

Abstract
Background and Purpose
Alternatively activated macrophages (AAMs) are important cells in the resolution of inflammation and tissue repair. We examined the impact of myofibroblasts, a vital cell in wound healing and tissue repair, on the development and function of AAMs.
Experimental Approach
The interaction between AAMs and myofibroblasts was tested using conditioned medium from murine dermal myofibroblasts and bone marrow‐derived macrophages. AAMs were differentiated with IL‐4 and IL‐13.
Key Results
Conditioned medium from myofibroblasts enhanced the expression of AAM markers, arginase 1 and Ym1 (chitinase‐3‐like 3) and the spontaneous production of IL‐10, while suppressing LPS‐induced nitric oxide production. IL‐6 from the myofibroblasts contributed to the amplification of the AAM phenotype; the selective COX‐2 inhibitor, NS‐398, significantly reduced the ability of myofibroblasts to promote an AAM phenotype. Pharmacological analyses indicated that myofibroblast‐derived IL‐6 enhanced arginase activity and spontaneous IL‐10 output, while PGE2, via the EP4 receptor, enhanced arginase expression and LPS‐evoked IL‐10 production. PGD2 suppressed LPS‐evoked nitric oxide via the DP1 receptor. Reciprocally, conditioned medium from macrophages treated with IL‐4 + IL‐13 and myofibroblast conditioned medium components, but not macrophages given IL‐4 + IL‐13 only, reduced myofibroblast migration, the expression of COX‐2, and the production of PGE2 and PGD2.
Conclusions and Implications
These findings define mechanisms by which myofibroblasts enhance an AAM phenotype, which can promote wound healing directly, and/or via feedback communication to the myofibroblast, subsequently down‐regulating its capacity to promote AAM function. This is an important homeostatic regulatory pathway in wound healing that can also limit unwanted fibrosis.
Abbreviations
- AAM
alternatively activated macrophage
- ECM
extracellular matrix
- MCM
macrophage conditioned medium
- MFbCM
myofibroblast conditioned medium
- M‐CSF
macrophage‐colony stimulating factor
- SMA
smooth muscle actin
Tables of Links
| TARGETS | |
| GPCRs a | Enzymes b |
| DP1 receptor | Arginase |
| DP2 receptor | Arginase 1 (ARG1) |
| EP1 receptor | COX‐1 |
| EP2 receptor | COX2 |
| NOS |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Macrophages are recognized as an important first line of defence as a result of their phagocytic activity. Furthermore, it is clear that macrophages serve many functions: as circumstances dictate, they can be active participants in pro‐inflammatory events and, conversely, participate in the suppression of inflammation, promote tissue recovery after injury, and return tissues to their homeostatic set points (Medzhitov, 2001; Gordon, 2003). Currently, macrophages are broadly classified as IFNγ ± bacterial product‐driven classically activated macrophages or anti‐inflammatory/pro‐resolution alternatively activated macrophages (AAMs), of which there are three major groups (Mantovani et al., 2004). However, these divisions do not denote terminally differentiated phenotypes (Galli et al., 2011), and the impact of the microenvironment is a major determinant of macrophage function. For example, an AAM phenotype is enhanced by IL‐33, IL‐10, PPARγ activation and oestrogen receptor ligation (Odegaard et al., 2007; Kurowska‐Stolarska et al., 2009; Schreiber et al., 2009; Fernando et al., 2014), whereas pro‐inflammatory signals (e.g. toll‐like receptor ligands and IFNγ) can inhibit AAM development and function (Chartouni El and Rehli, 2010; Nagy et al., 2013). While a variety of soluble signals influence AAM behaviour, much less is known of how other cell types may alter their function and phenotype: the myofibroblast could be of particular importance is this context.
Under baseline conditions, myofibroblasts exist as ‘inactive’ fibroblasts, whose primary role is to facilitate extracellular matrix (ECM) turnover through the synthesis of ECM proteins and degrading enzymes (Tomasek et al., 2002; Darby and Hewitson, 2007). Following tissue damage, and under the influence of cytokines, such as TGF‐β, fibroblasts differentiate into their activated myofibroblast form (Tomasek et al., 2002) and produce greater amounts of cytokines and ECM proteins to expedite wound closure. In addition, myofibroblasts can promote tissue recovery via interactions with other cells present at the site of injury (Werner et al., 2007). Recent studies indicate that myofibroblasts can promote the formation of inducible regulatory T‐cells necessary for the maintenance of intestinal homeostasis (Pinchuk et al., 2011), and that fibroblast‐mediated inhibition of arthritis was accompanied by the induction of regulatory and immunosuppressive T‐cells (Bouffi et al., 2011).
A myofibroblast–macrophage interaction is not unprecedented: myofibroblasts can promote monocyte‐to‐macrophage differentiation via the production of macrophage‐colony stimulating factor (M‐CSF) and recruit macrophages to sites of injury (Wynn and Barron, 2010; Barron and Wynn, 2011). However, there is a glaring gap in the knowledge of if, and how myofibroblasts affect AAMs, specifically IL‐4 + IL‐13 (from, for example, mast cells or innate lymphoid cells in health and disease) differentiated AAMs or M2a cells. Consequently, the current study was designed to assess the communication between myofibroblasts and AAMs mediated by the production of soluble factors. Our findings support the concept that myofibroblast‐derived IL‐6 and COX‐2‐derived PGE2 and PGD2 enhance the phenotype of an AAM. Moreover, AAMs differentiated in the presence of myofibroblast‐derived conditioned medium were, in turn, capable of down‐regulating myofibroblast activities, including cell migration and PG output. Thus, we present a novel and potentially crucial homeostatic feedback loop between myofibroblasts and IL‐4 + IL‐13‐differentiated AAMs that would be important for rapid and efficient wound repair, while preventing the development of fibrosis.
Methods
Mice
All experiments complied with the Canadian and Institutional guidelines for animal welfare. Male BALB/c and C57/Bl6 mice were purchased from Charles River (Quebec City, QB, Canada). C57/Bl6 IL‐6−/− mice and wild‐type controls were a gift from Dr M. Swain (University of Calgary). All studies complied with the ARRIVE guidelines (Killkenny et al., 2010) and the editorial on reporting animal studies (McGrath and Lilley, 2015), in which we sought to minimize the numbers of mice needed to derive robust data (see data presentation ).
Myofibroblast culture
Fibroblasts were isolated from mice using an established protocol (Singh and Sharma, 2011). Ear explants were minced and incubated in 0.25% trypsin‐EDTA (Invitrogen Canada Inc., Burlington, ON, Canada) for 1 h at 37°C to remove the epidermis. Explants were placed in tissue culture plates, and after 2–5 days, fibroblasts migrated out of the tissue. After 2 weeks, explants were removed, and cells were re‐seeded and cultured in medium [DMEM, 10% FBS and 2% penicillin‐streptomycin (pen/strep) (Invitrogen Canada Inc.)]. For conditioned medium experiments, fibroblasts were seeded at a density of 105 cells in a 6‐well plate and grown for 7 days. The culture process induced a myofibroblast phenotype (Tomasek et al., 2002) as assessed by α‐smooth muscle actin (SMA) expression (determined by immunocytochemistry). Medium was replaced on day 7 and collected 2 days later for use as myofibroblast conditioned medium (MFbCM) in experiments.
Culture of bone marrow‐derived macrophages
Bone marrow was isolated from murine femurs and tibias and cultured in medium (RPMI1640, 20% FBS, 1.2% GlutaMAX, 2.4% pen/strep (all from Invitrogen) supplemented with 20 ng·mL−1 mouse M‐CSF (R&D Systems Inc., Minneapolis, MN, USA) (Kurowska‐Stolarska et al., 2009; Fernando et al., 2014). Differentiated macrophages were determined by flow cytometry to be 95% F4/80+ and CD11b+ (data not shown). Following differentiation, bone marrow‐derived macrophages were stimulated for 48 h with 20 ng·mL−1 (each) of IL‐4 + IL‐13 (Cedarlane Laboratories, Burlington, ON, Canada) to induce AAMs as before (Fernando et al., 2014) ± reagents as defined in experiments. To assess nitric oxide (NO) production, cells were rinsed with PBS and challenged with LPS (1 μg·mL−1) for an additional 24 h after cytokine ± MFbCM stimulation.
