

Abstract
Background and Purpose
We investigated the hypothesis that elevated glucose increases contractile responses in vascular smooth muscle and that this enhanced constriction occurs due to the glucose‐induced PKC‐dependent inhibition of voltage‐gated potassium channels.
Experimental Approach
Patch‐clamp electrophysiology in rat isolated mesenteric arterial myocytes was performed to investigate the glucose‐induced inhibition of voltage‐gated potassium (Kv) current. To determine the effects of glucose in whole vessel, wire myography was performed in rat mesenteric, porcine coronary and human internal mammary arteries.
Key Results
Glucose‐induced inhibition of Kv was PKC‐dependent and could be pharmacologically dissected using PKC isoenzyme‐specific inhibitors to reveal a PKCβ‐dependent component of Kv inhibition dominating between 0 and 10 mM glucose with an additional PKCα‐dependent component becoming evident at concentrations greater than 10 mM. These findings were supported using wire myography in all artery types used, where contractile responses to vessel depolarization and vasoconstrictors were enhanced by increasing bathing glucose concentration, again with evidence for distinct and complementary PKCα/PKCβ‐mediated components.
Conclusions and Implications
Our results provide compelling evidence that glucose‐induced PKCα/PKCβ‐mediated inhibition of Kv current in vascular smooth muscle causes an enhanced constrictor response. Inhibition of Kv current causes a significant depolarization of vascular myocytes leading to marked vasoconstriction. The PKC dependence of this enhanced constrictor response may present a potential therapeutic target for improving microvascular perfusion following percutaneous coronary intervention after myocardial infarction in hyperglycaemic patients.
Abbreviations
- ATII
angiotensin II
- [Ca2+]i
intracellular calcium ion concentration
- ET1
endothelin‐1
- I/V
current–voltage relationship
- Kv
voltage‐gated potassium channel
- MASMC
Mesenteric arterial smooth muscle cell
- ROS
reactive oxygen species
- UTP
uridine‐5′‐triphosphate
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| AT receptors | PKCα |
| ET receptors | PKCβ |
| P2Y receptors | PKCδ |
| TP receptors | PKCε |
| Voltage‐gated ion channels b | |
| Kv channels | |
| L‐type voltage‐gated Ca2+ channels | |
| LIGANDS | |
|---|---|
| Angiotensin II (ATII) | L‐NAME |
| Endothelin‐1 (ET1) | Ruboxistaurin |
| Gö 6976 | U46619 |
| Isoprenaline | UTP |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
In arterial and arteriolar vessels, hyperglycaemia can mediate changes in both endothelial and smooth muscle cells, leading to changes in blood vessel diameter, BP and vascular dysfunction (Hink et al., 2001; Arun et al., 2004; Zamami et al., 2008). Significant fluctuations in plasma glucose concentration also occur physiologically through the diurnal cycle of feeding and fasting, and such changes can be exaggerated under certain pathophysiological circumstances (e.g. type 1 or type 2 diabetes). According to NICE guidelines, diabetes is often associated with cardiovascular complications, including coronary artery disease (leading to myocardial infarction and angina), peripheral artery disease (leg claudication and gangrene) and carotid artery disease (strokes and dementia). There are also microvascular complications caused by diabetes, such as diabetic retinopathy, kidney and nerve damage (NICE guidelines, 2014). Recent evidence suggests that the plasma concentration of blood glucose may also play a significant role in enhancing vasoconstriction and so have a deleterious effect on microvascular reperfusion following percutaneous coronary intervention (Iwakura et al., 2003). The risk associated with these complications can be minimized by tight glycaemic control, although there is a need for therapies to reduce the risk further. Acute hyperglycaemia (15 mM), in healthy human subjects, increases systolic and diastolic BP and heart rate and decreases leg blood flow and blood viscosity (Giugliano et al., 1997). Such acute glycaemic changes can cause rapid changes in vascular physiology, including alterations to ‘resting’ intracellular calcium concentration ([Ca2+]i) (Dunn and Nelson, 2010; Velmurugan and White, 2012), DAG levels (Lee et al., 1989; Miele et al., 2007; Descorbeth and Anand‐Srivastava, 2008), PKC localization/activity (Das Evcimen and King, 2007; Geraldes and King, 2010) and the generation of reactive oxygen species (ROS) (Tesfamariam and Cohen, 1992; Inoguchi et al., 2000; Hink et al., 2001).
In arterial smooth muscle, increases in glucose concentration often lead to vasoconstriction (Das Evcimen and King, 2007). We (Rainbow et al., 2006) and others (Li et al., 2003; Straub et al., 2009) have previously shown that this acute hyperglycaemic effect involves modulation of a voltage‐gated potassium channel (Kv) conductance, which is a key determinant of membrane potential and vascular tone (Chen et al., 2006b; Rainbow et al., 2006). This change in vascular tone occurred in vessels, or cells, isolated from a normoglycaemic animal with no reported diabetes. These findings demonstrate an effect of the elevated glucose concentration per se rather than a diabetes‐related change in contractile behaviour.
Vasoconstrictors modulate Kv through PKC isoenzymes (Rainbow et al., 2009), and there is accruing experimental evidence to support the proposition that glucose can also utilize this signal transduction mechanism (Lee et al., 1989; Miele et al., 2007; Descorbeth and Anand‐Srivastava, 2008). Such constriction may occur via a number of locally and somatically released constrictor mediators, such as endothelin‐1 (ET1) (Tamareille et al., 2013), neuropeptide Y (Clarke et al., 1987), 5‐HT (McFadden et al., 1991) and angiotensin II (ATII) (Arun et al., 2004).
In the present study, we have addressed this hypothesis by investigating the link between acute elevations in glucose concentration and changes in vascular function. We present data demonstrating for the first time that a glucose concentration‐dependent increase in vessel contraction is mediated by the DAG‐ and Ca2+‐sensitive PKCα and PKCβ isoenzymes, as also recently shown in cardiomyocytes (Sims et al., 2014). This mechanism appears to be common to rat mesentery, porcine coronary and human internal mammary arterial tissues. Crucially, we have shown that increases in glucose concentration within, or just beyond the normal physiological range, can significantly inhibit Kv current amplitude (Rainbow et al., 2006) and also alter the modulatory actions of vasoconstrictors, such as TxA2, UTP and ATII (Nobe et al., 2003, 2008; Arun et al., 2004; Ghatta and Ramarao, 2004). A glucose concentration‐dependent, PKC‐mediated modulation of Kv current provides a novel mechanism by which glucose can acutely modulate vasoconstriction within the physiological and pathophysiological glycaemic range. Such glucose‐induced constriction of vessels may provide a mechanism to help explain the worsened prognosis after acute myocardial infarction in patients with varying degrees of hyperglycaemia. Finally, inhibition of DAG‐ and Ca2+‐dependent PKC isoenzymes may pinpoint a therapeutic target for improving coronary microvascular perfusion in the heart after percutaneous coronary intervention and so potentially improve prognosis after acute myocardial infarction, improve perfusion in retinopathy and also improve systemic perfusion of the extremities.
Methods
Group sizes
For all experiments, the group size is provided in the corresponding figure legend. For myography experiments, the number of vessels and animals is reported, while for whole cell electrophysiological recordings, the number of cells and animals is reported.
Randomization
All rats used in this study were adult male Wistar rats bred in‐house at the University of Leicester. Animals were provided when they reached an appropriate size (over 300 g), and the researchers in this study had no input into the selection of animals.
Blinding
Experiments were not blinded in this study as the experimenter was responsible on each day for the cell preparation, solution making and analysis of each completed data set. To reduce any experimental bias, or anomalous results due to errors in experiments, only one cell (electrophysiology), or one pair of vessels (myography), was recorded from in 1 day for each data set. From previous experience with electrophysiology, in particular investigating changes in Kv current in the presence of glucose (Rainbow et al., 2006), it was calculated that six cells would be required for each data set. Data analysis was not performed until the complete data set had been collected to reduce experimental bias, and no complete recording was excluded. Similarly, duplicate data from three to four animals were required for myography experiments.
Normalization
Electrophysiology data in this study were normalized by two methods. (i) Data were normalized to maximum in control (normalized to 1). This allowed repeated measurements to be readily compared between cells within a data set. (ii) Data were normalized to cell capacitance in some experiments. Normalizing to cell capacitance allows comparison of currents by normalizing to cell size. Myography data were expressed as mN and normalized to the vessel diameter to allow comparison of contraction between arteries of different sizes.
Statistical comparison
All statistical analyses were performed in GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA); the statistical test used for each data set is reported in figure legends. Student's paired or unpaired t‐test and one‐way or two‐way anova with Bonferroni's post test were performed as indicated in the text. Significance was set at P < 0.05. The data and statistical analysis comply with British Journal of Pharmacology guidelines (Curtis et al., 2015).
Validity of animal species or model selection
Adult male Wistar rats have been widely used in the literature as a source of vascular tissue. The peptide‐based PKC inhibitor peptides were rat PKC isoform‐specific. The detailed electrophysiological and myography experiments would not have been possible in other species.
Finally, the validity of the data gathered in the rat model was confirmed by using porcine coronary arteries and human internal mammary arteries. These findings confirmed the same phenotypic findings in the rat.
Ethical statement
Rats were culled by stunning and cervical dislocation. The care and schedule 1 killing of animals conformed to the requirements of the United Kingdom Animals (Scientific Procedures) Act 1986 Amendment Regulations (SI 2012/3039). The ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath and Lilley, 2015) have been followed in this study. Human participants provided written, informed consent prior to tissue collection covered by the National Research Ethics Service (NHS) Ethics Committee approved study for the University of Leicester Hospitals entitled ‘Sample and Data Collection For Cardiovascular Research’. All was carried out in accordance with the Declaration of Helsinki.
Animals
Adult male Wistar animals (300–400 g) were used in this study. Porcine hearts for isolation of coronary arteries were sourced from a local abattoir.
