

Abstract
Background and Purpose
The Rho GTPase, Rac1, is involved in the pathogenesis of neuropathic pain induced by malformation of dendritic spines in the spinal dorsal horn (sDH) neurons. In the present study, the contribution of spinal Rac1 to peripheral inflammatory pain was studied.
Experimental Approach
Effects of s.c. bee venom (BV) injection on cellular localization of Rac1 in the rat sDH was determined with double labelling immunofluorescence. Activation of Rac1 and its downstream effector p21‐activated kinase (PAK), ERKs and p38 MAPK in inflammatory pain states was evaluated with a pull‐down assay and Western blotting. The preventive and therapeutic analgesic effects of intrathecal administration of NSC23766, a selective inhibitor of Rac1, on BV‐induced spontaneous nociception and pain hypersensitivity were investigated.
Key Results
Rac1 labelling was mainly localized within neurons in both the superficial and deep layers of the sDH in rats of naïve, vehicle‐treated and inflamed (BV injected) groups. GTP‐Rac1‐PAK and ERKs/p38 were activated following s.c. BV injection. Post‐treatment with intrathecal NSC23766 significantly inhibited GTP‐Rac1 activity and phosphorylation of Rac1‐PAK, ERKs and p38 MAPK in the sDH. Both pre‐treatment and post‐treatment with intrathecal NSC23766 dose‐dependently attenuated the paw flinches, primary thermal and mechanical hyperalgesia and the mirror‐image thermal hyperalgesia induced by BV injection, but without affecting the baseline pain sensitivity and motor coordination.
Conclusions and Implications
The spinal GTP‐Rac1‐PAK‐ERK/p38MAPK signalling pathway is involved in both the development and maintenance of peripheral inflammatory pain and can be used as a potential molecular target for developing a novel therapeutic strategy for clinical pain.
Abbreviations
- BV
bee venom
- FMMU
Fourth Military Medical University
- PAK
p21‐activated kinase
- PBD
p21 binding domain
- PBST
PBS with 0.05% Tween 20
- Rac1
Rho GTPase Ras‐related C3 botulinum toxin substrate 1
- SDS
sodium dodecyl sulfate
- WDR
wide dynamic range
Tables of Links
| TARGETS |
|---|
| Enzymes |
| ERK1/2 |
| p38 kinase |
| PAK, p21‐activated kinase |
| LIGANDS |
|---|
| Rac1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Rac1 is a member of the Rho GTPase family, which belongs to the Ras superfamily of low MW guanine nucleotide binding proteins. Rac1 plays an essential role in several cellular processes such as gene transcription (Hill et al., 1995), cell cycle progression (Olson et al., 1995) and cellular survival and death (Heasman and Ridley, 2008). Rac1 is also implicated in the regulation of neuronal development, neuronal survival and neurodegeneration (Stankiewicz and Linseman, 2014). Recently, Rac1 has been demonstrated to be involved in the pathogenesis of neuropathic pain. For instance, the dendritic spines of wide dynamic range (WDR) neurons located in the deep layers of the spinal dorsal horn can undergo malformation in several experimental neuropathic pain models (Melemedjian and Price, 2012; Tan et al., 2011, 2012, 2013, 2008; Tan and Waxman, 2012, 2014). Inhibition of Rac1, which is thought to play a pivotal role in the genesis and maturation of dendritic spines (Nakayama and Luo, 2000), with a selective Rac1 GTPase‐specific inhibitor NSC23766 (Gao et al., 2004), can restore malformation of the spines and attenuate pain hypersensitivity in animal models of chronic constriction injury (Tan et al., 2011), spinal cord injury (Tan et al., 2008; Melemedjian and Price, 2012; Tan and Waxman, 2012), diabetic neuropathic pain (Tan et al., 2012) and burn injury (Tan et al., 2013). However, whether activation of Rac1 signalling is involved in inflammatory pain remains unknown and warrants investigation.
Inflammatory pain (also referred to as nociceptive pain) is caused by tissue injury, trauma and diseases (e.g. arthritis) that are associated with ongoing activation of A‐δ and C nociceptors in response to internal and/or external noxious stimulus of somatic and visceral structures (Chen et al., 2013a). Neuropathic pain is newly defined as pain arising as a direct consequence of a lesion or disease affecting the somatosensory system that is likely to be driven by ectopic discharges from affected nerve fibers but without nociceptor activation and sensitization (Treede et al. 2008). Inflammatory pain differs from neuropathic pain in terms of aetiology, perceived location, affected anatomical system, pathogenesis, pathophysiological process and even therapeutic strategies (Chen et al., 2013a; Treede et al., 2015). Thus, elucidating the roles of Rac1 signalling in different aetiologies of pain may have major clinical implications and significance.
The bee venom (BV) test, which is produced by s.c. injection of BV, mimics the complex processes of tissue injury and inflammation. The BV test is a well‐established experimental pain model that can be used for studying the central and peripheral mechanisms underlying pathological pain (Chen, 2003, 2007, 2008; Chen and Lariviere, 2010; Chen and Guan, In press). It has many advantages over other inflammatory pain models, which use stimuli such as formalin, carrageenan and zymosan, because of it resembles a natural injury and exhibits clinical symptoms that are similar to those experienced by many patients (Chen and Lariviere, 2010; Chen and Guan, 2015). The s.c. injection of BV into the rat hindpaw induces persistent spontaneous pain‐related behaviours (for at least 1 h) and pain hypersensitivity (for 72–96 h) in a dose‐dependent manner (Lariviere and Melzack, 1996; Chen et al., 1999b; Chen and Lariviere, 2010; Chen and Guan, 2015). The spontaneous pain‐related behaviours consist of spinally organized flinch reflexes (You and Chen, 1999; You et al., 2003a, 2003b, 2003c; Li and Chen, 2004; Chen and Lariviere, 2010) and supraspinally mediated licking and lifting behaviours on the side of the injected paw (Ren et al., 2008). The pain hypersensitivity induced by BV injection includes primary heat and mechanical hyperalgesia in the injected paw and the mirror‐image thermal hyperalgesia in the contralateral paw (Chen et al., 1999b, 2000, 2006; Chen and Chen, 2000, 2001). Sensitization of WDR neurons is the cause of the paw flinch reflex and pain hypersensitivity, induced by BV injection (You and Chen, 1999; You et al., 2003c; Li and Chen, 2004; Zheng et al., 2004a, 2004b). As Rac1 has been shown to be involved in spine plasticity and inhibition of Rac1 can attenuate the hyperexcitability of WDR neurons in layers IV and V of the spinal cord and pain hypersensitivity under neuropathic pain state (Tan et al., 2011, 2012, 2013, 2008), it is highly possible that the activation of Rac1 may also contribute to the pain‐related behaviours induced by BV injection.
The aims of the present study were, therefore, (1) to identify the cellular distribution of Rac1 in neurons, astrocytes and microglial cells in the spinal dorsal horn of naïve, vehicle‐treated and BV‐inflamed rats; (2) to investigate whether Rac1 and its downstream effector p21‐activated kinase (PAK) (Johnson and D'Mello, 2005) and their downstream proteins ERK1/ERK2 and p38 MAPK (Shifrin et al., 2012; Wang et al., 2013) can be activated in the peripheral inflammatory pain state induced by s.c. BV injection; and (3) to examine whether intrathecal administration of an inhibitor of Rac1 has any preventive and therapeutic analgesic effects on the BV‐induced persistent spontaneous nociception and pain hypersensitivity.
Methods
Animals and ethical statement
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of Tangdu Hospital, Fourth Military Medical University (FMMU) and complied with the guidelines set by the International Association for the Study of Pain (Zimmermann, 1983). Studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male Sprague Dawley rats (180–220 g, 8–9 weeks old) were bought from the Laboratory Animal Center of the FMMU and Xi'an Jiaotong University, Xi'an, Shaanxi Province, China. Animals were maintained under standard laboratory conditions (12 h light/dark cycle, temperature 22–26°C, air humidity 40–60%) with free access to food and water. The animals were habituated to the testing environment 5 days before the experiment. The experimental animals were randomly assigned to different groups in each part of the experiment. The experimenters were unaware of the treatments given to the animals..
Double‐labelling immunofluorescence
After deep anaesthesia with chloralose hydrate (0.36 g·kg−1, i.p.), naïve rats, and rats that received an s.c. injection of saline vehicle or BV into a hindpaw were perfused transcardially with 4% paraformaldehyde. The lumbar spinal cord was removed and post‐fixed in 4% paraformaldehyde for 6 h, and then transferred to a 30% sucrose solution in 0.01 M phosphate buffer for cryoprotection until it sank to the bottom of the container. Transverse spinal cord sections of 40 μm were cut on a freezing microtome and were collected once for every 10 cuts and washed in 0.01 M PBS. Then spinal cord sections were incubated in 3% hydrogen peroxide for 10 min, followed by 1 h incubation in 1% bovine serum and 0.2% Triton X‐100 in 0.01 M PBS. Sections were incubated overnight with primary antibodies, and then washed in 0.01 M PBS for three times before being subjected to the incubation of secondary antibodies for 3 h. Sections were washed again for three times and mounted on a slide. Microphotographs were taken by a confocal microscope. The primary antibodies used in the present study were mouse monoclonal anti‐Rac1 (1:200, Cytoskeleton Inc., USA), rabbit anti‐NeuN (1:200, Abcam, UK), rabbit anti‐glia fibrillary acidic protein (GFAP) (1:200, Millipore, USA) and rabbit anti‐Iba1 (1:200, WAKO, Japan). The following secondary antibodies were used: rabbit anti‐mouse FITC (1:200, Sigma, USA) and Cy3‐conjugated sheep anti‐rabbit antibody (1:200, Sigma, USA). For details, see our previous reports (Yu et al., 2011; Yu et al., 2013). Each group contained five rats, and the immunoreactive cells were counted over 10–12 sections for each rat, the percentage of NeuN‐labelled neurons or GFAP‐labelled astrocytes or Iba1‐labelled microglia cells that co‐labelled with Rac1 was calculated in both the superficial (layers I and II) and the deep layers (layers III and VI) of the spinal dorsal horn respectively.