Arginase assay
Macrophage arginase activity was assessed by measuring urea production, a by‐product of the arginase reaction (Fernando et al., 2014). Arginase activity is expressed as units per 106 cells, where 1 unit equals the amount of enzyme needed to hydrolyse 1 μM of arginine min‐1.
Nitric oxide (Griess) assay
Nitric oxide production was determined by measuring nitrite (the stable breakdown product of NO metabolism) in cell supernatants. Supernatants were combined with an equal volume of 2% sulphanilamide and 0.1% N‐1‐naphthylethylenediamine dihydrochloride (both from Sigma‐Aldrich Canada, Ltd., Oakville, ON, Canada) to convert nitrite into a magenta coloured azo‐compound with a measurable absorbance at 540 nm (Fernando et al., 2014).
Neutralization of IL‐6
MFbCM was incubated for 1 h at 37°C with 10 μg·mL−1 goat anti‐IL‐6 antibody (R&D Systems Inc.) or normal goat serum (Invitrogen). These samples were then used to treat macrophages in the presence of IL‐4 + IL‐13, and arginase activity assessed 48 h later.
Immunoblotting
Cells were lysed in modified Radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl, 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate and 0.1% SDS) supplemented with protease inhibitor cocktail (Promega, Madison, WI, USA). Protein concentrations were determined using the Bradford assay (Bio‐Rad Laboratories Canada, Mississauga, ON, Canada). Samples (10–20 μg) were run by SDS‐PAGE (4% stacking, 8% separating) and transferred to a nitrocellulose membrane. Membranes were incubated with primary rabbit polyclonal antibodies to arginase 1 (ARG1) (1:1000, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), Ym1 (1:1000, Stem Cell Technologies, Vancouver, BC, Canada), α‐SMA (1:1000, Abcam Inc., Toronto, ON, Canada), vimentin (1:1000, Cell Signalling Technologies, Danvers, MA, USA) and a goat polyclonal antibody to β‐actin (1:1000, Santa Cruz). Subsequently, membranes were incubated with appropriate anti‐goat or anti‐rabbit secondary antibodies (all at 1:20000, Santa Cruz). Membranes were exposed to Western Lightning Plus Enhanced Chemiluminescence Solution (PerkinElmer, Woodbridge, ON, Canada) for 1 min, exposed to X‐Omat Blue film (PerkinElmer) for 5 s to 10 min and developed using an automatic film developer.
Quantitative polymerase chain reaction (qPCR)
RNA was isolated using TRIzol (Invitrogen) and quantified using a NanoDrop (Thermo Scientific, Waltham, MA, USA), as previously described (Fernando et al., 2014). Isolated RNA (1 ng) was used to generate cDNA via iScript RT kit (Bio‐Rad) in a MyCycler thermocycler (Bio‐Rad). cDNA was added to 300 nM gene‐specific primers (18S For 5′‐CGCGGTTCTATTTTGTTGGT‐3′, Rev. 5′‐AGTCGGCATCGTTTATGGTC‐3′; COX‐2 For 5′‐AGAAGGAAATGGCTGCAGAA‐3′, Rev. 5′‐GCTCGGCTTCCAGTATTGAG‐3′; α‐SMA For 5′‐CAGCCAGTCGCTGTCAGGAACC‐3′, Rev. 5′‐ACCAGCGAAGCCGGCCTTACA‐3′) and 1× SYBR green reaction mix (Bio‐Rad). Changes in gene expression were assessed using the Mastercycler Real Time RT‐PCR Thermocycler (Eppendorf Canada, Mississauga, ON, Canada), and data were analysed using the 2−ΔCT method using 18S as a housekeeper gene and normalized to expression in untreated controls (Schmittgen and Livak, 2008).
Inhibition of COX‐1 and ‐2
Myofibroblasts (isolated from IL‐6−/− mice in order to unequivocally assess the IL‐6‐independent effect on AAM development) were seeded at 105 cells per well and grown for 7 days as described above. Medium was changed, and cells were exposed to indomethacin (non‐selective COX inhibitor, 1 μM, Sigma‐Aldrich), SC‐560 (COX‐1 inhibitor, 1 μM, Sigma‐Aldrich) or NS‐398 (COX‐2 inhibitor, 20 μM, Cayman Chemicals, Ann Arbour, MI, USA) at 0 h and again at 24 h. Supernatants were then collected for AAM differentiation experiments.
Assessment of PG involvement
Macrophages were cultured with IL‐4 + IL‐13 ± PGE2 or PGD2 in various combinations and changes in arginase activity, NO, and IL‐10 production were assessed. To determine the PG receptor involved, IL‐4 + IL‐13‐treated macrophages were co‐treated with the selective EP4 receptor agonist, L‐902,688, the EP2 receptor agonist, ONO‐AE1‐259, the DP1 receptor agonist, BW 245C, or the DP2 receptor agonist, 13,14‐dihydro‐15‐keto PGD2 for 48 h, after which arginase activity and/or LPS‐evoked NO were assessed. In other experiments, macrophages were pre‐treated with varying concentrations of the EP4 receptor antagonist (L‐191,982), the DP1 receptor antagonist (BW A868C) or the DP2 receptor antagonist (CAY10471) for 1 h at 37°C, followed by treatment with IL‐6−/− MFbCM and IL‐4 + IL‐13 or PGD2 and IL‐4 + IL‐13. All PG receptor agonists and antagonists were from Cayman Chem.
MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assay
To determine the effect of conditioned medium from AAMs on myofibroblast mitochondrial activity, the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromid) (MTT) assay was used. Myofibroblasts (104 per well) were seeded in a 96‐well plate and cultured for 24 h. Macrophage conditioned medium (MCM) was added to wells at a 1:1 ratio with culture medium (DMEM + 10% FBS + 2% pen/strep). Cells were then cultured for an additional 24 h, MTT reagent (5 mg·mL−1, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) (Sigma Aldrich) was added to each well (100 μL) and incubated for 4 h at 37 C. The MTT reagent was then removed, and formazan crystals were solubilized in DMSO. Absorbance was measured at 595 nm (Niks and Otto, 1990).
Measurement of cytokines and PGs
Cytokine [IL‐10, IL‐6, IL‐33, TGFβ (measured before and after acidification, the former showing total pool of TGFβ and the latter levels of bioactive TGFβ)] production by fibroblasts was determined by sandwich ELISA (R&D Systems Inc.) and PGs (PGE2 and PGD2) by enzyme immunoassay (EIA) (Cayman Chem.), according to the manufacturer's instructions. Levels of LIF, VEGF, M‐CSF, PDGF‐BB, basic‐FGF, IL‐15, IL‐18, MIP‐2 (CXCL2) and MIG were measured by luminex assay (Eve Technologies, Univ. Calgary, AB, Canada).
Macrophage feedback experiment
To assess whether AAM‐derived mediators altered myofibroblast gene expression and PG production, MCM was prepared. Macrophages were seeded at 2 × 106 per well in a 6‐well plate. Cells were stimulated with IL‐4 + IL‐13 (20 ng·mL−1 each) ± IL‐6 (10 ng·mL−1) ± L‐902,688 (10 μM) for 24 h. After 24 h, supernatants were removed, cells were rinsed in sterile PBS and then re‐incubated with fresh low‐serum culture medium (DMEM + 2% FBS, 2.4% pen/strep) for an additional 24 h, after which MCM was collected. For co‐culture experiments with myofibroblasts, myofibroblasts were seeded at 105 per well in a 6‐well plate, grown to confluence and then incubated with MCM (diluted 1:1 in low serum (5%) fibroblast culture medium) for either 6 h (to assess gene expression via RT‐PCR) or 24 h (to assess supernatant levels of PGE2, PGD2 and IL‐6, changes in MTT activity and changes in migratory ability).
Chemotaxis assay
Migration of myofibroblasts in response to macrophage‐derived mediators was assessed by culturing 103 myofibroblasts on an 8 μm pore transwell (VWR International, Mississauga, ON, Canada) with conditioned medium from various macrophage treatments being added to the bottom of the transwell compartment. After 24 h incubation, transwells were removed, washed in PBS, fixed in methanol and stained with 0.1% crystal violet. Stained cells on the basolateral side of the transwell were imaged using an Olympus microscope, and the number of migrated cells was counted, in a blinded fashion, in 3 randomly selecting three fields of view.
