also appears important for KCC2 function and has implications for human diseases such as idiopathic generalized epilepsy, febrile seizures, and SE. Using the nomenclature and concepts of systems dynamics, we offered a model of kinase-KCC2 function as a rheostat of neuronal Cl− homeostasis, and suggest that the “tunability” of this mechanism is a two-edged sword that, while allowing for homeostatic adaptation, may be more vulnerable to genetically encoded or injury-induced set-point changes. Ultimately, some neuropsychiatric disorders associated with GABA disinhibition, or subtypes of these disorders, may fall into a category of diseases that result from failures of neuronal Cl− homeostasis. As such, an increased knowledge of the normal phosphoregulatory mechanisms of KCC2 (and NKCC1) during development and in disease models, and innovative strategies to pharmacologically target these mechanisms, could have real clinical benefit.
GRANTS
K. T. Kahle was supported by a Harvard-MIT Neuroscience Grant and the Manton Center for Orphan Disease Research at Harvard Medical School and Boston Children's Hospital. E. Delpire is supported by National Institutes of Health Grants GM-074771 and DK-093501.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.T.K. and E.D. conception and design of research; K.T.K. and E.D. prepared figures; K.T.K. and E.D. drafted manuscript; K.T.K. and E.D. edited and revised manuscript; K.T.K. and E.D. approved final version of manuscript.
REFERENCES
- Aguado F, Carmona MA, Pozas E, Aguilo A, Martinez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibanez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development 130: 1267–1280, 2003. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7: re3, 2014. [DOI] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Delpire E. Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Diseases. London: Academic, 2009. a. [Google Scholar]
- Alvarez-Leefmans FJ, Delpire E. Thermodynamics and kinetics of chloride transport in neurons: an outline. In: Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Diseases, edited by Alvarez-Leefmans FJ, Delpire E. London: Academic, 2009. b, p. 81–108. [Google Scholar]
- Arroyo JP, Kahle KT, Gamba G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol Aspects Med 34: 288–298, 2013. [DOI] [PubMed] [Google Scholar]
- Báldi R, Varga C, Tamás G. Differential distribution of KCC2 along the axo-somato-dendritic axis of hippocampal principal cells. Eur J Neurosci 32: 1319–1325, 2010. [DOI] [PubMed] [Google Scholar]
- Bateson P, Barker D, Clutton-Brock T, Deb D, D'Udine B, Foley RA, Gluckman P, Godfrey K, Kirkwood T, Lahr MM, McNamara J, Metcalfe NB, Monaghan P, Spencer HG, Sultan SE. Developmental plasticity and human health. Nature 430: 419–421, 2004. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416: 303–325, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Cation-chloride cotransporters in the nervous system: general features and clinical correlations. Neurology 80: 756–763, 2013. [DOI] [PubMed] [Google Scholar]
- Bernard C. Leçons sur les Phénomènes de la Vie: dans les Animaux et dans les Végétaux. Paris: Baillière, 1878. [Google Scholar]
- Bize I, Guvenc B, Robb A, Buchbinder G, Brugnara C. Serine/threonine protein phosphatases and regulation of K-Cl cotransport in human erythrocytes. Am J Physiol Cell Physiol 277: C926–C936, 1999. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron 61: 820–838, 2009. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Guillemin I, Schindler J, Schweizer M, Delpire E, Khiroug L, Friauf E, Nothwang HG. Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J Neurosci 26: 10407–10419, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortone D, Polleux F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62: 53–71, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med 16: 302–307, 2010. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem 278: 11303–11311, 2003. [DOI] [PubMed] [Google Scholar]
- Cabanac M. Adjustable set point: to honor Harold T. Hammel. J Appl Physiol 100: 1338–1346, 2006. [DOI] [PubMed] [Google Scholar]
- Cannon WB. Organization for physiological homeostasis. Physiol Rev 9: 399–431, 1929. [Google Scholar]
- Cellot G, Cherubini E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr 2: 70, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamma I, Heubl M, Chevy Q, Renner M, Moutkine I, Eugène E, Poncer JC, Lévi S. Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J Neurosci 33: 15488–15503, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell 54: 281–288, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolf JS, Smith RL. Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Brain Res Dev Brain Res 109: 281–292, 1998. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438: 1017–1021, 2005. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424: 938–942, 2003. [DOI] [PubMed] [Google Scholar]
- Darman RB, Flemmer A, Forbush BI. Modulation of ion transport by direct targeting of PP1 to the Na-K-Cl cotransporter. J Biol Chem 276: 34359–34362, 2001. [DOI] [PubMed] [Google Scholar]
- Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem 277: 37542–37550, 2002. [DOI] [PubMed] [Google Scholar]