Experimental procedures
All animals were culled using a schedule 1 procedure, stunning and cervical dislocation, that does not require prior anaesthesia. No in vivo experiments were carried out in this study, only animal‐excised tissue. Porcine coronary arteries were isolated from hearts collected from a local abattoir. Human internal mammary artery was collected from patients undergoing coronary bypass surgery.
Housing and husbandry
Rats are maintained in 1800 cm2 Tecniplast cages, using BED01/8 Datesand corn cob for bedding, housed in IVC caging (SPF free). The number of cage companions for the rats of size of 300–400 g is 5 based on a calculation of 350 cm2 being required for each rat of that size.
Interpretation
We have used the minimum possible animals to achieve statistical significance.
Further methods
Tissue dissection
Small rat mesenteric arteries were dissected as described previously (Rainbow et al., 2006, 2009). Rats were culled by stunning and cervical dislocation. The care and schedule 1 killing of animals conformed to the requirements of the United Kingdom Animals (Scientific Procedures) Act 1986 Amendment Regulations (SI 2012/3039). The ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath and Lilley, 2015) have been followed in this study. Coronary arteries were dissected from porcine hearts obtained from a local abattoir. Human internal mammary arteries were obtained from patients undergoing coronary artery surgery, immediately bathed in 5 mM glucose solution and transported on ice. The vessel was cleared of surrounding connective tissue and cut into rings. All vascular tissue was maintained on ice in 5 mM glucose solution until use. Human participants provided written, informed consent prior to tissue collection covered by the National Research Ethics Service (NHS) Ethics Committee approved study for the University of Leicester Hospitals entitled ‘Sample and Data Collection For Cardiovascular Research’. All was carried out in accordance with the Declaration of Helsinki.
Myography
Rat mesenteric (200–450 μm), porcine coronary (500–1000 μm) or human internal mammary arteries (600–1500 μm) were cut into 1.4 mm length rings and mounted in a Mulvany–Halpern wire myograph (Danish Myo‐Technology, Aarhus, Denmark) and maintained at a constant temperature of 37°C. After normalization, vessels were maintained at a tension equivalent to 90 mmHg and left to equilibrate in the test glucose solution for 30 min before experimentation. Solutions and vasoconstrictors were applied as indicated in the example traces provided in the figures. L‐NAME (20 μM) was included in all myography solutions unless stated otherwise. Data were normalized to tension after calibration. All vessels were incubated in the test glucose concentration for 30 min before experimentation unless otherwise indicated.
Patch‐clamp electrophysiology
Mesenteric arterial smooth muscle cells (MASMCs) were isolated as described previously (Rainbow et al., 2006, 2009) and maintained on ice in 5 mM glucose (0.1 mM Ca2+) solution until use. Currents were recorded in the conventional whole‐cell recording configuration. Isolated MASMCs were allowed to adhere to a glass coverslip mounted on a heated perfusion system for 15 min in a 0 mM glucose extracellular solution at 32 ± 2°C also containing penitrem A (100 nM) to limit BKCa channel interference. MASMCs were voltage clamped at −65 mV in the whole‐cell ruptured patch configuration, and an initial current–voltage relationship (I/V(1)) was recorded. After 9 min of perfusion with the test glucose concentration, a second I/V was recorded (I/V(2)) and expressed as a fraction of that in 0 mM glucose [I/V(2)/I/V(1)]. For experiments with cell‐permeant Tat peptide‐linked PKC isoenzyme‐specific inhibitor peptides, cells were pre‐incubated for 15 min in 0 mM glucose solution with 100 nM Tat‐PKC inhibitor. Pharmacological inhibitors Gö 6976 (PKCα/β inhibitor) and LY379196 (PKCβ‐selective inhibitor, a gift from Eli Lilly & Co., Indianapolis, IN, USA) (Wen et al., 2003) were used at 300 nM and pre‐incubated with the tissue for 10 min before other experimental manipulations. Concentration–response data were calculated from the fraction of current remaining after 9 min of perfusion with the test glucose at +60 mV [from I/V(2)] compared with current at +60 mV in the control I/V (0 mM glucose). In all electrophysiological recordings, the current was taken from the final 50 ms of the depolarizing step (protocol shown in Figure 1).
Figure 1.
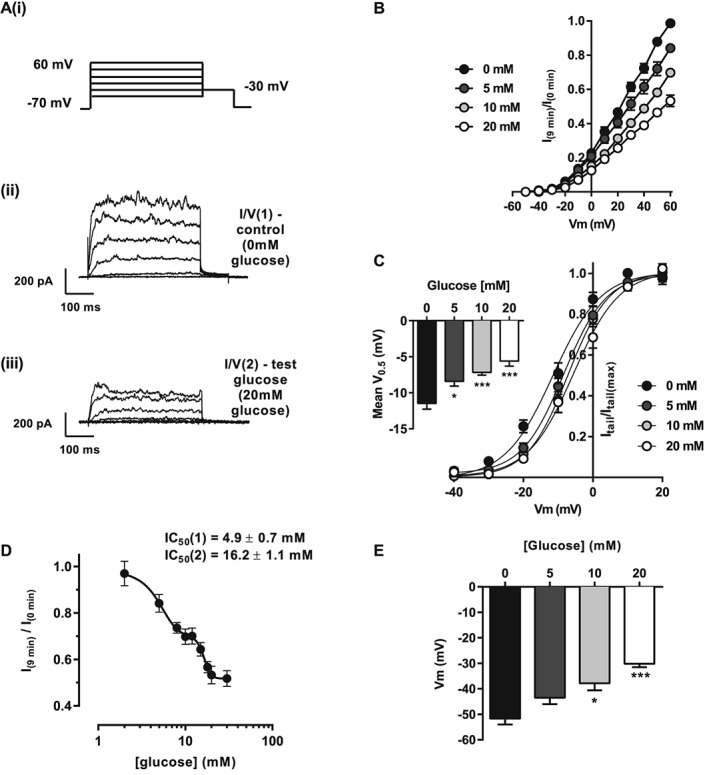
Kv currents are inhibited by glucose in a biphasic concentration‐dependent manner in rat mesenteric arterial smooth muscle cells. (A, panels i and ii) Kv current measured from isolated MASMCs in 0 mM glucose using a series of depolarizing steps from −70 to +60 mV in 10 mV increments; 20 mV steps are shown for clarity. The solution was exchanged for one containing the test glucose (e.g. 20 mM (A, panel iii)), and a second I/V recorded [I/V(2)] after 9 min perfusion. Mean data showing current–voltage relationships (B) and activation curves (C; V0.5 shown as insert) recorded from MASMCs incubated with 0, 5, 10 or 20 mM glucose. Data are expressed as I/V(2) normalized to the peak current in I/V(1) (at +60 mV). (D) Concentration‐dependent inhibitory effect of increasing glucose concentration on Kv current; two‐site analysis of the inhibition curve revealed IC50 values of 4.9 ± 0.7 and 16.2 ± 1.1 mM. (E) Histogram showing mean membrane potential after 9 min in solution containing the glucose concentration indicated (*P < 0.05; ***P < 0.001, anova with Bonferroni's post test).
Solutions
All myography solutions were based on the following solution; in mM, 135 NaCl, 6 KCl, 10 HEPES, 1.2 MgCl2, 1.8 CaCl2, 20 total monosaccharide (e.g. 5 glucose + 15 mannitol), pH 7.4 (adjusted with NaOH). Zero‐Ca2+ solution was as above with the omission of CaCl2. High K+ solution (60 mM; 60K) was as above except with 81 NaCl and 60 KCl. Bathing solutions for electrophysiology were as above with 30 mM total monosaccharide (e.g. 5 glucose + 25 mannitol) and 0.1 CaCl2. Pipette solution contained, in mM, 140 K+ (30 KOH and 110 KCl), 10 EGTA, 10 HEPES, 1 ATP, 0.5 GTP, 1 MgCl2 and 3.92 CaCl2 (equivalent to 100 nM free Ca2+). All chemicals were obtained from Sigma‐Aldrich (Poole, UK) unless stated otherwise.
Pharmacological agents and vasoconstrictors
Stock solutions of Gö 6976 and LY379196 (1 mM in DMSO) were diluted to the test concentration in the appropriate bathing solutions. DMSO concentration was never greater than 0.1%, which had no effect on the intact vessels, or in isolated MASMC patch clamp, experiments when applied (data not shown). L‐NAME, ATII, ET1, UTP, and the Tx‐mimetic U46619 (Tocris Bioscience, Bristol, UK) were dissolved in the appropriate bathing solution as stock solutions and diluted to the appropriate concentration indicated in the example traces seen in the figures. PKC isoenzyme‐selective, cell‐permeant and Tat peptide‐linked inhibitor peptides were used as previously described, pre‐incubated for 30 min before use at a concentration of 100 nM (Rainbow et al., 2009; Sims et al., 2014; Brennan et al., 2015).
Data analysis
All electrophysiology data were analysed using pCLAMP10 software, Microsoft Excel 2010 and GraphPad Prism 6. Myography data were analysed using either pCLAMP10 or Lab Chart 7.2.5, Microsoft Excel 2010 and GraphPad Prism 6. All statistical analyses were performed in GraphPad Prism 6; the statistical test used for each data set is reported in the figure legends.