Data presentation and statistical analysis
Data are presented as mean ± SEM, where n represents data from one mouse. Samples were not randomized; data were collected in a blinded fashion and analysed using graphpad prism 5 software (GraphPad Software, La Jolla, CA, USA) by one‐way anova, and when this returned P < 0.05, post hoc analysis using Tukey's test was performed, and P < 0.05 accepted as a level of statistical significance. In accordance with journal policy when n < 5, statistical analyses were not performed. In multi‐group studies where at least one group was n = 3, anova was performed, and the said group was not included in the post hoc comparisons. For some experiments, n = 3 was employed for ethical reasons to restrict animal numbers, that is, in the use of established inhibitors (e.g. indomethacin suppression of PG synthesis) and when multiple approaches were applied to test a hypothesis (e.g. synthetic PG, PG receptor agonist and antagonist used to identify receptor subtype). The data and statistical analysis comply with British Journal of Pharmacology guidelines (Curtis et al., 2015).
Results
Myofibroblast conditioned medium increases expression of hallmark AAM markers
Isolated dermal fibroblasts take on a myofibroblast phenotype in vitro in response to culture on plastic and growth factors present in culture medium (Tomasek et al., 2002), as confirmed by α‐SMA expression (data not shown). In the presence of IL‐4 + IL13, AAMs treated with MFbCM for 48 h displayed significant increases in Arg1 (expression and activity) and Ym1, compared with AAMs induced with IL‐4 + IL‐13 alone (Figure 1A–D). Following their initial induction, AAMs ± MFbCM were challenged for an additional 24 h with LPS (1 μg·mL−1) to stimulate NO and cytokine production. AAMs co‐treated with MFbCM showed suppression of nitrite (a surrogate measure of NO) production (Figure 1E). In contrast, macrophages stimulated with IL‐4 + IL‐13 alone showed elevated levels of NO in response to LPS, consistent with other studies (Raes et al., 2002; Varin et al., 2010; Whyte et al., 2011; Fernando et al., 2014). AAMs + MFbCM spontaneously produced IL‐10, which was not observed with untreated macrophages or AAMs (Figure 1F), and also displayed elevated levels of IL‐10 in response to LPS (Figure 1G).
Figure 1.
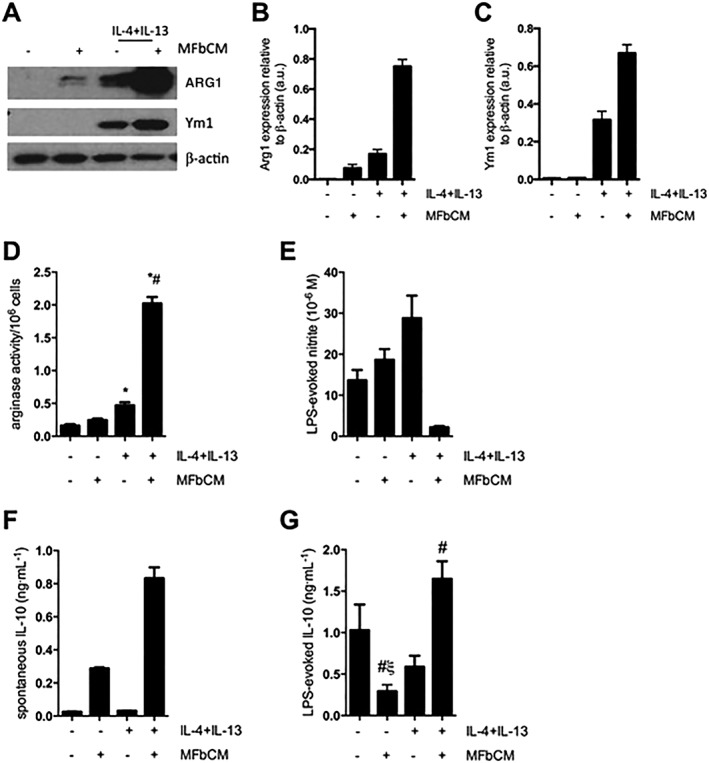
MFbCM enhances the expression of characteristic AAM markers. (A–C) Macrophages stimulated with IL‐4 + IL‐13 + MFbCM for 48 h displayed enhanced protein expression of ARG1 and Ym1 protein (A, data are representative of four experiments; (B and C) densitometric analysis of ARG1 and Ym1) and increased arginase activity (D, n = 10–12, except MFbCM only where n = 3). (E) Following cytokine exposure, macrophages were stimulated with LPS (1 μg·mL−1) for 24 h. Co‐treatment with IL‐4 + IL‐13 and MFbCM led to suppression of nitric oxide production (n = 3). (F–G) Macrophage treatment with IL‐4 + IL‐13 + MFbCM results in spontaneous IL‐10 production (48 h post‐cytokine/MFbCM treatment) and increased IL‐10 in response to LPS (n = 6–12, except negative controls and MFbCM only where n = 3). Data are mean ± SEM; * P < 0.05 versus untreated macrophages, #P < 0.05 versus IL‐4 + IL‐13, ξP < 0.05 compared with IL‐4 + IL‐13 + MFbCM‐treated macrophages; anova followed by Tukey's post hoc test.
Enhancement of AAM markers is partially dependent on IL‐6 production by dermal myofibroblasts
ELISAs and qPCR were used to identify dermal myofibroblast‐derived cytokines that could possibly enhance an AAM phenotype. As shown in Figure 2A, IL‐10, IL‐33 and bioactive TGFβ (i.e. from acidified samples), which can be produced by myofibroblasts and enhance markers of AAM polarization (Prasse et al., 2006; Kurowska‐Stolarska et al., 2009; Fernando et al., 2014), were not detected in MFbCM, which may reflect the cells dermal origin as typically these cytokines have been demonstrated in synovial or colonic myofibroblasts. However, myofibroblasts constitutively produced high levels of IL‐6 (Figure 2A). Treatment of macrophages with IL‐4 + IL‐13 + MFbCM in which IL‐6 had been depleted via a neutralizing antibody displayed a partial reduction in arginase activity compared with macrophages treated with an immune serum/irrelevant antibody (Figure 2B), supporting a role for myofibroblast‐derived IL‐6 in promoting an enhanced AAM phenotype, as previously demonstrated with recombinant IL‐6 (Fernando et al., 2014). Thus, the current findings underscore the potential in vivo significance of IL‐6 in enhancing an AAM phenotype.
Figure 2.
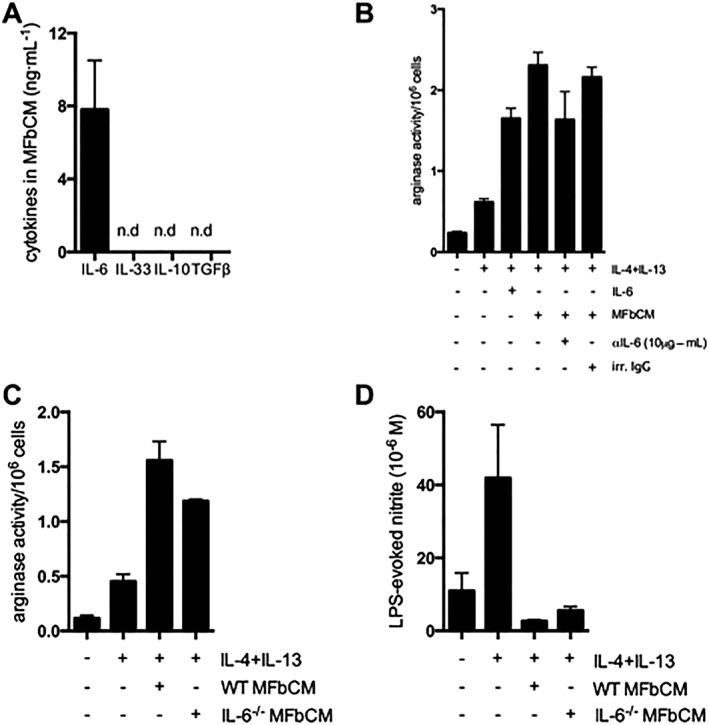
The effect of MFbCM is partially dependent on IL‐6 production. (A) Assessment of cytokine levels in conditioned medium revealed high levels of IL‐6 and no detectable levels of IL‐10, IL‐33 and TGFβ (n.d, not detected; n = 3). (B) Neutralization of IL‐6 in MFbCM via a neutralizing antibody against IL‐6 (10 μg·mL−1) significantly reduced the ability of MFbCM to enhance arginase activity following co‐treatment with IL‐4 + IL‐13 (n = 3). (C–D) Use of IL‐6−/− MFbCM in place of wild type (WT) MFbCM revealed that IL‐6−/− MFbCM was still able to enhance IL‐4 + IL‐13‐induced arginase activity and suppress LPS‐evoked nitric oxide (n = 5–6). Data are mean ± SEM, #P < 0.05 versus IL‐4 + IL‐13; anova followed by Tukey's post hoc test.