Intrathecal catheterization and drug delivery
Under anaesthesia (chloralose hydrate; 0.36 g·kg−1, i.p.), a laminectomy was performed on T2 or T3 and a PE‐10 tube was inserted into the subarachnoid space and threaded caudally to the lumbar enlargement, the rostral end of the tube was fixed to the paravertebral muscles and was sealed by heat (Zheng and Chen, 2001). The location of the tip was confirmed after the animals were killed and any rats with walking abnormalities were excluded. Behavioural tests were conducted 5 days post‐surgery. The animals that exhibited walking abnormalities were excluded from the behavioural test. For drug delivery, the rats were held gently and 5 μL volume of either vehicle (0.9% saline) or a Rac1 GTPase‐specific inhibitor, NSC23766 (1 mg·mL−1), which has no effects on the binding of Rac1 to PAK (Gao et al., 2004), was infused and followed by a flush of 20 μL sterile saline. Intrathecal pre‐administration of drugs means that the Rac1 inhibitor was given 5 min prior to s.c. BV injection. Post‐administration of drugs means that the drug was given 2 h after s.c. BV injection.
Western blot
Fresh spinal cord tissue of the lumbar enlargement was obtained from rats (n = 5 for each treatment) by decapitation 30 min after post‐administration of drug and vehicle treatment of the BV‐inflamed animals. Total protein was extracted in ice‐cold RIPA lysis buffer, which contained 150 mM NaCl, 1% NP‐40 and 0.1% sodium dodecyl sulphate (SDS). Protein quantification was performed with BCA™ protein assay kit (Thermo Scientific, Rockford, IL, USA), and then samples were heated for 10 min at 95°C with SDS‐PAGE sample buffer. For analysis, same amounts of proteins were subjected to electrophoresis (6% SDS‐PAGE separation gel), transferred to nitrocellulose membranes (Bio‐Rad, Hercules, CA, USA), incubated in 5% non‐fat milk powder in PBS with 0.05% Tween 20 (PBST) solution for 3 h at room temperature, and then subjected to overnight incubation of primary antibodies at 4°C. After washing, the blots were incubated in horseradish peroxidase (HRP)‐conjugated secondary antibodies. Blots were developed with ChemiGlow West chemiluminescent substrate kit, and signals were captured with FluorChem FC II (Alpha Innotech Corp.). AlphaImager EP Analysis Software (Cell Biosciences, Inc) was used for quantification of blot relative intensities of target bands. Tubulin staining intensity was set as the internal control, and proteins of interest were normalized to internal control. First, the control means of each group were calculated and then all the individual test values were expressed as fold of the control mean and appropriate statistical analysis was conducted on these normalized values (Curtis et al., 2015). The Western blot of each sample was replicated to test the reliability of single values, and only one value of each sample was subjected to statistical analysis. The following primary antibodies were used in the present study: rabbit anti‐phospho Rac1 plus Cdc42 (1:200, Santa Cruz, USA), mouse anti‐Rac1 (1:500, Millipore, USA), rabbit anti‐phospho PAK (1:500, Cell Signaling, USA), rabbit anti‐PAK (1:1000, Cell Signaling, USA), rabbit anti‐phospho‐p38 (1:200, Santa Cruz, USA), mouse anti‐p38 (1:1000, Abcam, UK), rabbit anti‐ERK1/anti‐ERK2 (1:200, Santa Cruz, USA) and mouse anti‐phospho‐ERK1/anti‐phospho‐ERK2 (1:200, Santa Cruz, USA). The following secondary antibodies were used: goat anti‐mouse HRP and goat anti‐rabbit HRP (1:3000, ZSGB‐Bio, Beijing, China). For details, see our previous reports (Yu et al., 2011; Yu et al., 2013).
Rac1‐activity assay
The GTP bound Rac1 was the active form of Rac1; thus, Rac1 activation was investigated with a pull‐down assay for Rac1‐GTP (BK035, Cytoskeleton Inc., USA). PAK, a Rac1 effector, which possesses a p21 binding domain (PBD), has high affinity for GTP‐Rac1 and was used to pull down the active form of Rac1. Briefly, fresh spinal cord tissue of the lumbar enlargement was homogenised in the presence of protease and phosphatase inhibitors and lysed with buffer provided by the manufacturer. After being centrifuged at 12,000 × g for 5 min at 4°C, the supernatants were collected and incubated with PAK‐PBD beads at 4°C on a rotator for 1 h and then the beads were pelleted through centrifugation at 5000 × g for 3 min at 4°C. The resulting pellet was resuspended in Laemmli buffer and boiled for 2 min. The bead samples were subjected to Western blot analysis as described above. Total Rac1 in each sample was also determined by Western blot analysis.
Measurement of spontaneous nociception
Five minutes after drug delivery, the rats received intraplantar injection of BV solution, containing 0.2 mg BV (Sigma, USA) in 50 μL physiological saline, into the left hind paw (Lariviere and Melzack, 1996; Chen et al., 1999b; Chen and Lariviere, 2010). After BV injection, the rat was put into a transparent plastic box (30 × 30 × 30 cm) placed on a supporting frame 30 cm above the experimental table. Persistent spontaneous nociception was determined by counting the numbers of flinching reflexes within a 5 min block, over 1 h.
Measurement of thermal sensitivity
For determination of the latency of paw withdrawal to a thermal stimulus, the rats were put into a transparent plastic box with a glass floor. The heat stimulation was generated from a TC‐1 radiant heat stimulator (new generation of RTY‐3, Bobang Technologies of Chemical Industry, China) and applied bilaterally to the centre of the hind paws with 10 min intervals for the same side and 5 min intervals for different sides. The latency of the paw withdrawal reflex in response to the radiant heat stimulation was recorded, the last three values were averaged and the mean was considered as the thermal latency for paw withdrawal. A cut‐off of 30 s was chosen to prevent tissue damage.
Measurement for mechanical sensitivity
For determination of the threshold of paw withdrawal after a mechanical stimulus, the rat was placed into a transparent plastic box located on a metal mesh floor. A series of Von Frey monofilaments of different forces (0.8, 2.0, 4.0, 6.0, 8.0, 10.0, 12.0, 14.0, 16.0, 18.0, 20.0, 25.0, 30.0, 45.0 and 60 g) was applied bilaterally to the centre of the hind paws of the animals at 10 s intervals with 10 repetitions. The forces able to elicit more than five paw withdrawal reflexes was considered as the mechanical threshold for paw withdrawal.
Measurement of sensory‐motor coordination function
A Rota‐Rod treadmill (Ugo Basile, Italy) was used to measure the effects of NSC23766 (1 mg·mL−1, intrathecal) on the sensory‐motor coordination function of naïve rats. The rod was set to accelerate from 6 to 30 r.p.m. in 2 min, and time was recorded automatically from the acceleration to the fall of the rat. A total of eight trials were conducted and the first three trials were considered as the training session. For details, see our previous report (Ren et al., 2008).
Data analysis
All experimental data are expressed as means ± SEM. One‐way ANOVA, with post hoc Fisher's least significant difference test, repeated measures and multivariate ANOVA were used when necessary. P < 0.05 was considered to be of statistical significance. Post hoc tests were run only if F achieved P < 0.05, and there was no significant variance inhomogeneity.
Materials
All drugs were dissolved in 0.9% saline, and the drugs used in the present study were obtained as follows: BV (0.2 mg·50·μL−1, Sigma, USA); choral hydrate (0.36 g·kg−1, Shanghai Shanpu Chemical Co., Ltd., China); NSC23766 (N 6‐[2‐[[4‐(diethylamino)‐1‐methylbutyl]amino]‐6‐methyl‐4‐pyrimidinyl]‐2‐methyl‐4,6‐quinolinediamine trihydrochloride;1 mg·mL−1, Tocris, UK).
Results
Cellular localization of Rac1 labelling in the spinal dorsal horn
Rac1 labelling was broadly distributed in both the superficial (I and II) and the deep (III and VI) layers of the spinal dorsal horn in naïve rats (Supporting Information Figs [Link], [Link]). Double labelling of Rac1 with NeuN, GFAP or Iba1 showed that Rac1 labelling was predominantly localized within the cytoplasm of NeuN‐labelled neurons (Supporting Information Fig. S1), whereas it was less co‐localized with GFAP labelling (Supporting Information Fig. S2) and Iba1 labelling (Supporting Information Fig. S3), which are markers for activated astrocytes and microglia respectively. The averaged counts of Rac1‐positive profiles co‐localized with NeuN‐positive, GFAP‐positive or Iba1‐positive profiles in the superficial, and the deep layers of the spinal dorsal horn is shown in Supporting Information Fig. S4. Replacement of primary antibody against Rac1 with 1% bovine serum in 0.01 M PBST did not show any significant positive immunoreactivity within the spinal dorsal horn in rats (Supporting Information Fig. S9).