Results
Kv current inhibited by extracellular glucose in a biphasic manner
We (Rainbow et al., 2006), and others (Li et al., 2003; Straub et al., 2009), have reported that glucose inhibits the Kv current in freshly isolated smooth muscle cells. Initial experiments in this study were performed to further characterize the glucose‐induced inhibition of the Kv current. Extracellular glucose inhibited Kv current in freshly isolated rat MASMC in a concentration‐dependent manner (Figure 1A, panels ii and iii, and B). Further to this, there was a glucose concentration rightward shift in the voltage dependence of activation calculated from the tail current (Figure 1C, −11.5 ± 0.8 vs. −5.6 ± 0.6 mV in 0 and 20 mM glucose respectively, P < 0.001). This inhibitory response of increasing extracellular glucose was most accurately modelled by a two‐site analysis (IC50 H 4.9 ± 0.7 mM; IC50 L 16.2 ± 1.1 mM), indicative of a biphasic response, with a maximal inhibitory effect on Kv current of around 50% (Figure 1D). Kv inhibition by increasing extracellular glucose concentration resulted in graded depolarization of membrane potential (Figure 1E, P < 0.001 at 20 mM glucose).
Glucose‐mediated inhibition of Kv current is attenuated by inhibition of Ca2 +/DAG‐sensitive PKC isoenzymes
We have recently reported that glucose causes action potential prolongation, presumably via Kv inhibition, via conventional PKCs in freshly isolated cardiomyocytes (Sims et al., 2014). To determine if a similar mechanism occurs in vascular smooth muscle, we investigated the role of PKC in glycaemic modulation of the Kv current in MASMCs. Initial experiments used the PKC inhibitor, Tat‐PKC 20‐28 peptide, which was able to prevent glucose‐induced suppression of Kv current (Figure 2A–C). These data suggest that PKC played a role in inhibiting the amplitude of Kv current in MASMCs.
Figure 2.
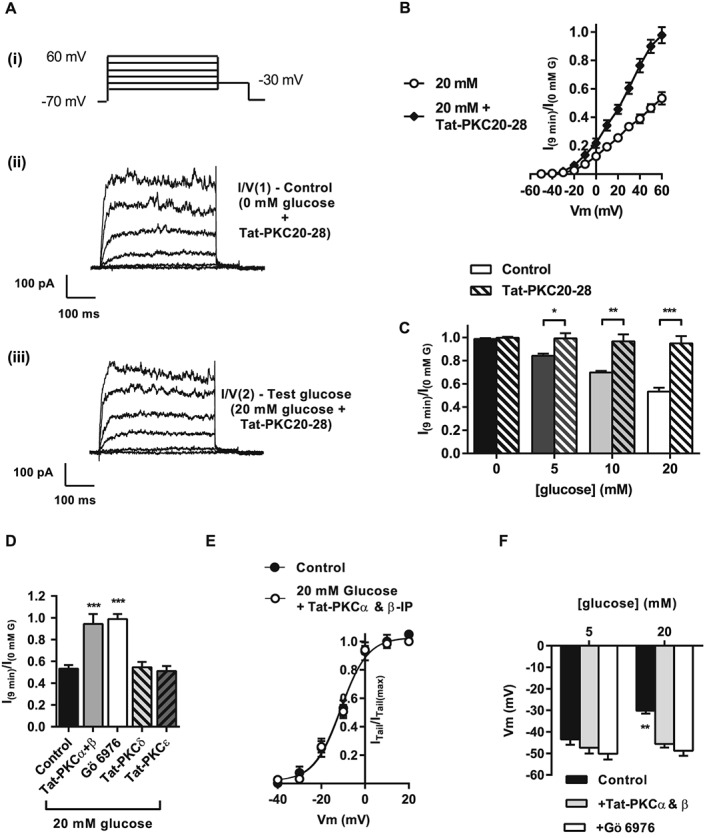
Tat‐PKC20‐28 attenuates the inhibition of the voltage‐dependent potassium (Kv) current by glucose in rat mesenteric arterial smooth muscle cells. (A, panels i and ii) Kv current measured from isolated MASMCs in 0 mM glucose + Tat‐PKC20‐28 using a series of depolarizing steps from −70 to +60 mV in 10 mV increments. The solution was exchanged for one containing the test glucose Tat‐PKC20‐28 (e.g. 20 mM (A, panel iii)), and a second I/V recorded [I/V(2)] after 9 min perfusion. (B) Mean data for I/V plots recorded from cells incubated with 20 mM glucose with and without Tat‐PKC20‐28. Data expressed as I/V(2) normalized to the peak current in I/V(1) (at +60 mV). (C) Histogram showing attenuation by Tat‐PKC20‐28 of Kv current inhibition via 0, 5, 10 or 20 mM glucose (*P < 0.05; **P < 0.05; ***P < 0.001, anova with Bonferroni's post test). (D) Histogram showing effects of Tat‐PKC isoenzyme‐specific inhibitor peptides (PKCα + β, PKCδ or PKCε, each at 100 nM), or pharmacological inhibition of PKCα + PKCβ with Gö 6976 (300 nM) on the Kv current in the presence of 20 mM glucose (***P < 0.001, one‐way anova with Bonferroni's post test). (E) Mean data showing activation curves from MASMCs incubated with 0 or 20 mM glucose in the presence of Tat‐PKC20‐28. Data are expressed as I/V(2) normalized to the peak current in I/V(1) (at +60 mV). (F) Gö 6976 or Tat‐PKCα + Tat‐PKCβ inhibition prevented the depolarized membrane potential observed in the presence of 20 mM compared with 5 mM glucose (**P < 0.01, two‐way anova with Bonferroni's post test). Data in the absence of Tat‐PKC or pharmacological inhibitors (‘control’ B–F) are taken from Figure 1 to allow direct comparison.
The PKC isoenzyme selectivity of Kv inhibition by elevated glucose was investigated using a selective inhibitor of conventional, DAG/Ca2 +‐dependent PKCs, Gö 6976 (300 nM), or a combination of two isoenzyme‐specific peptide inhibitors Tat‐PKCα and Tat‐PKCβ (Figure 2D–F). Both small molecule and peptide inhibitors caused a complete reversal of current suppression by high (20 mM) glucose and reversed the shift in the voltage dependence of activation (Figure 2E). In contrast, treatment with Tat‐PKCδ or Tat‐PKCε inhibitory peptides was without effect (Figure 2D). High glucose‐induced membrane depolarization was also abolished by combined inhibition by Tat‐PKCα plus Tat‐PKCβ, or Gö 6976 (Figure 2F). These data suggest that the PKCα and β isoenzymes are responsible for the inhibition of the Kv current imparted by high glucose in MASMCs.
Dissecting the contribution of PKCα and β
To determine the relative contributions of PKCα and β to the Kv current inhibition caused by glucose, electrophysiological experiments were performed using Tat‐PKCα or Tat‐PKCβ. The Tat‐PKCβ inhibitor peptide completely prevented the suppression of Kv current by glucose concentrations below 10 mM but not by higher glucose concentrations (Figure 3A, C and E). In the presence of Tat‐PKCβ, increasing extracellular glucose caused a monophasic inhibition of Kv current (IC50 16.3 ± 1.8 mM; Figure 3E). The Tat‐PKCα peptide also caused a monophasic inhibition of current. Conversely to Tat‐PKCβ, Tat‐PKCα only prevented the current suppression at glucose concentrations above 10 mM (IC50 4.9 ± 1.3 mM; Figure 3F). These data suggest that PKCα and β play distinct and complementary roles in the actions of glucose on Kv current with PKCβ modulating activity between 0 and 10 mM and PKCα modulating the current between 10 and ~20 mM.
Figure 3.
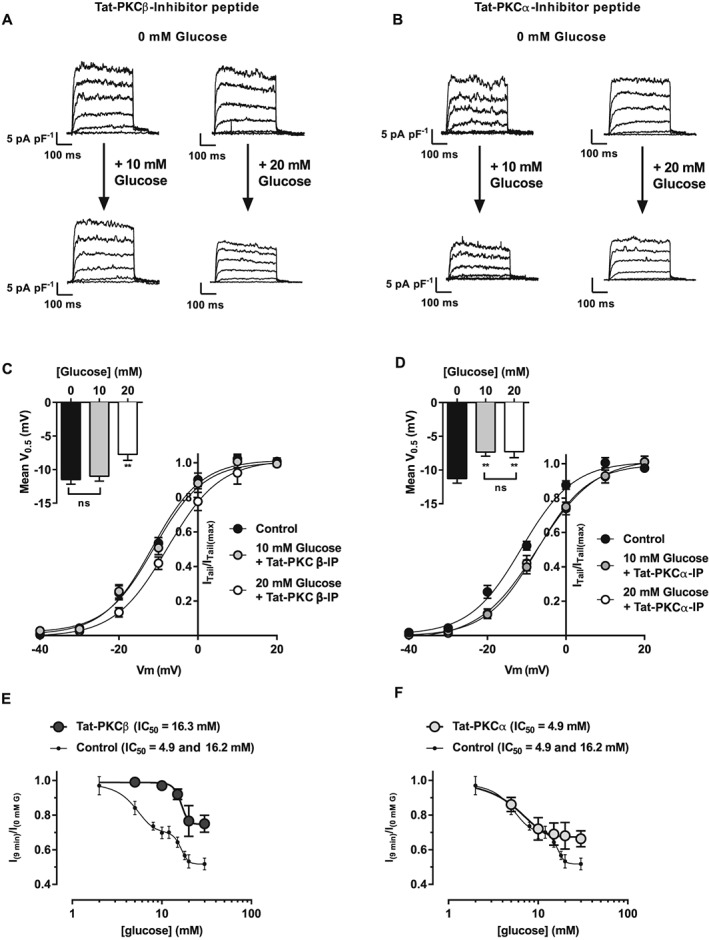
Kv current inhibited by glucose in a monophasic manner in rat mesenteric arterial smooth muscle cells in the presence of Tat‐PKCβ and Tat‐PKCα inhibitor peptides. Kv current measured from isolated MASMCs in 10 or 20 mM glucose in the presence of (A) Tat‐PKCβ or (B) Tat‐PKCα in a series of depolarizing steps from −70 to +60 mV in 10 mV increments. Mean data showing activation curves (V0.5 shown as insert) recorded from MASMCs incubated with 0, 10 or 20 mM glucose in the presence of (C) Tat‐PKCβ or (D) Tat‐PKCα. Data are expressed as I/V(2) normalized to the peak current in I/V(1) (at +60 mV). Tat‐PKCβ (E) and Tat‐PKCα (F) each altered the maximal inhibitory response by glucose to Kv current and converted the biphasic effect into a monophasic curves. Each data point was obtained from >6 cells from >3 animals. In (E) and (F), concentration–response curves are repeated from Figure 1 to allow direct comparison with control.
