

Abstract
Small molecules are indispensable to modern medical therapy. However, their use may lead to unintended, negative medical outcomes commonly referred to as adverse drug reactions (ADRs). These effects vary widely in mechanism, severity, and populations affected, making ADR prediction and identification important public health concerns. Current methods rely on clinical trials and post-market surveillance programs to find novel ADRs; however, clinical trials are limited by small sample size, while post-market surveillance methods may be biased and inherently leave patients at risk until sufficient clinical evidence has been gathered. Systems pharmacology, an emerging interdisciplinary field combining network and chemical biology, provides important tools to uncover and understand ADRs and may mitigate the drawbacks of traditional methods. In particular, network analysis allows researchers to integrate heterogeneous data sources and quantify the interactions between biological and chemical entities. Recent work in this area has combined chemical, biological, and large-scale observational health data to predict ADRs in both individual patients and global populations. In this review, we explore the rapid expansion of systems pharmacology in the study of ADRs. We enumerate the existing methods and strategies and illustrate progress in the field with a model framework that incorporates crucial data elements, such as diet and comorbidities, known to modulate ADR risk. Using this framework, we highlight avenues of research that may currently be underexplored, representing opportunities for future work.
Keywords: Systems biology, Drug-Related Side Effects and Adverse Reactions, Precision Medicine, Pharmacogenetics, Transcriptional Networks
1. Introduction
Adverse drug reactions (ADRs) continue to be a major burden on healthcare systems across the world, accounting for millions of hospitalizations each year.1,2 Their severity may range from the relatively minor (e.g. itchiness) to the life-threatening (e.g. liver failure).3 Many ADRs occur from known and preventable causes, such as CYP-based interactions; however, less predictable mechanisms, such as genetic susceptibility, can also cause these harmful events. The scope of pre-clinical and clinical trials cannot account for every source of therapeutic variance, meaning that unexpected interactions, such as uncommon drug-drug interactions, may not be explored during this phase of drug development. Further, clinical trials are unable to identify rare side effects due to their small sample sizes.
In response, global drug regulating agencies (like the FDA and WHO) have relied on the submission and analysis of adverse event reports by doctors, pharmaceutical companies, and patients. These pharmacovigilance programs have helped identify many dangerous effects of drugs, such as Vioxx and Avandia. However, they have some important limitations. The most obvious is the delay between evidence collection and detection of an ADR, which puts those taking the drug immediately after release at risk of serious and unexpected harm. In addition, stimulated reporting (i.e., increased reporting rates for a drug receiving a lot of media coverage—these reports tend to include a lot of false positives) can cause what appears to be increased ADR risk; for example, dabigatran was heavily covered by the media during post-marketing surveillance, and the FDA Adverse Event Reporting System received numerous reports of bleeding.4
In addition to the dangers ADRs pose for patients, these events contribute to growing costs of drug development and decreasing numbers of drug approvals.5 Up to 30% of experimental drug failures can be attributed to safety concerns,6 and each such failure comes at significant cost to pharmaceutical companies. A priori prediction of ADRs (prediction of an ADR before it happens—this is typically done using knowledge regarding at-risk patient populations) may increase the efficiency of the drug development process. Computational methods that are grounded in biological mechanisms, such as those developed and used in systems biology, are a particularly promising tool for pre-clinical drug safety assessment.
Systems biology is the study of groups of interacting components, such as genes, proteins, or drugs. Often, these systems are represented in network form, facilitating topological analyses that can identify emergent relationships among these entities.7 These methods allow us to visualize larger contexts and complement experimental methods that often consider only very specific interactions. Systems biology has many subfields,8 including systems pharmacology – the application of systems biology methods to pharmacological inquiries, such as drug effects and interactions.9
Recent, extensive characterization of human protein-protein interactions 10 and large repositories of drug-target and drug-effect data11,12 have enabled the development of systems pharmacology analyses to predict and understand ADRs.13 In this review, we will first provide a brief overview covering the traditional clinical methods for detecting ADRs. We will then delve into systems pharmacology approaches for predicting ADRs and elucidating their mechanisms in both the general population and individuals. Finally, we will describe new integrative approaches that combine clinical, biological, and chemical data to better predict ADRs. We conclude with suggestions for future inquiry.
2. Background
Detecting ADRs during post-market surveillance has relied on disproportionality analysis of adverse event reports through pharmacovigilance. These methods quantify the degree to which a drug-event combination co-occurs disproportionately compared to the occurrence of the event for other drugs. Methods are based on both frequentist and Bayesian statistics. Frequentist approaches estimate associations and implement statistical tests, whereas Bayesian approaches deal with the uncertainty of the disproportionality measure associated with small observations and counts by comparing to the “no-association” baseline case. In order to analyze ADRs in realistic scenarios that include comorbidities and polypharmacology, statistical extensions of disproportionality analysis can be used.
Emerging trends in the domain of ADR analytics employ new information sources to facilitate ADR detection. These include using biomedical literature as a complementary strategy to prioritize ADR associations.14 User-generated health web forums are also becoming popular information sources for such analyses.15
Methods that employ item-set, or association rule, mining have also been used for temporal data analytics in biomedical data.16-18 In these methods, frequent temporal patterns (also known as temporal association rules19) are discovered and extended for more expressive association rules mining.20-22
Many data sources exist for detecting ADRs, including both publicly available datasets and proprietary datasets (Table 1). Other important data sources exist for detecting ADRs, including survey3 and retrospective analysis methods. Retrospective analyses use data sources such as electronic health records (EHRs),23-25 federal repositories,4,26,27 clinical trial data,28 and clinical narratives.29-31 Novel resources, such as genome wide-association studies (GWAS), are a budding resource for the study of pharmacogenetics and pharmacovigilance. A search in the Database of Genotypes and Phenotypes (dbGAP) for “adverse drug reaction” reveals 29 available studies. For more detail on clinical methods in detecting ADRs, we direct the reader to the review by Harpaz et al.32; for additional information regarding use of GWAS studies in ADR prediction please see a review by Motsinger-Reif.33 A summary of relevant data sources, with source reference and appropriate links, is provided in Table 1.
Table 1. Useful Data Sources for Detecting ADRs and Mechanisms Behind ADRs.
| Resource | Description | Resource URL |
|---|---|---|
| Drug-Target DB: | ||
| DrugBank | Knowledgebase for drugs, drug actions and drug targets | http://www.drugbank.ca |
| STITCH | Database of chemical-protein interactions containing information on chemical association networks | http://stitch.embl.de |
| Chembl | Resource containing drug targets, and compounds. Contains information on compound activity and potential therapeutic uses | https://www.ebi.ac.uk/chembl |
| Matador | Manually Annotated Targets and Drugs Online Resource | http://matador.embl.de |
| PDSP | Psychoactive Drug Screening Program | http://pdspdb.unc.edu |
|
| ||
| Drug-ADR DB: | ||
| Offsides | Database of significant drug-effect associations | http://tatonettilab.org/resources/tatonetti-stm.htm |
| SIDER | Drug side effect reference with ADR information extracted from public information and product labels | http://sideeffects.embl.de |
| CTD | Comparative Toxicogenomics Database | http://ctdbase.org |
|
| ||
| ATC | Anatomical Therapeutic Chemical (ATC) Classification | http://www.whocc.no/atc_ddd_index |
|
| ||
| Biological Pathway DB: | ||
| KEGG | Database of pathways involved in various cellular functions. Each pathway is linked to the well-known genes involved in those pathways | http://www.kegg.jp |
| REACTOME | Open-source, curated, and peer-reviewed pathway database | http://www.reactome.org |
| PharmGKB | Pharmacogenomics knowledgebase containing information on drug mechanism pathways | https://www.pharmgkb.org |
|
| ||
| Chemical Structure DB: | ||
| PubChem Compound | Repository of validated chemical information | https://pubchem.ncbi.nlm.nih.gov |
| ZINC | Database of commercially-available compounds for virtual screening | http://zinc.docking.org |
|
| ||
| Connectivity Map | Collection of transcriptional expression data from drug-treated cultured human cells | https://www.broadinstitute.org/cmap |
|
| ||
| Drug-Drug Interaction (DDI) DB: | ||
| Twosides | Database of significant drug-drug-effect associations | http://tatonettilab.org/resources/tatonetti-stm.htm |
| INDI | INferring Drug Interactions | http://www.cs.tau.ac.il/∼bnet/software/INDI |
| Drugs.com | Database of drug-drug interactions | http://www.drugs.com/drug_interactions.php |
|
| ||
| Gene Function DB: | ||
| GO | Gene Ontology | http://geneontology.org |
