

Significance
How RING-E3s, several of which are required for cell division, differentiation, or survival, achieve E2 activation at the right time and place is poorly understood. Here we used the essential RING-E3 anaphase-promoting complex (APC/C) and its E2 Ube2S to dissect the mechanism of E2 recognition and activation by RING-E3s. Our work reveals a dynamic interplay between E2 and E3 that is regulated by reversible phosphorylation; while phosphorylation of Cdc20 inhibits Ube2S binding to the APC/C, dephosphorylation by PP2AB56 allows for rapid Ube2S activation. Given that the kinetochore-bound PP2AB56 also silences the spindle checkpoint signal, this work suggests that cells coordinate Ube2S activation with other events in human cell cycle control.
Keywords: ubiquitin, anaphase-promoting complex, APC/C, Ube2S, phosphorylation
Abstract
Most metazoan E3 ligases contain a signature RING domain that promotes the transfer of ubiquitin from the active site of E2 conjugating enzymes to lysine residues in substrates. Although these RING-E3s depend on E2 enzymes for catalysis, how they turn on their E2s at the right time and place remains poorly understood. Here we report a phosphorylation-dependent mechanism that ensures timely activation of the E2 Ube2S by its RING-E3, the anaphase-promoting complex (APC/C); while phosphorylation of a specific serine residue in the APC/C coactivator Cdc20 prevents delivery of Ube2S to the APC/C, removal of this mark by PP2AB56 allows Ube2S to bind the APC/C and catalyze ubiquitin chain elongation. PP2AB56 also stabilizes kinetochore–microtubule attachments to shut off the spindle checkpoint, suggesting that cells regulate the E2–E3 interplay to coordinate ubiquitination with critical events during cell division.
By promoting the ubiquitination and proteasomal degradation of anaphase inhibitors, the anaphase-promoting complex (APC/C) triggers sister chromatid separation and mitotic exit (1–5). The APC/C also targets kinases and microtubule-binding proteins that ensure accurate assembly of the mitotic spindle. Misregulation of the APC/C has dramatic consequences for cell cycle control; whereas APC/C inhibition causes mitotic arrest and cell death, its untimely activation results in aneuploidy, a common feature of human cancer cells (6).
As a RING-dependent E3 ligase, the APC/C stimulates the transfer of ubiquitin from the catalytic cysteine of E2 conjugating enzymes to lysine residues in substrates. In most cases, the APC/C initiates chain formation by using a specific E2, Ube2C (7–10). Once the first ubiquitin molecules have been attached to substrates, another conserved E2, Ube2S, extends K11-linked chains that are recognized by the proteasome for degradation (11–16). Ube2S frequently acts on short chains rather than on single ubiquitin subunits, thereby producing branched conjugates that impart high affinity for proteasomal receptors (13). Consistent with an important role in cell division, activation of Ube2S during mitosis results in a dramatic increase in the abundance of K11 linkages (17, 18), a chain topology required for APC/C-dependent substrate degradation (19).
As with many key cell cycle regulators, the APC/C and Ube2S need to be under tight control, and overexpression of Ube2S can promote tumor growth and metastasis in mice (20). The correct timing of APC/C activation is ensured by the spindle checkpoint, a signaling cascade turned on by kinetochores that have not achieved bipolar attachment to the spindle (4, 21, 22). Spindle checkpoint signaling leads to formation of the mitotic checkpoint complex (MCC), composed of Mad2, BubR1, Bub3, and Cdc20. When bound to the APC/C, the MCC competes for recognition of substrate KEN boxes and puts the APC/C coactivator Cdc20 in a position where it is unable to engage another degron, the D box (23–25). In contrast, the MCC does not occupy the binding sites for APC/C E2s or impede the ability of the APC/C to stimulate ubiquitin transfer by Ube2S (12). Thus, although overexpression of Ube2S has been associated with tumorigenesis, the mechanisms that restrict its activity during mitosis have remained elusive.
RING-E3s, such as the APC/C, engage their E2 enzymes in a dynamic manner (26). On binding a charged E2, the RING domain stabilizes a closed conformation between the E2 and its donor ubiquitin (14, 27–30). Once this ubiquitin is transferred to a target lysine, the E2 dissociates from the RING domain to allow for its recharging by the E1 (31). For most RING-E3s, the cycles of E2 engagement and dissociation are thought to occur constitutively (32), and only a few examples of controlled E2 activation are known. Access of Cdc34 to its specific RING-E3, the Skp1-Cul1-F box (SCF) complex, can be regulated by phosphorylation or competition with the inhibitory protein glomulin (33, 34). Reminiscent of this situation, Ube2S interacts with the APC/C in a cell cycle-dependent manner, and depletion of Cdc20 prevents Ube2S from stably binding to the APC/C in cells (12, 15). However, as part of the MCC, Cdc20 already associates with the APC/C during prometaphase, when APC/C activity must be low to allow sufficient time for chromosome alignment. How the ability of Ube2S to build ubiquitin chains is restricted during early stages of mitosis to safeguard cells against premature APC/C activation remains unknown.
In this study, we identified a mechanism that establishes how the RING-E3 APC/C activates Ube2S at the right time and place. In early mitosis, phosphorylation of a specific serine residue in the APC/C coactivator Cdc20 prevents the stable association of Ube2S with Cdc20 and the APC/C. Conversely, removal of the inhibitory mark on Cdc20 by the phosphatase PP2AB56 allows Ube2S to engage the APC/C and catalyze ubiquitin chain elongation. PP2AB56 also stabilizes the kinetochore–microtubule interface to silence the spindle checkpoint (35, 36), suggesting that cells regulate the interplay between RING-E3s and their E2s to coordinate ubiquitination with important events in cell division.
Results
Cdc20 Associates with PP2AB56 During Prometaphase.
Based on the observation that Ube2S is delivered to the APC/C by Cdc20 (12), we hypothesized that regulators of Ube2S activation might already be recognized by Cdc20 itself. To identify such proteins, we synchronized HeLa cells expressing FLAGCdc20 in prometaphase using S-trityl-L-cysteine (STLC), a chemical inhibitor of the mitotic motor Eg5 that allows the formation of kinetochore–microtubule attachments but interferes with formation of a bipolar spindle (37). During the last 2 h of synchronization, we treated these cells with the proteasome inhibitor MG-132, a condition that stabilizes Cdc20 interactions with Ube2S and the APC/C (12). Finally, we purified FLAGCdc20 and determined high-confidence interactors by CompPASS mass spectrometry (38), using a database of immunoprecipitation analyses performed with mitotic HeLa cells.
As expected based on previous reports (12, 39–41), Cdc20 bound the APC/C, Ube2S, spindle checkpoint proteins, cyclin A2-Cdk2 complexes, and CCT chaperone (Fig. 1A). With lower coverage, we observed an association between Cdc20 and multiple APC/C substrates, including geminin, cyclin B3, Skp2, Tpx2, Nek2A, Plk1, and Kif14, which was dependent on proteasome inhibition. In addition, the stabilization of Cdc20 complexes by MG-132 treatment allowed us to detect an abundant interaction between Cdc20 and the PP2AB56 phosphatase (composed of catalytic subunit PPP2CA or PPP2CB, regulatory subunit PPP2R1A, and substrate targeting subunits PPP2R5A–E), as well as the kinetochore anchor of PP2AB56, Knl1. In contrast, PP2AB55, an isoform of PP2A that is active during mitotic exit (42), was not registered in these affinity purifications, and PP1, a phosphatase also recruited to kinetochores by Knl1, was detected only with low total spectral counts.
Fig. 1.