Effect of extracellular glucose concentration on arterial contractility
Myography experiments were performed to determine the effect of high glucose on contraction. The contractile response of rat mesenteric arteries to addition of 60 mM K+ (60K) increased (by ≥30%) when extracellular glucose concentration was raised from 5 to 20 mM (Figure 4A and B). Additionally, there was a significant increase in the Ca2 + response to 60K concomitantly with tension in isolated vessels loaded with Fura‐2 (Supporting Information Fig. S1). Consistent with our previous findings of the reversible nature of the Kv current inhibition by elevated glucose (Rainbow et al., 2006), an initial application of 60K in 20 mM glucose, followed by washout with 5 mM glucose, yielded a smaller second 60K response (Figure 4C).
Figure 4.
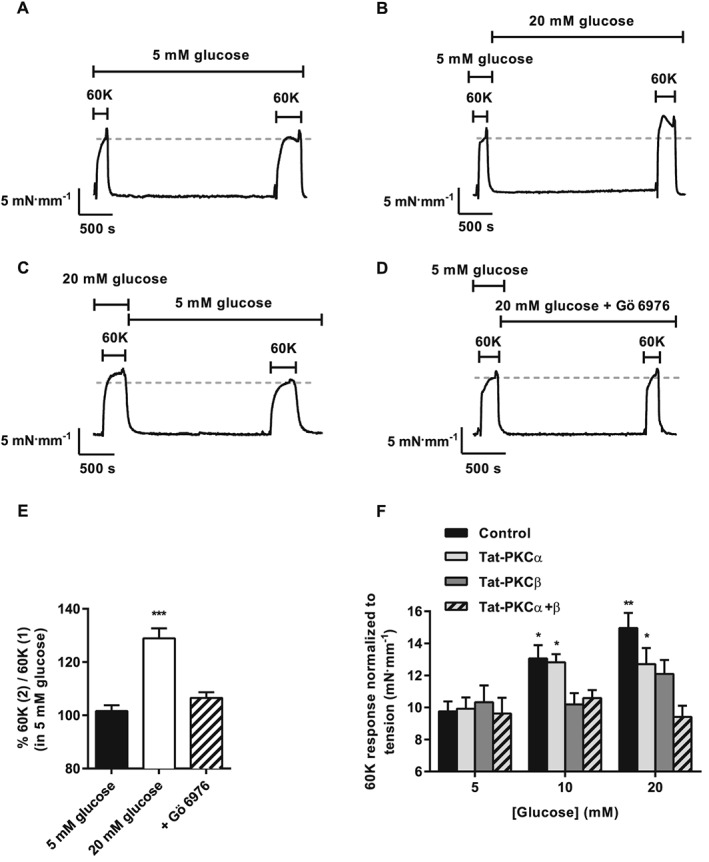
K+‐induced vasoconstriction of rat mesenteric artery is enhanced by high extracellular glucose and reversed by PKCα + β inhibition. Example traces of tension produced to two applications of 60K (60K(1) and 60K(2)) with 30 min incubation in (A) 5 mM glucose and (B) 20 mM glucose before 60K(2). (C) Example trace shows that enhancement of 60K contraction by 20 mM glucose is reversed by 30 min incubation in 5 mM glucose. (D) Experiment identical to (B) except for addition of Gö 6976 (300 nM) after 60K(1). (E) Mean data showing increases in tension for 60K(2) relative to 60K(1) (%) and reversal of the high glucose potentiation by 300 nM Gö 6976 (**P < 0.01; ***P < 0.001, repeated measures anova with Bonferroni's post test, or unpaired t‐test). (F) Histogram showing the mean contractile response to 60K (mN·mm−1) for control vessels and vessels pre‐incubated with the indicated Tat‐PKC isoenzyme inhibitors before switching to solution containing 5, 10 or 20 mM glucose (*P < 0.05; **P < 0.01, two‐way anova with Bonferroni's post test).
Using 125 mM K+ (125K) to abolish the K+ gradient completely, the effects of 20 mM glucose were investigated using the same protocol; 125K abolished the potentiation of contraction in 20 mM glucose, demonstrating that in 60K there remains a small hyperpolarizing K+ flux that can be modulated by glucose, presumably with PKC dependence (Supporting Information Fig. S2). In the absence of a complete molecular characterization of Kv expression in MASMC, the Kv1‐family channel blocker Psora‐4 (Marzian et al., 2013) was used to probe the contribution of Kvs. Pre‐incubation with the Kv1‐inhibitor Psora‐4 enhanced the vasoconstrictor response to 60K in 5 mM glucose, and no further increase was observed when glucose concentration was raised from 5 to 20 mM. Furthermore, Psora‐4 (100 nM) elicited a marked inhibition of Kv and membrane depolarization, confirming a substantial role for Kv1 subunits as components of the voltage‐gated K+ current seen in MASMC (control, −49 ± 2 mV; +Psora‐4, −25 ± 2 mV). Together, these findings suggest that inhibition of channels containing Kv1‐family subunits plays a role in the enhanced vasoconstrictor response to depolarization after acute treatment with high glucose.
Consistent with our previous findings (Rainbow et al., 2006), replacement of d‐glucose with non‐metabolizable l‐glucose did not yield a potentiation of contractile response after incubation with 20 mM l‐glucose. Furthermore, replacement of extracellular glucose with fructose (20 mM) yielded a potentiated second 60K response (Supporting Information Fig. S3), consistent with a glycolytic metabolism‐dependent mechanism.
The enhanced vasoconstriction and Ca2 + response to 60K in rat mesenteric arteries exposed to 20 mM glucose were abolished by the DAG/Ca2 +‐dependent PKC inhibitor Gö 6976 (Figure 4D–E and Supporting Information Fig. S1). These data suggest that the response imparted by high extracellular glucose is mediated via conventional PKC isoenzymes.
Glucose‐mediated enhancement of depolarization‐induced vasoconstriction is reversed by selective PKCα and PKCβ inhibition
Tat‐PKCα and β peptides were used to determine the relative contributions of PKCα and β, respectively, to the enhanced contraction caused by high glucose in the presence of 60K. At an intermediate glucose concentration (10 mM), isoenzyme‐selective PKCα inhibition had no effect, while PKCβ inhibition completely abolished the enhanced contraction (Figure 4F). At a higher glucose concentration (20 mM), selective PKCα or PKCβ inhibition each partially prevented the enhanced constriction, which was abolished in the presence of both inhibitors (Figure 4F). These contractile data are in accordance with the electrophysiological data shown in Figure 3. Combined, these findings suggest that DAG/Ca2 +‐dependent PKC isoenzymes play distinct and complementary roles in the actions of glucose on rat mesenteric artery contraction with PKCβ modulating activity between 0 and 10 mM and PKCα modulating the contraction between 10 and ~20 mM.
Contractile response to angiotensin II, but not endothelin‐1, is enhanced by elevated glucose and attenuated by selective PKCα or PKCβ inhibition
To investigate if elevated glucose concentrations could alter the contractile response to vasoconstrictors, ATII and ET1 were applied to rat mesenteric arteries in the presence of 5 and 20 mM glucose. In rat mesenteric arteries, increasing extracellular glucose concentration to 20 mM markedly enhanced (by ~2.5 mN·mm−1) the vasoconstrictor response to ATII (100 nM; Figure 5A, panels i and ii). This effect was abolished by pre‐incubation with Tat‐PKCα‐plus‐Tat‐PKCβ inhibitor peptides (Figure 5A, panel iii), or Gö 6976 (300 nM; Figure 5B). The constrictor response to ATII was completely abolished by selective inhibition of PKCε by a Tat‐conjugated inhibitor peptide, consistent with the known dependence of ATII vasoconstrictor activity on this novel PKC isoenzyme (Figure 5A, panel iv) (Rainbow et al., 2009). Consistent with the glucose concentration‐dependent inhibition of Kv, selective inhibition of PKCβ, but not PKCα, abolished the hyperglycaemia‐mediated increase in ATII‐induced vasoconstriction at 10 mM glucose (Figure 5C); at higher glucose concentrations (20 mM), the enhanced response was fully attenuated by combined inhibition of PKCα and PKCβ (Figure 5C).
Figure 5.