To address this issue further, MFbCM from IL‐6−/− myofibroblasts was used, to unequivocally determine if other factors in the MFbCM promoted an AAM‐phenotype (i.e. one could argue that the efficiency of the IL‐6 neutralization was responsible for the effect seen in Figure 2B rather than other mediators). MFbCM from IL‐6−/− myofibroblasts increased AAM arginase activity and suppressed NO production in response to LPS (Figure 2C–D), indicating the presence of one or more additional factors capable of promoting an AAM phenotype. In subsequent experiments, conditioned medium was used from IL‐6−/− mice to allow analysis of the promotion of an AAM phenotype in the absence of IL‐6, which is known to promote this phenotype (Fernando et al., 2014).
COX‐2 in MFbCM is required for MFbCM to amplify AAM polarization
In addition to cytokines and growth factors, dermal myofibroblasts produce lipid mediators, such as PGs. Dermal myofibroblasts from both wild type and IL‐6−/− mice constitutively expressed COX‐1 and COX‐2 protein (data not shown). Indomethacin (1 μM), a non‐selective inhibitor of both COX isoforms, suppressed the ability of MFbCM derived from IL‐6−/− myofibroblasts to enhance AAM arginase activity and suppress LPS‐evoked NO (Figure 3A–B). However, indomethacin, added directly to macrophages, did not promote polarization to an alternatively activated phenotype (data not shown) indicating that this reduction in MFbCM effect was not due to residual indomethacin acting on AAMs. Selective inhibitors of COX‐1 (SC‐560, 1 μM) and COX‐2 (NS‐398, 20 μM) revealed that the enhancement of an AAM phenotype by IL‐6−/− MFbCM was dependent on COX‐2 activity in the myofibroblast (Figure 3A–B). It has been reported that the anti‐inflammatory and pro‐resolving properties of COX‐2 are primarily associated with the production of PGD2 and PGE2 (Rajakariar et al., 2006). Both wild type and IL‐6−/− dermal myofibroblasts constitutively produced PGE2 and PGD2 (hence, IL‐6 is not needed to autocrinely induce macrophage PGs), which were reduced by indomethacin and NS‐398 (Figure 3C–D). The involvement of PGE2 in macrophage polarization was further indicated by the finding that a selective EP4 receptor antagonist (L‐161982) reversed the effects of IL‐6−/− MFbCM on arginase activity in a concentration‐dependent manner (Figure 3E).
Figure 3.
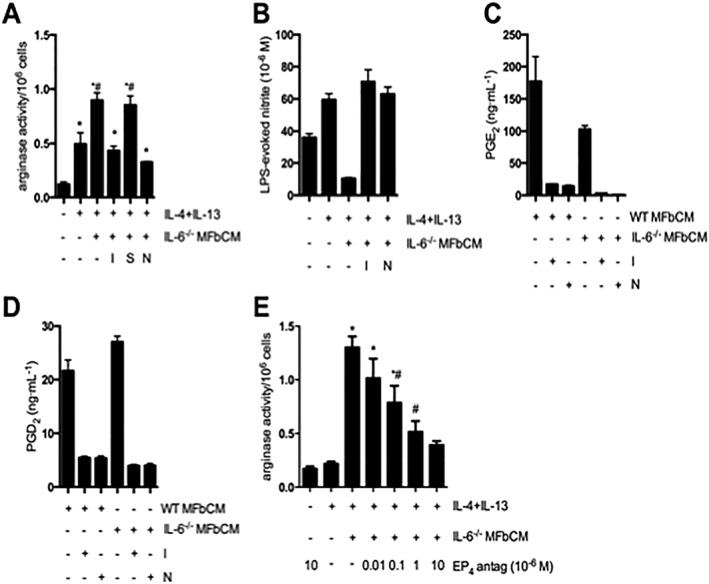
COX‐2‐derived products from dermal myofibroblasts enhance AAM function. (A–B) Myofibroblast conditioned medium from IL‐6−/− myofibroblasts treated with indomethacin (COX‐1 and 2 inhibitor, 1 μM), SC‐560 (COX‐1 inhibitor, 1 μM) and NS‐398 (COX‐2 inhibitor, 20 μM) at 0 and 24 h was used to stimulate macrophages (+IL‐4 + IL‐13). MFbCM from myofibroblasts exposed to indomethacin (I) and NS‐398 (N), but not SC‐560 (S), lost their ability to increase arginase activity in IL‐4 + IL‐13‐treated macrophages. MFbCM from myofibroblasts exposed to indomethacin (I) and NS‐398 (N), lost their ability to suppress nitric oxide production in IL‐4 + IL‐13‐treated macrophages (A; n = 5–11 except negative control where n = 3; B; n = 3;). (C–D) Production of PGE2 and PGD2 by wild type (WT) and IL‐6−/− myofibroblasts was suppressed by treatment with indomethacin and NS‐398 (n = 3,). (E) Enhanced arginase activity in IL‐4 + IL‐13 + MFbCM‐treated macrophages was dose‐dependently inhibited by the EP4 receptor antagonist, L161982 (EP4 antag) (n = 6, except IL‐4 + IL‐13 + MFbCM + EP4 antag (10 × 10−6 M) where n = 3). Data are mean ± SEM; * P < 0.05 versus untreated macrophages, #P < 0.05 versus IL‐4 + IL‐13 + IL‐6−/− MFbCM; anova followed by Tukey's post hoc test.
Activation of EP 4 and DP 1 receptors on macrophages mediates the effects of MFbCM on AAM function
Activation of the EP4 receptor using a selective agonist (L‐902,688: 10 μM), in the presence of IL‐4 + IL‐13, led to an enhancement in arginase activity, similar to that observed in AAMs co‐treated with MFbCM (Figure 4A). AAMs induced in the presence of the EP4 receptor agonist produced less nitrite in response to LPS (Figure 4B), did not show appreciable spontaneous IL‐10 production (Figure 4C), although there was increased LPS‐evoked IL‐10 in AAMs induced with the EP4 receptor (Figure 4D). Activation of the EP2 receptor with ONO‐AE1‐259 (10 μM) did not enhance arginase activity (data not shown) in AAMs, but marginally suppressed nitrite production (although not to the same extent as IL‐4 + IL‐13 and the EP4 receptor agonist) (Figure 4B). The EP2 receptor agonist did not augment spontaneous IL‐10 production (Figure 4C), or increase LPS‐evoked IL‐10 production compared with IL‐4 + IL‐13 alone (Figure 4D). Co.‐stimulation of macrophages with IL‐4 + IL‐13 and IL‐6 suppressed NO production (Figure 4B) and was the only treatment that enhanced spontaneous production of IL‐10 (Figure 4C). There was no effect of IL‐6 co‐treatment on LPS‐evoked IL‐10 (Figure 4D). AAMs induced with PGE2 showed up‐regulations in arginase activity and LPS‐induced IL‐10 production but were unable to suppress LPS‐induced nitrite production (Figure 4E–G).
Figure 4.