| GeneCards | Comprehensive database on genes and gene function | http://www.genecards.org |
|
| ||
| OMIM | Reference for Human Genes and Genetic Disorders (Mendelian and Non-Mendelian Diseases) | http://www.omim.org |
|
| ||
| Protein Structure/Sequence: | ||
| PDB | Protein Data Bank | http://www.rcsb.org/pdb/home/home.do |
| Uniprot | UniProtKB/Swiss-Prot | http://www.uniprot.org |
|
| ||
| Protein-ADR DB: | ||
| DART | Developmental and Reproductive Toxicology | http://toxnet.nlm.nih.gov/newtoxnet/dart.htm |
| DITOP | Drug-Induced TOxicology related Proteins | bioinf.xmu.edu.cn/databases/DITOP |
|
| ||
| Protein-gene Interaction DBs: | ||
| TRED | Transcriptional Regulatory Element Database | https://cb.utdallas.edu/cgi-bin/TRED/tred.cgi?process=home |
| ChEA | Chip Enrichment Analysis Database | http://amp.pharm.mssm.edu/lib/chea.jsp |
|
| ||
| Protein-Protein Interaction DBs: | ||
| BioGRID | Biological General Repository for Interaction Datasets | http://thebiogrid.org/ |
| DIP | Database of Interacting Proteins. | http://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| HPRD | Human Protein Reference Standard (HPRD) | |
| IntAct | Molecular interaction database with contributions from MINT, InnateDB, MatrixDB, and others | http://www.ebi.ac.uk/intact/ |
| STRING | Functional protein-association networks. Contains information from literature, genomic context, high-throughput experiments, and co-expression data | http://string-db.org |
|
| ||
| Post-market reporting systems (PMRS): | ||
| FAERS | Registry of adverse event and medication error reports sent to the FDA and CDC | http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ |
| VAERS | Registry of vaccine-related adverse events sent to the FDA and CDC | https://vaers.hhs.gov/index |
| MedEffect Canada | Registry of adverse event and medication error reports from Canada | http://www.hc-sc.gc.ca/dhp-mps/medeff/index-eng.php |
|
| ||
| Disease terminologies: | ||
| ICD | International Classification for Diseases, a hierarchical disease classification for billing purposes | http://www.who.int/classifications/icd/en/ |
| MedDRA | Specialized medical terminology designed for the pharmaceutical industry | http://www.meddra.org |
| UMLS | Unified Medical Language System, an integrated terminology system | http://www.nlm.nih.gov/research/umls/ |
| MeSH | Medical Subject Headings vocabulary | http://www.nlm.nih.gov/mesh/ |
| SNOMED | Systemized Nomeclature for Medicine, an ontology of medical terms | http://ihtsdo.org/snomed-ct/ |
|
| ||
| Clinical Trials | Registry and Results Repository for Clinical Trials | ClinicalTrials.gov |
|
| ||
| Electronic Health Records (EHRs) | ||
| Billing Data | Medical information collected at the point-of-care by healthcare providers. Data collected primary for insurance and billing purposes. Data can be reused for pharmacovigiliance. | - |
| Clinical Narratives | Clinical notes taken at the point-of-care by clinicians and healthcare providers. Data can be reused for pharmacovigiliance. | - |
|
| ||
| GWAS | Genome-wide association studies typically conducted on individuals who have had a serious ADR, or who have serious ADRs within their family. | Database of Genotypes and Phenotypes (dbGAP): http://www.ncbi.nlm.nih.gov/gap |
|
| ||
| Surveys | Questionnaires sent to providers regarding medications given that results in ADRs, and other comorbidities that patients were on. | National Electronic Injury Surveillance System-Cooperative Adverse Drug Event Surveillance Project: http://www.healthindicators.gov/Resources/DataSources/NEISS-CADES_86/Profile |
|
| ||
| Metabolic Data | Metabolic studies that find specific metabolic profiles associated with serious ADRs, and ADR outcomes | The Human Metabolome Database (HMDB): http://www.hmdb.ca |
3. Methods for Predicting Population-Level ADRs
Each year, approximately 20 new drugs are released into the market in the United States (fda.gov); each will have both anticipated and unexpected side effects. In addition, more patients are simultaneously taking multiple drugs (polypharmacotherapy) as a consequence of increased life expectancy.34 During polypharmacotherapy, drug-drug interactions (DDIs) can result from the interplay of drug mechanisms or metabolism, leading to ADRs that do not occur when each drug is taken individually.35 These factors underscore the importance of developing accurate methods to identify ADRs, both for individual drugs and DDIs. We begin by addressing methods that have been used to predict drug targets, as any protein-protein interaction network-based method for predicting drug safety must rely on knowing all of the expected and unexpected biological processes a drug is perturbing. We then proceed to describe approaches that utilize drug target data as well as other data sources to predict drug safety.
3.1 Predicting Drug Targets Using Systems Pharmacology
The enumeration of all protein targets of a drug is an important step in predicting ADEs, including both those the drug was designed to hit (“on-targets”) and unintended interactions (“off-targets”). These data facilitate systems-level analyses of a drug's cellular effects and toxicities. In addition to the growth of curated databases of drug-target interactions,11 systems pharmacology and chemical informatics approaches have enabled the large-scale prediction of drug off-targets.
Some methods of target prediction involve similarity analyses. For example, Campillos et al.36 predicted new drug targets using side effect similarity. Drugs with similar side effects were predicted to share targets, and their final model combined both side effect and chemical structural similarity. However, their approach requires the side effects of a new drug to be known, limiting the avenues of inquiry for drugs in the clinical pipeline.
Other methods of target prediction utilize the chemical similarity of the drugs themselves. Keiser et al.37,38 developed a BLAST-derived, fingerprint-based algorithm called the similarity ensemble approach (SEA), wherein drugs with comparable chemical structures to a given protein's ligand set are also predicted to bind that protein. Although this method does not depend on protein structure for target prediction, it relies on a reference of experimentally derived drug-protein sets. SEA was used to successfully predict that paroxetine (Paxil) and fluoxetine (Prozac), both selective serotonin reuptake inhibitors, are also beta-blockers. The authors concluded that these results helped to explain the ADRs of patients taking either drug.38 However, while paroxetine was experimentally determined to bind β adrenergic receptors with a Ki of 1μM, the mean Cmax for the maximum dosage of paroxetine has been found to be 105ng/ml (280 nM) (https://www.gsk.com/media/389890/pharma_715.pdf). While this clinical perspective does not discount the possibility of drug-drug interactions or genetic predispositions affecting CYP2D6 activity and, therefore, the serum concentration of paroxetine, such examples highlight the need for not only experimental validation but also cross-referencing with clinical data to determine the translatability of these predictions.
In another method, Yamanishi et al. used a bipartite graph and supervised learning to predict new drug-protein pairs by combining chemical space (chemical structure similarity), genomic space (amino acid similarity), and pharmacological effect (keywords from package inserts).39 In another integrative approach, Zhao et al. combined drug therapeutic similarity (ATC classification), chemical similarity, and gene proximity in a human PPI network to predict new targets.40 Further work that combines chemical similarity with additional techniques can help to realize the potential of structure-free approaches to ADR identification.
3.2 Predicting Drug Safety Using Systems Pharmacology
Many systems biology methods use the human protein-protein interaction (PPI) network as a basis for models. For example, Jiang et al.41 used a network biology approach to identify proteins involved in specific ADRs based on the topological properties of the PPI. Among several properties, every protein was described with its average shortest path length to known ADR-related proteins. This method highlights the robustness and ‘customizability’ of network-based methods, allowing authors to define novel topological measurements in order to create query-specific models. The authors of this paper found that they were able to identify ADR-related proteins (ADRPs) with a relatively high degree of sensitivity and specificity, and observed that ADRPs tended to be much closer to each other on average than non-ADRPs are to such drugs. However, this may simply be a reflection of the higher centrality of ADRPs; that is, more central genes tend to be closer to the rest of the network, and central proteins are more likely to cause ADRs because the effects of drug-induced inhibition or activation propagate more easily through the network.
In another PPI-based study, Huang et al. combined human PPI network expansion with drug target data and Gene Ontology (GO) terms to generate SVM and logistic regression models for predicting cardiotoxic adverse drug effects.42 The authors systematically annotated drug targets with GO terms at varying hierarchical levels and were able to demonstrate that these annotations can improve model performance.
Kuhn et al.43 approached the identification of ADRs for particular drugs by using a network based on non-experimental data. The authors created networks with three types of nodes: drugs, targets, and side effects, and identified side effect causality predictors. The authors considered overrepresented protein-side effect pairs, and hypothesized that such overrepresentation could be indicative of causality. Of the 116 high-confidence predictions, 72 were supported by the literature. The authors also used a mouse model to perform in vivo validation of the predicted link between serotonin receptor 1 family protein activation and increased pain sensitivity, which is a side effect of triptans. While this method could be very useful in providing expected ADRs for well-studied drugs, the authors note that statistical power is limited when considering proteins that are off-targets for a small number of drugs, and also for very common side effects (such as headaches).