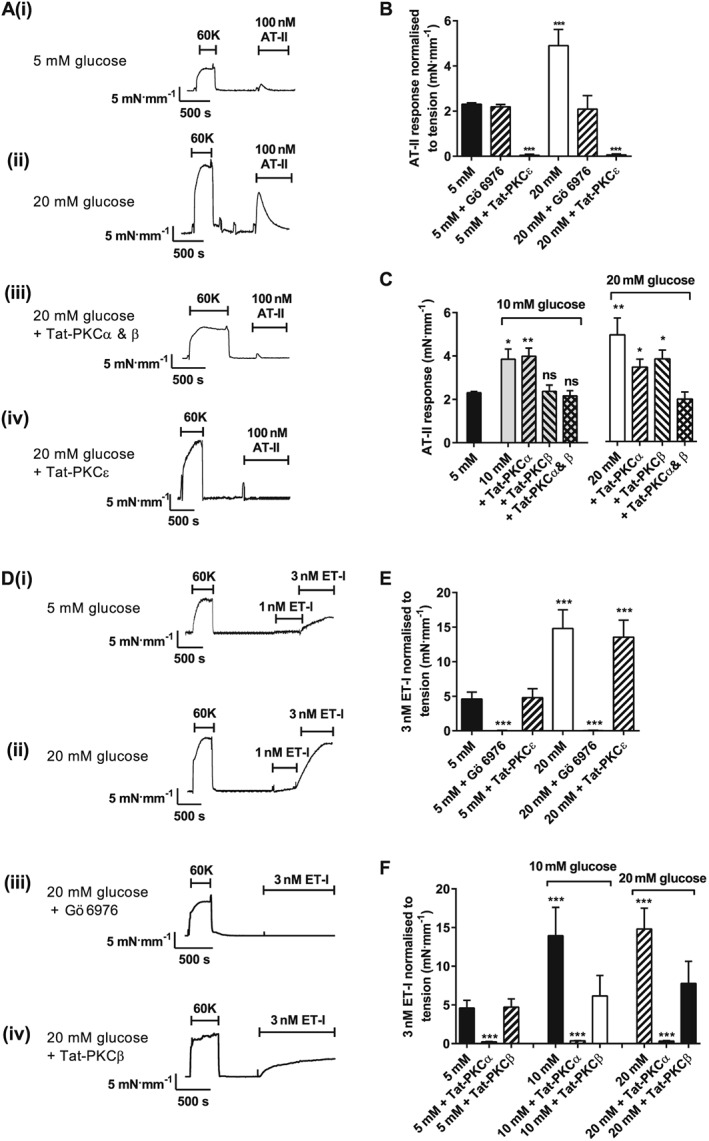
Agonist‐induced constriction of rat mesenteric artery is enhanced by high extracellular glucose in a conventional PKC isoenzyme manner. Example traces of 60K and ATII (100 nM)‐induced constrictor responses for rat mesenteric arteries in 5 (A, panel i) or 20 (A, panel ii) mM glucose, 20 mM glucose + Tat‐PKCα + Tat‐PKCβ (A, panel iii) or Tat‐PKCε (A, panel iv). (B) Mean contractile responses to ATII in 5 or 20 mM glucose with Gö 6976 or Tat‐PKCε. (C) Mean contractile responses to ATII in 10 or 20 mM glucose with Tat‐PKCα, Tat‐PKCβ and Tat‐PKCα + Tat‐PKCβ (≥12 vessels per condition from ≥4 animals: *P < 0.05; **P < 0.01, one‐way anova with Bonferroni's post hoc test). (D) Example traces of 60K and ET1 (1 and 3 nM)‐induced constrictor responses for rat mesenteric arteries in 5 (D, panel i) or 20 (D, panel ii) mM glucose, 20 mM glucose + Tat‐PKCα + Tat‐PKCβ (D, panel iii) or Tat‐PKCε (D, panel iv). (E) Mean contractile responses to ET1 (3 nM) in 5 and 20 mM glucose normalized to tension (mN·mm−1) in control vessels and vessels pretreated with Gö 6976 or Tat‐PKCε inhibitor. (F) Mean contractile responses to ET1 (3 nM) with 10 or 20 mM glucose in the absence and presence of Tat‐PKCα or Tat‐PKCβ inhibitory peptides (≥10 vessels per condition from ≥5 animals: ***P < 0.001, anova with Bonferroni's post test).
Contractile responses to ET1 were abolished in small rat mesenteric arteries by PKC inhibition with Gö 6976 (Figure 5D, panel iii, and E), consistent with the predominantly PKCα‐mediated mechanism of action reported for this vasoconstrictor (Rainbow et al., 2009). ET1‐mediated vasoconstriction was increased by increasing extracellular glucose to 10 mM and largely prevented by selective PKCβ inhibition (Tat‐PKCβ inhibitor peptide). No further enhancement was observed by increasing glucose concentration from 10 to 20 mM (Figure 5F). These data are consistent with high glucose and ET1 both exerting their contractile effects through PKCα and so showing no further potentiation of contraction at glucose concentrations greater than 10 mM. This was supported by experiments investigating Kv current inhibition by ATII and ET1 in rat MASMC in 20 mM glucose, where ATII was able to further inhibit the current, whereas ET1 was ineffective (Supporting Information Fig. S4). Additionally, the enhanced contractile responses in the presence of 20 mM glucose of rat mesenteric arteries to UTP and U46619 were also prevented by Gö 6976 (Supporting Information Fig. S5). Despite there being a common glucose‐induced PKC‐mediated enhancement of vasoconstrictor activity, the PKA‐dependent dilatory pathway, via β‐adrenoceptor stimulation, was unaffected by elevated glucose (Supporting Information Fig. S6). Collectively, these data suggest that there is a common mechanism by which enhanced concentrations of glucose cause inhibition of Kv current, membrane depolarization, calcium influx and contraction.
Glucose‐mediated PKC‐dependent enhancement of vasoconstriction in porcine coronary and human internal mammary arteries
To assess the generality of these findings, similar experiments were performed in porcine coronary arteries (Figure 6) and human internal mammary arteries (Figure 7). These data from porcine coronary and human internal mammary arteries were analogous to the findings in rat mesenteric arteries. In both vascular beds, the contractile response to 60K (Figure 6A and B and Figure 7A and B) was increased in the presence of elevated glucose. There was no further enhancement of ET1 porcine coronary vasoconstrictor activity above 10 mM extracellular glucose (Figure 6C). ET1 contractions were abolished in porcine coronary arteries by PKC inhibition with Gö 6976 but not with the selective PKCβ inhibitor LY379196 (300 nM) (Figure 6A, panels iii and iv, and C). This further supports the predominantly PKCα‐mediated mechanism of action reported for this vasoconstrictor (Rainbow et al., 2009). Contractions of porcine coronary arteries and human internal mammary arterioles induced by U46619 (Figure 6D and E and Figure 7D and E) were markedly enhanced in high extracellular glucose. For each contractile stimulus tested on porcine coronary and human internal mammary arterioles, the presence of Gö 6976 prevented the enhanced contraction caused by high glucose (Figure 6B, C and F and Figure 7B, C and F). Selective PKCβ inhibition (LY379196; 300 nM) also decreased the magnitude of contraction to each contractile stimulus tested. The data collected from these three different vascular beds (across three different species) suggest that PKCα‐ and PKCβ‐mediated enhancement of contractions by high extracellular glucose is a common mechanism throughout the cardiovascular system.
Figure 6.
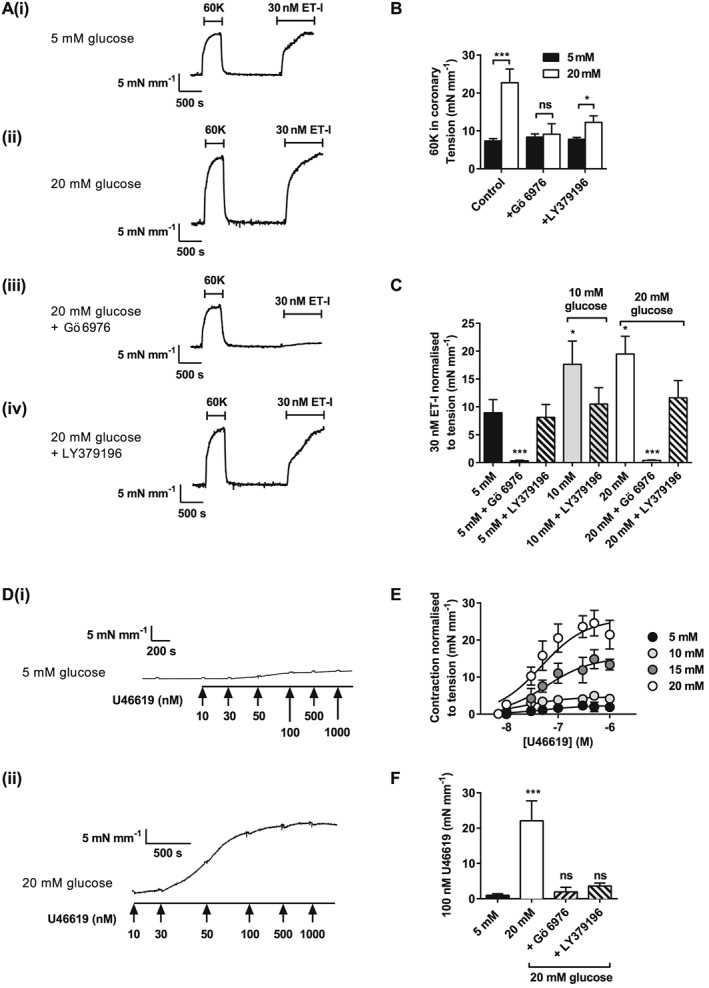
Agonist‐induced constriction of porcine coronary artery is enhanced by high extracellular glucose in a conventional PKC isoenzyme manner. (A) Example traces of 60K and ET1 (30 nM)‐induced constrictor responses from porcine coronary arterial rings in 5 (A, panel i) or 20 (A, panel ii) mM glucose, and in 20 mM glucose with Gö 6976 (A, panel iii), or PKCβ‐selective inhibitor LY379196 (A, panel iv). (B) Mean contractile responses to ET1 in 5 or 20 mM glucose in the absence or presence of Gö 6976 or LY379196. (C) Mean contractile responses to KCl in 5, 10 or 20 mM glucose in the presence or absence of PKCα and/or PKCβ inhibitors (≥9 vessels per condition from ≥4 pigs: *P < 0.05; ***P < 0.001, two‐way anova with Bonferroni's post test). (D) Example traces of U46619 (10–1000 nM)‐induced constrictor responses from porcine coronary arterial rings in 5 (D, panel i) or 20 (D, panel ii) mM glucose. (E) Concentration–response curve to U46619 in the presence of 5, 10, 15 and 20 mM glucose. (F) Mean contractile responses to U46619 (100 nM) in 5 and 20 mM glucose in the absence or presence of Gö 6976 or LY379196.
Figure 7.