Cdc20 recruits Ube2S and PP2AB56 to the APC/C. (A) Identification of high-confidence interactors of Cdc20 during protemetaphase. FLAGCdc20 was purified from prometaphase HeLa cells synchronized with STLC and MG-132, and associated proteins were determined by CompPASS mass spectrometry. Only abundant interactors detected with high TSCs in five independent experiments are shown. (B) Endogenous Cdc20 interacts with PP2AB56. Cdc20 was affinity-purified from STLC-synchronized HeLa cells using monoclonal αCdc20 antibodies, and bound proteins were identified by Western blot analysis. MG-132 was added as indicated. (C) Cdc20 delivers Ube2S and PP2AB56 to the APC/C. HeLa cells were transfected with siRNAs targeting Cdc20, synchronized in prometaphase using STLC, and subjected to αAPC3 affinity purification. Proteins bound to the APC/C were identified by Western blot analysis using specific antibodies.
We performed several experiments to further test for an interaction between Cdc20 and PP2AB56. By mass spectrometry, we observed the binding between FLAGCdc20 and all subunits of PP2AB56 if we impaired mitotic protein degradation by expressing a dominant-negative version of the APC/C-E2 Ube2C, Ube2CC114S (Fig. S1A). Moreover, we found that FLAGCdc20 bound to endogenous PP2AB56 using Western blot analysis (Fig. S1B), and obtained similar results in reciprocal experiments in which we observed endogenous Cdc20 and Ube2S in affinity purifications of a substrate-targeting factor of PP2AB56, FLAGB56α (Fig. S1C).
Fig. S1.

Cdc20 binds PP2AB56. (A) Inhibition of mitotic protein degradation by expression of a dominant negative version of the APC/C-specific E2, Ube2C (Ube2CDN), stabilizes binding of Cdc20 to the PP2AB56 phosphatase. The efficiency of binding to FLAGCdc20 in prometaphase cells exposed to Ube2CDN was compared with control experiments in which no Ube2CDN was expressed. TSC, total spectral count. (B) Cdc20 binds endogenous PP2AB56. HeLa cells expressing FLAGCdc20 were synchronized in prometaphase; if indicated, MG-132 was added during the last 2 h of synchronization. FLAGCdc20 was affinity-purified over αFLAG-agarose, and bound proteins were detected by Western blot analysis using specific antibodies. (C) Cdc20 binds PP2AB56. HeLa cells were transfected with FLAGB56α, synchronized with STLC, and analyzed for specific binding partners by αFLAG affinity purification and Western blot analysis. Proteasome inhibitors were added as indicated. (D) Endogenous Cdc20 associates with an active phosphatase. Cdc20 was affinity-purified from prometaphase HeLa cells, and copurifying phosphatase activity was measured in an enzymatic assay using a phospho-Thr model peptide. Cells were exposed to proteasome inhibitors before lysis as indicated. (E) Phosphatase activity purification with Cdc20 is likely mediated by PP2A. Cdc20 was affinity-purified from prometaphase cells treated with proteasome inhibitors and then subjected to the enzymatic phosphatase assay described above. Increasing concentrations of the phosphatase inhibitor okadaic acid were added as indicated.
To determine whether Cdc20 and PP2AB56 interact at the endogenous level, we purified Cdc20 from lysates of prometaphase HeLa cells using a monoclonal αCdc20 antibody. Similar to Ube2S, PP2AB56 was readily detected in Cdc20 immunoprecipitates when MG-132 was present (Fig. 1B). PP1 or PP2AB55 was seen much less readily, if at all, in purifications of endogenous Cdc20.
To evaluate these findings by an orthogonal method, we used an enzymatic assay to ask whether phosphatase activity copurified with Cdc20. Consistent with our earlier results, we were able to measure phosphatase activity in Cdc20 immunoprecipitations in the presence, but not in the absence, of MG-132 (Fig. S1D). This activity was inhibited by okadaic acid at an IC50 of ∼5 nM (Fig. S1E), close to the IC50 of PP2A inhibition (35, 36). Taken together, these experiments demonstrate that Cdc20 interacts with PP2AB56 in a very similar manner as it binds to Ube2S (12).
Cdc20 Delivers PP2AB56 to the APC/C.
Our finding of equivalent interactions between Cdc20 and Ube2S or PP2AB56 raised the possibility that Cdc20 might recruit PP2AB56 to the APC/C. To test this hypothesis, we depleted Cdc20 from HeLa cells using validated siRNAs (12), synchronized these cells in mitosis by treating them with STLC and MG-132, and purified the APC/C using monoclonal αAPC3 antibodies. As reported previously (12), depletion of Cdc20 prevented the stable APC/C binding of Ube2S, as well as that of the spindle checkpoint proteins BubR1 and Mad2 (Fig. 1C). In addition, the loss of Cdc20 interfered with the association of PP2AB56 with the APC/C, indicating that Cdc20 delivers PP2AB56 to the prometaphase APC/C.
PP2AB56 Activates Phospho-Inhibited APC/C Toward Ube2S.
PP2AB56 has frequently been observed to counteract phosphorylation events that control protein interactions, including those occurring at kinetochores (35, 36). Based on these findings, we asked whether phosphorylation impacts the binding of Ube2S to Cdc20 or the APC/C, and whether PP2AB56 could overcome such regulation. As an initial test for this hypothesis, we synchronized HeLa cells in prometaphase, treated these cells with proteasome inhibitors to enrich them for Cdc20 complexes, and supplemented cell lysates with okadaic acid to shut off both PP1 and PP2A phosphatases. We then affinity-purified endogenous Cdc20 and used Western blot analysis to identify proteins that engaged Cdc20 more or less efficiently when phosphatases were turned off. Notably, phosphatase inhibition led to a striking decrease in the binding of Ube2S to Cdc20 (Fig. 2A). Persistent phosphorylation also reduced the interaction of Cdc20 with the checkpoint protein BubR1, as well as with PP2AB56 itself.
Fig. 2.

Phosphorylation inhibits Ube2S-activation by the APC/C. (A) Endogenous Cdc20 was purified from lysates of prometaphase cells treated with the proteasome inhibitor MG-132 or the phosphatase inhibitor okadaic acid, as indicated. Bound proteins were identified by Western blot analysis using specific antibodies. (B) Phosphatase inhibition alters the interaction of Ube2S with the APC/C. APC/C purified from okadaic acid-treated extracts was incubated with Ube2SC118A, a variant of Ube2S that contains the active site Cys95 as the only cysteine. Reactions were supplemented with the bifunctional cysteine reactive BMB and analyzed for cross-links by Western blotting against Cdc20, Apc2, and Apc11. Green asterisks mark previously validated cross-links (12). (C) Active Ube2S copurifies with prometaphase APC/C. HeLa cells treated with control- or Ube2S-siRNAs were synchronized in prometaphase using STLC and MG-132, as indicated. APC/CCdc20 was purified from these cells using monoclonal αAPC3-antibodies and incubated with E1, ubiquitin, and ATP. Formation of K11-linked ubiquitin dimers was monitored by αK11-linkage specific Western blot analysis. (D) Phosphatase inhibition blocks the ability of the APC/C to catalyze formation of K11-linkages. APC/CCdc20 was purified from prometaphase cells treated with MG-132 or okadaic acid, as indicated, before incubation with E1, ubiquitin, and ATP. Formation of K11-linked dimers was determined by αK11-linkage specific Western blot analysis.