Following s.c. injection of saline or BV into a hindpaw, Rac1 labelling was still predominantly localized within the cytoplasm of NeuN‐labelled neurons (Figures 1, 2) and was less co‐localized with GFAP labelling (Supporting Information Figs S5 and S6) and Iba1 labelling (Supporting Information Figs S7 and S8) in the spinal dorsal horn of the injection side. Although GFAP‐labelled astrocytes and Iba1‐labelled microglia increased significantly following s.c. BV injection, the co‐localization of Rac1 labelling with GFAP labelling or Iba1 labelling did not show any increase between the vehicle or BV groups (Figure 3).
Figure 1.
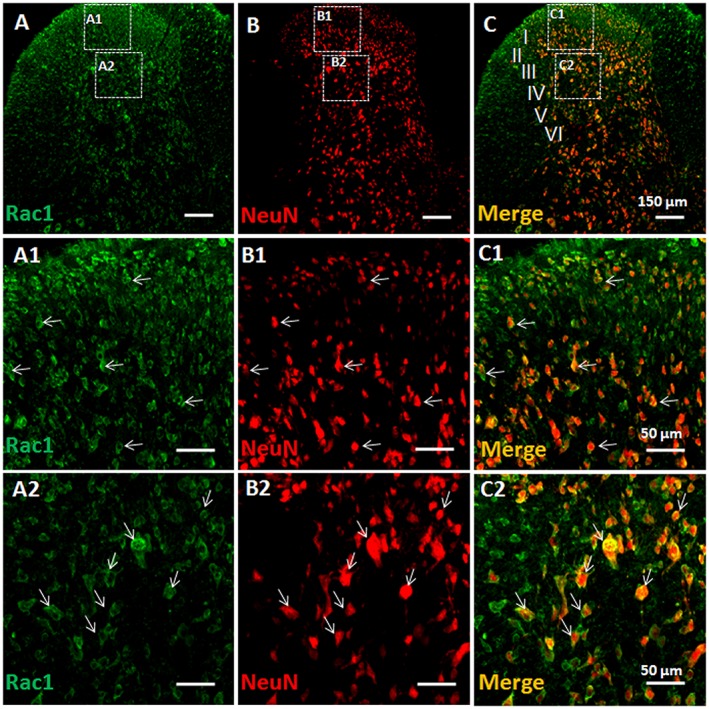
Double immunofluorescent labelling of Rac1 and NeuN in vehicle‐treated rats. Immunofluorescent labelling of Rac1 (A) and NeuN (B) in the injection side of spinal dorsal horn of rats that received s.c. saline (vehicle) into a hind paw. (C) Merged images of A and B. A1–A2, B1–B2 and C1–C2 show enlarged images of the insets in A, B and C respectively. Scale bars, 150 μm (A–C), 50 μm (A1–C1, A2–C2).
Figure 2.
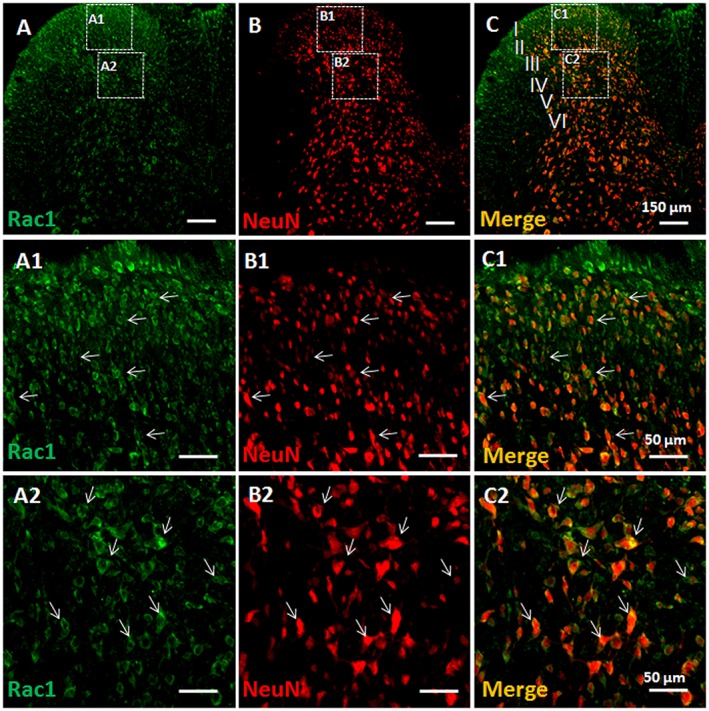
Double immunofluorescent labelling of Rac1 and NeuN in BV‐treated rats. Immunofluorescent labelling of Rac1 (A) and NeuN (B) in the injection side of spinal dorsal horn of rats that received s.c. injection of BV into a hindpaw. (C) Merged images of A and B. A1–A2, B1–B2 and C1–C2 show enlarged images of the insets in A, B and C respectively. Scale bars, 150 μm (A–C), 50 μm (A1–C1 and A2–C2).
Figure 3.
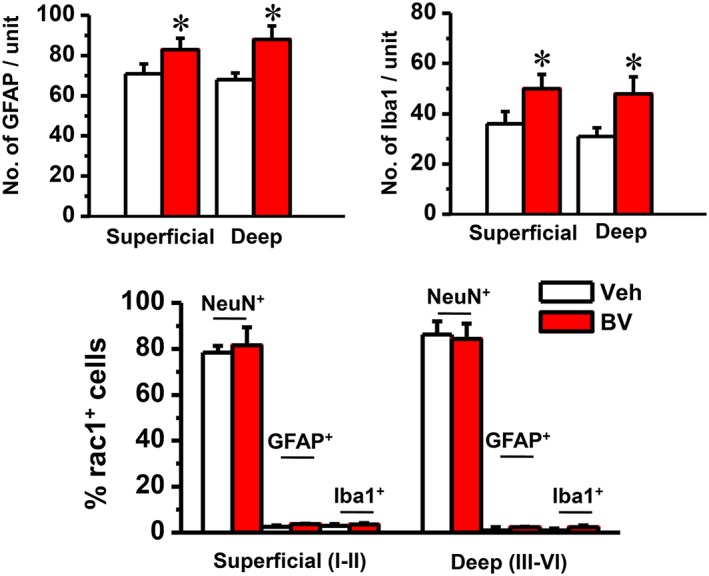
Quantification of immunofluorescent labelling of GFAP‐positive and Iba1‐ positive profiles and double immunofluorescent labelling of Rac1 with NeuN‐positive, GFAP‐positive‐ and Iba1‐positive profiles. The upper panel shows the average numbers of GFAP‐labelled astrocytes and Iba1‐labelled microglia cells in the injection side of both superficial (layers I and II) and deep layers (layers III and VI) of spinal dorsal horn of rats that received s.c. injection of saline (Veh) or BV into a hind paw respectively. The lower panel shows that the percentage of NeuN‐labelled neurons, GFAP‐labelled astrocytes and Iba1‐labelled microglial cells in Rac1‐positive profiles was calculated in both superficial (layers I and II) and deep layers (layers III and VI) of the spinal dorsal horn respectively. Data are expressed as mean ± SEM. *P < 0.05, significantly different from Veh.
Reversal of BV‐induced activation of GTP‐Rac1‐PAK signalling and downstream activities by NSC23766, a selective inhibitor of Rac1
As shown in Figure 4A and B, there was no change in the total protein expression level of Rac1 and PAK between the animals receiving s.c. saline (Control) and those receiving either s.c. BV injection plus intrathecal vehicle or s.c. BV injection plus intrathecal NSC23766. However, the phosphorylated form of both Rac1 (p‐Rac1) and PAK (p‐PAK) was significantly increased in animals receiving s.c. BV injection plus intrathecal vehicle relative to s.c. saline control (P < 0.05, Figure 4A and B). In a parallel manner, the increased level of both p‐Rac1 and p‐PAK was reversed by intrathecal post‐administration of NSC23766 (P < 0.05, Figure 4A and B).
Figure 4.