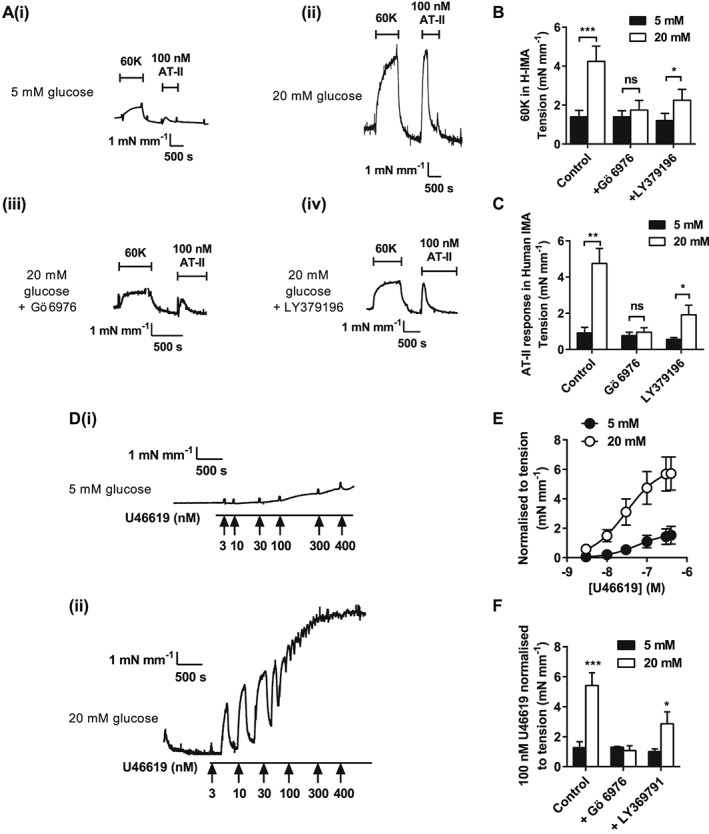
Agonist‐induced constriction of human mammary artery is enhanced by high extracellular glucose in a conventional PKC isoenzyme manner. Example traces of 60K and ATII (100 nM)‐induced constrictor responses human mammary artery in 5 (A, panel i) or 20 (A, panel ii) mM glucose, 20 mM glucose + Gö 6976 (A, panel iii) or LY379196 (A, panel iv). (B) Mean contractile responses to 60K (B) and ATII (C) in 5 or 20 mM glucose with Gö 6976 or LY379196 (≥4 vessels per condition from ≥4 patients, *P < 0.05; ***P < 0.001, two‐way anova with Bonferroni's post test). (D) Example traces of U46619 (3–400 nM)‐induced constrictor responses from human mammary artery in 5 (panel i) or 20 (panel ii) mM glucose. (E) Concentration–response curve to U46619 in the presence of 5 and 20 mM glucose. (F) Mean contractile responses to U46619 (100 nM) in 5 and 20 mM glucose in the absence or presence of Gö 6976 or LY379196 (≥4 vessels per condition from ≥4 patients, *P < 0.05; ***P < 0.001, two‐way anova with Bonferroni's post test).
Discussion
We have presented novel experimental data indicating that acute changes in extracellular glucose concentration within the physiological/pathophysiological range increase basal tone and vasoconstrictor responses in small arteries from three different species. This occurs through glucose concentration‐dependent recruitment of conventional PKC activities and modulation of specific K+ currents. In particular, we report that acutely increasing extracellular glucose concentration leads to a biphasic inhibition of the voltage‐gated K+ current and membrane depolarization. In addition, this inhibitory effect on Kv is dependent on at least two DAG/Ca2 +‐dependent PKC isoenzymes, with PKCβ playing a key role at lower glucose concentrations (≤10 mM) and the PKCα isoenzyme contributing at higher glucose concentrations. Furthermore, we demonstrate that this glucose‐mediated PKC isoenzyme‐dependent inhibition of Kv results in increased vascular tone and increased vasoconstrictor responsiveness through a mechanism that is conserved across a number of different vasoconstrictors, species and vascular beds.
Previous reports have shown that inhibition of the Kv current occurs within minutes of elevating extracellular glucose (Li et al., 2003; Rainbow et al., 2006; Straub et al., 2009). We hypothesized that increasing glucose concentration causes activation of PKC isoenzymes leading to inhibition of the Kv current, presumably by phosphorylation of Kv subunits. The exact molecular identity of the Kv current in arterial smooth muscle is complex and currently unclear making direct measurement of subunit phosphorylation difficult; however, a number of subunits have been identified, with particular importance being placed on the Kv1 family (Knot and Nelson, 1995; Cole et al., 1996; Albarwani et al., 2003). Indeed, our data using Psora‐4 (Marzian et al., 2013; Wulff and Yarov‐Yarovoy, 2013), and previous findings by others using Kv1.5 dominant‐negative channel subunits (Chen et al., 2006a), indicate an important role for this Kv family subgroup in vasoregulation. A number of other Kv subtypes have been described in systemic vascular smooth muscle, including Kv2, 4 and 7 (Ko et al., 2008; Stott et al., 2014). Given the hetero‐multimerization possible within expressed Kv complexes, a role for other Kv subfamilies cannot be ruled out.
Elevated glucose concentration is known to increase cellular levels of the endogenous PKC activator, DAG in a variety of tissues, including arterial smooth muscle (Inoguchi et al., 1994; Nobe et al., 2003, 2004). The increase in DAG concentration provides a potential mechanism for glucose‐induced PKC activation, specifically activation of the DAG/Ca2 +‐dependent isoenzymes PKCα and PKCβ. Basal cytoplasmic Ca2 + concentrations have also been reported to increase in rat tail artery smooth muscle cells across a similar range of extracellular glucose concentrations (5–20 mM) to that used in the present study (Barbagallo et al., 1995). However, we did not observe an increase in basal Ca2 + in whole vessels during 30 min incubation with solution containing 20 mM glucose. However, we did not observe an increase in basal Ca2 + in whole vessels during 30 min incubation with solution containing 20 mM glucose. Despite this, there was a small constriction in response to the increased glucose during this time (0.95 ± 0.13 mN·mm−1). It is plausible that any changes in intracellular basal Ca2 + were too small to be detected by the recording system used. Additionally, there may be a glucose‐induced PKC‐dependent sensitization of contractile machinery giving a small constrictor response for little or no apparent increase in [Ca2 +]i. Further to this, Kvs are one component of the hyperpolarizing response to depolarization that limits Ca2 + influx. BKCa channels, sensitive to both intracellular Ca2 + and voltage, together with KATP channel current could act to limit constrictor responses in the short term with a subtle depolarization of basal membrane potential. Furthermore, the changes in membrane potential seen in whole‐cell recordings of isolated cells may not occur as rapidly in the intact tissue, where cells are electrically coupled to adjacent cells. Basal intracellular Ca2 + increases in the whole vessel would not be as large in this scenario, and so increases in [Ca2 +]i may not be detected. An enhanced response to 60K‐induced depolarization under hyperglycaemic conditions was observed, suggesting an increase in Ca2 + influx and/or enhanced Ca2 + release. It would seem likely that an enhanced Ca2 + influx can account for this elevated Ca2 + response, consistent with our hypothesis of a reduced Kv current. Reducing the Kv‐induced hyperpolarizing current, resulting in further depolarization, would enhance L‐type voltage‐gated Ca2 + channel activity.
Collectively, our electrophysiology and myography data demonstrate that extracellular glucose increases in the 5–10 mM range lead to an increased vascular tone and vasoconstrictor responsiveness mediated by PKCβ/Kv current inhibition, while a PKCα/Kv‐dependent component is evident at higher glucose concentrations. The importance of these signal transduction mechanisms is further emphasized by assessment of the response to specific vasoconstrictors with known mechanisms of action. The potentiation of vasoconstrictor responsiveness in the presence of increased extracellular glucose occurs via additive PKCα‐ plus PKCβ‐dependent mechanisms (Figure 8). For example, the glucose‐induced PKCβ‐ and PKCα‐mediated mechanism leads to further vasoconstriction of ATII, a primarily PKCε‐dependent pathway, revealing the additive effect of the different PKC isoenzymes on Kv current inhibition and vasoconstriction. These findings of enhanced PKCα‐ and PKCβ‐dependent vasoconstriction in high glucose are seen consistently using other vasoconstrictors, such as U46619 in porcine coronary and UTP in rat mesenteric arteries. Hence, it appears that glucose‐mediated PKC‐dependent inhibition of the Kv current not only increases basal tone but also potentiates vasoconstrictor response to G‐protein‐activated signalling pathways, as well as (60K) depolarization‐induced vasoconstriction. In contrast, the glucose‐mediated potentiation of ET1‐induced vasoconstriction, a predominantly PKCα‐dependent pathway (Rainbow et al., 2009), is only observed in the 5–10 mM (PKCβ‐dependent) range, with no increase in vasoconstrictor activity seen in the 10–20 mM (PKCα‐dependent) range. In summary, the glucose‐mediated potentiation of constriction is greater for agonists that do not act via PKCβ or PKCα (e.g. ATII) compared with agonists that constrict via PKCβ or PKCα (e.g. ET1).
Figure 8.