Huang et al.44 also used PPIs to identify pharmacodynamic (PD) DDIs by first mapping drugs to PPI nodes. Unlike Jiang et al, the authors of this paper integrated expression data into their model by weighting PPI network edges with the encoding genes' Pearson correlation coefficient of coexpression across 79 tissues. They integrated data from SIDER12 in order to cross-validate their method, then used a Bayesian probabilistic model to validate, explain, and compare predictions.
Guimera and Sales-Pardo45 used a similar network approach to identify novel DDIs. They constructed network models such that nodes represent drugs, while edges represent interactions (antagonistic, additive, and synergistic). Unknown drug interactions were predicted from these data using stochastic block models. At first glance, such analyses seem superficial due to the lack of experimental or detailed biological data; however, they are often accurate and manage to grasp global patterns that would not necessarily be visible when considering the mechanistic details of each drug pair.
Networks can also be represented as adjacency matrices. Taking this into consideration, Cobanoglu et al.46 predicted drug-target interactions using a filtering algorithm called probabilistic matrix factorization. The authors first modeled drug-target interactions from DrugBank as a bipartite network, defined as having two independent sets of nodes (in this case drugs and targets) where edges can only be drawn between drugs and targets. The authors then used these known interactions to train a model that represents each drug and target as a vector of latent variables and used these attributes to assign probabilities to missing edges in the network. Edges receiving high probabilities represent new interactions between drugs and targets. In this case, the identification of novel targets can lead to potential insights on previously unidentified drug side effects for that particular drug.
A number of systems pharmacology approaches have utilized chemical similarity to predict ADRs. Lounkine et al. used SEA to screen a panel of 656 approved drugs for binding to 73 known side effect targets.47 The predicted results were then compared to experimental activity assays. While close to half of the predictions were disproved, the study still highlighted the high degree of drug target promiscuity. Other chemical fingerprinting-based approaches extracted chemical features with high correlation to each ADR and compared ADR-ADR pairs using these features to compute ADR similarity metrics.48,49
Other studies have combined chemical similarity with additional analyses. For example, Atias et al. combined chemical structure similarity with drug-ADR data from SIDER and side effect similarity data to perform i) a canonical correlation analysis that maximizes the correlation between drug characteristics (e.g. chemical structure) and side effects, and ii) network-based diffusion to prioritize side effects using a side effect similarity network.50 Liu et al. combined chemical (substructures and fingerprints), biological (protein targets and pathway), and phenotypic (indications and other known ADRs) properties of drugs to predict ADRs.51 A follow-up study to Huang et al.42 combined drug structures with PPI networks in their predictive models.52
Computationally intensive and large-scale studies can also fall into systems pharmacology. LaBute et al. predicted off-target drug effects using protein-docking simulation. This study circumnavigates the inherent bias in systems biology approaches that integrate experimental data, which is limited by funding and research interest. LaBute et al.53 hypothesized that it is most valuable to predict ADRs during lead identification in drug development. The authors trained in silico docking models on compounds and their associated ADRs (such as those found in SIDER) to demonstrate a pipeline for automated evaluation of drug safety.
Nonetheless, docking approaches remain limited by the availability of protein structures. Furthermore, even with available structures the binding affinities predicted by docking approaches can diverge wildly from experimental results.54 Many of these issues stem from the use of rigid structures and therefore being unable to sample the range of conformations a protein and ligand can occupy.55 New chemoinformatics approaches such as molecular dynamics simulations (MDS) have been used to circumvent these problems by simulating atomic motions on micro- to millisecond timescales.56 While MDS has yielded improvements in prediction accuracy for both correct ligand-protein poses and binding affinity,56-58 these simulations have high CPU demands and can therefore only be run on multi-core clusters or with cloud computing.56 The ability to screen large libraries of ligands against large libraries of potential targets with both accuracy and efficiency thus remains a present impossibility.
3.3 Understanding Mechanisms of ADRs Using Systems Pharmacology
Efforts to characterize mechanisms of drug side effects rely on databases of side effects (SIDER) and biological pathways (e.g. KEGG). To date, they have additionally integrated chemical structure,59 drug-target interactions,43,60,61 drug-induced differential gene expression,62,63 or a combination thereof.64-68
Scheiber et al. predicted biological pathways related to ADRs using chemical structure data to develop an in silico workflow.59 Beginning with known drug-ADR pairs, the authors then used chemical fingerprints to train Bayesian models predict protein targets for each drug. After mapping these predicted targets to biological pathways, they ranked the pathways to prioritize those containing predicted drug targets for a given ADR's drugs while simultaneously de-prioritizing pathways that were also targeted by drugs not causing the ADR. Using rhabdomyolysis and hypotension as examples, the authors found that top-scoring pathways implicated by their method are supported in the literature.
In contrast, some papers use protein structures to predict ADR mechanisms. Xie et al.'s pioneering work identified mechanistic explanations for the hypertensive side effects of the cholesteryl ester transfer protein (CETP) inhibitor torcetrapib.61 To do so, the authors first identified off-targets of CETP inhibitors by searching for protein structures or homology models with similar ligand binding sites to the primary target. Putative off-targets were further evaluated using protein-ligand docking. The authors then mapped the off-targets to a mechanistic network incorporating metabolic, signal transduction, and gene regulation pathways and found that these predicted protein-ligand networks could differentiate between CETP inhibitors that caused hypertension and those that did not. The study elegantly demonstrated how each inhibitor's safety could not be derived from the predicted strength of binding to different regulators of the renin-angiotensin-aldosterone system implicated in ADR-associated hypertension.
In a similar vein, Wallach et al. associated ADRs with biological pathways by applying multiple stages of logistic regression to drug-protein docking profiles mapped to side effect data from SIDER and pathway data from KEGG.60 In doing so, the authors identified almost 200 ADR-pathway associations, of which 22 were supported by literature review. The model offers advantages over those used by Xie et al.61 in that it can link cases where two drugs may target different proteins but affect the same biological pathway. However, the authors note several examples were the associations are not causative, and the method is still limited to drug targets with known structure and the accuracy of virtual docking algorithms.
In a drug-target study relying solely on known interactions, Mizutani et al. used sparse canonical correlation analysis to correlate proteins with ADRs based on the co-occurrence of drugs in drug-target and drug-ADR profiles.69 They then searched for KEGG pathways enriched for these predicted ADR proteins.
Other approaches rely on drug-induced changes in gene expression to identify mechanisms of ADRs. Lee et al. systematically evaluated relationships between biological processes (Gene Ontology (GO) terms) and side effects (SIDER) by using the Connectivity Map to generate a multi-level process-drug-side effect network combining 2209 biological processes, 74 drugs, and 168 side effects.63 Another approach mapped drug-induced microarray changes to “principal response networks” by first parsing KEGG pathways into unique “sub-pathways” in an attempt to better account for the high degree of redundancy and cross-talk between biological pathways.62 While this subdivision allows for improved granularity in determining the mechanisms of drug action, both methods assume that differentially expressed genes belong to the same biological pathway, and do not consider compensatory pathways as an option. Further, these studies are limited to known and curated pathways through the use of biological databases.
To address some of these limitations, Silberberg et al. combined protein-protein and protein-DNA interactions in the context of drug-target data and drug-induced gene expression changes to identify drug-specific subnetworks connecting drug targets to differentially expressed genes. The authors used over-represented short paths within these subnetworks to construct a panel of pathways that were then checked for overlap with the drug-specific subnetworks.67 They found that close to 90% of these inferred pathways did not overlap significantly with KEGG pathways. Furthermore, the inferred pathways achieved better performance in predicting side effects than KEGG pathways. In related work, Gottlieb et al. generated drug-specific pathways by linking drug targets, disease genes, and pharmacogenes (genes modulating drug response) within a pathway-annotated human PPI network to predict drug mechanisms of action and adverse effects.65 Such approaches demonstrate that curated biological process databases represent a good starting point but should not be considered definitive sources for pathway elucidation.
In an effort to bridge chemical and biological approaches to evaluate drug side effects, Duran-Frigola et al. performed a top-down enrichment analysis for each ADR in SIDER to identify over-represented chemical and biological features (e.g. chemical fragments, therapeutic targets, and pathways).64 These enriched molecular features were then used to build simple decision tree classifiers. While the approach is innovative, the authors note that they could only achieve their performance cut-off for 6% of the ADRs investigated. While it is clear that larger data sets will improve such analyses, future work must achieve a more challenging compromise must be made between model simplicity, interpretability, and performance.
4. Systems Biology of ADRs and Precision Medicine
Many ADRs are extremely rare, occurring infrequently among the general population. However, some ADRs are familial suggesting a genetic component that can increase ADR risk.70,71 In response, researchers and physicians must decide whether a particular treatment will help or harm a given patient and this decision is often mired with difficulties. The field of precision medicine is focused on understanding the differing reactions of patients to the same treatment regimen and aims to provide physicians with optimal therapy options for each patient. ADRs that seem to occur in certain individuals or families are termed idiosyncratic ADRs. Because precision medicine is focused on the individual, we focus here on idiosyncratic ADRs.