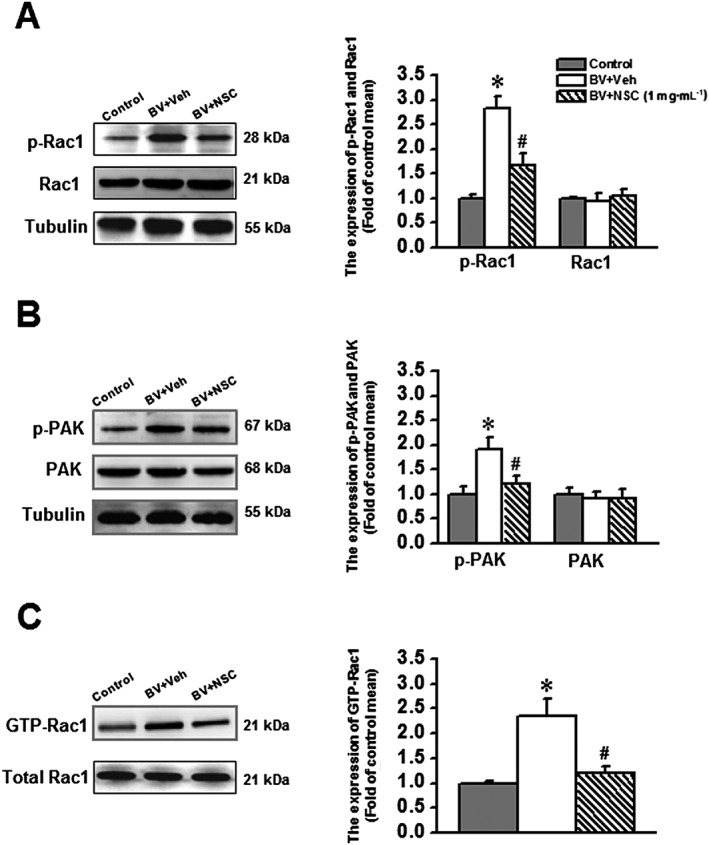
Effects of intrathecal NSC23766 (1 mg·mL−1), a selective inhibitor of Rac1, on the BV‐induced phosphorylation of Rac1 and its downstream effector PAK and enhancement of GTP‐bound Rac1 activity. The left panels of A, B and C show representative immunoblotting and pull‐down assay bands of phosphorylated form and total protein level of Rac1 and PAK and GTP‐bound Rac1. The right panels of A, B and C show quantitative analysis of the level of phosphorylated form and total protein expression for Rac1 (n = 5) and PAK (n = 5) and that of GTP bound Rac1 activity (n = 5) in group of animals receiving s.c. saline injection (Control), s.c. BV injection followed by intrathecal vehicle (saline, BV + Veh) and s.c. BV injection followed by intrathecal NSC23766 (BV + NSC). The relative density for bands of phosphorylated Rac1 or PAK was normalized to the value of total Rac1 or PAK. The relative density for bands of GTP‐Rac1 activity was normalized to the total Rac1. The relative density for bands of total Rac1 and PAK was normalized to Tubulin. Data are expressed as mean ± SEM. *P < 0.05, significantly different from Control. #P < 0.05, significantly different from BV + Veh. p‐Rac1, phosphorylated Rac1; p‐PAK, phosphorylated PAK.
Because the GTP‐bound Rac1 is the active form of Rac1, the GTP‐Rac1 activity was measured through a pull‐down assay to confirm whether the increased p‐Rac1 and p‐PAK reflect the active state of Rac1. As shown in Figure 4C, the GTP‐bound Rac1 activities were increased significantly in animals receiving s.c. BV injection and intrathecal vehicle (P < 0.05), whereas intrathecal NSC23766 completely reversed the increase of GTP‐bound Rac1, induced by s.c. BV (P < 0.05).
The MAPKs are downstream targets of GTP‐Rac1‐PAK signalling; thus, assessment of the activated MAPK levels, critically involved in the BV‐induced inflammatory pain state (Chen and Lariviere 2010; Chen and Guan, 2015) is of particular importance for understanding the functions of Rac1 signalling. As shown in Figure 5, the phosphorylated levels of ERK1/ERK2 (p‐ERK) and p38 MAPK (p‐p38), two isoforms of MAPKs, were significantly increased in animals receiving s.c. BV injection and intrathecal vehicle compared with the s.c. saline control; however, the increased level of both p‐ERK and p‐p38 was reversed by intrathecal NSC23766 in animals receiving s.c. BV. Compared with s.c. saline control, the total protein expression level of ERK1/ERK2 and p38 MAPK was not changed in rats either receiving a combined BV injection and intrathecal vehicle or BV injection and intrathecal NSC23766 (Figure 5).
Figure 5.
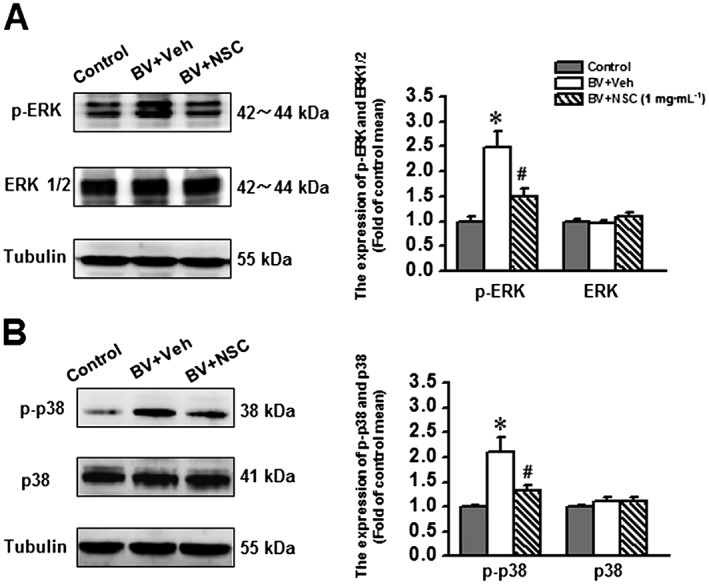
Effects of intrathecal intrathecal administration of NSC23766 (1 mg·mL−1), a selective inhibitor of Rac1, on the bee venomBV‐induced phosphorylation of ERK1/ERK2 and p38 MAPK. The left panels of A and B show representative immunoblotting bands of phosphorylated form and total protein level of ERK1/ERK2 and p38 MAPK. The right panels of A and B show quantitative analysis of the level of phosphorylated form and total protein expression for ERK1/ERK2 and p38 MAPK in group of animals receiving s.c. saline injection (Control, n = 5), s.c. BV injection followed by intrathecal administration of vehicle (saline, BV + Veh, n = 5) and s.c. BV injection followed by intrathecal NSC23766 (BV + NSC, n = 5).The relative density for bands of phosphorylated ERK1/ERK2 and p38 MAPK was normalized to the value of total ERK1/ERK2 and p38 MAPK. The relative density for bands of total ERK1/ERK2 and p38 MAPK was normalized to Tubulin. Data are expressed as mean ± SEM. *P < 0.05, significantly different from Control. #P < 0.05, significantly different from BV + Veh. p‐ERK1/p‐ERK2, phosphorylated ERK; p‐p38, phosphorylated p38 MAPK;
Preventive and therapeutic effects of intrathecal NSC23766 on BV‐induced persistent spontaneous nociception and pain hypersensitivity
To see whether blockade of Rac1 activity has preventive and/or therapeutic analgesic effects on peripheral inflammatory pain, behavioural tests were performed in a set of separate experiments. As shown in Figure 6A, intrathecal pre‐treatment with NSC23766, 5 min prior to s.c. BV, blocked the pain‐related paw flinches, in a dose‐related manner, relative to intrathecal vehicle control (P < 0.05, n = 5–6 animals). The averaged total number of the BV‐induced paw flinches during 1 h is shown in Figure 6B. Pre‐administration of intrathecal NSC23766 could also prevent the primary thermal and mechanical pain hypersensitivity (injection paw) as well as the mirror‐image thermal pain hypersensitivity (contralateral paw), in a dose‐dependent manner (P < 0.05 Veh + BV vs. Control, n = 14–18 animals for each group; P < 0.05 NSC + BV vs. Veh + BV; n = 6–7 animals for each dose; Figure 7). Because s.c. BV is known not to induce mirror‐image mechanical pain hypersensitivity (Chen and Chen, 2000; Chen and Lariviere, 2010; Chen and Guan, 2015), the failure to change the mechanical sensitivity of the contralateral paw by either of the three doses of intrathecal NSC23766 implied that activation of Rac1 signalling was not involved in the physiological process of pain sensation.
Figure 6.
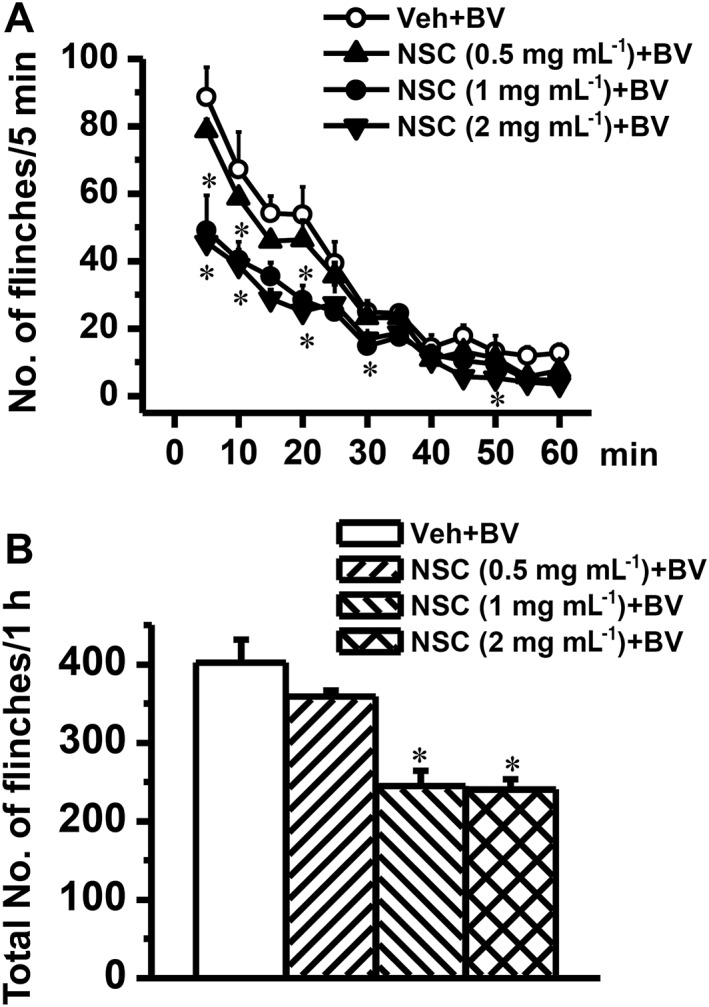
Effects of intrathecal pre‐administration of NSC23766 on BV‐induced paw flinch reflex. Graph A shows time courses of dose‐related effects of intrathecal NSC23766 (NSC) on the BV‐induced paw flinches. Graph B shows dose‐related effects of intrathecal NSC23766 on the total number of BV‐induced paw flinches. The drug was dissolved in physiological saline (vehicle; Veh) and administered 5 min prior to s.c. BV injection. Data are expressed as mean ± SEM. *P < 0.05, significantly different from Veh + BV.