The reduced association of Cdc20 with Ube2S caused by phosphorylation should interfere with the delivery of Ube2S to its correct position on the APC/C. To investigate whether this in fact was the case, we used an established cross-linking assay built on a single Cys variant of Ube2S (Ube2SC118A) and a bifunctional cysteine-reactive cross-linker, 1,4-bismaleimidobutane (BMB) (12). We incubated Ube2SC118A and BMB with prometaphase APC/C that was purified in the presence or absence of phosphatase inhibitors and probed for known cross-links of Ube2S with Apc2, Apc11, and Cdc20. These experiments revealed that phosphorylation impaired the association of Ube2S with Cdc20 and Apc11, but had less of an effect on the binding of Ube2S with Apc2 (Fig. 2B). The reduced interaction of Ube2S with Cdc20 confirms our results from Cdc20 affinity purifications, whereas the lower efficiency of cross-linking between Ube2S and Apc11 suggests that phosphorylation might interfere with Ube2S activation by the APC/C (27, 43).
Consistent with the cross-linking results, phosphatase inhibition prevented the ubiquitination of endogenous Cdc20 and geminin, two known Ube2S substrates, in mitotic extracts (Fig. S2 A and B). Moreover, purification of the APC/C under conditions of phosphatase inhibition diminished its ability to activate Ube2S toward its model substrate, Ub-cyclin A (Fig. S2C). To separate the effects of phosphatase inhibition on Ube2S from those on APC/C substrate binding or chain initiation, we developed an assay that specifically monitored APC/C-dependent activation of endogenous Ube2S. We purified APC/C from prometaphase cells that were synchronized in the presence or absence of MG-132, with the former condition known to stabilize the association of Ube2S with the APC/C (12, 14). We incubated this APC/C with E1, ubiquitin, and ATP, but no E2s, and monitored the formation of K11-linked ubiquitin dimers using linkage- specific antibodies. Under these conditions, the formation of K11-linked dimers is strictly dependent on E2 enzymes that copurify with the APC/C. Prometaphase APC/C supported formation of K11-linked ubiquitin dimers (Fig. 2C), which was lost on depletion of Ube2S and improved by proteasome inhibition. Thus, this assay monitors the activity of endogenous Ube2S bound to the APC/C. Importantly, phosphatase inhibition prevented formation of K11-linked ubiquitin dimers by endogenous APC/C and Ube2S (Fig. 2D), indicating that one or more phosphorylation events interfere with the APC/C-dependent activation of Ube2S.
Fig. S2.

Persistent phosphorylation inhibits catalytic activation of the APC/C. (A) Phosphorylation inhibits the ability of prometaphase APC/C to catalyze the ubiquitination of endogenous Cdc20. Extracts of prometaphase HeLa cells were supplemented with His-tagged ubiquitin and treated with the phosphatase inhibitor okadaic acid. After incubation at room temperature, ubiquitin conjugates were purified under denaturing conditions using NiNTA-agarose and analyzed for endogenous Cdc20 modification by Western blot analysis. (B) Phosphorylation inhibits the ability of APC/CCdc20 to catalyze K11-dependent ubiquitination of the Ube2S-substrate geminin. Extracts of prometaphase HeLa cells were supplemented with His-tagged ubiquitin or ubiquitinK11R, and ubiquitin conjugates were purified under denaturing purification as described above. Modification of endogenous geminin was detected by Western blot analysis using specific antibodies. (C) Phosphorylation inhibits activation of Ube2S by the APC/C. APC/C was purified from mitotic extracts treated with okadaic acid, as indicated, and its ability to promote Ube2S-dependent ubiquitin chain elongation toward the model substrate Ub-cyclin A was measured by Western blot analysis.
To determine whether PP2AB56 reverts the inhibitory effects of phosphorylation on Ube2S, we purified PP2AB56 from human 293T cells and added it to APC/C that was immunoprecipitated under conditions of phosphatase inhibition. In binding assays, we found that PP2AB56 restored the cross-links between Ube2S and Apc11 and partially rescued the cross-links between Ube2S and Cdc20 (Fig. 3A), indicating that PP2AB56 can revert specific, but not all, phosphorylation events on the APC/CCdc20. To test whether reinstating the interaction between Apc11/Cdc20 and Ube2S allows for activation of E2, we tested the ability of purified PP2AB56 to regulate ubiquitination of Ub-cyclin A. Indeed, PP2AB56 was able to partially restore the ability of phosphorylated APC/C to activate Ube2S toward Ub-cyclin A (Fig. 3B). Taken together, these experiments show that phosphorylation inhibits the ability of the APC/C to activate Ube2S, which can be overcome by PP2AB56, a phosphatase delivered to the APC/C by Cdc20.
Fig. 3.

PP2AB56 controls APC/C recruitment and activation of Ube2S. (A) PP2AB56 restores cross-linking of Ube2S to Apc11 on phosphatase-inhibited APC/C purified and treated with BMB as described above. (B) PP2AB56 restores the activity of phosphatase-inhibited APC/C toward Ube2S. APC/C was purified from prometaphase extracts treated with okadaic acid, as indicated, and its activity to promote Ube2S-dependent ubiquitin chain elongation on Ub-cyclin A was measured by Western blot analysis. Where indicated, purified PP2AB56α was added to the reactions.
PP2AB56 Targets Ser92 in Cdc20.
We next wished to understand how reversible phosphorylation controls the activation of Ube2S, and thus identified the phosphorylation sites on Cdc20 that are regulated by PP2AB56 and affect the APC/C binding or activation of Ube2S. We purified FLAGCdc20 from prometaphase cells and used mass spectrometry to identify phosphorylated peptides that were enriched on phosphatase inhibition. These experiments revealed that phosphatase inhibition resulted in a striking increase in the phosphorylation of Ser92 of Cdc20 (Fig. 4A), a residue in the amino terminal domain of Cdc20 known to engage the APC/C. Using a phospho-specific antibody, we confirmed phosphorylation of endogenous Cdc20-Ser92, which occurred during prometaphase and was increased on phosphatase inhibition (Fig. 4B). Suggesting that this phosphorylation event occurs with high efficiency, Phos-tag gels revealed an abundant phosphorylated Cdc20 species during mitosis that was lost on mutation of Ser92 (Fig. S3A).
Fig. 4.

PP2AB56 regulates Cdc20 phosphorylation to control Ube2S recruitment to the APC/C. (A) Ser92 in Cdc20 is a phosphatase-sensitive site during prometaphase. FLAGCdc20 was purified from prometaphase cells in the presence or absence of okadaic acid, and the abundance of phosphorylated peptides was determined by mass spectrometry. (B) Phosphatase inhibition increases phosphorylation of Cdc20S92, as determined by Western blot analysis using a phospho-S92 specific antibody. Endogenous Cdc20 was purified from prometaphase extracts using monoclonal αCdc20-antibodies, and okadaic acid was added as indicated. (C) PP2AB56α targets Ser92 of Cdc20 in vitro. Cdc20 was purified from prometaphase extracts treated with okadaic acid, it was and incubated with purified PP2AB56α or with PP2AB56 that had been inactivated with okadaic acid. Phosphorylation of Cdc20 was analyzed by Western blot analysis using the pS92-specific antibody. (D) PP2AB56 targets Ser92 of Cdc20 in cells. HeLa cells were depleted of all B56 substrate targeting factors using validated siRNAs. After cells were synchronized in prometaphase, Cdc20 was affinity-purified, and Ser92 phosphorylation and Ube2S binding were measured by Western blot analysis. (E) A phosphomimetic mutation in Cdc20-Ser92 obliterates binding to Ube2S, but not the MCC. Wild type (WT), Ser92Ala, or Ser92Asp mutants of Cdc20 were immunoprecipitated from lysates of prometaphase cells and analyzed for Ube2S, APC/C, or MCC binding (as indicated by BubR1) by Western blot analysis. The asterisk marks a nonspecific band on the BubR1 Western blot. (F) Cdc20 phosphorylation prevents delivery of Ube2S to the APC/C. Cells were depleted of endogenous Cdc20 and reconstituted with either WT or phosphomimetic Cdc20S92D. APC/C was purified with αAPC3 antibodies and analyzed for bound proteins by Western blot analysis.