4.1 Identifying and Explaining Idiosyncratic ADRs
Idiosyncratic ADRs are patient-specific reactions that occur without a known biological mechanism and exhibit dose-dependency among those who experience it.72 These reactions place a significant burden on public health, as they represent approximately 20% of all ADRs.72 Because their underlying mechanism is unknown, idiosyncratic ADRs are challenging to predict a priori. Understanding the true mechanistic etiologies of idiosyncratic drug reactions would enable personalized ADR risk assessment.
4.1.1 Genetic ADR Susceptibility
Related individuals can be at an increased risk for developing certain ADRs, which was found via familial studies.71,72 These initial results prompted additional research into the relationship between ethnicity and ADR risk. For example, hypersensitivity to anticonvulsants, specifically phenytoin and carbamazepine, was originally thought to follow a recessive, autosomal pattern of inheritance.70,71 However, additional investigation revealed that population stratification was a major issue. For example, the allele HLA-B*1502 is associated with carbamazepine-induced ADRs among both Thai73 and Han Chinese74 populations, while HLA-A*3101 occurs in Japanese populations75 and no HLA-B alleles are associated in European populations.76 Stratification by ethnicity has been a crucial tool for understanding the genetic underpinnings of ADR susceptibility.77
Several systems biology methods have been developed to uncover genes associated with ADR susceptibility. Berger et al.78 investigated long-QT syndrome (LQTS), which can be drug-induced. They used GWAS-identified seed proteins associated with congenital LQTS risk to identify a LQTS neighborhood in the human PPI network. The authors found that drugs connected with identified LQTS disease genes in their network also resulted in QT prolongation being reported in the FDA Adverse Event Reporting System (FAERS).78 Their results demonstrate that network-based approaches can be used to find additional genes and SNPs related to an ADR. Consequently, mutations in those genes may increase the likelihood of an ADR in some individuals.
4.1.2 Lifestyle-Induced ADR Susceptibility
Lifestyle factors, such as diet, can also play a critical role in ADR susceptibility by affecting gut microflora, which alter metabolite absorption rates and vary widely based on diet.79 Lifestyle-induced ADR susceptibility can be investigated using metabolomics, the systematic study of all physiological metabolites.8 For example, Winnike et al. showed that urine metabolite profiles taken after the start of therapy can predict Drug-Induced Liver Injury (DILI) before the onset of clinical signs.80 This enables early intervention and can improve patient outcomes. Similarly, Cunningham et al. found a metabolite signature response to isoniazid in urine that could be used to determine an individual's risk for certain ADRs.81 However, a drug's effect on urine metabolites can be mitigated or exacerbated by other factors, such as diet, culture and ethnicity.82 In summary, understanding a patient's precise microbiome, metabolome, and diet can help researchers understand the underlying cause of an ADR in a population subset.
Understanding the effect of lifestyle on ADR susceptibility can help elucidate the underlying mechanisms behind individual responses to drug therapy.83 Systems biology approaches are ideal because they can be used to integrate and analyze data to provide a global picture of the underlying biological processes.84 Recently, techniques have been developed to understand the gut microbiome, and its relationship with drug metabolism.85 Others investigated lifestyle and environment factors termed “environmental exposures,” these exposures were then mapped to biological networks containing genes and gene pathways.86 Therefore, it becomes possible to link environmental exposures to specific genes and pathways they modulate.87 These ‘systems exposure event networks’ have implications for uncovering personalized drug-ADR effects.86 Kasarskis et al. propose an integrative network approach that combines genetic, clinical, and environmental data to help predict drug outcomes and ADRs.88 Such approaches enable clinicians to determine if a particular drug would be efficacious for an individual patient given that individual patient's diet containing reduced sodium and their exercise routine of bicycle riding. Integrative approaches for understanding ADRs should also take into account pertinent environmental exposures that can modulate ADR risk.
4.1.3 Comorbidity-Induced ADR Susceptibility
Increased ADR risk is associated with certain disease comorbidities, such as congestive cardiac failure, peripheral vascular disease, and diabetes.89 Likewise, certain comorbidities are associated with decreased ADR risk, including cerebrovascular disease, dementia, and paraplegia.89 Pre-systems biology methods of identifying such ADRs involved retrospective analysis of health data.89,90 Onder et al. had physicians who suspected an ADR fill out a questionnaire detailing the drugs hypothesized to be causative.90 They found that each point increase in a patient's Charlson Comorbidity Index, a common tool used to assess patient comorbidity profiles, was associated with increased risk for a serious ADR. The authors also found that the number of prescribed drugs is positively correlated with an increased risk for serious ADRs.
The correlation between number of disease comorbidities and ADR risk may be the result of adverse polypharmacology, where a drug binds to an off-target in the target tissue, or to the therapeutic target in a non-target tissue.91 For example, Human Epidermal Growth Factor Receptor 2 (HER2) inhibitors used in the treatment of breast cancer, which may cause cardiac toxicity in off-target cardiac tissue.91-93 These types of ADRs are more likely to occur if a patient is on multiple prescription drugs, which may explain the epidemiological findings.90
Systems biology methods have been applied to understand the etiologies of adverse polypharmacology effects.91 Specifically, systems biology can be used to probe the effects of a drug and its interaction with a non-target regulatory network, or signal propagation within the regulatory network, that leads to the ADR.91 Signaling networks are vital in understanding the complex interplay among diseases, their various comorbidities and the therapeutic effect of the drug target of interest.91
Another important facet of comorbidity-induced ADR susceptibility involves DDIs resulting from the treatment of comorbidities. Currently, data-driven approaches predict ADRs from drugs administered alone or in combination.94 These statistical approaches generally do not incorporate systems biology techniques to account for polypharmacological effects95 and the biological aspects of the drug targets.96 Systems biology methods to reveal and understand the mechanism responsible for DDIs and their resulting ADRs would significantly benefit this field of inquiry.
4.2 Systems Biology Approaches and Personalized ADR Prediction
Many factors can affect an individual's response to a given drug therapy: genetics, lifestyle (e.g. diet), and comorbidities. Each of these factors may mask, exacerbate, or otherwise change an individual's personal ADR risk. Several of these factors are shown in Figure 1. For example, genetic/microbiomic factors can disrupt the absorption of a drug (Figure 1A), while lifestyle choices such as dietary factors can disrupt the absorption of certain drugs (Figure 1B). In some cases, the primary disease may cause a decrease in a key metabolite, and a drug may be administered to increase this metabolite in the bloodstream. However, if a given patient has a comorbidity that also increases this metabolite, the administration of the drug could exacerbate the comorbid condition, depending on the drug's specific mechanism of action (Figure 1C). The ADR in this particular case would be the result of an exacerbation of the patient's comorbid condition. Personalized approaches are required that take into account the entire patient state (i.e. diet, lifestyle, comorbidities, disease). Systems biology methods have been developed to probe the interaction of ADR risk and genetic/ethnicity, lifestyle, and comorbidity factors. However, each of these methods focuses principally on one area. Currently, there is a paucity of research that integrates all of these methods and directly ties the result to personalized medical outcomes. More research is needed to harness these data in interesting, novel ways to realize the potential of personalized medicine.97
Figure 1. Adverse Drug Reactions (ADRs) Can Occur Among Certain Individuals Due to Diverse Disruptions in the Drug's Mechanism of Action.
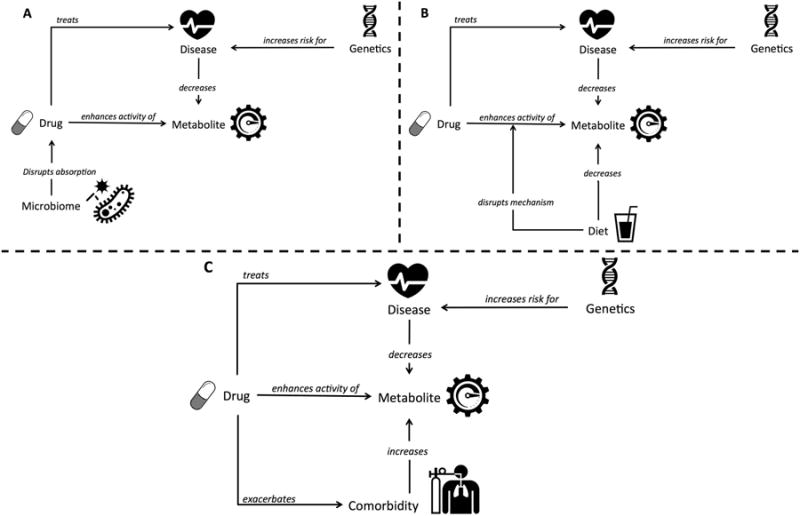
Some examples of these disruptions include, genetic/microbiome related (Figure 1A), dietary or lifestyle dependent (Figure 1B) or driven by a patient's comorbidities (Figure 1C).