Figure 7.

Effects of intrathecal pre‐administration of NSC23766 on BV‐induced pain hypersensitivity. Graph A shows dose‐related inhibitory effects of NSC23766 (NSC) on the BV‐induced primary (injection paw) and mirror‐image (contralalteral (Contl) paw) thermal hyperalgesia. Graph B shows partial inhibitory effects of NSC23766 on the BV‐induced primary mechanical hyperalgesia. The drug was dissolved in physiological saline (vehicle; Veh) and administered 5 min prior to s.c. BV injection. Data are expressed as mean ± SEM. #P < 0.05, significantly different from Control; *P < 0.05, significantly different from Veh + BV.
Post‐administration of intrathecal NSC23766, at a single concentration (1 mg·mL−1), not only completely reversed the BV‐induced bilateral thermal pain hypersensitivity (n = 13, P < 0.05) but also partly reversed the primary mechanical pain hypersensitivity (n = 14, P < 0.05) (Figure 8). Post‐administration of intrathecal NSC23766, at the same concentration, had no effects on the baseline bilateral thermal and mechanical pain sensitivity (Figure 8).
Figure 8.
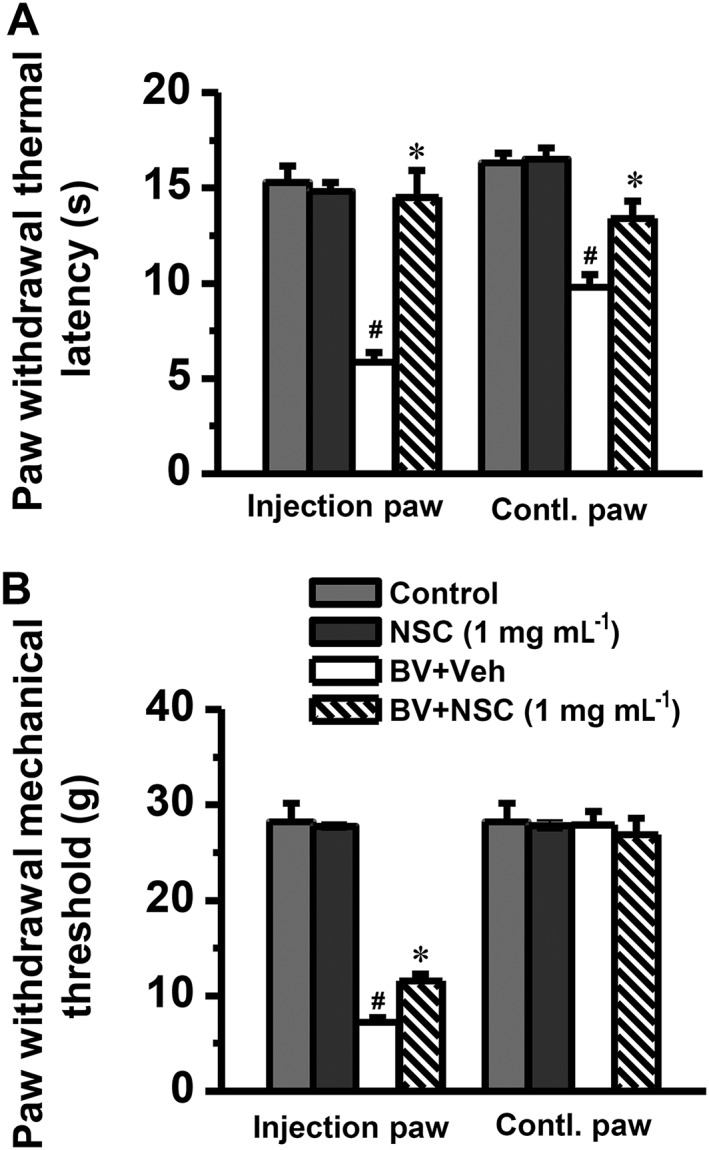
Effects of i.t. post‐administration of NSC23766 on BV‐induced pain hypersensitivity. Graph A shows complete reversal by NSC23766 (NSC) of the BV‐induced primary (injection paw) and mirror‐image contralalteral (Contl) paw thermal hyperalgesia. Graph B shows partial reversal effect of NSC23766 on the BV‐induced primary mechanical hyperalgesia. The drug was dissolved in physiological saline (vehicle; Veh) and administered 2 h after s.c. BV injection. Data are expressed as mean ± SEM. ## P < 0.05, significantly different from Control; *P < 0.05, significantly different from BV + Veh.
Lack of side effects on motor‐coordinating performance of rats following intrathecal NSC23766
During the training courses of the Rota‐Rod test, the time spent on the treadmill by both naïve rats and those rats receiving intrathecal saline or NSC23766 (1 mg·mL−1) was increased linearly from the first to the third trial but remained almost unchanged (plateau effect) for the later five trials (Figure 9). There was no difference in the time spent on the treadmill between naïve rats and rats receiving intrathecal saline and NSC23766 (n = 8 for each group, P > 0.05; Figure 9).
Figure 9.
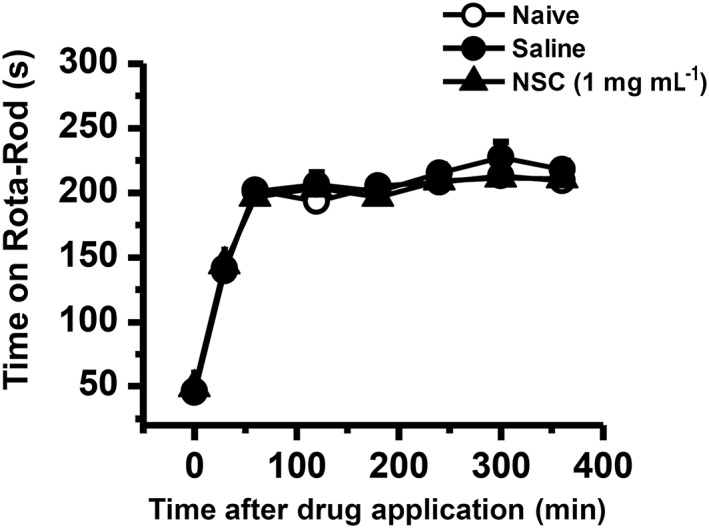
Effects of intrathecal NSC23766 on motor coordination. Graph shows effects of NSC23766 on motor coordination of naïve rats and rats receiving intrathecal saline or NSC23766. The drug was injected 5 min before the initiation of the test. A total of eight trials were conducted, and the first three trials were considered as the training session. NSC, NSC23766. Data are expressed as mean ± SEM.
Discussion
The present study identified an essential role of GTP‐Rac1‐PAK‐ERK/p38 MAPK signalling activities in the development and maintenance of peripheral inflammatory pain induced by s.c. BV injection. Inhibition of Rac1 with intrathecal NSC23766 inhibited the activation of GTP‐Rac1 and Rac1‐PAK signalling pathway and the phosphorylation of its downstream targets ERK1/ERK2 and p38 MAPK. The phosphorylation of ERK1/ERK2 and p38 MAPK are known to be involved in the BV‐induced and melittin‐induced pain‐related behaviours (Yu and Chen, 2005; Chen et al., 2006; Cao et al., 2007; Hao et al., 2008; Chen and Lariviere, 2010). Moreover, targeting the Rac1 signalling with intrathecal NSC23766 had both preventive and therapeutic analgesic effects upon the BV‐induced primary thermal and mechanical pain hypersensitivity as well as mirror‐image thermal pain hypersensitivity. Pre‐treatment with NSC23766 also attenuated BV‐induced persistent spontaneous pain‐related behaviours. Moreover, intrathecal NSC23766 exerted no effects on baseline pain sensitivity and motor coordination, suggesting that activation of GTP‐Rac1‐PAK signalling was only involved in the pathogenesis of pathological pain.