Fig. S3.

Characterization of Cdc20 phosphorylation. (A) Ser92-phosphorylated Cdc20 is abundant during mitosis. HeLa cells expressing FLAGCdc20 or FLAGCdc20S92A were synchronized by double-thymidine arrest and release, and lysates were analyzed by Phos-tag gels and Western blot analysis using specific antibodies. The double asterisk (**) denotes the S92-dependent phosphorylated Cdc20 species. (B) Cdc20 phosphorylation affects degradation of the APC/C substrate geminin. Cells depleted of endogenous Cdc20 were reconstituted with either WT or phosphomimetic Cdc20S92D. After synchronization in prometaphase, cells were released into fresh medium, and degradation of the APC/C substrate geminin was monitored over time by Western blot analysis.
Purified PP2AB56, but not a version of the phosphatase that had been inactivated by okadaic acid treatment, efficiently removed the phosphorylation mark from Cdc20-Ser92 in vitro (Fig. 4C). As indicated by the increased mobility in gel electrophoresis, PP2AB56 also was able to dephosphorylate BubR1, a known PP2AB56 substrate. To test whether this regulation occurs in vivo, we depleted PP2AB56 from HeLa cells using five validated siRNAs that target all known B56 isoforms (44). We synchronized these cells in prometaphase, purified endogenous Cdc20, and probed for Cdc20-Ser92 phosphorylation using our phospho-specific antibody. Importantly, the loss of all B56 substrate-targeting factors of PP2A increased the phosphorylation of Cdc20-Ser92 (Fig. 4D), demonstrating that PP2AB56 regulates the modification status of Cdc20 during mitosis.
Consistent with our experiments based on chemical phosphatase inhibition, we noticed that depletion of PP2AB56 reduced the association of Ube2S with Cdc20 (Fig. 4D). This finding suggested that phosphorylation of Ser92 might affect the interaction of Cdc20 with Ube2S. To test this hypothesis, we exchanged Ser92 of Cdc20 with Ala or Asp, thereby either interfering with or mimicking phosphorylation at this site. We purified the Cdc20 variants from prometaphase cells and probed for Ube2S binding by Western blot analysis. Strikingly, mutation of Ser92 to either Ala or Asp strongly impaired the binding of Cdc20 with Ube2S, but did not affect the binding of Cdc20 to BubR1 or the APC/C (Fig. 4E). These observations indicate that phosphorylation of Ser92 specifically interferes with a role of the hydroxyl group of this residue in mediating the binding of Cdc20 to Ube2S.
Based on these results, phosphorylation of Cdc20-Ser92 should impair the delivery of Ube2S to the APC/C in cells, a reaction that is dependent on the recognition of Ube2S by Cdc20. To test this assumption, we purified the APC/C from prometaphase cells that were depleted of Cdc20. As reported previously (12), the loss of Cdc20 prevented the stable binding of Ube2S or BubR1 to the APC/C, and this effect of Cdc20 depletion could be rescued by expression of siRNA-resistant Cdc20 (Fig. 4F). When phosphomimetic Cdc20S92D was used to reinstate Cdc20 levels, cells failed to deliver Ube2S to the APC/C, yet were still able to mediate the binding of BubR1 (Fig. 4F). Cells expressing Cdc20S92D as their sole source of Cdc20 could accordingly mount a spindle checkpoint-dependent prometaphase arrest, but were delayed in mediating the degradation of the Ube2S-substrate geminin upon release into a new cell cycle (Fig. S3B). Thus, phosphorylation of Ser92 of Cdc20, a mark regulated by PP2AB56, specifically regulates APC/C binding and activation of Ube2S.
PP2AB56 and Ube2S Binding to the APC/C Is Sensitive to Kinetochore Status.
Finally, we wished to determine how the activity of PP2AB56 toward Cdc20 is integrated into the cell cycle program. We had observed that Ser92 of Cdc20 was phosphorylated more efficiently when cells were synchronized under conditions that disrupt proper spindle formation (Fig. 4B). In addition, we found that depletion of Kif18A, a kinesin that regulates the dynamics of kinetochore–microtubule interactions (45), led to a strong enrichment of Cdc20-Ser92 phosphorylation (Fig. 5A). Similar to STLC, Kif18A depletion also increases the fraction of cells in the prometaphase state of the cell cycle (45). These findings suggested that the binding of PP2AB56 to Cdc20 might be linked to the status of kinetochore–microtubule attachments during early stages of mitosis.
Fig. 5.

Kinetochore function is required for PP2AB56 recognition by Cdc20. (A) Depletion of Kif18A increases phosphorylation of Ser92 of Cdc20. Asynchronous HeLa cells were treated with indicated siRNAs, and endogenous Cdc20 was purified using monoclonal αCdc20 antibodies. Phosphorylation of Cdc20S92 was determined by Western blot analysis. (B) Depletion of Knl1 interferes with Cdc20 binding to BubR1, PP2AB56, and Ube2S. HeLa cells were depleted of Knl1, synchronized in prometaphase, and treated with MG-132 as indicated. Cdc20 was purified using specific antibodies and analyzed for binding partners by Western blot analysis. (C) Model of Ube2S regulation by reversible phosphorylation.
To test this hypothesis, we depleted HeLa cells of Knl1, a kinetochore protein that provides binding sites for spindle microtubules as well as for PP2AB56 (36). The depletion of Knl1 also resulted in a loss of Cdc20 from prometaphase kinetochores (Fig. S4A) and diminished levels of BubR1 (Fig. 5B), a known binding partner of PP2AB56 at kinetochores (36). Strikingly, depletion of Knl1 prevented both PP2AB56 and Ube2S from stably binding to Cdc20 (Fig. 5B). To independently disturb kinetochore function, we reduced levels of Ndc80. Ndc80 also provides microtubule-binding sites for kinetochore–microtubule attachments, but its loss had less dramatic consequences on BubR1 or Mad2 levels during prometaphase (Fig. S4B). Importantly, as observed on depletion of Knl1, a reduction in Ndc80 levels impaired the binding of PP2AB56 and Ube2S to Cdc20 (Fig. S4B). These results underscore the notion that PP2AB56 and Ube2S are coregulated, and that both proteins are recruited to Cdc20 in a manner dependent on a functional kinetochore. Given that Cdc20 delivers PP2AB56 and Ube2S to the APC/C, our results suggest that phosphorylation-dependent regulation of Ube2S is coordinated with the microtubule-binding status of kinetochores.
Fig. S4.