5. Systems Pharmacology of Biologically Sourced Compounds
In addition to commercially available pharmaceuticals, there are many naturally occurring plant- and animal-produced compounds that were recently rediscovered for their clinical validity in treating disease.98 Typically, these take three forms: small-molecule metabolites,99 peptides, and immunologic components. Together, these pharmaceutically active biomolecules are called ‘biopharmaceuticals’ (colloquially, ‘biologics’). Peptides and antibodies often have an incredibly high specificity for a distinct molecular target,100 making them ideal candidates for therapeutic use. Large libraries of potentially useful biologically active compounds are actively being developed.101,102
Despite their potential, there are substantial barriers that prevent the discovery and widespread adoption of clinically useful biologics. The staggering diversity and structural complexity of biological molecules presents a nearly intractable problem with regards to their identification, classification, and isolation from a living organism.103 Auxiliary compounds that coexist with the compound of interest may themselves be biologically active with the ability to interact with other drugs in unexpected ways. For example, St. John's-wort, a common herbal supplement,104 often interacts with traditional synthetic drugs (e.g. cyclosporine) and results in ADRs (e.g., transplant rejection).104,105 Some of these effects may be due to synergistic perturbations to the underlying biological system. Modern computational techniques are enabling the automated identification and classification of novel biopharmaceuticals,106 but the complexity of their behavior in a diverse biological system still poses a daunting challenge.
6. Integrative approaches to identifying ADRs (Basic Biology and Clinical Medicine)
Most efforts to predict ADRs have relied either on clinical data mining or systems pharmacology analyses. However, several groups have begun to combine these two data types with the promise of generating models that account for lingering biases and limitations and are better able to identify and understand drug-ADR pairs. In contrast to using clinical data as a sole validation of the systems pharmacology approach, these studies incorporate clinical data into the model itself.78 We illustrate these integrative approaches in Figure 2, where edge thickness between pairs of data sources (nodes) corresponds to the number of publications that have combined the two (Figure 2).
Figure 2. Bipartite graph of data sources used in the literature for systems biology of adverse drug reactions.
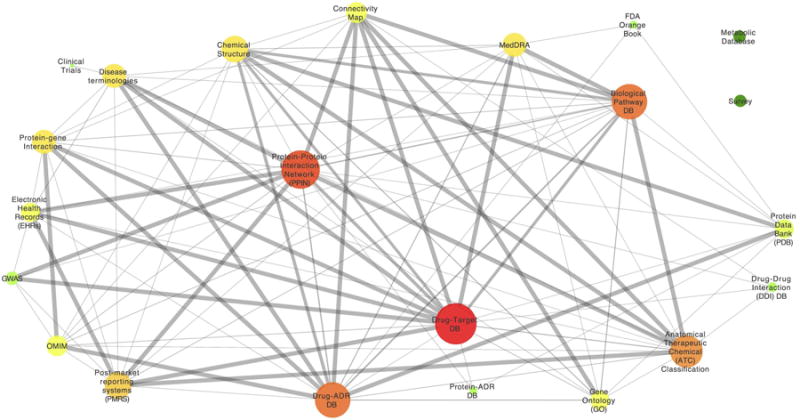
We surveyed data sources used in previous studies and whether or not those datasets were used in combination or not (singleton nodes in Figure 2). Edges in the graph represent datasets used in combination by the same publication. Edge-thickness indicates the number of publications using that particular dataset combination. Node size is based on the degree of the node, and color indicates the closeness centrality. Figure 2 illustrates that some datasets are used together often, while others are rarely used in combination. This helps indicate areas of opportunity for future systems biology researchers interested in using novel or under-utilized data sources.
Cami et al. generated predictive pharmacosafety networks that integrated clinical drug-ADR data (a 2005 snapshot of the Lexicomp database), taxonomic data (the ATC taxonomy of drugs and the MedDRA taxonomy of ADRs), and biological data (DrugBank and PubChem).107 The authors used these data to train a logistic regression classifier to predict new ADRs and performed a prospective method evaluation by comparing their predicted results to the 2010 version of the same drug safety database. While the method is highly novel and achieved high specificity (0.95), sensitivity was limited to 0.42, and the method did not account for the reporting biases present in drug safety databases. Nonetheless, the prospective method employed by the authors provides a valuable template for how to use clinical data both for model creation and realistic validation.
While the previous study relied on a proprietary drug-ADR database, other methods have been developed to utilize publically available data. In a study combining clinical data mining, network analysis, and experimental validation, Zhao et al. sought to identify drugs taken concurrently with rosiglitazone that could reduce the incidence of rosiglitazone-associated myocardial infarction (MI) in type II diabetic patients.108 To do this, the authors mined the FDA Adverse Event Reporting System (FAERS) for such safe combinations, finding that concurrent use of exenatide led to decreased occurrence of MI. The authors replicated these results using EHRs. Beginning with peroxisome proliferator–activated receptor γ (PPARγ), the target of rosiglitazone, the authors used GO term annotations and DrugBank targets to identify a subnetwork in the human interactome associated with rosiglitazone-induced MI and mechanistically investigate the positive effects of concurrent exenatide use. Their hypothesis was finally validated in a mouse model. While the method does not account for potential off-target effects, it highlights a powerful pipeline leveraging clinical data as well as both in silico and in vivo biology.
To address the inability of many clinical data mining approaches to eliminate false positives and false negatives, Lorberbaum et al. developed the Modular Assembly of Drug Safety Subnetworks (MADSS), a network analysis-based algorithm that identifies adverse event neighborhoods within the human interactome.109 Drugs targeting proteins within this neighborhood are predicted to be more likely to cause the ADR than drugs targeting proteins outside the neighborhood. Beginning with a small “seed” set of highly interconnected proteins with a direct genetic link to an ADR of interest, the authors then scored every protein in the human PPI network on how well-connected it was to the seed set using multiple network connectivity functions including shortest path and shared neighbors. They then trained a random forest classifier using each of the connectivity metrics as features to generate drug safety subnetwork (SubNet) models. The authors then evaluated drug safety using both known and predicted drug targets. Combining SubNet and the results of a clinical data medication-wide association study (MWAS)110 using logistic regression led to significant improvements in drug safety predictions, and at a false positive rate of 10%, sensitivity increased from 0.32 (MWAS alone) to 0.59 when systems pharmacology approaches were combined with pharmacovigilance statistics.
The success of each of the above studies to accurately predict ADRs suggests that future work will greatly benefit from the continued integration of biological, chemical, and clinical data. This work is required to realize the vision of precision medicine. Therefore, we provide a data model framework (Figure 3) to illustrate how integrating diverse data types can allow for unique views of individual ADR risk. The microbiome, metabolomics, genetics, and nutritional information are all necessary to provide precise treatment regimens for individual patients. Nutrition can affect gene expression; genetic mutations may render genes non-functional, and the microbiome can affect biological interactions. Finally, metabolomics studies are important to understanding rate-change relationships between compounds, which may affect the speed of interactions in turn. Systems biology methods may be able to integrate these disparate data sources, and research in this area is of utmost importance.
Figure 3. A Data Model Framework Illustrates How Diverse Data Types can form a Complete Profile of an Individual's Adverse Drug Reaction Risk.
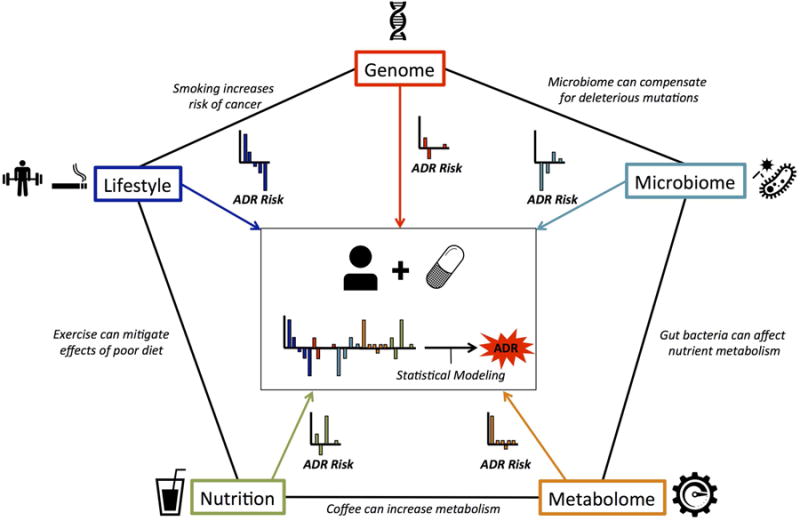
Many types of data form various fields investigate the individual aspects of ADR risk including: microbiome, metabolome, lifestyle, nutrition and the genome. Each of these contributes important information on an individual's adverse drug reaction (ADR) risk. Achieving precision medicine requires integrating these diverse data and the application of statistical modeling techniques to predict an individual's overall ADR risk for a given drug.