- de Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, Kahle KT, Zhang J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem J 458: 559–573, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda G, Bozarth IF, Cancedda L. Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front Cell Neurosci 8: 119, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E. Cation-chloride cotransporters in neuronal communication. News Physiol Sci 15: 309–312, 2000. [DOI] [PubMed] [Google Scholar]
- Delpire E, Baranczak A, Waterson AG, Kim K, Kett N, Morrison RD, Daniels JS, Weaver CD, Lindsley CW. Further optimization of the neuronal K-Cl cotransporter KCC2 antagonist ML077: development of a highly selective and more potent in vitro probe. Bioorg Med Chem Lett 22: 4532–4535, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Days E, Mi D, Lewis M, Kim K, Lindsley C, Weaver CD. Small molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci USA 106: 5383–5388, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Staley KJ. Novel determinants of the neuronal Cl− concentration. J Physiol 592: 4099–4114, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Ponce-Coria J, Delpire E. A trafficking-deficient mutant of KCC3 reveals dominant-negative effects on K-Cl cotransport function. PLoS One 8: e61112, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnan FG. Theorie der Membrangleichgewichte und Membranpotentiale bei Vorhandensein von nicht dialysierenden Elektrolyten. Ein Beitrag zur physikalisch-chemischen Physiologie (The theory of membrane equilibrium and membrane potential in the presence of a non-dialyzable electrolyte. A contribution to physical-chemical physiology). Z Elektrochem Angewandte Phys Chem 17: 572–581, 1911. [Google Scholar]
- Doyon N, Ferrini F, Gagnon M, De Koninck Y. Treating pathological pain: is KCC2 the key to the gate? Expert Rev Neurother 13: 469–471, 2013. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med 11: 1205–1213, 2005. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Briner A, Puskarjov M, Blaesse P, Belem BJ, Dayer AG, Kaila K, Martin JL, Vutskits L. An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb Cortex 23: 378–388, 2013. [DOI] [PubMed] [Google Scholar]
- Friedel P, Kahle KT, Zhang J, Hertz N, Pisella LI, Buhler E, Schaller F, Duan J, Khanna AR, Bishop PN, Shokat KM, Medina I. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. J Cell Sci 8: ra65, 2015. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, Delpire E. Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function. The N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for Ste20-related proline/alanine-rich kinase (SPAK) and PP1. J Biol Chem 285: 14115–14121, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, Delpire E. Molecular physiology of SPAK and OSR1: two Ste20-related protein kinases regulating ion transport. Physiol Rev 92: 1577–1617, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, Delpire E. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am J Physiol Cell Physiol 304: C693–C714, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, England R, Delpire E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol Cell Biol 26: 689–698, 2006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, England R, Delpire E. A single binding motif is required for SPAK activation of the Na-K-2Cl cotransporter. Cell Physiol Biochem 20: 131–142, 2007a. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, England R, Delpire E. Volume sensitivity of cation-chloride cotransporters is modulated by the interaction of two kinases: SPAK and WNK4. Am J Physiol Cell Physiol 290: C134–C142, 2006b. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, England R, Diehl L, Delpire E. Apoptosis-associated tyrosine kinase scaffolding of protein phosphatase 1 and SPAK reveals a novel pathway for Na-K-2Cl cotransporter regulation. Am J Physiol Cell Physiol 292: C1809–C1815, 2007b. [DOI] [PubMed] [Google Scholar]
- Gagnon M, Bergeron MJ, Lavertu G, Castonguay A, Tripathy S, Bonin RP, Perez-Sanchez J, Boudreau D, Wang B, Dumas L, Valade I, Bachand K, Jacob-Wagner M, Tardif C, Kianicka I, Isenring P, Attardo G, Coull JA, De Koninck Y. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med 19: 1524–1528, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105: 521–532, 2001. [DOI] [PubMed] [Google Scholar]
- Glykys J, Dzhala V, Egawa K, Balena T, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ. Local impermeant anions set the GABAA reversal potential. Science 343: 670–675, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JD. The “set-point” concept in physiological temperature regulation. In: Physiological Controls and Regulations, edited by Yamamoto WS, Brobeck JR. Philadelphia, PA: Saunders, 1965, p. 98–116. [Google Scholar]
- Hartmann AM, Nothwang HG. Molecular and evolutionary insights into the structural organization of cation chloride cotransporters. Front Cell Neurosci 8: 470, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori N, Kitagawa K, Higashida T, Yagyu K, Shimohama S, Wataya T, Perry G, Smith MA, Inagaki C. Cl−-ATPase and Na+/K+-ATPase activities in Alzheimer's disease brains. Neurosci Lett 254: 141–144, 1998. [DOI] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. Mutations in the K+/Cl− cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci 26: 8943–8954, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30: 515–524, 2001. [DOI] [PubMed] [Google Scholar]
- Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, Straub RE, Ye T, Colantuoni C, Herman MM, Bigelow LB, Weinberger DR, Kleinman JE. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci 31: 11088–11095, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Furukawa T, Kumada T, Yamada J, Wang T, Inoue R, Fukuda A. Taurine inhibits K+-Cl− cotransporter KCC2 to regulate embryonic Cl− homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J Biol Chem 287: 20839–20850, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 41: e151–e159, 2010. [DOI] [PubMed] [Google Scholar]
- Jennings ML, Adame MF. Direct estimate of 1:1 stoichiometry of K+-Cl− cotransport in rabbit erythrocytes. Am J Physiol Cell Physiol 281: C825–C832, 2001. [DOI] [PubMed] [Google Scholar]
- Jennings ML, al-Rohil N. Kinetics of activation and inactivation of swelling-stimulated K+/Cl− transport. The volume-sensitive parameter is the rate constant for inactivation. J Gen Physiol 95: 1021–1040, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings ML, Schultz RK. Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either swelling or N-ethylmaleimide. J Gen Physiol 97: 799–817, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci 36: 726–737, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Khanna A, Clapham DE, Woolf CJ. Therapeutic restoration of spinal inhibition via druggable enhancement of potassium-chloride cotransporter KCC2-mediated chloride extrusion in peripheral neuropathic pain. JAMA Neurol 71: 640–645, 2014a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Khanna AR, Alper SL, Adragna NC, Lauf PK, Sun D, Delpire E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol Med 21: 513–523, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, Shang Y, Lachance-Touchette P, Bourassa C, Levert A, Dion PA, Walcott B, Spiegelman D, Dionne-Laporte A, Hodgkinson A, Awadalla P, Nikbakht H, Majewski J, Cossette P, Deeb TZ, Moss SJ, Medina I, Rouleau GA. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep 15: 766–774, 2014b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochem Biophys Acta 1802: 1150–1158, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Ring AM, Lifton RP. Molecular physiology of the WNK kinases. Annu Rev Physiol 70: 329–355, 2008a. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol 4: 490–503, 2008b. [DOI] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15: 637–654, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkman HO. Alterations in the expression of neuronal chloride transporters may contribute to schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 35: 410–414, 2011. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Chazal G, Chudotvorova I, Pellegrino C, Corby S, Ferrand N, Gubkina O, Nardou R, Tyzio R, Yamamoto S, Jentsch TJ, Hübner CA, Gaiarsa JL, Ben-Ari Y, Medina I. Enhanced synaptic activity and epileptiform events in the embryonic KCC2 deficient hippocampus. Front Cell Neurosci 5: 23, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci 30: 12028–12035, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell 160: 816–827, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf PK, Bauer J, Adragna NC, Fujise H, Zade-Oppen AM, Ryu K, Delpire E. Erythrocyte K-Cl cotransport: properties and regulation. Am J Physiol Cell Physiol 263: C917–C932, 1992. [DOI] [PubMed] [Google Scholar]
- Lee HA, Hong SH, Kim JW, Jang IS. Possible involvement of DNA methylation in NKCC1 gene expression during postnatal development and in response to ischemia. J Neurochem 114: 520–529, 2010a. [DOI] [PubMed] [Google Scholar]
- Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci 14: 736–743, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Jurd R, Moss SJ. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci 45: 173–179, 2010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HL, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem 282: 29777–29784, 2007. [DOI] [PubMed] [Google Scholar]
- Llinas R, Baker R, Precht W. Blockage of inhibition by ammonium acetate action on chloride pump in cat trochlear motoneurons. J Neurophysiol 37: 522–532, 1974. [DOI] [PubMed] [Google Scholar]
- Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol 39: 558–568, 1999. [PubMed] [Google Scholar]
- Lux HD. Ammonium and chloride extrusion: hyperpolarizing synaptic inhibition in spinal motoneurons. Science 173: 555–557, 1971. [DOI] [PubMed] [Google Scholar]
- Lytle C, Forbush BI. The Na-K-Cl cotransport protein of shark rectal gland. II. Regulation by direct phosphorylation. J Biol Chem 267: 25438–25443, 1992. [PubMed] [Google Scholar]
- MacKenzie G, Maguire J. Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res 109: 13–27, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan V, Pressey JC, Acton BA, Huang MY, Puchalski A, Ivakine EA, Delpire E, McInnes RR, Woodin MA. KCC2 and kainate receptors coexist in a functional complex. Cell Rep 7: 1762–1770, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick JA, Ellison DH. The WNKs: atypical protein kinases with pleiotropic actions. Physiol Rev 91: 177–219, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P, Pellegrino C. Current view on the functional regulation of the neuronal K+-Cl− cotransporter KCC2. Front Cell Neurosci 8: 27, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU, Lux HD. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science 232: 1413–1415, 1986. [DOI] [PubMed] [Google Scholar]