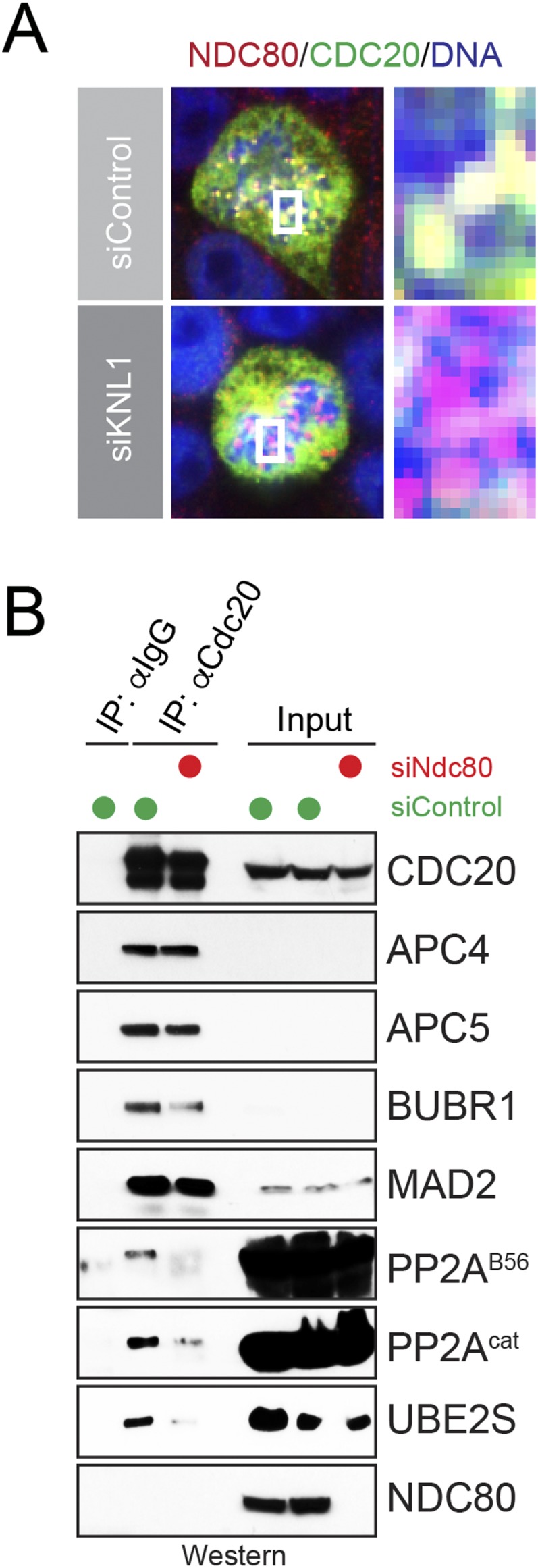
Kinetochore function regulates the interaction of Cdc20 with Ube2S and PP2AB56. (A) Depletion of Knl1 from HeLa cells displaces Cdc20 from kinetochores. HeLa cells were transfected with siRNA against Knl1, and localization of Cdc20 was determined in prometaphase by immunofluorescence microscopy against Cdc20 (green), Ndc80 (red), and DNA (blue). (B) Depletion of Ndc80 impairs binding of Cdc20 to PP2AB56 and Ube2S. HeLa cells were depleted of Ndc80, synchronized in prometaphase with STLC and MG-132, and subjected to Cdc20 affinity purification and Western blot analysis.
Discussion
Mitotic cells ensure faithful distribution of the genetic material by delaying activation of the APC/C until all kinetochores have been attached to the spindle. Once this has been accomplished, the APC/C is turned on rapidly and completely to guarantee synchronous sister chromatid separation. If the APC/C is activated too early or too slowly, sister chromatid separation occurs erroneously, and aneuploidy arises. Whereas the spindle checkpoint and its effector, the MCC, have long been recognized to inhibit the APC/C by blocking substrate recognition, we now report that cells also ensure timely activation of the APC/C-specific E2 Ube2S.
Our results identify phosphorylation of Cdc20 as a gatekeeper for the delivery of Ube2S to the APC/C. Modification of Ser92 in Cdc20 prevents binding to Ube2S, but not the MCC or APC/C, thereby blocking the formation of a complex important for the recruitment of Ube2S to the APC/C. Accordingly, phosphorylation impairs the ability of the APC/C to activate Ube2S, which could provide a basis for previous observations that hypophosphorylated APC/C displays higher activity than the heavily phosphorylated APC/C present in early mitotic cells (46). Ser92 of Cdc20 is modified by a kinetochore-bound complex composed of a Bub1 scaffold and Plk1 kinase (47). Consistent with this idea, the loss of Ube2S and persistent Cdc20 phosphorylation cause phenotypes that are the opposite of Bub1 depletion; whereas the former events delay APC/C activation at the metaphase–anaphase transition and increase the sensitivity of cells to spindle toxins (12), the loss of Bub1 results in premature APC/C activation and anaphase entry (39).
The inhibitory phosphorylation on Cdc20 is removed by PP2AB56, a kinetochore-bound phosphatase. Because depletion of all B56 substrate-targeting factors stabilized Ser92 phosphorylation less dramatically than okadaic acid, other phosphatases, including PP1, could target this Cdc20 residue as well. PP2AB56 is recruited to kinetochores by BubR1, and a D464R mutation in Cdc20 that disrupts binding to BubR1 (12) also interferes with recognition of PP2AB56 (Fig. S1B). In addition, depletion of Knl1, the kinetochore anchor of BubR1, prevents incorporation of PP2AB56 and Ube2S into Cdc20 complexes. These findings suggest that Cdc20 coordinates the formation of a kinetochore-bound complex that contains BubR1, Ube2S, and PP2AB56. Notably, the arrival of PP2AB56 at kinetochores stabilizes their binding to spindle microtubules, which turns off the spindle checkpoint and allows substrate binding to the APC/C (36). Thus, we propose that PP2AB56 improves Ube2S recruitment to the APC/C on stable attachment of mitotic chromosomes to the spindle apparatus (Fig. 5C). If correct, this model will provide a molecular basis for synchronizing spindle checkpoint silencing with catalytic activation of the APC/C.
Our work sheds new light onto the role of Cdc20 as a coactivator of the APC/C. In addition to its ability to recruit substrates to the APC/C (48–52), Cdc20 has long been known to increase the catalytic activity of the APC/C, yet how this occurs is less well understood. Structural studies have revealed that Cdc20 changes the conformation of the APC/C, which increases the ability of Apc11 to stimulate ubiquitin release from the initiating E2 Ube2C (2, 24, 50). Our present study and previous work suggest that Cdc20 also controls the APC/C-specific chain elongating E2 Ube2S; Cdc20 delivers Ube2S to the APC/C and places it in proximity to its partners Apc2 and Apc11 (12, 27, 53). As shown here, Cdc20 also coordinates recruitment of Ube2S with that of PP2AB56, which in turn permits the APC/C to simulate Ube2S. We therefore propose that the activator function of Cdc20 originates in part from its role in recruiting and activating Ube2S, providing a striking example of how RING-E3s exploit multiple layers of regulation to activate their cognate E2 enzymes at the right time and place.
Materials and Methods
Details of the materials and methods used in this study, including antibodies and siRNA sequences, are provided in SI Materials and Methods. Protein purification, APC/C activity assays, immunoprecipitation, immunofluorescence microscopy, and CompPASS mass spectrometry were performed as described previously (12, 14, 54). Phosphatase assays were performed using the PP2A Immunoprecipitation Phosphatase Assay Kit (EMD Millipore) in accordance with the manufacturer’s directions.
SI Materials and Methods
Antibodies and siRNA Oligos.
The following antibodies were used in our experiments: α-tubulin (mouse monoclonal; Calbiochem; DM1A); APC2, cyclin B1, vinculin (rabbit polyclonal; Cell Signaling Technologies; 12301, 4138, 4650S); BubR1, Hec1, Kif18A, PPP1CC (rabbit polyclonal; Bethyl Laboratories; A301-080A, A300-906A); Mad2, PP2A catalytic α subunit (mouse monoclonal; BD Transduction Laboratories; 610679, 610555); Ube2S, PPP2R5A, APC11 (rabbit polyclonal; Novus Biologicals; 22570002, 100–41412, NBP1-78050); β-actin (mouse monoclonal; MP Biomedicals; 08691001); and APC4, APC7, APC5, CDC27, cyclin B1, Cdc20, geminin, PP2A-B55(h300), and cyclin A (C-19) (rabbit polyclonal; Santa Cruz Biotechnology; 20985, 20987, 20986, 5618, 594, H-175/8358, FL209, 33191, 596). For immunoprecipitation, the following antibodies were used: Cdc27 and Cdc20 (mouse monoclonal; Santa Cruz Biotechnology; 9972, 13162), α-FLAG (rabbit polyclonal; Sigma-Aldrich; F7425), normal rabbit IgG (Santa Cruz Biotechnology; 2027), and normal mouse IgG (Santa Cruz Biotechnology; 2025). The antibody for detecting phosphorylated Ser92 of Cdc20 was produced by H.Y.