Involvement of Rac1 signalling in spinal neuronal plasticity in a peripheral inflammatory state
In the current study, Rac1‐like immunoreactivities were mainly localized within NeuN‐labelled neuronal profiles in both the superficial and the deep layers of rat spinal dorsal horn. However, Rac1 proteins were less expressed in astrocytic and microglial cells, even in rats with peripheral inflammatory pain. Although astrocytic and microglial cells increased markedly following s.c. BV injection, the cellular distribution of Rac1 proteins did not show any changes. These results strongly suggest that Rac1 is predominantly expressed in neuronal cell bodies but not in astrocytic and microglial cells in the dorsal horn. The expression of Rac1 in oligodendrocytes could not be completely excluded in the present study because there were many Rac1‐positive cellular profiles that did not label for NeuN. Rac1 is involved in the genesis and maturation of neuronal dendritic spines (Nakayama and Luo, 2000). It has been shown previously that spinal malformation of dendritic spines occurred 10 days after peripheral nerve injury (Tan et al., 2011) and 28 days after spinal cord injury or burn injury (Tan et al., 2008, 2013; Tan and Waxman, 2012). Chronic inhibition of Rac1 could attenuate the spine dysgenesis and spinal WDR neuronal hyperexcitability and alleviate pain hypersensitivity ((Tan et al., 2011, 2012, 2013, 2008). In the present study, Rac1 and its effector PAK were activated very quickly following s.c. BV injection (within 2 h). A single application of intrathecal NSC23766, at doses similar to that used in neuropathic pain models, was effective in attenuating BV‐induced persistent spontaneous pain‐related behaviours and thermal or mechanical hypersensitivity at the primary injury site and mirror‐image thermal hypersensitivity at the contralateral side. Whether spine remodelling also occurs in the spinal dorsal horn following s.c. BV injection remains to be studied.
It is believed that dendritic spine remodelling may reflect morphological changes of synaptic plasticity that contribute to spinal WDR neuronal sensitization and chronicity of pain under neuropathic conditions (Melemedjian and Price, 2012; Tan et al., 2011, 2012, 2013, 2008; Tan and Waxman, 2012, 2014). It is interesting to note that s.c. BV injection can activate spinal WDR neurons as well that give rise to long‐term spike discharges and increased responsiveness to various stimuli applied to their cutaneous receptive field (Chen et al., 1999a, 1998; You and Chen, 1999; You et al., 2003c; Li and Chen, 2004; Zheng et al., 2004a, 2004b; see also Chen and Lariviere, 2010; Chen and Guan, 2015). Taken together, it is possible that BV‐induced long‐term plastic changes of spinal WDR neurons may be caused by dendritic spine remodelling via activation of GTP‐Rac1‐PAK‐ERK/p38 MAPK signalling. This presumption can be supported by our previous results showing significant suppression of the BV‐induced spinal WDR neuronal spike discharges and enhancement of evoked responsiveness by cord dorsum application of ERK and p38 MAPK inhibitors (Yu and Chen, 2005; Li et al., 2008; see also Chen and Guan, 2015) and inhibition of the BV‐induced persistent spontaneous nociception and pain hypersensitivity by a p38 MAPK inhibitor (Cao et al., 2007). Whether dendritic spines of spinal dorsal horn neurons also undergo malformation and contribute to different behavioural phenotypes following s.c. BV injection warrants further studies.
Because s.c. BV injection also activates spinal astrocytic and microglial cells (Cui et al., 2008; Chen et al., 2013b) and Rac1‐like immunoreactivity was also shown to be co‐localized with GFAP and Iba1 in a few spinal cellular profiles, the involvement of glial cell Rac1 signalling in the BV‐induced spinal neuronal changes could not be completely excluded. Because p38 MAPK is activated in neuronal cell bodies earlier within the superficial layers (1 h after s.c. BV), followed by late activation in microglial cells within the deep layers of dorsal horn (24 h after s.c. BV) (Cui et al., 2008), the link between Rac1 and p38 MAPK signalling in microglial cells might be underestimated due to lack of long‐term time course observation in the present study. The action of Rac1‐PAK‐ERK/p38 MAPK in astrocytic cells is unlikely to be involved in the BV‐induced spinal neuronal plasticity, because neither ERKs nor p38 MAPK was seen in astrocytic cells in the spinal cord dorsal horn after s.c. BV injection (Cui et al., 2008).
Activation of GTP‐bound Rac1‐PAK‐ERK/p38 MAPK signalling in the spinal dorsal horn contributes to BV‐induced peripheral inflammatory pain
The GTP‐bound Rac1 is the active form of Rac1 and represents the activity of Rac1 in the living cells. Previous studies show that the phosphorylation of Rac1 can inhibit the GTP‐binding activity of Rac1 in clonal cell lines (Kwon et al., 2000; Tong et al., 2013). However, in the present study, we showed that the GTP‐bound Rac1 and the phosphorylated Rac1 both increased significantly following s.c. BV injection. Moreover, intrathecal NSC23766 could reverse the BV‐induced elevation of both GTP‐bound Rac1 and phosphorylated Rac1. One possible explanation concerning this discrepancy may be that the phosphorylation of Rac1 functions as a negative feedback loop to turn off Rac1 signalling following Rac1 activation. Another possibility underling the difference between our in vivo and in vitro results may be that Rho GTPases may execute diverse cell functions through multiple pathways. Nonetheless, by now, it is still difficult to identify the specific pathway activated by a given Rho GTPase (Wang and Zheng, 2007). It may also function differentially between clonal cell lines and mammalian neuronal cells between in vivo and in vitro experiments.
Although NSC23766 can block NMDA receptor function (Hou et al., 2014), this possibility is unlikely to underlie its analgesic effects in this experiment, because BV‐induced primary heat and mechanical hyperalgesia are known to be independent of NMDA receptors (Chen and Lariviere, 2010). The Rac1 inhibitor, NSC23766, exerted greater analgesic effects on BV‐induced thermal hyperalgesia than on mechanical hyperalgesia, which may be attributed to their different underlying mechanisms (see Chen and Lariviere, 2010). The underlying molecular mechanism for the analgesic effects of Rac1 inhibitors may be due to a disruption of the interaction between the Rac1‐PAK signalling pathway and the MAPK pathways − the downstream target signalling pathway of Rac1‐PAK (Stankiewicz et al., 2012). Our previous studies have shown that ERKs, p38 MAPK and JNK in the spinal dorsal horn can be phosphorylated by s.c. BV injection (Cui et al., 2008). Moreover, the spinal or peripheral inhibition of ERKs, p38 MAPK and JNK could differentially modulate BV‐induced or melittin‐induced pain‐related behaviours (Yu and Chen, 2005; Cao et al., 2007; Cui et al., 2008; Hao et al., 2008; Chen et al., 2009; see also Chen and Lariviere, 2010; Chen and Guan, 2015). Peripheral inhibition of ERK could suppress melittin‐induced long‐lasting spike discharges and enhancement of thermally nociceptive responsiveness of WDR neurons (Li et al., 2008; Yu et al., 2009). Our results here further showed that spinal ERK1/ERK2 and p38 MAPK were activated by s.c. BV injection, and that intrathecal NSC23766 could eliminate the activation of ERK1/ERK2 and p38 MAPK. Thus, it is possible that Rac1‐PAK activation may further activate the MAPK pathways and contribute to BV‐induced spinal dorsal horn neuronal plasticity and behavioural phenotypes of pain. As supporting evidence, the BV‐induced phosphorylation of ERK1/ERK2 was exclusively expressed in NeuN‐labelled neurons within the superficial layers of the dorsal horn, while the phosphorylated p38 MAPK was seen in both NeuN‐labelled neurons and Iba1‐labelled microglial cells within the deep layers of the dorsal horn (Cui et al., 2008). Taken together, it is proposed that GTP‐Rac1‐PAK‐ERK/p38 MAPK signalling may be involved in spinal dorsal horn neuronal plasticity that contribute to persistent spontaneous nociception and pain hypersensitivity induced by s.c. BV injection.
In summary, our current study provided another line of evidence showing that GTP‐bound Rac1‐PAK‐ERK/p38 MAPK signalling pathways were activated by peripheral inflammatory pain, and inhibition of Rac1 signalling by intrathecal NSC23766 resulted in preventive and therapeutic analgesic efficacy, implicating a potential therapeutic target for treatment of chronic pain by targeting Rac1 signalling.
Author contributions
J. C. designed the research work. Y. W., W. S. and C.‐L. L. performed the experiments. R.‐R. W., T. H., F. Y., Y. Y. and X.‐L. W. contributed partly to the behavioural tests. Y. W., Y.‐F. L., Z. L. and J. C. analysed the data. Y. W., Y. ‐F. L, S.‐M. G. and J. C. wrote the paper.
Conflicts of interest
The authors declare they have no conflict of interests.
Supporting information
Figure S1 Double immunofluorescent labeling of Rac1 and NeuN in naïve rats. Immunofluorescent labeling of Rac1 (A) and NeuN (B) in the spinal dorsal horn of naïve rats. C shows merged images of A and B. A1‐A2, B1‐B2, and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S2 Double immunofluorescent labeling of Rac1 and GFAP in naïve rats. Immunofluorescent labeling of Rac1 (A) and GFAP (B) in the spinal dorsal horn of naïve rats. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S3 Double immunofluorescent labeling of Rac1 and Iba1 in naïve rats. Immunofluorescent labeling of Rac1 (A) and Iba1 (B) in the spinal dorsal horn of naïve rats. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S4 Quantification of double immunofluorescent labeling of Rac1 with NeuN‐, GFAP‐ and Iba1‐positive profiles in naïve rats. The percentage of NeuN‐labeled neurons, GFAP‐labeled astrocytes and Iba1‐labeled microglial cells in Rac1‐positive profiles was calculated in both superficial (layers I‐II) and deep layers (layers III‐VI) of the spinal dorsal horn, respectively. Data are expressed as mean ± SEM.