7. Conclusion
In this review, we discuss the recent surge in systems pharmacology publications related to ADR discovery. Since the publication of a 2011 review also exploring these approaches,9 the number of publications has more than doubled. The works highlighted in this review integrate a variety of data types and methodologies to uncover population-wide and precision-medicine-specific ADRs, and underscore the applications and benefits of systems pharmacology to ADR discovery. For example, network parameters are highly customizable; Jiang et al. created a novel network measure in order to identify proteins associated with specific ADRs,41 and a number of methods define other novel, application-specific, network-based measures for use in their research. In addition, systems biology methods are highly integrative, allowing researchers to stack various data sources. This is exemplified in Duran-Frigola's 2013 study,64 where the authors were able to combine numerous chemical and biological features to predict ADRs.
Although data integration can provide many novel insights, authors must bear in mind the balance of data integration versus specificity. For example, a 2013 Zhao et al. paper focused only on diabetes;108 further data integration may make the model more generalizable at the expense of decreased performance for particular ADRs.
The application of systems biology techniques to the problem of ADR detection and prevention continues to grow rapidly.111 However, several caveats must be stated. The first is the overuse of the same datasets. For example, multiple papers integrate post-market surveillance systems and PPI networks in order to identify ADRs using various algorithms. This creates a case of information saturation, and we have reached a point where identifying the optimal algorithm using these data sources is much more important than developing novel algorithms integrating the two. In this vein, Kuang et al. compared several existing methods for predicting ADRs112 and found that integrated approaches combining both intrinsic features of drugs and topological features of drug-ADR association networks tended to perform better in predicting side effects. More of these comparative analyses should be performed to assess the most accurate predictive methods given up-to-date datasets.
A second caveat is that of poor follow-up in confirming in silico predictions experimentally. At present, systems pharmacology approaches provide a list of hypothetical relationships between drug and ADR. These predictions can and should be integrated into everyday drug safety research, but experimental validation in a binding experiment, cellular assay, or animal model is critical for not only distilling an initial list of candidate drugs, but also for identifying false positives that can be used to refine the algorithm.
Finally, experimental results should – when possible – be validated in a clinical context. Issues with translating results from animal models to humans are well-known;113 integration of systems pharmacology predictions with EHRs therefore presents researchers with an opportunity to evaluate drug safety using retrospective “experiments” performed on humans during clinical care. Most importantly, validation using clinical data will help build confidence amongst clinicians and regulatory bodies. While such validation may not always be possible (e.g. during early drug development) and presents its own share of challenges,114 post-market studies of drug safety should leverage these data to fulfill the translatable promise of the field.
In surveying the selection of data sources used in previous studies (Figure 2), some dataset combinations occur more frequently in the literature than others. For example, drug-target datasets such as Drug Bank have been combined with drug-ADR databases (e.g. SIDER) far more often than with the Connectivity Map. We encourage future work to identify dataset combinations that have not yet been sufficiently explored as a strategy to focus new study design. The combination of rigorous algorithm comparison and dataset integration should pave the way for not only increased number of publications but also truly transformative productivity in systems pharmacology.
Acknowledgments
Funding Sources: The National Library of Medicine training grant T15 LM00707 (MRB), and R01 GM107145 (NPT, AJ, TL, JR, RM, MRB) supported this work.
List of Abbreviations
- ADR
Adverse Drug Reaction
- DDI
Drug-Drug Interaction
- DILI
Drug-Induced Liver Injury
- HER
Electronic Health Records
- FAERS
FDA Adverse Event Reporting System
- GWAS
Genome Wide Association Studies
- LQTS
Long-QT syndrome
- MDS
Molecular dynamics simulation
- PPI
Protein-Protein Interaction
References
- 1.Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med. 2007;147:755–765. doi: 10.7326/0003-4819-147-11-200712040-00006. [DOI] [PubMed] [Google Scholar]
- 2.Barker KN, Flynn EA, Pepper GA, Bates DW, Mikeal RL. Medication errors observed in 36 health care facilities. Arch Intern Med. 2002;162:1897–1903. doi: 10.1001/archinte.162.16.1897. [DOI] [PubMed] [Google Scholar]
- 3.Gandhi TK, et al. Adverse Drug Events in Ambulatory Care. N Engl J Med. 2003;348:1556–1564. doi: 10.1056/NEJMsa020703. [DOI] [PubMed] [Google Scholar]
- 4.Southworth MR, Reichman ME, Unger EF. Dabigatran and Postmarketing Reports of Bleeding. N Engl J Med. 2013;368:1272–1274. doi: 10.1056/NEJMp1302834. [DOI] [PubMed] [Google Scholar]
- 5.Paul SM, Mytelka DS, Dunwiddie CT. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nature reviews Drug …. 2010 doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 6.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 7.Jacunski A, Tatonetti NP. Connecting the dots: applications of network medicine in pharmacology and disease. Clin Pharmacol Ther. 2013;94:659–669. doi: 10.1038/clpt.2013.168. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson JK, Lindon JC. Systems biology: Metabonomics. Nature. 2008;455:1054–1056. doi: 10.1038/4551054a. [DOI] [PubMed] [Google Scholar]
- 9.Berger SI, Iyengar R. Role of systems pharmacology in understanding drug adverse events. WIREs Syst Biol Med. 2010;3:129–135. doi: 10.1002/wsbm.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franceschini A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Research. 2012;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Law V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Research. 2013;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol. 2010;6:343. doi: 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tatonetti NP, Liu T, Altman RB. Predicting drug side-effects by chemical systems biology. Genome Biology. 2009;10:238. doi: 10.1186/gb-2009-10-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shetty KD, Dalal SR. Using information mining of the medical literature to improve drug safety. Journal of the American Medical Informatics Association. 2011;18:668–674. doi: 10.1136/amiajnl-2011-000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leaman R, et al. Proceedings of the …. Association for Computational Linguistics; 2010. Towards internet-age pharmacovigilance: extracting adverse drug reactions from user posts to health-related social networks; pp. 117–125. [Google Scholar]
- 16.Moskovitch R, Walsh C, Hripsack G, Tatonetti N. Prediction of Biomedical Events via Time Intervals Mining. ACM KDD Workshop on Connected Health in Big Data Era. 2014 [Google Scholar]
- 17.Moskovitch R, Shahar Y. Classification-driven temporal discretization of multivariate time series. Data Min Knowl Disc. 2014:1–43. doi: 10.1007/s10618-014-0380-z. [DOI] [Google Scholar]
- 18.Moskovitch R, Shahar Y. Fast time intervals mining using the transitivity of temporal relations. Knowl Inf Syst. 2013;42:21–48. [Google Scholar]
- 19.Sacchi L, Dagliati A, Bellazzi R. Analyzing complex patients' temporal histories: new frontiers in temporal data mining. Methods Mol Biol. 2015;1246:89–105. doi: 10.1007/978-1-4939-1985-7_6. [DOI] [PubMed] [Google Scholar]
- 20.Harpaz R, Chase HS, Friedman C. Mining multi-item drug adverse effect associations in spontaneous reporting systems. BMC Bioinformatics. 2010;11 Suppl 9:S7. doi: 10.1186/1471-2105-11-S9-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouane-Hacene M, Toussaint Y, Valtchev P. Mining safety signals in spontaneous reports database using concept analysis. Artificial Intelligence in …. 2009 [Google Scholar]
- 22.Zorych I, Madigan D, Ryan P, Bate A. Disproportionality methods for pharmacovigilance in longitudinal observational databases. Stat Methods Med Res. 2013;22:39–56. doi: 10.1177/0962280211403602. [DOI] [PubMed] [Google Scholar]
- 23.Overby CL, Hripcsak G, Shen Y. Estimating heritability of drug-induced liver injury from common variants and implications for future study designs. Sci Rep. 2014;4 doi: 10.1038/srep05762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haerian K, et al. Detection of pharmacovigilance-related adverse events using electronic health records and automated methods. Clin Pharmacol Ther. 2012;92:228–234. doi: 10.1038/clpt.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boland MR, Tatonetti NP. Are All Vaccines Created Equal? Using Electronic Health Records to Discover Vaccines Associated With Clinician-Coded Adverse Events. AMIA Summits on Translational Science Proceedings. 2015:196–200. [PMC free article] [PubMed] [Google Scholar]