The following siRNAs were used: Ube2S (3′ UTR: GGCACUGGGACCUGGAUUUUU, Dharmacon; ORF: CCCGATGGCATCAAGGTCTTT, Qiagen FlexiTube #5), Kif18A (ORF: Qiagen FlexiTubes #1, 3, and 5; ON-TARGETplus siRNA-SMARTpool; 3′ UTR: two distinct oligos designed using Dharmacon tools), All Stars negative control (CAGGGTATCGACGATTACAAA; Qiagen), Cdc20 (ORF: ON-TARGETplus siRNA-SMARTpool), KNL1/CASC5 (ON-TARGETplus siRNA-SMARTpool), and Hec1/Knct2/Ndc80 (ORF: Qiagen FlexiTube #6). Pooled PP2A B56 oligos were used as described previously (44).
Cell Culture and Synchronizations.
Cells were maintained at 37 °C and 5% (vol/vol) CO2 in DMEM containing 10% (wt/vol) FBS. For mitotic synchronization, cells at 75–90% confluency were treated with 2 mM thymidine (Sigma-Aldrich) for 20–24 h, incubated in fresh medium with no drug for 3 h, and then arrested in early mitosis with 100 ng/mL nocodazole (Sigma-Aldrich) or 5 μg/mL STLC (Sigma-Aldrich) for 12 h. MG-132 (UBPBio) was used at 20 μM for 2 h when indicated.
siRNA Depletion Experiments.
For siRNA transfection experiments performed by forward transfection, cells were plated at ∼50% cell density and then incubated until ∼70% confluency was reached or for ∼18 h. Transfections were performed using the standard protocol for Lipofectamine RNAiMAX (Life Technologies), with each siRNA added at a final concentration of 4–20 nM. For double-depletion experiments, siRNAs were combined at equimolar concentrations, with the final siRNA concentration per treatment kept constant within an experiment.
Production of Lentiviruses and Transduction.
Cdc20 (WT or an S92D variant) was cloned into a pENTR1A vector and recombined into pLenti XI DEST by LR recombination. Viruses were produced by 293T cells that had been transfected with packaging plasmids (Addgene) and lentiviral constructs for 48–72 h, as described previously (55). Viral supernatant was collected, filtered through a 0.2-μM filter, and frozen at −80 °C until use. HeLa cells were infected with virus in the presence of 6 μg/mL Polybrene. The medium was changed at 24 h postinfection.
Immunofluorescence.
Cells were plated on coverslips at ∼40–60% density. 48 h after transfection or infection, cells were fixed for immunofluorescence imaging at room temperature with one of the following methods: cold methanol for 4–7 min, PTEMF buffer [20 mM Pipes pH 6.8, 10 mM EGTA, 1 mM MgCl2, 0.2% Triton X-100, and 4% (vol/vol) formaldehyde] for 10 min, TBS with 4% (vol/vol) formaldehyde and 0.1% Triton X-100, or PBS with 4% (vol/vol) formaldehyde and 0.1% Triton X-100. Fixed cells were rinsed with wash buffer (0.1% Triton X-100 in TBS or PBS). Methanol- and formaldehyde-fixed cells were permeabilized in TBS with 0.4% Triton X-100. Cells were blocked in Abdil buffer [2% (wt/vol) BSA and 0.1% Triton X-100 in TBS] or 5% (vol/vol) normal donkey serum in PBS and then stained with primary antibodies diluted in Abdil buffer or 5% (vol/vol) normal donkey serum in PBS overnight at 4 °C. The next day, cells were moved to room temperature, washed five times, and stained with secondary antibodies coupled to Alexa Fluor 488, Alexa Fluor 549, and/or Alexa Fluor 568 (Molecular Probes) and Hoechst in Abdil buffer or 5% (vol/vol) normal donkey serum in PBS for 45 min in the dark. After washing, coverslips were mounted with ProLong Gold Antifade reagent (Invitrogen) and then dried at room temperature overnight in the dark. Cells were visualized at 60× or 100× using MetaMorph on an Olympus IX81 epifluorescence microscope or an Olympus Revolution XD spinning disk confocal microscope equipped with a CCD camera (Andor Technology) and a Yokogawa spinning disk.
Phos-Tag Gel Electrophoresis.
Cell lysates from HeLa cells expressing FLAGCdc20 or FLAGCdc20S92A and synchronized with double-thymidine treatment were run on a gel containing 40 μM Phos-tag Acrylamide (Wako Chemicals; AAL-107) according to the manufacturer's protocol, and then transferred for 90 min at 110 V onto methanol-activated PVDF membrane (Bio-Rad; 162-0175). Western blot analyses were performed using anti-FLAG antibody.
Immunoprecipitation.
HeLa cells were treated with siRNA and grown asynchronously, or synchronously with a thymidine/nocodazole or STLC protocol enacted 4–6 h after transfection as described (14). Cells were resuspended in 50 mM Hepes (pH 7.5), 1.5 mM MgCl2, and 5 mM KCl, and then lysed by freeze/thaw cycles in liquid nitrogen and multiple passages through a 25 G × 5/8” needle. Cell debris was pelleted by centrifugation, and the remaining lysates were normalized by their absorption at 280 nm. When indicated, normalized cell lysates were incubated with 9 μM okadaic acid (Cell Signaling Technology; 5934) for 30 min at 30 °C and 400 rpm. Normalized lysates were incubated with mouse IgG, αCdc20 antibody, or αCdc27 antibody coupled to protein G agarose with 150 mM NaCl and 0.05% Tween at 4 °C for 3–5 h. Beads were washed carefully, and bound proteins were eluted by the addition of SDS buffer and incubation for 10 min at 95 °C. Western blot analyses were performed using specific antibodies. For the Cdc20 rescue experiment, cells were transfected with FLAG-Cdc20 constructs using TransIT LT-1 reagent (Mirus Bio) and then, on the next day, transfected using Lipofectamine RNAiMAX with Cdc20 siRNA against the 3′ UTR to deplete endogenous Cdc20. The FLAG immunoprecipitation protocol is described in Mass Spectrometry Sample Preparation.
Phosphatase Assay.
Endogenous CDC20 immunoprecipitation from mitotic HeLa cells was performed as described previously (12). pThr peptide (K-R-pT-I-R-R) was added to the washed Cdc20-bound fraction, and dephosphorylation was allowed to proceed at room temperature. The dephosphorylated samples were incubated with phosphate detection dye and then read at A630nM using a fluorescence plate reader. The protocol for detection and dephosphorylation was obtained from the PP2A Immunoprecipitation Phosphatase Assay Kit (EMD Millipore). When indicated, okadaic acid was added before addition of the pThr peptide.
Mass Spectrometry Sample Preparation.