- Mykoniatis A, Shen L, Fedor-Chaiken M, Tang J, Tang X, Worrell RT, Delpire E, Turner JR, Matlin KS, Bouyer P, Matthews JB. Phorbol 12-myristate 13-acetate-induced endocytosis of the Na-K-2Cl cotransporter in MDCK cells is associated with a clathrin-dependent pathway. Am J Physiol Cell Physiol 298: C85–C97, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687–698, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole KK, Hooper A, Wakefield S, Maguire J. Seizure-induced disinhibition of the HPA axis increases seizure susceptibility. Epilepsy Res 108: 29–43, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MBE, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci 16: 6414–6423, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol Cell Physiol 273: C1516–C1525, 1997. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem 271: 16245–16252, 1996. [DOI] [PubMed] [Google Scholar]
- Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechotta K, Garbarini NJ, England R, Delpire E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl− cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J Biol Chem 278: 52848–52856, 2003. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Lu J, Delpire E. Cation-chloride cotransporters interact with the stress-related kinases SPAK and OSR1. J Biol Chem 277: 50812–50819, 2002. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol 33: 781–795, 1997. [DOI] [PubMed] [Google Scholar]
- Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia 55: 806–818, 2014a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO Rep 15: 723–729, 2014b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, Lifton RP. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138: 525–536, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251–255, 1999. [DOI] [PubMed] [Google Scholar]
- Sachs JR. Volume-sensitive K influx in human red cell ghosts. J Gen Physiol 92: 685–711, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar J, Wakefield S, MacKenzie G, Moss SJ, Maguire J. Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J Neurosci 31: 18198–18210, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekarabi M, Girard N, Riviere JB, Dion P, Houle M, Toulouse A, Lafreniere RG, Vercauteren F, Hince P, Laganiere J, Rochefort D, Faivre L, Samuels M, Rouleau GA. Mutations in the nervous system-specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest 118: 2496–2505, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silayeva L, Deeb TZ, Hines RM, Kelley MR, Munoz MB, Lee HH, Brandon NJ, Dunlop J, Maguire J, Davies PA, Moss SJ. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc Natl Acad Sci USA 112: 3523–3528, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Mercado A, Desai R, George AL Jr, Gamba G, Mount DB. Characterization of hKCC2, the human neuronal-specific K-Cl cotransporter (Abstract). FASEB J 15: A440, 2001. [Google Scholar]
- Tanis JE, Bellemer A, Moresco JJ, Forbush B, Koelle MR. The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J Neurosci 29: 9943–9954, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett 89: 49–54, 1988. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH. Activity-dependent dishinibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl in hippocampal CA3 neurons. J Neurophysiol 61: 512–523, 1989. [DOI] [PubMed] [Google Scholar]
- Titz S, Sammler EM, Hormuzdi SG. Could tuning of the inhibitory tone involve graded changes in neuronal chloride transport? Neuropharmacology 95: 321–331, 2015. [DOI] [PubMed] [Google Scholar]
- Toda T, Ishida K, Kiyama H, Yamashita T, Lee S. Down-regulation of KCC2 expression and phosphorylation in motoneurons, and increases the number of in primary afferent projections to motoneurons in mice with post-stroke spasticity. PLoS One 9: e114328, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, Khalilov I, Tsintsadze V, Brouchoud C, Chazal G, Lemonnier E, Lozovaya N, Burnashev N, Ben-Ari Y. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343: 675–679, 2014. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Pruunsild P, Kaila K, Delpire E, Timmusk T, Rivera C, Airaksinen MS. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J Biol Chem 282: 30570–30576, 2007. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Soni S, Hübner CA, Rivera C, Airaksinen MS. Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J Biol Chem 284: 13696–13704, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J 397: 223–231, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hartmann AM, Beyer T, Ripperger A, Nothwang HG. A novel regulatory locus of phosphorylation in the C terminus of the potassium chloride cotransporter KCC2 that interferes with N-ethylmaleimide or staurosporine-mediated activation. J Biol Chem 289: 18668–18679, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001. [DOI] [PubMed] [Google Scholar]
- Yeo M, Berglund K, Hanna M, Guo J, Kitture J, Abramowitz J, Busciglio J, Gao Y, Birnbaumer L, Liedtke W. Bisphenol A delays the perinatal chloride shift in cortical neurons by epigenetic effects on the Kcc2/Slc12a5 promoter. Proc Natl Acad Sci USA 110: 4315–4320, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol 93: 1557–1568, 2005. [DOI] [PubMed] [Google Scholar]
- Zhu L, Polley N, Mathews GC, Delpire E. NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res 79: 201–212, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]