- 26.Casey CG, et al. Adverse Events Associated With Smallpox Vaccination in the United States, January-October 2003. JAMA. 2005;294:2734–2743. doi: 10.1001/jama.294.21.2734. [DOI] [PubMed] [Google Scholar]
- 27.LaRussa PS, et al. Understanding the Role of Human Variation in Vaccine Adverse Events: The Clinical Immunization Safety Assessment Network. PEDIATRICS. 2011;127:S65–S73. doi: 10.1542/peds.2010-1722J. [DOI] [PubMed] [Google Scholar]
- 28.Luo Zhihui, Z GQ, X R. Mining Patterns of Adverse Events Using Aggregated Clinical Trial Results. AMIA Summits on Translational Science Proceedings. 2013;2013:112. [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Hripcsak G, Markatou M, Friedman C. Active Computerized Pharmacovigilance Using Natural Language Processing, Statistics, and Electronic Health Records: A Feasibility Study. Journal of the American Medical Informatics Association. 2009;16:328–337. doi: 10.1197/jamia.M3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melton GB, Hripcsak G. Automated Detection of Adverse Events Using Natural Language Processing of Discharge Summaries. Journal of the American Medical Informatics Association. 2005;12:448–457. doi: 10.1197/jamia.M1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eriksson R, Jensen PB, Frankild S, Jensen LJ, Brunak S. Dictionary construction and identification of possible adverse drug events in Danish clinical narrative text. Journal of the American Medical Informatics Association. 2013;20:947–953. doi: 10.1136/amiajnl-2013-001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harpaz R, et al. Novel data-mining methodologies for adverse drug event discovery and analysis. Clin Pharmacol Ther. 2012;91:1010–1021. doi: 10.1038/clpt.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Motsinger-Reif AA, et al. Genome-wide association studies in pharmacogenomics: successes and lessons. Pharmacogenet Genomics. 2013;23:383–394. doi: 10.1097/FPC.0b013e32833d7b45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hajjar ER, Cafiero AC, Hanlon JT. Polypharmacy in elderly patients. The American Journal of Geriatric Pharmacotherapy. 2008;5:345–351. doi: 10.1016/j.amjopharm.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 35.Sun X, Vilar S, Tatonetti NP. High-throughput methods for combinatorial drug discovery. Science Translational Medicine. 2013;5:205rv1. doi: 10.1126/scitranslmed.3006667. [DOI] [PubMed] [Google Scholar]
- 36.Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Drug Target Identification Using Side-Effect Similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- 37.Keiser MJ, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 38.Keiser MJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamanishi Y, Kotera M, Kanehisa M, Goto S. Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework. Bioinformatics. 2010;26:i246–i254. doi: 10.1093/bioinformatics/btq176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao S, Li S. PLOS ONE: Network-Based Relating Pharmacological and Genomic Spaces for Drug Target Identification. 2010;5:e11764. doi: 10.1371/journal.pone.0011764. journals.plos.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, et al. Predicting putative adverse drug reaction related proteins based on network topological properties. Analytical Methods. 2014;6:2692–2698. [Google Scholar]
- 42.Huang LC, Wu X, Chen JY. Predicting adverse side effects of drugs. BMC Genomics. 2011;12:S11. doi: 10.1186/1471-2164-12-S5-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuhn M, et al. Systematic identification of proteins that elicit drug side effects. Mol Syst Biol. 2013;9 doi: 10.1038/msb.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang J, et al. Systematic prediction of pharmacodynamic drug-drug interactions through protein-protein-interaction network. PLoS Comput Biol. 2013;9:e1002998. doi: 10.1371/journal.pcbi.1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guimerà R, Sales-Pardo M. A network inference method for large-scale unsupervised identification of novel drug-drug interactions. PLoS Comput Biol. 2013;9:e1003374. doi: 10.1371/journal.pcbi.1003374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cobanoglu MC, Liu C, Hu F, Oltvai ZN, Bahar I. Predicting drug-target interactions using probabilistic matrix factorization. J Chem Inf Model. 2013;53:3399–3409. doi: 10.1021/ci400219z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lounkine E, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486:361–367. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheiber J, et al. Mapping Adverse Drug Reactions in Chemical Space. J Med Chem. 2009;52:3103–3107. doi: 10.1021/jm801546k. [DOI] [PubMed] [Google Scholar]
- 49.Duran-Frigola M, Rossell D, Aloy P. A chemo-centric view of human health and disease. Nat Comms. 2014;5:5676. doi: 10.1038/ncomms6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atias N, Sharan R. An Algorithmic Framework for Predicting Side Effects of Drugs. journal of computational Biology. 2011;18 doi: 10.1089/cmb.2010.0255. [DOI] [PubMed] [Google Scholar]
- 51.Liu M, et al. Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. Journal of the American Medical Informatics Association. 2012;19:e28–e35. doi: 10.1136/amiajnl-2011-000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang LC, Wu X, Chen JY. Predicting adverse drug reaction profiles by integrating protein interaction networks with drug structures. Proteomics. 2013;13:313–324. doi: 10.1002/pmic.201200337. [DOI] [PubMed] [Google Scholar]
- 53.LaBute MX, et al. Adverse drug reaction prediction using scores produced by large-scale drug-protein target docking on high-performance computing machines. PLoS ONE. 2014;9:e106298. doi: 10.1371/journal.pone.0106298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith RD, et al. CSAR benchmark exercise of 2010: combined evaluation across all submitted scoring functions. J Chem Inf Model. 2011;51:2115–2131. doi: 10.1021/ci200269q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sousa SF, et al. Protein-ligand docking in the new millennium--a retrospective of 10 years in the field. Curr Med Chem. 2013;20:2296–2314. doi: 10.2174/0929867311320180002. [DOI] [PubMed] [Google Scholar]
- 56.Kohlhoff KJ, et al. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat Chem. 2014;6:15–21. doi: 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stjernschantz E, Oostenbrink C. Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys J. 2010;98:2682–2691. doi: 10.1016/j.bpj.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campbell AJ, Lamb ML, Joseph-McCarthy D. Ensemble-based docking using biased molecular dynamics. J Chem Inf Model. 2014;54:2127–2138. doi: 10.1021/ci400729j. [DOI] [PubMed] [Google Scholar]
- 59.Scheiber J, et al. Gaining Insight into Off-Target Mediated Effects of Drug Candidates with a Comprehensive Systems Chemical Biology Analysis. J Chem Inf Model. 2009;49:308–317. doi: 10.1021/ci800344p. [DOI] [PubMed] [Google Scholar]
- 60.Wallach I, Jaitly N, Lilien R. A structure-based approach for mapping adverse drug reactions to the perturbation of underlying biological pathways. PLoS ONE. 2010;5:e12063. doi: 10.1371/journal.pone.0012063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie L, Li J, Xie L, Bourne PE. Drug discovery using chemical systems biology: identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput Biol. 2009;5:e1000387. doi: 10.1371/journal.pcbi.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen X, et al. A sub-pathway-based approach for identifying drug response principal network. Bioinformatics. 2011;27:649–654. doi: 10.1093/bioinformatics/btq714. [DOI] [PubMed] [Google Scholar]
- 63.Lee S, Lee KH, Song M, Lee D. Building the process-drug–side effect network to discover the relationship between biological Processes and side effects. BMC Bioinformatics. 2011;12:S2. doi: 10.1186/1471-2105-12-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duran-Frigola M, Aloy P. Analysis of Chemical and Biological Features Yields Mechanistic Insights into Drug Side Effects. Chemistry & Biology. 2013;20:594–603. doi: 10.1016/j.chembiol.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 65.Gottlieb A, Altman RB. Integrating systems biology sources illuminates drug action. Clin Pharmacol Ther. 2014;95:663–669. doi: 10.1038/clpt.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ivanov SM, Lagunin AA, Pogodin PV, Filimonov DA, Poroikov VV. Identification of drug-induced myocardial infarction-related protein targets through the prediction of drug-target interactions and analysis of biological processes. Chem Res Toxicol. 2014;27:1263–1281. doi: 10.1021/tx500147d. [DOI] [PubMed] [Google Scholar]
- 67.Silberberg Y, Gottlieb A, Kupiec M, Ruppin E, Sharan R. Large-scale elucidation of drug response pathways in humans. journal of computational Biology. 2012;19:163–174. doi: 10.1089/cmb.2011.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song M, Yan Y, Jiang Z. Drug-pathway interaction prediction via multiple feature fusion. Mol BioSyst. 2014;10:2907–2913. doi: 10.1039/c4mb00199k. [DOI] [PubMed] [Google Scholar]