Samples for mass spectrometry analysis were prepared from 25–35 15-cm tissue culture dishes (Corning) of transfected HeLa cells per condition. Transfections of cDNA5/TO-FLAGCDC20 was performed using calcium phosphate [HBS: Hepes, NaCl, dextrose, KCl, Na2HPO4(7 H2O) with CaCl2] at 60–80% cell density. When indicated, cells were also transfected with pCS2-Ube2CC114S. The morning after transfection, fresh medium was added to stimulate uptake of the transfected DNA. Approximately 8 h later, thymidine was added to 2 mM. At 20–24 h after thymidine addition, cells were washed in fresh medium and then incubated for 3 h, after which STLC was added to 5 μM for 12 h. When indicated, MG-132 (20 μM; UBPBio) or DMSO was added for an additional 3 h. Cells were harvested by scraping, washed with PBS, and resuspended on ice in swelling buffer [50 mM Hepes pH 7.5, 1.5 mM MgCl2, 5 mM KCl, 0.1% Triton X-100 (Thermo Fisher), and a protease inhibitor mixture tablet (Roche)] for 30 min. Cells were lysed by freeze/thaw cycles in liquid nitrogen and multiple passages through a 25 G × 5/8” needle. Cell debris was pelleted by centrifugation, and NaCl was added to 150 mM. Extract was precleared with protein G agarose for 30 min at 4 °C. Cleared cell lysate was incubated with FLAG M2 agarose resin (Sigma-Aldrich) and then incubated for 3–5 h at 4 °C. FLAG resin was washed in swelling buffer with 150 mM NaCl and 0.1% Triton X-100, and further with swelling buffer with 150 mM NaCl only. Proteins bound to FLAG resin were eluted with FLAG peptide (500 μg/mL in PBS plus 0.1% Triton X-100; Sigma-Aldrich) in three steps. Eluates were pooled, and TCA (Thermo Fisher Scientific) was added to 20% for overnight precipitation at 4 °C. Pellets were washed three times with ice-cold 0.01 M HCl/90% (vol/vol) acetone solution, centrifuged, and allowed to air-dry. The pellets were resuspended directly in 100 mM Tris (pH 8.5) with 8 M urea for trypsin digestion. Samples were reduced by 100 mM Tris(2-carboxyethyl)phosphine (Sigma-Aldrich), alkylated by iodoacetamide (Sigma-Aldrich), and trypsin-digested in the presence of 100 mM CaCl2. Samples were trypsinized (Thermo Fisher Scientific) overnight at 37 °C in the dark under continuous shaking. Tryspinization was completed by the addition of formic acid (Thermo Fisher Scientific) to 5%.
Raw mass spectrometry interaction data were filtered through the CompPASS statistical software platform (38), in which a given interaction set was assigned a variety of confidence values based on a database from previous mass spectrometry experiments. When indicated, cleared lysates were incubated with okadaic acid or DMSO as described above, before FLAG resin pulldown.
APC/C Purification and In Vitro Assays.
Human APC/C was purified from extracts of synchronized HeLa S3 cells using protein G agarose resin (Roche) and αCdc27 monoclonal antibodies in the presence of 500 mM NaCl and 0.1% Tween to remove endogenous Ube2S. For control reactions, extracts were incubated with protein G beads and mouse IgG (Santa Cruz Biotechnology). The beads were washed and divided between reactions; 1 mL of concentrated extract was used for 12 reactions.
For cross-linking experiments, immunopurified APC/C was incubated with 2 μM Ube2SC118A or Ube2SC95A/C118A and 10 μM BMB cross-linker (Pierce) for 30 min at 22 °C and 1,200 rpm. Bound proteins were eluted with SDS sample buffer, separated by SDS/PAGE, and analyzed by Western blot analysis.
For in vitro ubiquitination assays, immunopurified APC/C was incubated with 1 μM recombinant substrate, 1 μM ubiquitin E1, 1 μM Ube2S, 100 μM ubiquitin, 10 mM DTT, 3 mM ATP, and 22.5 mM creatine phosphate in 25 mM Tris⋅HCl (pH 7.5), 50 mM NaCl, and 10 mM MgCl2 at 30 °C for the indicated times. Samples were resolved by SDS/PAGE, and substrate ubiquitination was detected by Western blot analysis with substrate-specific antibodies.
For ubiquitin dimer assays monitoring Ube2S activity copurifying with APC/C, synchronized HeLa cells were treated with MG-132 when indicated for 2 h, followed by APC/C purification as described above, except that immunoprecipitations were done under less-stringent conditions (150 mM NaCl and 0.05% Tween) to preserve the interaction with endogenous Ube2S. Where indicated, okadaic acid was added to extracts before pull-down as described above.
Protein Purification.
All proteins except ubiquitin E1 were expressed and purified from BL21/DE3 (RIL) cells. Cells were grown in LB medium to an OD600 of 0.5, followed by induction of protein production overnight in 0.5 mM isopropyl β-D-1-thiogalactopyranoside (LabScientific). 6×HisE1 was expressed from pFASTBac in Sf9 cells and purified using NiNTA (Qiagen). Ube2S6×His, Ube2C6×His, 6×Hisubiquitin, and Ub-cyclin AHA-6×His were purified as described previously (13). Ube2S6×His constructs used in cross-linking (C118A and C95A/C118A) were purified as described previously, but were dialyzed into buffer containing 50 mM Tris (pH 7.2) and 100 mM NaCl (12). Ubiquitin was purchased from UBPBio and Boston Biochem. To purify PP2AB56 complexes, catalytic subunit alpha (PPP2CA), regulatory subunit PR65 beta (PPP2R1B), and FLAG-tagged regulatory subunit B56 alpha (PPP2R5A) were expressed in 293T cells, purified on FLAG-M2 affinity resin (Sigma-Aldrich), and eluted with FLAG peptide (500 μg/mL in PBS).
Acknowledgments
We thank Vishva Dixit and Marissa Matsumoto (Genentech) for the gift of αK11-antibodies, and Rebecca Heald for the gift of mCherry-histone H2B/GFP-tubulin HeLa cells. We also thank Julia Schaletzky, Rebecca Heald, Iain Cheeseman, Julie Welburn, and members of the M.R. laboratory for advice and stimulating discussions. This work was performed using the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory at University of California, Berkeley (supported by National Institutes of Health Grants S10RR029668, S10RR027303, and S10RR025622). A.C. was funded in part by a fellowship from the National Science Foundation. This work was funded by a grant from the National Institutes of Health (to M.R.). M.R. is the K. Peter Hirth Chair of Cancer Biology at University of California, Berkeley. H.Y. and M.R. are investigators with the Howard Hughes Medical Institute.
Footnotes
Conflict of interest statement: M.R. is a cofounder of and consultant to Nurix, a company working in the ubiquitin space. This work does not overlap with any work performed at Nurix.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1522423113/-/DCSupplemental.