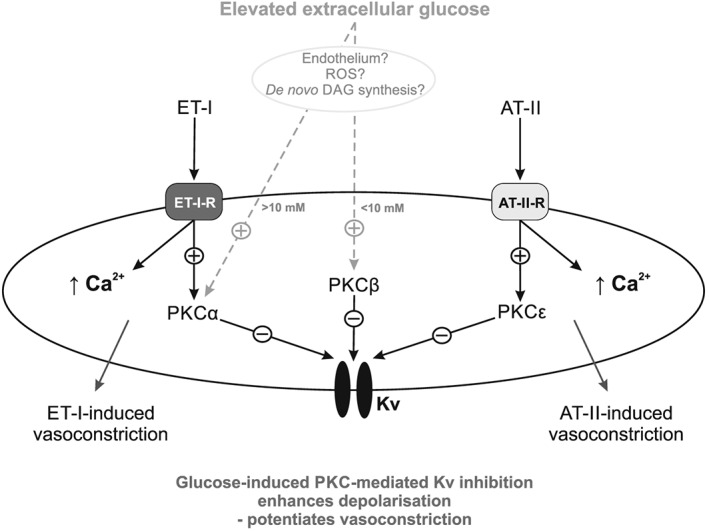
A representation of the proposed mechanism of glycaemia‐enhanced vasoconstrictor signalling in vascular smooth muscle. Activation of PKCα and PKCβ with increasing glucose has a cumulative inhibitory effect on the Kv current so enhancing vasoconstriction by depolarizing stimuli (such as 60K), or enhancing contraction to ATII, which activates an alternate PKC isoenzyme. ET1‐induced vasoconstriction is only potentiated by glucose concentrations up to 10 mM, due to it acting via a PKCα‐dependent pathway. The mechanism by which glucose activates PKC is currently unclear with ROS generation, de novo DAG synthesis, or a presently unidentified endothelial‐derived modulator being potential candidates.
In contrast to a previous study in rat coronary arteries (Li et al., 2003), we also provide experimental evidence that high glucose‐induced DAG/Ca2 +‐dependent PKC‐Kv‐mediated potentiation of vasoconstriction does not significantly alter the vasodilator activity of the β‐adrenoceptor agonist, isoprenaline (Supporting Information Fig. S6).
Our previous findings, and our current data in isolated MASMCs, give added credence to our hypothesis that there is a direct effect of glucose, or a glucose metabolite(s), on PKC activation in vascular smooth muscle independent of the endothelium. The electrophysiology data in this current study and others (Li et al., 2003; Rainbow et al., 2006; Straub et al., 2009) show that elevated glucose decreases Kv current in isolated vascular smooth muscle cells. Furthermore, enhanced arterial contractility in elevated glucose was also maintained in the presence of the NOS inhibitor, L‐NAME. This is a potentially important finding, as in hyperglycaemia endothelial dysfunction has been hypothesized to reduce the activity of NOS‐dependent vasodilator pathways (Bohlen and Nase, 2001; Beckman et al., 2002). Our data suggest that the effects of acute hyperglycaemia occur independently of any vascular endothelial dysfunction.
The involvement of ROS, and enhanced generation in hyperglycaemic conditions, is a further potential mechanism for altered signalling. In our previous study, however, use of a free radical scavenger (N‐(2‐mercaptopropionyl)glycine) did not have a significant effect on the glucose‐induced inhibition of Kv current in isolated MASMCs (Rainbow et al., 2006). However, in another study, the use of superoxide dismutase and catalase partially reversed a high glucose‐induced inhibition of Kv current, suggesting a role for superoxide radicals (Liu et al., 2001). In addition, ROS has been suggested to have effects on other channels involved in vasoconstriction, including L‐type voltage‐gated Ca2 + channels (Shaifta et al., 2015). In this recently published study, the authors demonstrated an enhanced vasoconstriction, and an enhanced Ca2 + current, in response to ROS generation in a number of arterial beds, including mesenteric. Given that elevated glucose has been demonstrated to increase ROS in vascular tissue, further investigations will be required to characterize their effects on the enhanced constrictor responses seen in the present study and whether other currents are modulated by this pathway.
Acute elevations of extracellular glucose have also been shown to have deleterious effects on the electrical properties of isolated cardiomyocytes, causing a marked prolongation of action potential duration (Hreiche et al., 2009) and subsequent QT prolongation, which is suggested to be pro‐arrhythmic (Marfella et al., 2001; Gordin et al., 2008). Our own recent findings suggest a similar DAG/Ca2 +‐PKC isoenzyme‐dependent dysregulation of electrical signalling in cardiac tissue (Sims et al., 2014). The findings of the current study provide evidence for a common potential mechanism through which elevated glucose may exert direct deleterious effects on cardiovascular function with similar DAG/Ca2 +‐PKC isoenzyme‐dependent mechanisms being evident in both heart and vasculature. The present study also demonstrates that hyperglycaemia exacerbates vasoconstriction both directly and by exacerbating the response to circulating vasoconstrictors.
Hyperglycaemia has been previously shown to cause adverse effects on leg bloodflow (Giugliano et al., 1997). Additionally, there is ample evidence that acute hyperglycaemia is a predictor of poor outcome in several vascular diseases, such as acute myocardial infarction, ischaemic stroke (Parsons et al., 2002) and vascular (mesenteric) ischaemia (Schaffler et al., 1998). It is presently unclear whether acute hyperglycaemia, observed under these conditions, has a cause and effect relationship. The need for further research into potential underlying mechanisms is clear; however, here we provide compelling evidence that specific PKC isoenzymes are implicated in a process that could contribute to this phenomenon. Our findings suggest the selective inhibition of PKCα and PKCβ isoenzymes as a potential therapeutic target in patients with hyperglycaemia‐complicating acute syndromes, such as myocardial infarction and ischaemic stroke. In clinical practice, inhibition of both PKCα and PKCβ may not be well tolerated, due to the ubiquitous expression and multiple roles of these PKC isoenzymes (Serova et al., 2006; Rosse et al., 2010). However, our demonstration of primarily PKCβ isoenzymic involvement in Kv current modulation at 5–10 mM glucose, a level of hyperglycaemia associated with adverse prognosis following acute myocardial infarction (Goyal et al., 2006; Kosiborod et al., 2008; Squire et al., 2010), suggests that PKCβ inhibition alone should have significant therapeutic potential in this context. The PKCβ inhibitor ruboxistaurin has been shown to be well tolerated in clinical trials for other indications but has not to date been investigated in the context of such cardiovascular indications (Idris and Donnelly, 2006; Geraldes and King, 2010). Therefore, isoenzyme‐specific PKC inhibition may represent a potential therapeutic target in the management of acute syndromes complicated by hyperglycaemia‐potentiated vasoconstriction.
Author contributions
R. J. and S. B. performed experimental work, conducted data analysis and contributed to the writing and revision of the manuscript. P. F. and M. W. S. performed experimental work and conducted data analysis. R. A. J. C. initiated the research, contributed to experimental design and contributed to the writing and revisions of the manuscript. D. A. contributed to the writing and revisions of the manuscript. I. B. S. initiated the research, contributed to experimental design and contributed to the writing and revisions of the manuscript. R. D. R. initiated and led the research and experimental design, performed experimental work and conducted data analysis, and led the writing and revision of the manuscript.
Funding information
This work was partially supported by the British Heart Foundation (grant no. RG06/008/22062). R. J., P. F., M. W. S. and R. D. R. acknowledge financial support from the Department of Cardiovascular Sciences, University of Leicester. R. D. R. and S. B. acknowledge financial support from the van Geest Heart and Cardiovascular Diseases Research Fund.
Conflict of interest
Authors declare that they have no conflict of interest.
Supporting information
Figure S1 K+‐induced increases in [Ca2+]i and vasoconstriction are increased in the presence of high glucose and reversed by Gö6976. Isolated rat mesenteric arteries were stimulated to contract by two applications of 60 mM KCl (60K) separated by a 30 min washout period. Example traces showing 60K(1) and a second response to 60K (60K(2)) after 30 min incubation in (A) 5 mM glucose, (B) 20 mM glucose and (C) 20 mM glucose in the presence of the cPKC inhibitor Gö6976 (300 nM) immediately after washout of the first 60K addition. (D) Mean data showing increases in tension (mN/mm) and Fura‐2 ratio for 60K(2) relative to 60K(1) (%) and the reversal of the high glucose potentiation in the presence of Gö6976 (**P<0.01; ***P<0.001, repeated measures ANOVA with Bonferroni's post‐hoc test, or unpaired t‐test).
Figure S2 Glucose‐mediated increases in high extracellular [K+]‐induced vasoconstriction of mesenteric smooth muscle are mimicked by Kv1 channel blockade. (A) Example I/V relationships from MASMCs in 5 mM glucose before (i) and after (ii) perfusion for 9 min with the Kv1 family channel inhibitor Psora‐4 (100 nM). (B) Mean I/V relationship from MASMCs before and after Psora‐4 (n=6 cells). (C) Mean data for membrane potential in the absence and presence of Psora‐4 (***P<0.001, paired t‐test: 6 cells from ≥3 animals). (D) Example trace showing 60K(1) and second 60K response (60K(2)) after 30 min incubation with Psora‐4 and 5 mM glucose with a third 60K addition in the presence of Psora‐4 and 20 mM glucose. (E) Mean data showing increases in 60K(2) relative to 60K(1) (%) in the presence of Psora‐4 (***P<0.001, repeated measures ANOVA with Bonferroni's post‐hoc test, ≥ 6 vessels from ≥ 3 animals). (F) Example trace showing the response of mesenteric arteries to 125K‐induced contraction in 5 and 20 mM glucose. (G) Mean data showing a lack of potentiation of the 125K‐response in 20 mM glucose suggesting that the small hyperpolarizing effect from K+ currents seen in 60K is abolished by further decreasing the K+ gradient (≥ 8 vessels from ≥ 3 animals for each dataset).
Figure S3 Enhanced K+‐induced contraction by monosaccharides is dependent of glycolytic metabolism. Isolated rat mesenteric arteries were stimulated to contract by two applications of 60 mM KCl (60K(1) and 60K(2)) separated by a period of 30 min. (A) 60K(1) contraction was in solution containing the non‐metabolized monosaccharide L‐glucose (5 mM) with 60K(2) added after 30 min incubation with the L‐glucose (20 mM). (B) Mean data showing the % of 60K(2) in L‐ and D‐glucose for comparison. (C) 60K(1) contraction was in solution containing fructose (5 mM) with 60K(2) added after 30 min incubation with 20 mM fructose. (D) Mean data showing the % of 60K(2) in 20 mM fructose to 20 mM D‐glucose (≥ 6 vessels from ≥ 3 animals for each dataset).