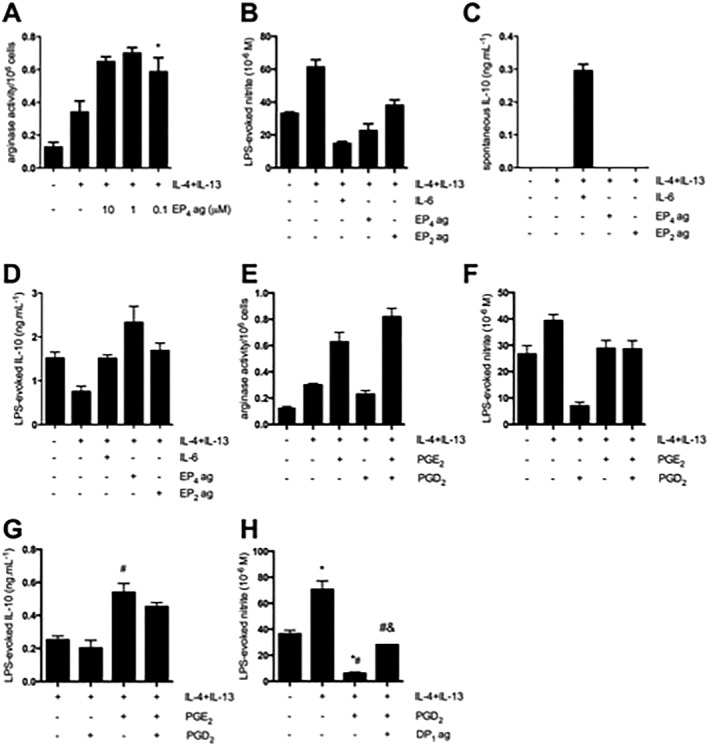
The macrophage EP4 and DP1 prostanoid receptors mediate the effects of PGE2 and PGD2 on AAMs. (A–B) Activation of the EP4 receptor using L902,688 (EP4 agonist) in the presence of IL‐4 + IL‐13 increases arginase activity (n = 3) and also suppresses LPS‐evoked nitric oxide production. Subtle suppression of nitric oxide was also observed following co‐treatment with an EP2 agonist (ONO‐AE1‐259, 10 μM, n = 3). (C–D) Activation of the EP4 receptor in the presence of IL‐4 + IL‐13 has no effect on the spontaneous production of IL‐10. In response to LPS, IL‐10 production is enhanced by the combination of IL‐4 + IL‐13 + L902,688 (n = 3). (E) Stimulation of macrophages with IL‐4 + IL‐13 and PGE2 resulted in increased arginase activity, while IL‐4 + IL‐13 + PGD2 did not increase arginase activity relative to IL‐4 + IL‐13‐treated macrophages (n = 6). (F) Stimulation of macrophages with IL‐4 + IL‐13 + PGD2 suppressed nitric oxide production in response to LPS, which was not observed with IL‐4 + IL‐13 + PGE2 (n = 3). (G) Stimulation of macrophages with IL‐4 + IL‐13 + PGE2 enhanced IL‐10 production in response to LPS (n = 6). (H) Macrophages exposed to IL‐4 + IL‐13 and a DP1 receptor agonist showed suppression of nitric oxide compared with IL‐4 + IL‐13 alone, but not to the same extent as IL‐4 + IL‐13 + PGD2 (n = 6). Data are mean ± SEM, * P < 0.05 versus control, #P < 0.05 versus IL‐4 + IL‐13, &P < 0.05 versus IL‐4 + IL‐13 + PGD2; anova followed by Tukey's post hoc test.
Macrophages co‐treated with IL‐4 + IL‐13, and PGD2 did not up‐regulate arginase activity or LPS‐induced IL‐10 production (Figure 4E–G). In contrast, PGD2 co‐treatment suppressed LPS‐evoked nitrite generation (Figure 4H), although this was not apparent when PGE2 was also present (Figure 4F). The use of DP1 and DP2 receptor agonists and antagonists revealed that treatment of macrophages with a DP1 receptor agonist (+IL‐4 + IL‐13) was able to partially mimic the effect of synthetic PGD2 (Figure 4H). Interestingly, while PGE2 inhibited the ability of PGD2 to suppress AAM NO production in response to LPS, the reverse was not seen, the PGE2 effects on AAMs were unchanged by PGD2 exposure (Figure 4E, G).
MFbCM‐induced changes in AAM phenotype drive a feedback loop that alters myofibroblast characteristics
Of the cytokines measured (Methods), only IL‐6 production by myofibroblasts was increased by exposure to AAM CM, AAM + IL‐6 CM or AAM + L‐902,688 CM (IL‐6 production in response to CM from naïve macrophages was intermediate between naïve myofibroblasts and those treated with AAM CM with post hoc statistic revealing it was not significantly different from either group) (Table 1). The production of PGE2 by myofibroblasts treated with conditioned medium from AAM and IL‐6 or L902,688 while still greater than that of non‐treated myofibroblasts was less than that of cells treated with AAM CM only (Table 1). In addition, the increased PGD2 produced by myofibroblasts in response to AAM CM was not apparent in cells treated with conditioned medium from AAM + IL‐6 or AAM + L902,688 (Table 1).
Table 1.
Cytokine and mRNA levels of murine dermal myofibroblasts are altered by exposure to conditioned medium from AAMs
| Untreated mΦ CM | AAM CM | AAM + IL‐6 CM | AAM + EP4ag CM | AAM + EP4ag + IL‐6 CM | |
|---|---|---|---|---|---|
| mIL‐6 | 0.32 ± 0.03 | 0.56 ± 0.02 | 0.65 ± 0.1 | 0.63 ± 0.02 | 0.71 ± 0.07 |
| PGE2 | 9.8 ± 0.9 | 27.5 ± 2.9 | 33.4 ± 2.9 | 22.1 ± 1.7 | 21.9 ± 2.2 |
| PGD2 | 0.36 ± 0.07 | 0.74 ± 0.1 | 1.95 ± 0.29 | 0.81 ± 0.1 | 1.02 ± 0.24 |
| α‐SMA mRNA | 2.6 ± 0.44 | 4.9 ± 0.71 | 3.7 ± 0.3 | 2.78 ± 1.1 | 4.3 ± 2.2 |
| COX‐2 mRNA | 33.9 ± 5.4 | 70.0 ± 15.1 | 64.2 ± 9.9 | 13.8 ± 1.5 | 22.3 ± 9.2 |
Data are mean ± SEM; n = 3; IL‐6, PGE2, PGD2 are ng·mL−1; α‐smooth muscle actin (α‐SMA) and COX‐2 are fold change relative to control; ag, agonist.
Assessment of changes in myofibroblast gene expression following 6 h exposure to the various macrophage conditioned media showed no changes in COX‐1 expression (data not shown), while COX‐2 gene expression was increased by treatment with AAM conditioned medium and conditioned medium from AAMs + IL‐6, and in both instances, this was prevented by co‐treatment of the macrophages with the EP4 receptor agonist (L‐902,688). There was no change in expression of the myofibroblast marker α‐SMA following exposure to any of the MCM groups (Table 1), indicative of a lack of de‐differentiation in the myofibroblasts. Mitochondria activity (gauged by MTT assay) in myofibroblasts (Figure 5A) was, on average, increased, and their chemotaxis across a porous (8 μm) filter (Figure 5B) was enhanced by exposure to AAM (±IL‐6) CM. When the macrophages were co‐treated with the EP4 receptor agonist reduced mitochondrial activity and chemotaxis were observed with AAM CM, while the EP4 agonist did prevent the AAM‐IL‐6 CM enhanced myofibroblast mitochondrial activity, the increased chemotaxis was unaffected by L‐902,688 (Figure 5A–B).
Figure 5.
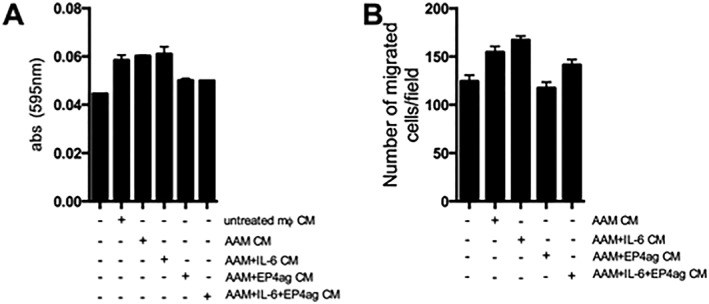
Alternatively activated macrophage conditioned medium (AAM CM) alters dermal myofibroblast phenotype. Myofibroblast mitochondrial activity (i.e. MTT cleavage) was enhanced by CM from all macrophage groups ( A; n = 3,). (B) Migration of myofibroblasts across an 8 μm pore transwell in response to macrophage‐derived mediators was increased by AAM CM and suppressed by conditioned medium from AAM + EP4ag CM (n = 4–5, except negative control where n = 3). Data are mean ± SEM.