References
- 1.Sivakumar S, Gorbsky GJ. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat Rev Mol Cell Biol. 2015;16(2):82–94. doi: 10.1038/nrm3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang L, Barford D. Insights into the anaphase-promoting complex: A molecular machine that regulates mitosis. Curr Opin Struct Biol. 2014;29:1–9. doi: 10.1016/j.sbi.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Jia L, Kim S, Yu H. Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem Sci. 2013;38(6):302–311. doi: 10.1016/j.tibs.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Primorac I, Musacchio A. Panta rhei: The APC/C at steady state. J Cell Biol. 2013;201(2):177–189. doi: 10.1083/jcb.201301130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters JM. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7(9):644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- 6.Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015;16(8):473–485. doi: 10.1038/nrm4025. [DOI] [PubMed] [Google Scholar]
- 7.Yu H, King RW, Peters JM, Kirschner MW. Identification of a novel ubiquitin-conjugating enzyme involved in mitotic cyclin degradation. Curr Biol. 1996;6(4):455–466. doi: 10.1016/s0960-9822(02)00513-4. [DOI] [PubMed] [Google Scholar]
- 8.Williamson A, et al. Regulation of ubiquitin chain initiation to control the timing of substrate degradation. Mol Cell. 2011;42(6):744–757. doi: 10.1016/j.molcel.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirkpatrick DS, et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8(7):700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y, Wang W, Kirschner MW. Specificity of the anaphase-promoting complex: A single-molecule study. Science. 2015;348(6231):1248737. doi: 10.1126/science.1248737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garnett MJ, et al. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol. 2009;11(11):1363–1369. doi: 10.1038/ncb1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly A, Wickliffe KE, Song L, Fedrigo I, Rape M. Ubiquitin chain elongation requires E3-dependent tracking of the emerging conjugate. Mol Cell. 2014;56(2):232–245. doi: 10.1016/j.molcel.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Meyer HJ, Rape M. Enhanced protein degradation by branched ubiquitin chains. Cell. 2014;157(4):910–921. doi: 10.1016/j.cell.2014.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell. 2011;144(5):769–781. doi: 10.1016/j.cell.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williamson A, et al. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci USA. 2009;106(43):18213–18218. doi: 10.1073/pnas.0907887106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu T, et al. UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc Natl Acad Sci USA. 2010;107(4):1355–1360. doi: 10.1073/pnas.0912802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto ML, et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell. 2010;39(3):477–484. doi: 10.1016/j.molcel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 18.Min M, Mevissen TE, De Luca M, Komander D, Lindon C. Efficient APC/C substrate degradation in cells undergoing mitotic exit depends on K11 ubiquitin linkages. Mol Biol Cell. 2015;26(24):4325–4332. doi: 10.1091/mbc.E15-02-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133(4):653–665. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung CR, et al. E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat Med. 2006;12(7):809–816. doi: 10.1038/nm1440. [DOI] [PubMed] [Google Scholar]
- 21.London N, Biggins S. Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol. 2014;15(11):736–747. doi: 10.1038/nrm3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S, Yu H. Mutual regulation between the spindle checkpoint and APC/C. Semin Cell Dev Biol. 2011;22(6):551–558. doi: 10.1016/j.semcdb.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chao WC, Kulkarni K, Zhang Z, Kong EH, Barford D. Structure of the mitotic checkpoint complex. Nature. 2012;484(7393):208–213. doi: 10.1038/nature10896. [DOI] [PubMed] [Google Scholar]
- 24.Herzog F, et al. Structure of the anaphase-promoting complex/cyclosome interacting with a mitotic checkpoint complex. Science. 2009;323(5920):1477–1481. doi: 10.1126/science.1163300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian W, et al. Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc Natl Acad Sci USA. 2012;109(45):18419–18424. doi: 10.1073/pnas.1213438109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10(11):755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown NG, et al. RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase-promoting complex. Proc Natl Acad Sci USA. 2015;112(17):5272–5279. doi: 10.1073/pnas.1504161112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott DC, et al. Structure of a RING E3 trapped in action reveals a ligation mechanism for the ubiquitin-like protein NEDD8. Cell. 2014;157(7):1671–1684. doi: 10.1016/j.cell.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plechanovová A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489(7414):115–120. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol. 2012;19(9):876–883. doi: 10.1038/nsmb.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eletr ZM, Huang DT, Duda DM, Schulman BA, Kuhlman B. E2 conjugating enzymes must disengage from their E1 enzymes before E3-dependent ubiquitin and ubiquitin-like transfer. Nat Struct Mol Biol. 2005;12(10):933–934. doi: 10.1038/nsmb984. [DOI] [PubMed] [Google Scholar]
- 32.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 33.Cocklin R, Goebl M. Nutrient-sensing kinases PKA and Sch9 phosphorylate the catalytic domain of the ubiquitin-conjugating enzyme Cdc34. PLoS One. 2011;6(11):e27099. doi: 10.1371/journal.pone.0027099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duda DM, et al. Structure of a glomulin-RBX1-CUL1 complex: Inhibition of a RING E3 ligase through masking of its E2-binding surface. Mol Cell. 2012;47(3):371–382. doi: 10.1016/j.molcel.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nijenhuis W, Vallardi G, Teixeira A, Kops GJ, Saurin AT. Negative feedback at kinetochores underlies a responsive spindle checkpoint signal. Nat Cell Biol. 2014;16(12):1257–1264. doi: 10.1038/ncb3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suijkerbuijk SJ, Vleugel M, Teixeira A, Kops GJ. Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev Cell. 2012;23(4):745–755. doi: 10.1016/j.devcel.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM. Correcting improper chromosome-spindle attachments during cell division. Nat Cell Biol. 2004;6(3):232–237. doi: 10.1038/ncb1102. [DOI] [PubMed] [Google Scholar]
- 38.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138(2):389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang Z, Shu H, Oncel D, Chen S, Yu H. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell. 2004;16(3):387–397. doi: 10.1016/j.molcel.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 40.Hutchins JR, et al. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 2010;328(5978):593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Camasses A, Bogdanova A, Shevchenko A, Zachariae W. The CCT chaperonin promotes activation of the anaphase-promoting complex through the generation of functional Cdc20. Mol Cell. 2003;12(1):87–100. doi: 10.1016/s1097-2765(03)00244-2. [DOI] [PubMed] [Google Scholar]
- 42.Schmitz MH, et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol. 2010;12(9):886–893. doi: 10.1038/ncb2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown NG, et al. Mechanism of polyubiquitination by human anaphase-promoting complex: RING repurposing for ubiquitin chain assembly. Mol Cell. 2014;56(2):246–260. doi: 10.1016/j.molcel.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foley EA, Maldonado M, Kapoor TM. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol. 2011;13(10):1265–1271. doi: 10.1038/ncb2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stumpff J, Wagenbach M, Franck A, Asbury CL, Wordeman L. Kif18A and chromokinesins confine centromere movements via microtubule growth suppression and spatial control of kinetochore tension. Dev Cell. 2012;22(5):1017–1029. doi: 10.1016/j.devcel.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sivakumar S, Daum JR, Tipton AR, Rankin S, Gorbsky GJ. The spindle and kinetochore-associated (Ska) complex enhances binding of the anaphase-promoting complex/cyclosome (APC/C) to chromosomes and promotes mitotic exit. Mol Biol Cell. 2014;25(5):594–605. doi: 10.1091/mbc.E13-07-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia L, Li B, Yu H. The Bub1-Plk1 kinase complex promotes spindle checkpoint signaling through Cdc20 phosphorylation. Nat Commun. 2016 doi: 10.1038/ncomms10818. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, Peters JM. The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol Cell. 2005;18(5):543–553. doi: 10.1016/j.molcel.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 49.Buschhorn BA, et al. Substrate binding on the APC/C occurs between the coactivator Cdh1 and the processivity factor Doc1. Nat Struct Mol Biol. 2011;18(1):6–13. doi: 10.1038/nsmb.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D. Molecular architecture and mechanism of the anaphase-promoting complex. Nature. 2014;513(7518):388–393. doi: 10.1038/nature13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.da Fonseca PC, et al. Structures of APC/C(Cdh1) with substrates identify Cdh1 and Apc10 as the D-box co-receptor. Nature. 2011;470(7333):274–278. doi: 10.1038/nature09625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Fiore B, et al. The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev Cell. 2015;32(3):358–372. doi: 10.1016/j.devcel.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown N, et al. Mechanism of polyubiquitination by human anaphase-promoting complex: RING repurposing for ubiquitin chain assembly. Mol Cell. 2014;56(2):246–260. doi: 10.1016/j.molcel.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Werner A, et al. Cell fate determination by ubiquitin-dependent regulation of translation. Nature. 2015;525(7570):523–527. doi: 10.1038/nature14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song L, Craney A, Rape M. Microtubule-dependent regulation of mitotic protein degradation. Mol Cell. 2014;53(2):179–192. doi: 10.1016/j.molcel.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]