Figure S5 Double immunofluorescent labeling of Rac1 and GFAP in vehicle‐treated rats. Immunofluorescent labeling of Rac1 (A) and GFAP (B) in the injection side of spinal dorsal horn of rats that received s.c. injection of saline vehicle into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S6 Double immunofluorescent labeling of Rac1 and GFAP in BV‐treated rats. Immunofluorescent labeling of Rac1 (A) and GFAP (B) in the injection side of spinal dorsal horn of rats that received s.c. injection of BV into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S7 Double immunofluorescent labeling of Rac1 and Iba1 in vehicle‐treated rats. Immunofluorescent labeling of Rac1 (A) and Iba1 (B) in the injection side of spinal dorsal horn of rats that received a s.c. injection of saline vehicle into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S8 Double immunofluorescent labeling of Rac1 and Iba1 in BV‐treated rats. Immunofluorescent labeling of Rac1 (A) and Iba1 (B) in the injection side of spinal dorsal horn of rats that received a s.c. injection of BV into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S9 Negative control for immunofluorescent labeling of Rac1. The immunofluorescence was conducted as described in the methods except that the primary antibody against Rac1 was replaced with 1% bovine serum in 0.01 M PBST. A1 and B1 (magnified) showed images of spinal dorsal horn that incubated with secondary antibody (FITC); A2 and B2 (magnified) showed the background; A and B (magnified) showed merged images of A1 and A2, and B1 and B2. Scale bars, 150 μm (A1, A2, A), 100 μm (B1, B2, B).
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
This work was supported by grants from National Basic Research Program of China (2013CB835100, 2011CB504100), National Key Technology R&D Program (2013BAI04B04), the NSFC (81171049), the Twelfth Five‐Year project (AWS12J004) to J. C., the NSFC (31500854) and the Social Development Project of Shaanxi Province (2015SF003) to Y. W.
Wang, Y. , Lu, Y.‐F. , Li, C.‐L. , Sun, W. , Li, Z. , Wang, R.‐R. , He, T. , Yang, F. , Yang, Y. , Wang, X.‐L. , Guan, S.‐M. , and Chen, J. (2016) Involvement of Rac1 signalling pathway in the development and maintenance of acute inflammatory pain induced by bee venom injection. British Journal of Pharmacology, 173: 937–950. doi: 10.1111/bph.13413.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao FL, Liu MG, Hao J, Li Z, Lu ZM, Chen J (2007). Different roles of spinal p38 and c‐Jun. N‐terminal kinase pathways in bee venom‐induced multiple pain‐related behaviors. Neurosci Lett 427: 50–54. [DOI] [PubMed] [Google Scholar]
- Chen HS, Chen J (2000). Secondary heat, but not mechanical, hyperalgesia induced by subcutaneous injection of bee venom in the conscious rat: effect of systemic MK‐801, a non‐competitive NMDA receptor antagonist. Eur J Pain 4: 389–401. [DOI] [PubMed] [Google Scholar]
- Chen HS, Chen J, Sun YY (2000). Contralateral heat hyperalgesia induced by unilaterally intraplantar bee venom injection is produced by central changes: a behavioral study in the conscious rat. Neurosci Lett 284: 45–48. [DOI] [PubMed] [Google Scholar]
- Chen HS, He X, Qu F, Kang SM, Yu Y, Liao D, et al (2009). Differential roles of peripheral mitogen‐activated protein kinase signal transduction pathways in bee venom‐induced nociception and inflammation in conscious rats. J Pain 10: 201–207. [DOI] [PubMed] [Google Scholar]
- Chen HS, Wang JX, Zhang JH, Li FP, Qu F, Liu BJ, et al (2013b). Contribution of the spinal microglia to bee venom‐induced inflammatory pain in conscious rats. Neurosci Lett 534: 301–305. [DOI] [PubMed] [Google Scholar]
- Chen J (2003). The bee venom test: a novel useful animal model for study of spinal coding and processing of pathological pain information In: {0} (eds)Chen J, Chen ACN, Han JS, Willis WD. Experimental pathological pain: from molecules to brain functions. Science Press: Beijing, pp. 77–110. [Google Scholar]
- Chen J (2007). Processing of different ‘phenotypes’ of pain by different spinal signaling pathways In: {0} (ed)Kumamoto K. Cellular and molecular mechanisms for the modulation of nociceptive transmission in the peripheral and central nervous systems, recent research development series. Research Sign Post: Kerala, pp. 147–165. [Google Scholar]
- Chen J (2008). Spinal processing of bee venom‐induced pain and hyperalgesia. Sheng Li Xue Bao: Shanghai, PR China: 645–652. [PubMed] [Google Scholar]
- Chen J, Chen HS (2001). Pivotal role of capsaicin‐sensitive primary afferents in development of both heat and mechanical hyperalgesia induced by intraplantar bee venom injection. Pain 91: 367–376. [DOI] [PubMed] [Google Scholar]
- Chen J, Guan SM (2015). Bee venom and pain In: {0} (ed)Gopalakrishnakone P. Toxinology: Toxins and Drug Discovery. Springer Netherlands: Dordrecht. [Google Scholar]
- Chen J, Han JS, Zhao ZQ, Wei F, Hsieh JC, Bao L, et al (2013a). Pain In: {0} (ed)Pfaff DW. Neuroscience in the 21st Century: From Basic to Clinical. New York. Springer: New York, pp. 965–1023. [Google Scholar]
- Chen J, Lariviere WR (2010). The nociceptive and anti‐nociceptive effects of bee venom injection and therapy: a double‐edged sword. Prog Neurobiol 92: 151–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li HL, Luo C, Li Z, Zheng JH (1999a). Involvement of peripheral NMDA and non‐NMDA receptors in development of persistent firing of spinal wide‐dynamic‐range neurons induced by subcutaneous bee venom injection in the cat. Brain Res 844: 98–105. [DOI] [PubMed] [Google Scholar]
- Chen J, Luo C, Li HL (1998). The contribution of spinal neuronal changes to development of prolonged, tonic nociceptive responses of the cat induced by subcutaneous bee venom injection. Eur J Pain 2: 359–376. [DOI] [PubMed] [Google Scholar]
- Chen J, Luo C, Li H, Chen H (1999b). Primary hyperalgesia to mechanical and heat stimuli following subcutaneous bee venom injection into the plantar surface of hindpaw in the conscious rat: a comparative study with the formalin test. Pain 83: 67–76. [DOI] [PubMed] [Google Scholar]
- Chen YN, Li KC, Li Z, Shang GW, Liu DN, Lu ZM, et al (2006). Effects of bee venom peptidergic components on rat pain‐related behaviors and inflammation. Neuroscience 138: 631–640. [DOI] [PubMed] [Google Scholar]
- Cui XY, Dai Y, Wang SL, Yamanaka H, Kobayashi K, Obata K, et al (2008). Differential activation of p38 and extracellular signal‐regulated kinase in spinal cord in a model of bee venom‐induced inflammation and hyperalgesia. Mol Pain 4: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA, et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 2671–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y (2004). Rational design and characterization of a Rac GTPase‐specific small molecule inhibitor. Proc Natl Acad Sci U S A 101: 7618–7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Liu MG, Yu YQ, Cao FL, Li Z, Lu ZM, et al (2008). Roles of peripheral mitogen‐activated protein kinases in melittin‐induced nociception and hyperalgesia. Neuroscience 152: 1067–1075. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ (2008). Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev. Mol Cell Biol 9: 690–701. [DOI] [PubMed] [Google Scholar]
- Hill CS, Wynne J, Treisman R (1995). The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81: 1159–1170. [DOI] [PubMed] [Google Scholar]
- Hou H, Chávez AE, Wang CC, Yang H, Gu H, Siddoway BA, et al (2014). The Rac1 inhibitor NSC23766 suppresses CREB signaling by targeting NMDA receptor function. J Neurosci 34: 140006–140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, D'Mello SR (2005). p21‐Activated kinase‐1 is necessary for depolarization‐mediated neuronal survival. J Neurosci Res 79: 809–815. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T, Kwon DY, Chun J, Kim JH, Kang SS (2000). Akt protein kinase inhibits Rac1‐GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem 275: 423–428. [DOI] [PubMed] [Google Scholar]
- Lariviere WR, Melzack R (1996). The bee venom test: a new tonic‐pain test. Pain 66: 271–277. [DOI] [PubMed] [Google Scholar]
- Li KC, Chen J (2004). Altered pain‐related behaviors and spinal neuronal responses produced by s.c. injection of melittin in rats. Neuroscience 126: 753–762. [DOI] [PubMed] [Google Scholar]
- Li MM, Yu YQ, Fu H, Xie F, Xu LX, Chen J (2008). Extracellular signal‐regulated kinases mediate melittin‐induced hypersensitivity of spinal neurons to chemical and thermal but not mechanical stimuli. Brain Res Bull 77: 227–232. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Price TJ (2012). Dendritic spine plasticity as an underlying mechanism of neuropathic pain: commentary on Tan et al . Exp Neurol 233: 740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama AY, Luo L (2000). Intracellular signaling pathways that regulate dendritic spine morphogenesis. Hippocampus 10: 582–586. [DOI] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A (1995). An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269: 1270–1272. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP et al, NC‐IUPHAR (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren LY, Lu ZM, Liu MG, Yu YQ, Li Z, Shang GW, et al (2008). Distinct roles of the anterior cingulate cortex in spinal and supraspinal bee venom‐induced pain behaviors. Neuroscience 153: 268–278. [DOI] [PubMed] [Google Scholar]