Discussion
Tissue recovery after damage and the termination of inflammation are active events, involving many different cell types and soluble signals (Buckley et al., 2013). The important roles that myofibroblasts play in wound healing and tissue remodelling are well known (Tomasek et al., 2002; Werner et al., 2007; Flavell et al., 2008; Pinchuk et al., 2011), yet while they are likely active participants in the resolution of inflammation, there are few data demonstrating this. Thus, the effect of myofibroblasts on the development of the anti‐inflammatory and pro‐resolving AAM was investigated. We opted to use a co‐culture model composed of bone marrow‐derived macrophages and dermal fibroblasts as recruitment of bone marrow monocytes that differentiate into macrophages is important in wound healing (Shi and Pamer, 2011), and dermal fibroblasts are necessary for cutaneous wound healing (Bristic‐Dujmovic et al., 2010). We found that myofibroblast‐derived IL‐6, PGE2 and PGD2 amplify the development of AAMs, which in turn feedback to the myofibroblast to limit/shut‐off PG production. Bi‐directional communication was identified where myofibrobalsts reinforce an AAM phenotype that would suppress inflammation and promote tissue recovery after injury, which, in turn, can inhibit myofibroblast activity to avoid unwanted fibrosis.
Myofibroblast‐derived conditioned medium enhanced the expression of markers associated with AAM polarization, and this was critically dependent on co‐treatment with IL‐4 + IL‐13, because MFbCM alone did not affect expression of canonical features of the IL‐4‐driven AAM. This may represent a regulatory checkpoint, preventing the aberrant induction of AAMs as these cells have been associated with the exacerbation of fibrotic disease (Prasse et al., 2006; Kurowska‐Stolarska et al., 2009; Li et al., 2014). ARG1 is important in wound healing and resolution via the production of l‐ornithine, a precursor of prolines and polyamines that are required for collagen deposition and cell proliferation respectively. While over‐production of ARG1 would be expected to contribute to fibrosis (McLarren et al., 2011; Moura et al., 2013), a role for AAM‐associated ARG1 in fibrosis has been disputed (Pesce et al., 2009), and, furthermore, its absence has been shown to impair cutaneous wound healing (Campbell et al., 2013): AAM ARG1 has been implicated in the anti‐colitic effect of these cells (McLarren et al., 2011), and ARG1 levels are reduced in patients with atopic dermatitis (Dimitriades et al., 2014). NO produced by the constitutive NOS has important roles in wound healing (Witte and Barbul, 2002). In contrast, NO derived from inducible NOS is considered cytostatic and cytotoxic (Witte and Barbul, 2002). Thus, the enhanced ARG1 expression/activity coupled to suppressed LPS‐evoked NO in MFbCM‐treated AAMs indicates a functional shift to facilitate tissue restitution after damage.
The IL‐4 + IL‐13 differentiated AAM is not a significant source of spontaneous IL‐10 (Kurowska‐Stolarska et al., 2009; Fernando et al., 2014; Murray et al., 2014) and so the increased spontaneous and LPS‐stimulated production of IL‐10 by MFbCM‐treated AAMs could be a crucial component of their regulatory activity. IL‐10 is well known as an anti‐inflammatory cytokine, capable of inducing regulatory T‐cells (Gregori et al., 2010) and deactivating macrophages (Mantovani et al., 2004). IL‐10 can promote wound healing in animals and humans (Peranteau et al., 2008; Kieran et al., 2013), and reverse or inhibit the conversion of fibroblasts to myofibroblasts (Barron and Wynn, 2011). In the correct scenario, IL‐10 could have autocrine effects and, in a paracrine fashion, drive a negative feedback loop to turn‐off myofibroblasts, for the completion of repair processes and to prevent progression to fibrosis (Barron and Wynn, 2011). These data showing that myofibroblast‐derived products can enhance the development and function of an AAM are comparable with those of myofibroblast induction of regulatory T‐cells (which have similar function to AAMs) (Bouffi et al., 2011; Pinchuk et al., 2011).
A variety of myofibroblast‐derived mediators are excellent candidate molecules to promote an AAM phenotype, and cytokine measurements revealed a significant amount of IL‐6 in the MFbCM. Subsequent IL‐6 immuno‐neutralization in MFbCM and use of MFbCM from IL‐6−/− cells revealed that the increased ARG1 activity was partially dependent on IL‐6, whereas the suppression of LPS‐stimulated nitrate was not, and likewise AAM spontaneous PGE2 and PGD2 production were not IL‐6 dependent. These findings are consistent with recombinant IL‐6 promoting murine and human AAM phenotypes (Fernando et al., 2014; Mauer et al., 2014). IL‐6 is often considered to be pro‐inflammatory, but it can participate in the resolution of inflammation through mediation of the switch between neutrophil and monocyte recruitment (Scheller et al., 2011), and, in the context of tissue recovery, IL‐6−/− mice display defects in wound healing (Gallucci et al., 2000). Indeed, case studies have shown that patients treated with anti‐IL‐6 neutralizing antibodies can develop ulceration that only resolves when the anti‐IL6 treatment is withdrawn (Iwasa et al., 2011). While myofibroblast‐derived IL‐6 contributed to the enhancement of an AAM phenotype, it was not an absolute requirement as notably defined by the suppression of nitrate production by IL‐6−/− MFbCM. Recombinant IL‐6 suppresses LPS‐stimulated nitrite production from AAMs (Fernando et al., 2014), and other mediators in the MFbCM were capable of producing this outcome.
Myofibroblasts are major sources of PGs (Kendall and Feghali‐Bostwick, 2014) that affect many aspects of macrophage biology and promote the development and/or function of regulatory macrophage subtypes (Kunkel et al., 1988; Heusinkveld et al., 2011; Ylöstalo et al., 2012; MacKenzie et al., 2013; Na et al., 2013). For example, PGE2 from human mesenchymal stem cells can convert pro‐inflammatory M1 macrophages to a more anti‐inflammatory M2 phenotype (Ylöstalo et al., 2012). Despite these precedents, there are few data on how PGs can affect the development of IL‐4 (±IL‐13)‐induced AAMs.
Conditioned medium from IL‐6−/− myofibroblasts treated with indomethacin to target both COX isoforms failed to amplify an AAM phenotype. Use of selective COX inhibitors revealed that the non‐IL‐6 effect of MFbCM was dependent on a COX‐2‐derived molecule. Older dogma on COX‐2 being an exclusively inducible enzyme with pro‐inflammatory functions has been eroded by awareness that it can be constitutively expressed (Rouzer and Marnett, 2009) and can perform anti‐inflammatory, wound healing and pro‐resolving roles (Gilroy et al., 1999; Rajakariar et al., 2006; Buckley et al., 2013). These data implicating both IL‐6 and COX‐2 in the promotion of an anti‐inflammatory macrophage phenotype aptly illustrate the dichotomous nature of inflammatory mediators, and the need for contextual understanding, rather than strict classifications of these mediators.
MFbCM contained substantial amounts of PGE2 and PGD2, the production of which was almost virtually abolished by the selective COX‐2 inhibitor, NS‐398. These data are in accordance with the finding that when both COX‐1 and COX‐2 are present, the latter is principally responsible for PGE2 and PGD2 synthesis (Gilroy et al., 1999). Mechanistic studies with PGE2, PGD2 and selective EP and DP receptor agonists and antagonists indicated differential roles for these PGs in the up‐regulation of an AAM phenotype and enhanced function: PGE2 via EP4 receptor signalling drove the increase in arginase activity and LPS‐evoked IL‐10 production, while PGD2, via the DP1 receptor, was responsible for the suppression of LPS‐induced NO production. These findings are noteworthy because (i) this is the first time these mediators and associated receptors have been shown to enhance an IL‐4‐driven AAM phenotype (PGE2 increases in IL‐10 regulatory macrophages have been reported (Strassmann et al., 1994; MacKenzie et al., 2013)); (ii) they refute the notion that differences in arginase activity and NO production are simply due to competition for the arginine precursor; (iii) they provide mechanistic insight on why selective COX‐2 inhibition can be detrimental under certain circumstances (Fairweather et al., 2014); and (iv) the fact that PGD2 did not alter arginase activity is intriguing, especially in light of the fact that its spontaneous metabolite, 15‐d‐PGJ2, acts as a PPARγ agonist in the mmol concentration range (Powell, 2003) to facilitate AAM polarization (Odegaard et al., 2007). We suggest that under the present experimental conditions, the degradation of PGD2 and production of PGJ2 is insufficient to significantly enhance the PPARγ pathway.