- 69.Mizutani S, Pauwels E, Stoven V, Goto S, Yamanishi Y. Relating drug-protein interaction network with drug side effects. Bioinformatics. 2012;28:i522–i528. doi: 10.1093/bioinformatics/bts383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shear NH, Spielberg SP. Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk. J Clin Invest. 1988;82:1826–1832. doi: 10.1172/JCI113798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gennis MA, et al. Familial occurrence of hypersensitivity to phenytoin. Am J Med. 1991;91:631–634. doi: 10.1016/0002-9343(91)90216-k. [DOI] [PubMed] [Google Scholar]
- 72.Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587–1591. doi: 10.1016/S0140-6736(00)03137-8. [DOI] [PubMed] [Google Scholar]
- 73.Tassaneeyakul W, Tiamkao S, Jantararoungtong T. Association between HLA-B* 1502 and carbamazepine-induced severe cutaneous adverse drug reactions in a Thai population. …. 2010 doi: 10.1111/j.1528-1167.2010.02533.x. [DOI] [PubMed] [Google Scholar]
- 74.Hung SI, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics. 2006;16:297–306. doi: 10.1097/01.fpc.0000199500.46842.4a. [DOI] [PubMed] [Google Scholar]
- 75.Ozeki T, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20:1034–1041. doi: 10.1093/hmg/ddq537. [DOI] [PubMed] [Google Scholar]
- 76.Lonjou C, et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet Genomics. 2008;18:99–107. doi: 10.1097/FPC.0b013e3282f3ef9c. [DOI] [PubMed] [Google Scholar]
- 77.Lonjou C, et al. A marker for Stevens-Johnson syndrome…: ethnicity matters. The pharmacogenomics journal. 2006;6:265–268. doi: 10.1038/sj.tpj.6500356. [DOI] [PubMed] [Google Scholar]
- 78.Berger SI, Ma'ayan A, Iyengar R. Systems pharmacology of arrhythmias. Science signaling. 2010;3:ra30. doi: 10.1126/scisignal.2000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gibney MJ, et al. Metabolomics in human nutrition: opportunities and challenges. Am J Clin Nutr. 2005;82:497–503. doi: 10.1093/ajcn.82.3.497. [DOI] [PubMed] [Google Scholar]
- 80.Winnike JH, et al. Use of pharmaco-metabonomics for early prediction of acetaminophen-induced hepatotoxicity in humans. Clin Pharmacol Ther. 2010;88:45–51. doi: 10.1038/clpt.2009.240. [DOI] [PubMed] [Google Scholar]
- 81.Cunningham K, et al. Pharmacometabonomic characterization of xenobiotic and endogenous metabolic phenotypes that account for inter-individual variation in isoniazid-induced toxicological response. J Proteome Res. 2012;11:4630–4642. doi: 10.1021/pr300430u. [DOI] [PubMed] [Google Scholar]
- 82.Lenz EM, et al. Metabonomics, dietary influences and cultural differences: a 1H NMR-based study of urine samples obtained from healthy British and Swedish subjects. J Pharm Biomed Anal. 2004;36:841–849. doi: 10.1016/j.jpba.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 83.Sun J, Beger RD, Schnackenberg LK. Metabolomics as a tool for personalizing medicine: 2012 update. Personalized Medicine. 2013 doi: 10.2217/pme.13.8. [DOI] [PubMed] [Google Scholar]
- 84.Nicholson JK, Wilson ID. Understanding ‘global’ systems biology: metabonomics and the continuum of metabolism. Nat Rev Drug Discov. 2003 doi: 10.1038/nrd1157. [DOI] [PubMed] [Google Scholar]
- 85.Li H, Jia W. Cometabolism of microbes and host: implications for drug metabolism and drug-induced toxicity. Clin Pharmacol Ther. 2013;94:574–581. doi: 10.1038/clpt.2013.157. [DOI] [PubMed] [Google Scholar]
- 86.Pleil JD, Sheldon LS. Adapting concepts from systems biology to develop systems exposure event networks for exposure science research. Biomarkers. 2011;16:99–105. doi: 10.3109/1354750X.2010.541565. [DOI] [PubMed] [Google Scholar]
- 87.Gourley M, Miller FW. Mechanisms of disease: Environmental factors in the pathogenesis of rheumatic disease. Nat Clin Pract Rheumatol. 2007;3:172–180. doi: 10.1038/ncprheum0435. [DOI] [PubMed] [Google Scholar]
- 88.Kasarskis A, Yang X, Schadt E. Integrative genomics strategies to elucidate the complexity of drug response. Pharmacogenomics. 2011;12:1695–1715. doi: 10.2217/pgs.11.115. [DOI] [PubMed] [Google Scholar]
- 89.Zhang M, et al. Comorbidity and repeat admission to hospital for adverse drug reactions in older adults: retrospective cohort study. BMJ. 2009;338:a2752. doi: 10.1136/bmj.a2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Onder G, et al. Adverse drug reactions as cause of hospital admissions: results from the Italian Group of Pharmacoepidemiology in the Elderly (GIFA) J Am Geriatr Soc. 2002;50:1962–1968. doi: 10.1046/j.1532-5415.2002.50607.x. [DOI] [PubMed] [Google Scholar]
- 91.Boran ADW, Iyengar R. Systems approaches to polypharmacology and drug discovery. Curr Opin Drug Discov Devel. 2010;13:297–309. [PMC free article] [PubMed] [Google Scholar]
- 92.Perez EA. Cardiac toxicity of ErbB2-targeted therapies: what do we know? Clin Breast Cancer. 2008;8 Suppl 3:S114–20. doi: 10.3816/cbc.2008.s.007. [DOI] [PubMed] [Google Scholar]
- 93.Tan-Chiu E, Yothers G, Romond E. Assessment of cardiac dysfunction in a randomized trial comparing doxorubicin and cyclophosphamide followed by paclitaxel, with or without trastuzumab as adjuvant …. Journal of Clinical …. 2005 doi: 10.1200/JCO.2005.02.4091. [DOI] [PubMed] [Google Scholar]
- 94.Tatonetti NP, Ye PP, Daneshjou R, Altman RB. Data-Driven Prediction of Drug Effects and Interactions. Science Translational Medicine. 2012;4:125ra31. doi: 10.1126/scitranslmed.3003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 96.He Z, et al. Predicting drug-target interaction networks based on functional groups and biological features. PLoS ONE. 2010;5:e9603. doi: 10.1371/journal.pone.0009603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Turner RM, Park BK, Pirmohamed M. Parsing interindividual drug variability: an emerging role for systems pharmacology. WIREs Syst Biol Med. 2015 doi: 10.1002/wsbm.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sharma V, Sarkar IN. Leveraging concept-based approaches to identify potential phyto-therapies. Journal of Biomedical Informatics. 2013;46:602–614. doi: 10.1016/j.jbi.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Staniek A, et al. Natural products - modifying metabolite pathways in plants. Biotechnol J. 2013;8:1159–1171. doi: 10.1002/biot.201300224. [DOI] [PubMed] [Google Scholar]
- 100.Vargas HM, Amouzadeh HR, Engwall MJ. Nonclinical strategy considerations for safety pharmacology: evaluation of biopharmaceuticals. Expert Opin Drug Saf. 2013;12:91–102. doi: 10.1517/14740338.2013.745851. [DOI] [PubMed] [Google Scholar]
- 101.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaiser M, Wetzel S, Kumar K, Waldmann H. Biology-inspired synthesis of compound libraries. Cell Mol Life Sci. 2008;65:1186–1201. doi: 10.1007/s00018-007-7492-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yue Y, et al. TMDB: a literature-curated database for small molecular compounds found from tea. BMC Plant Biol. 2014;14:243. doi: 10.1186/s12870-014-0243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Howland RH. Update on St. John's Wort. J Psychosoc Nurs Ment Health Serv. 2010;48:20–24. doi: 10.3928/02793695-20100930-99. [DOI] [PubMed] [Google Scholar]
- 105.Huang SM, Lesko LJ. Drug-drug, drug-dietary supplement, and drug-citrus fruit and other food interactions: what have we learned? J Clin Pharmacol. 2004;44:559–569. doi: 10.1177/0091270004265367. [DOI] [PubMed] [Google Scholar]
- 106.Zhou X, Li Y, Chen X. Computational identification of bioactive natural products by structure activity relationship. J Mol Graph Model. 2010;29:38–45. doi: 10.1016/j.jmgm.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 107.Cami A, Arnold A, Manzi S, Reis B. Predicting Adverse Drug Events Using Pharmacological Network Models. Science Translational Medicine. 2011;3:114ra127–114ra127. doi: 10.1126/scitranslmed.3002774. [DOI] [PubMed] [Google Scholar]
- 108.Zhao S, et al. Systems pharmacology of adverse event mitigation by drug combinations. Science Translational Medicine. 2013;5:206ra140. doi: 10.1126/scitranslmed.3006548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lorberbaum T, et al. Systems Pharmacology Augments Drug Safety Surveillance. Clin Pharmacol Ther. 2014;97:151–158. doi: 10.1002/cpt.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ryan PB, Madigan D, Stang PE, Schuemie MJ. Medication-Wide Association Studies. CPT: pharmacometrics …. 2013 doi: 10.1038/psp.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perkins JR, Sanak M, Canto G, Blanca M, Cornejo-García JA. Unravelling adverse reactions to NSAIDs using systems biology. Trends in Pharmacological Sciences. 2015;36:172–180. doi: 10.1016/j.tips.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 112.Kuang Q, et al. A systematic investigation of computation models for predicting Adverse Drug Reactions (ADRs) PLoS ONE. 2014;9:e105889. doi: 10.1371/journal.pone.0105889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Seok J, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ryan PB, et al. Empirical assessment of methods for risk identification in healthcare data: results from the experiments of the Observational Medical Outcomes Partnership. Stat Med. 2012;31:4401–4415. doi: 10.1002/sim.5620. [DOI] [PubMed] [Google Scholar]