- Shifrin Y, Pinto VI, Hassanali A, Arora PD, McCulloch CA (2012). Force‐induced apoptosis mediated by Rac/Pak/p38 signaling pathway is regulated by filamin A. Biochem J 445: 57–67. [DOI] [PubMed] [Google Scholar]
- Stankiewicz TR, Linseman DA (2014). Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci 8: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz TR, Loucks FA, Schroeder EK, Nevalainen MT, Tyler KL, Aktories K, et al (2012). Signal transducer and activator of transcription‐5 mediates neuronal apoptosis induced by inhibition of Rac GTPase activity. J Biol Chem 287: 16835–16848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AM, Chang YW, Zhao P, Hains BC, Waxman SG (2011). Rac1‐regulated dendritic spine remodeling contributes to neuropathic pain after peripheral nerve injury. Exp Neurol 232: 222–233. [DOI] [PubMed] [Google Scholar]
- Tan AM, Samad OA, Fischer TZ, Zhao P, Persson AK, Waxman SG (2012). Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci 32: 6795–6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AM, Samad OA, Liu S, Bandaru S, Zhao P, Waxman SG (2013). Burn injury‐induced mechanical allodynia is maintained by Rac1‐regulated dendritic spine dysgenesis. Exp Neurol 248: 509–519. [DOI] [PubMed] [Google Scholar]
- Tan AM, Stamboulian S, Chang YW, Zhao P, Hains AB, Waxman SG, et al (2008). Neuropathic pain memory is maintained by Rac1‐regulated dendritic spine remodeling after spinal cord injury. J Neurosci 28: 13173–13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AM, Waxman SG (2012). Spinal cord injury, dendritic spine remodeling, and spinal memory mechanisms. Exp Neurol 235: 142–151. [DOI] [PubMed] [Google Scholar]
- Tan AM, Waxman SG (2014). Dendritic spine dysgenesis in neuropathic pain. Neurosci Lett 601: 54–60. [DOI] [PubMed] [Google Scholar]
- Tong J, Li L, Ballermann B, Wang Z (2013). Phosphorylation of Rac1 T108 by extracellular singal‐regulated kinase in response to epidermal growth factor: a novel mechanisms to regulate Rac1 function. Mol Cell Biol 33: 4538–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, et al (2015). A classification of chronic pain for ICD‐11. Pain 156: 1003–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al (2008). Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 70: 1630–1635. [DOI] [PubMed] [Google Scholar]
- Wang L, Zheng Y (2007). Cell type‐specific functions of Rho GTPases revealed by gene targeting in mice. Trends Cell Biol 17: 58–64. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fu M, Wang L, Liu J, Li Y, Brakebusch C, et al (2013). P21‐activated kinase 1 (PAK1) can promote ERK activation in a kinase‐independent manner. J Biol Chem 288: 20093–20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You HJ, Chen J (1999). Differential effects of subcutaneous injection of formalin and bee venom on responses of wide‐dynamic‐range neurons in spinal dorsal horn of the rat. Eur J Pain 3: 177–180. [Google Scholar]
- You HJ, Morch CD, Chen J, Arendt‐Nielsen L (2003a). Differential antinociceptive effects induced by a selective cyclooxygenase‐2 inhibitor (SC‐236) on dorsal horn neurons and spinal withdrawal reflexes in anesthetized spinal rats. Neuroscience 121: 459–472. [DOI] [PubMed] [Google Scholar]
- You HJ, Morch CD, Chen J, Arendt‐Nielsen L (2003b). Role of central NMDA versus non‐NMDA receptor in spinal withdrawal reflex in spinal anesthetized rats under normal and hyperexcitable conditions. Brain Res 981: 12–22. [DOI] [PubMed] [Google Scholar]
- You HJ, Morch CD, Chen J, Arendt‐Nielsen L (2003c). Simultaneous recordings of wind‐up of paired spinal dorsal horn nociceptive neuron and nociceptive flexion reflex in rats. Brain Res 960: 235–245. [DOI] [PubMed] [Google Scholar]
- Yu YQ, Zhao F, Guan SM, Chen J (2011). Antisense‐mediated knockdown of Nav1. 8, but not Nav1. 9, generates inhibitory effects on complete Freund's adjuvant‐induced inflammatory pain in rat. PLoS One 6: e19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YQ, Zhao ZY, Chen XF, Xie F, Yang Y, Chen J (2013). Activation of tetrodotoxin‐resistant sodium channel NaV1. 9 in rat primary sensory neurons contributes to melittin‐induced pain behavior. Neuromolecular Med 15: 209–217. [DOI] [PubMed] [Google Scholar]
- Yu YQ, Chen J (2005). Activation of spinal extracellular signaling‐regulated kinases by intraplantar melittin injection. Neurosci Lett 381: 194–198. [DOI] [PubMed] [Google Scholar]
- Yu YQ, Zhao F, Chen J (2009). Activation of ERK1/2 in the primary injury site is required to maintain melittin‐enhanced wind‐up of rat spinal wide‐dynamic‐range neurons. Neurosci Lett 459: 137–141. [DOI] [PubMed] [Google Scholar]
- Zheng JH, Chen J (2001). Differential roles of spinal neurokinin 1/2 receptors in development of persistent spontaneous nociception and hyperalgesia induced by subcutaneous bee venom injection in the conscious rat. Neuropeptides 35: 32–44. [DOI] [PubMed] [Google Scholar]
- Zheng JH, Chen J, Arendt‐Nielsen L (2004a). Complexity of tissue injury‐induced nociceptive discharge of dorsal horn wide dynamic range neurons in the rat, correlation with the effect of systemic morphine. Brain Res 1001: 143–149. [DOI] [PubMed] [Google Scholar]
- Zheng JH, Feng W, Jian Z, Chen J (2004b). Age‐related changes in deterministic behaviors of nociceptive firing of rat dorsal horn neurons. Sheng Li Xue Bao 56: 178–182. [PubMed] [Google Scholar]
- Zimmermann M (1983). Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16: 109–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Double immunofluorescent labeling of Rac1 and NeuN in naïve rats. Immunofluorescent labeling of Rac1 (A) and NeuN (B) in the spinal dorsal horn of naïve rats. C shows merged images of A and B. A1‐A2, B1‐B2, and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S2 Double immunofluorescent labeling of Rac1 and GFAP in naïve rats. Immunofluorescent labeling of Rac1 (A) and GFAP (B) in the spinal dorsal horn of naïve rats. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S3 Double immunofluorescent labeling of Rac1 and Iba1 in naïve rats. Immunofluorescent labeling of Rac1 (A) and Iba1 (B) in the spinal dorsal horn of naïve rats. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S4 Quantification of double immunofluorescent labeling of Rac1 with NeuN‐, GFAP‐ and Iba1‐positive profiles in naïve rats. The percentage of NeuN‐labeled neurons, GFAP‐labeled astrocytes and Iba1‐labeled microglial cells in Rac1‐positive profiles was calculated in both superficial (layers I‐II) and deep layers (layers III‐VI) of the spinal dorsal horn, respectively. Data are expressed as mean ± SEM.
Figure S5 Double immunofluorescent labeling of Rac1 and GFAP in vehicle‐treated rats. Immunofluorescent labeling of Rac1 (A) and GFAP (B) in the injection side of spinal dorsal horn of rats that received s.c. injection of saline vehicle into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S6 Double immunofluorescent labeling of Rac1 and GFAP in BV‐treated rats. Immunofluorescent labeling of Rac1 (A) and GFAP (B) in the injection side of spinal dorsal horn of rats that received s.c. injection of BV into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S7 Double immunofluorescent labeling of Rac1 and Iba1 in vehicle‐treated rats. Immunofluorescent labeling of Rac1 (A) and Iba1 (B) in the injection side of spinal dorsal horn of rats that received a s.c. injection of saline vehicle into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S8 Double immunofluorescent labeling of Rac1 and Iba1 in BV‐treated rats. Immunofluorescent labeling of Rac1 (A) and Iba1 (B) in the injection side of spinal dorsal horn of rats that received a s.c. injection of BV into a hindpaw. C shows merged images of A and B. A1‐A2, B1‐B2 and C1‐C2 show enlarged images of the insets in A, B and C, respectively. Scale bars, 150 μm (A‐C), 50 μm (A1‐C1, A2‐C2).
Figure S9 Negative control for immunofluorescent labeling of Rac1. The immunofluorescence was conducted as described in the methods except that the primary antibody against Rac1 was replaced with 1% bovine serum in 0.01 M PBST. A1 and B1 (magnified) showed images of spinal dorsal horn that incubated with secondary antibody (FITC); A2 and B2 (magnified) showed the background; A and B (magnified) showed merged images of A1 and A2, and B1 and B2. Scale bars, 150 μm (A1, A2, A), 100 μm (B1, B2, B).
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item