Collectively, the data demonstrate the ability and the mechanisms involved, whereby, myofibroblasts can enhance the expression of canonical markers of the IL‐4 + IL‐13‐driven AAM, and impart additional immuno‐regulatory, anti‐inflammatory effects that could be crucial in tissue restitution after injury. Postulating the myofibroblast‐AAM as a functional unit, the issue of reciprocity arises, and we determined whether the AAM would affect the behaviour of the myofibroblast. Conditioned medium from IL‐4 + IL‐13‐exposed AAMs increased myofibroblast mitochondrial activity, supporting studies showing that conditioned medium from AAMs increases fibroblast proliferation (Ploeger et al., 2013) via platelet‐derived growth factor (Song et al., 2000), CCL18 (Prasse et al., 2006) and insulin‐like growth factor‐1 (Wynes et al., 2004). However, when an EP4 receptor agonist was used as a proxy for PGE2 or IL‐6 was added with IL‐4 + IL‐13 to differentiate AAMs, the cells had a reduced capacity to stimulate myofibroblast mitochondrial activity. Moreover, activation of the EP4 receptor on AAMs allowed them to suppress (relatively) myofibroblast migration, COX‐2 expression and PGE2 and PGD2 (but not IL‐6) production to prevent a feed‐forward loop to the AAMs; together, these events would be predicted to promote the resolution of inflammation and limit the ability of either AAM or myofibroblasts to drive fibrosis. The suppression of COX‐2 mRNA and PGE2 and PGD2 output by myofibroblasts exposed to conditioned medium from AAMs treated concomitantly with IL‐6 or the EP4 agonist did not exactly mirror each other, and this could reflect temporal differences in their expression, or that substantial amounts of PGs can be produced in the face of diminishing COX‐2 mRNA (also a role for COX‐1 in the overall PG output is worthy of consideration). It has been proposed that PGE2‐induced M2 macrophages can promote airways allergic inflammation; however, the macrophages in that study were not IL‐4 derived (Kim et al., 2014). Also, culturing human dermal fibroblasts with classically activated macrophages elicited an inflammatory phenotype in the fibroblasts (Ploeger et al., 2013), attesting to the contextual nature of macrophage–fibroblast interactions and how these can affect the balance between homeostatic recovery from an insult or the development of the pathophysiology of acute, and potentially, chronic inflammatory disease (Fiocchi, 1997).
In summary, myofibroblasts were shown to produce mediators that were identified to direct specific functions in IL‐4 + IL‐13‐evoked AAMs and a pathway of bi‐directional communication between the two cells was defined (Figure 6); the interplay, or lack thereof, of these mediators in terms of intracellular signalling and the control of macrophage function remain to be determined. Given the central roles that myofibroblasts/fibroblasts and AAMs can play in wound healing, we suggest that this interaction represents a novel mechanism by which AAMs and myofibroblasts are able to facilitate tissue repair, as well as maintain tissue homeostasis in vivo, and that dysregulation of these processes may contribute to the development of fibrotic disease.
Figure 6.
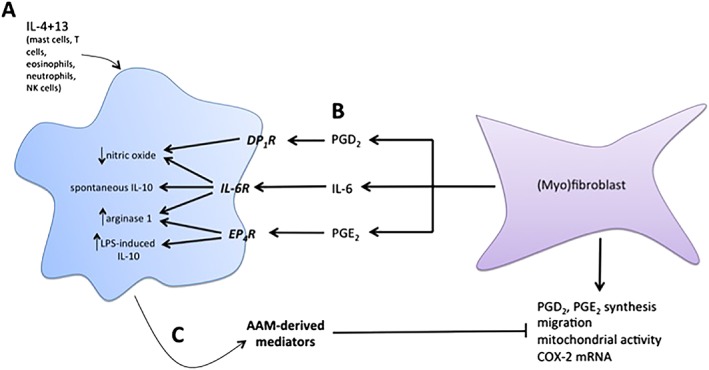
Model of AAM–myofibroblast interactions. (A) Exposure of macrophages to IL‐4 and IL‐13 induces expression of canonical AAM markers, arginase 1 and Ym1. In the presence of LPS, these AAMs also produce substantial amounts of nitric oxide. (B) Conditioned medium from myofibroblasts, containing IL‐6, PGD2 and PGE2, interacts with specific receptors on macrophages during IL‐4 and IL‐13 priming. In the presence of IL‐6, AAMs display enhanced expression of ARG1 and Ym1, spontaneously produce IL‐10 and suppress LPS‐induced nitric oxide. PGE2 increases ARG1 expression and also enhances LPS‐evoked IL‐10 production, and PGD2 suppresses LPS‐induced NO. (C) Conditioned medium from AAMs induced with MFbCM components suppress PGE2 and PGD2 production from myofibroblasts, and also suppresses migration, mitochondrial activity and COX‐2 gene expression. This indicates that AAMs induced with MFbCM can ‘turn off’ the activity of myofibroblasts, thus demonstrating an important feedback loop that will prevent aberrant activation of both cell types.
Author contributions
M. F. designed and performed experiments, analysed the data and wrote the manuscript. M. G. provided input on experimental design, reviewed and edited the manuscript. D. M. provided input on experimental design, reviewed and edited the manuscript.
Conflict of interest
The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors have no financial or other conflicts of interest to disclose.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was funded by an operating grant from the Crohn's and Colitis Foundation of Canada to D. M. M. M. F. is a recipient of Graduate Studentships from the Canadian Institutes for Health Research (CIHR)/Canadian Digestive Health Foundation (CDHF) and Alberta Innovates‐Health Solutions (AI‐HS). D. M. M. is a recipient of a Canada Research Chair (CRC) (Tier 1) in Intestinal Immunophysiology, M. A. G. is a recipient of a CRC (Tier 1) in Pulmonary Pharmacology.
Fernando, M. R. , Giembycz, M. A. , and McKay, D. M. (2016) Bidirectional crosstalk via IL‐6, PGE2 and PGD2 between murine myofibroblasts and alternatively activated macrophages enhances anti‐inflammatory phenotype in both cells. British Journal of Pharmacology, 173: 899–912. doi: 10.1111/bph.13409.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron L, Wynn TA (2011). Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol 300: G723–G728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristic‐Dujmovic T, Bohan I, Clark SH (2010). Fibroblasts/myofibroblasts that participate in cutaneous wound healing are not derived from circulating progenitor cells. J Cell Physiol 222: 703–712. [DOI] [PubMed] [Google Scholar]
- Bouffi C, Bony C, Jorgensen C, Noël D (2011). Skin fibroblasts are potent suppressors of inflammation in experimental arthritis. Ann Rheum Dis 70: 1671–1676. [DOI] [PubMed] [Google Scholar]
- Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP (2013). The resolution of inflammation. Nat Rev Immunol 13: 59–66. [DOI] [PubMed] [Google Scholar]
- Campbell L, Saville CR, Murray PJ, Cruickshank SM, Hardman MJ (2013). Local arginase 1 activity is required for cutaneous wound healing. J Invest Dermatol 133: 2461–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartouni El C, Rehli M (2010). Comprehensive analysis of TLR4‐induced transcriptional responses in interleukin 4‐primed mouse macrophages. Immunobiology 215: 780–787. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby IA, Hewitson TD (2007). Fibroblast differentiation in wound healing and fibrosis. Int Rev Cytol 257: 143–179. [DOI] [PubMed] [Google Scholar]
- Dimitriades V, Rodriguez PC, Zabaleta J, Ochoa AC (2014). Arginase I levels are decreased in the plasma of pediatric patients with atopic dermatitis. Ann Allergy Asthma Immunol 113: 271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairweather M, Heit YI, Buie J, Rosenberg LM, Briggs A, Orgill DP, et al. (2014). Celecoxib inhibits early cutaneous wound healing. J Surg Res 194: 717–724. [DOI] [PubMed] [Google Scholar]
- Fernando MR, Reyes JL, Iannuzzi J, Leung G, McKay DM (2014). The pro‐inflammatory cytokine, interleukin‐6, enhances the polarization of alternatively activated macrophages. PLoS One. 9: e94188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiocchi C (1997). Intestinal inflammation: a complex interplay of immune and nonimmune cell interactions. A J Physiol 273: G769–G775. [DOI] [PubMed] [Google Scholar]
- Flavell SJ, Hou TZ, Lax S, Filer AD, Salmon M, Buckley CD (2008). Fibroblasts as novel therapeutic targets in chronic inflammation. Br J Pharmacol 153: S241–S246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli SJ, Borregaard N, Wynn TA (2011). Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 12: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci RM, Simeonova PP, Matheson JM, Kommineni C, Guriel JL, Sugawara T, et al. (2000). Impaired cutaneous wound healing in interleukin‐6‐deficient and immunosuppressed mice. FASEB J 14: 2525–2531. [DOI] [PubMed] [Google Scholar]
- Gilroy DW, Colville‐Nash PR, Willis D, Chivers J, Paul‐Clark MJ, Willoughby DA (1999). Inducible cyclooxygenase may have anti‐inflammatory properties. Nat Med 5: 698–701. [DOI] [PubMed] [Google Scholar]
- Gordon S (2003). Alternative activation of macrophages. Nat Rev Immunol 3: 23–35. [DOI] [PubMed] [Google Scholar]
- Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, et al. (2010). Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC‐10 requires the IL‐10‐dependent ILT4/HLA‐G pathway. Blood 116: 935–944. [DOI] [PubMed] [Google Scholar]
- Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJP, et al. (2011). M2 macrophages induced by prostaglandin E2 and IL‐6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol 187: 1157–1165. [DOI] [PubMed] [Google Scholar]
- Iwasa T, Nakamura K, Ogino H, Itaba S, Akiho H, Okamoto R, et al. (2011). Multiple ulcers in the small and large intestines occurred during tocilizumab therapy for rheumatoid arthritis. Endoscopy 43: 70–72. [DOI] [PubMed] [Google Scholar]
- Kendall RT, Feghali‐Bostwick CA (2014). Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 5: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran I, Knock A, Bush J, So K, Metcalfe A, Hobson R, et al. (2013). Interleukin‐10 reduces scar formation in both animal and human cutaneous wounds: results of two preclinical and phase II randomized control studies. Wound Repair Regen 21: 428–436. [DOI] [PubMed] [Google Scholar]
- Killkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Udayanga KGS, Totsuka N, Weinberg JB, Núñez G, Shibuya A (2014). Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi‐induced PGE2 . Cell Host Microbe 15: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D (1988). Prostaglandin E2 regulates macrophage‐derived tumor necrosis factor gene expression. J Biol Chem 263: 5380–5384. [PubMed] [Google Scholar]
- Kurowska‐Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. (2009). IL‐33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 183: 6469–6477. [DOI] [PubMed] [Google Scholar]
- Li D, Guabiraba R, Besnard AG, Komai‐Koma M, Jabir MS, Zhang L, et al. (2014). IL‐33 promotes ST2‐dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol 134: 1422–1432.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie KF, K C, S N, Va MG, G N, Kristariyanto Y, et al. (2013). PGE2 induces macrophage IL‐10 production and a regulatory‐like phenotype via a protein kinase A‐SIK‐CRTC3 pathway. J Immunol 190: 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25: 677–686. [DOI] [PubMed] [Google Scholar]
- Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. (2014). Signaling by IL‐6 promotes alternative activation of macrophages to limit endotoxemia and obesity‐associated resistance to insulin. Nat Immunol 15: 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarren KW, Cole AE, Weisser SB, Voglmaier NS, Conlin VS, Jacobson K, et al. (2011). SHIP‐deficient mice develop spontaneous intestinal inflammation and arginase‐dependent fibrosis. Am J Pathol 179: 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R (2001). Toll‐like receptors and innate immunity. Nat Rev Immunol 1: 135–145. [DOI] [PubMed] [Google Scholar]
- Moura VBL, Silva MM, Batista LF, Gomes CM, Leenen PJM, Lino RS, et al. (2013). Arginase activity is associated with fibrosis in experimental infection with Taenia crassiceps, but does not play a major role in resistance to infection. Exp Parasitol 135: 599–605. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na Y‐R, Yoon Y‐N, Son D‐I, Seok S‐H (2013). Cyclooxygenase‐2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS One 8: e63451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy ZS, Czimmerer Z, Szanto A, Nagy L (2013). Pro‐inflammatory cytokines negatively regulate PPARγ mediated gene expression in both human and murine macrophages via multiple mechanisms. Immunobiology 218: 1336–1344. [DOI] [PubMed] [Google Scholar]
- Niks M, Otto M (1990). Towards an optimized MTT assay. J Immunol Methods 130: 149–151. [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo‐Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. (2007). Macrophage‐specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. , NC‐IUPHAR(2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peranteau WH, Zhang L, Muvarak N, Badillo AT, Radu A, Zoltick PW, et al. (2008). IL‐10 overexpression decreases inflammatory mediators and promotes regenerative healing in an adult model of scar formation. J Invest Dermatol 128: 1852–1860. [DOI] [PubMed] [Google Scholar]
- Pesce JT, Ramalingam TR, Mentink‐Kane MM, Wilson MS, Kasmi El KC, Smith AM, et al. (2009). Arginase‐1‐expressing macrophages suppress Th2 cytokine‐driven inflammation and fibrosis. PLoS Pathog 5: e1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinchuk IV, Beswick EJ, Saada JI, Boya G, Schmitt D, Raju GS, et al. (2011). Human colonic myofibroblasts promote expansion of CD4+ CD25high Foxp3+ regulatory T cells. Gastroenterology 140: 2019–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploeger DT, Hosper NA, Schipper M, Koerts JA, de Rond S, Bank RA (2013). Cell plasticity in wound healing: paracrine factors of M1/M2 polarized macrophages influence the phenotypical state of dermal fibroblasts. Cell Commun Signal 11: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WS (2003). 15‐Deoxy‐delta12,14‐PGJ2: endogenous PPARgamma ligand or minor eicosanoid degradation product? J Clin Invest 112: 828–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. (2006). A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med 173: 781–792. [DOI] [PubMed] [Google Scholar]
- Raes G, Noël W, Beschin A, Brys L, de Baetselier P, Hassanzadeh GHG (2002). FIZZ1 and Ym1 as tools to discriminate between differentially activated macrophages. Dev Immunol 9: 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakariar R, Yaqoob MM, Gilroy DW (2006). COX‐2 in inflammation and resolution. Mol Interv 6: 199–207. [DOI] [PubMed] [Google Scholar]
- Rouzer CA, Marnett LJ (2009). Cyclooxygenases: structural and functional insights. J Lipid Res 50: S29–S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller J, Chalaris A, Schmidt‐Arras D, Rose‐John S (2011). The pro‐ and anti‐inflammatory properties of the cytokine interleukin‐6. Biochim Biophys Acta 1813: 878–888. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008). Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schreiber T, Ehlers S, Heitmann L, Rausch A, Mages J, Murray PJ, et al. (2009). Autocrine IL‐10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J Immunol 183: 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Pamer EG (2011). Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11: 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Sharma AK (2011). Outgrowth of fibroblast cells from goat skin explants in three different culture media and the establishment of cell lines. In Vitro Cell Dev Biol Anim 47: 83–88. [DOI] [PubMed] [Google Scholar]
- Song E, Ouyang N, Hörbelt M, Antus B, Wang M, Exton MS (2000). Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol 204: 19–28. [DOI] [PubMed] [Google Scholar]
- Strassmann G, Patil‐Koota V, Finkelman F, Fong M, Kambayashi T (1994). Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2 . J Exp Med 180: 2365–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002). Myofibroblasts and mechano‐regulation of connective tissue remodelling. Nat Rev. Mol Cell Biol 3: 349–363. [DOI] [PubMed] [Google Scholar]
- Varin A, Mukhopadhyay S, Herbein G, Gordon S (2010). Alternative activation of macrophages by IL‐4 impairs phagocytosis of pathogens but potentiates microbial‐induced signalling and cytokine secretion. Blood 115: 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Krieg T, Smola H (2007). Keratinocyte–fibroblast interactions in wound healing. J Invest Dermatol 127: 998–1008. [DOI] [PubMed] [Google Scholar]
- Whyte CS, Bishop ET, Rückerl D, Gaspar‐Pereira S, Barker RN, Allen JE, et al. (2011). Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol 90: 845–854. [DOI] [PubMed] [Google Scholar]
- Witte MB, Barbul A (2002). Role of nitric oxide in wound repair. Am J Surg 183: 406–412. [DOI] [PubMed] [Google Scholar]
- Wynes MW, Frankel SK, Riches DWH (2004). IL‐4‐induced macrophage‐derived IGF‐I protects myofibroblasts from apoptosis following growth factor withdrawal. J Leukoc Biol 76: 1019–1027. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Barron L (2010). Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 30: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylöstalo JH, Bartosh TJ, Coble K, Prockop DJ (2012). Human mesenchymal stem/stromal cells cultured as spheroids are self‐activated to produce prostaglandin E2 that directs stimulated macrophages into an anti‐inflammatory phenotype. Stem Cells 30: 2283–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]