Figure S4 Additive and non‐additive effects of high glucose for ATII and ET1 on Kv current inhibition in rat mesenteric artery smooth muscle cells. Representative whole‐cell current recordings of Kv current from MASMC recorded in 0 mM glucose, after 9 min of perfusion with 20 mM glucose, and after a further 9 min of perfusion with ATII (100 nM) or ET1 (10 nM). Mean current (normalized to cell capacitance) in 0 mM glucose, after 9 min of perfusion with 20 mM glucose and after a further 9 min of perfusion with either ATII or ET1. Kv current was reduced on switching to 20 mM glucose (P<0.001, repeated measures ANOVA with Bonferroni's post hoc test) with a further inhibition on co‐addition of ATII (***P<0.001), but not ET1 (P>0.05, repeated measures ANOVA with Bonferroni's post hoc test, n=3 animals (6 cells) for each dataset).
Figure S5 U46619‐ and UTP‐induced constrictions of rat mesenteric artery are enhanced by high extracellular glucose in a conventional PKC isoenzyme‐dependent manner. (A) Example traces of U46619 (3‐1000 nM)‐induced constrictor responses from rat mesenteric arterial rings in 5 (i) or 20 (ii) mM glucose. (B) U46619 concentration‐response curves in the presence of 5 and 20 mM glucose. (C) Mean contractile responses to U46619 (100 nM) in 5 and 20 mM glucose in the absence or presence of Gö6976 or Tat‐PKC α&β. (D) Example traces of UTP (3‐1000 nM)‐induced constrictor responses from rat mesenteric arterial rings in 5 (i) or 20 (ii) mM glucose. Addition of 100 nM AngII to rat mesenteric arterial rings caused contraction in both 5 mM (i) and 20 mM (ii) glucose but was substantially enhanced in 20 mM as shown in Figure 5. (E) UTP concentration‐response curves in the presence of 5 and 20 mM glucose. (F) Mean contractile responses to UTP (100 μM) in 5 and 20 mM glucose in the absence or presence of Gö6976 or Tat‐PKC α&β.
Figure S6 Vasodilator responses to a β‐adrenoceptor agonist are unaffected by elevated extracellular glucose concentrations in isolated rat mesenteric artery. (A) Representative traces showing vasodilator responses of agonist (100 μM UTP) pre‐constricted small rat mesenteric arteries by cumulative additions of isoprenaline in either 5 mM (i) and 20 mM (ii) glucose. (B) Mean contractile response to 60K and UTP in 5 and 20 mM glucose. Consistent with previous figures both vasoconstrictor activities are potentiated in elevated glucose (**P<0.01, ***P<0.001, unpaired Student's t‐test). (C) Isoprenaline concentration‐response curves normalized to the peak contraction induced by UTP in 5 or 20 mM glucose (mean IC50 values 11.5 ± 2.6 and 12.3 ± 1.9 nM at 5 and 20 mM glucose, respectively; P>0.05, n ≥ 3 animals (8 vessels) for each data‐point). (D) Representative traces of vasodilator responses of agonist (U46619, 100 nM) pre‐constricted porcine coronary arterioles by cumulative additions of isoprenaline in 5 (i) and 20 mM (ii) glucose. (E) Mean contractile response to 60K and 100 nM U46619 in 5 and 20 mM glucose demonstrating an enhanced vasoconstrictor response in elevated glucose (***P<0.001, unpaired Student's t‐test). (F) Isoprenaline concentration‐response curves normalized to the peak contraction induced by U46619 in 5 or 20 mM glucose (mean IC50 values 6.3 ± 2.1 and 6.1 ± 1.9 nM in 5 and 20 mM glucose respectively; P>0.05, n≥4 animals (16 vessels) for each data‐point).
Supporting info item
Acknowledgements
We gratefully acknowledge the support and encouragement offered by Prof. N. B. Standen and Dr N. W. Davies during the early stages of this project. We thank Eli Lilly & Co. Research Laboratories (Indianapolis, USA) for the gift of the selective PKCβ inhibitor, LY379196.
Jackson, R. , Brennan, S. , Fielding, P. , Sims, M. W. , Challiss, RA. J. , Adlam, D. , Squire, I. B. , and Rainbow, R. D. (2016) Distinct and complementary roles for α and β isoenzymes of PKC in mediating vasoconstrictor responses to acutely elevated glucose. British Journal of Pharmacology, 173: 870–887. doi: 10.1111/bph.13399.
References
- Albarwani S, Nemetz LT, Madden JA, Tobin AA, England SK, Pratt PF, et al. (2003). Voltage‐gated K+ channels in rat small cerebral arteries: molecular identity of the functional channels. J Physiol 551: 751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun KH, Kaul CL, Ramarao P (2004). High glucose concentration augments angiotensin II‐mediated contraction via AT1 receptors in rat thoracic aorta. Pharmacol Res 50: 561–568. [DOI] [PubMed] [Google Scholar]
- Barbagallo M, Shan J, Pang PK, Resnick LM (1995). Glucose‐induced alterations of cytosolic free calcium in cultured rat tail artery vascular smooth muscle cells. J Clin Invest 95: 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Creager MA (2002). Inhibition of protein kinase Cβ prevents impaired endothelium‐dependent vasodilation caused by hyperglycemia in humans. Circ Res 90: 107–111. [DOI] [PubMed] [Google Scholar]
- Bohlen HG, Nase GP (2001). Arteriolar nitric oxide concentration is decreased during hyperglycemia‐induced betaII PKC activation. Am J Physiol Heart Circ Physiol 280: H621–H627. [DOI] [PubMed] [Google Scholar]
- Brennan S, Jackson R, Patel M, Sims MW, Hudman D, Norman RI, et al. (2015). Early opening of sarcolemmal ATP‐sensitive potassium channels is not a key step in PKC‐mediated cardioprotection. J Mol Cell Cardiol 79: 42–53. [DOI] [PubMed] [Google Scholar]
- Chen TT, Luykenaar KD, Walsh EJ, Walsh MP, Cole WC (2006a). Key role of Kv1 channels in vasoregulation. Circ Res 99: 53–60. [DOI] [PubMed] [Google Scholar]
- Chen Y, Budd RC, Kelm RJ, Sobel BE, Schneider DJ (2006b). Augmentation of proliferation of vascular smooth muscle cells by plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol 26: 1777–1783. [DOI] [PubMed] [Google Scholar]
- Clarke JG, Davies GJ, Kerwin R, Hackett D, Larkin S, Dawbarn D, et al. (1987). Coronary artery infusion of neuropeptide Y in patients with angina pectoris. Lancet 1: 1057–1059. [DOI] [PubMed] [Google Scholar]
- Cole WC, Clement‐Chomienne O, Aiello EA (1996). Regulation of 4‐aminopyridine‐sensitive, delayed rectifier K+ channels in vascular smooth muscle by phosphorylation. Biochem Cell Biol 74: 439–447. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Evcimen N, King GL (2007). The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res 55: 498–510. [DOI] [PubMed] [Google Scholar]
- Descorbeth M, Anand‐Srivastava MB (2008). High glucose increases the expression of Gq/11α and PLC‐β proteins and associated signaling in vascular smooth muscle cells. Am J Physiol Hear. Circ Physiol 295: H2135–H2142. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Nelson MT (2010). Calcium and diabetic vascular dysfunction. Focus on ‘Elevated Ca2 + sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells’. Am J Physiol Cell Physiol 298: C203–C205. [DOI] [PubMed] [Google Scholar]
- Geraldes P, King GL (2010). Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 106: 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatta S, Ramarao P (2004). Increased contractile responses to 5‐hydroxytryptamine and angiotensin II in high fat diet fed rat thoracic aorta. Lipids Health Dis 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giugliano D, Marfella R, Coppola L, Verrazzo G, Acampora R, Giunta R, et al. (1997). Vascular effects of acute hyperglycemia in humans are reversed by L‐arginine. Evidence for reduced availability of nitric oxide during hyperglycemia. Circulation 95: 1783–1790. [DOI] [PubMed] [Google Scholar]
- Gordin D, Forsblom C, Ronnback M, Groop PH (2008). Acute hyperglycaemia disturbs cardiac repolarization in type 1 diabetes. Diabet Med 25: 101–105. [DOI] [PubMed] [Google Scholar]
- Goyal A, Mahaffey KW, Garg J, Nicolau JC, Hochman JS, Weaver WD, et al. (2006). Prognostic significance of the change in glucose level in the first 24 h after acute myocardial infarction: results from the CARDINAL study. Eur Heart J 27: 1289–1297. [DOI] [PubMed] [Google Scholar]
- Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, et al. (2001). Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88: E14–E22. [DOI] [PubMed] [Google Scholar]
- Hreiche R, Plante I, David LP, Simard C, Turgeon J, Drolet B (2009). Impact of glucose concentration on cardiac ventricular repolarization under IKr/IKs blocking agents. J Mol Cell Cardiol 47: 210–220. [DOI] [PubMed] [Google Scholar]
- Idris I, Donnelly R (2006). Protein kinase Cβ inhibition: a novel therapeutic strategy for diabetic microangiopathy. Diab Vasc Dis Res 3: 172–178. [DOI] [PubMed] [Google Scholar]
- Inoguchi T, Xia P, Kunisaki M, Higashi S, Feener EP, King GL (1994). Insulin effects on protein kinase C and diacylglycerol induced by diabetes and glucose in vascular tissues. Am J Physiol 267: E369–E379. [DOI] [PubMed] [Google Scholar]
- Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. (2000). High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C‐dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49: 1939–1945. [DOI] [PubMed] [Google Scholar]
- Iwakura K, Ito H, Ikushima M, Kawano S, Okamura A, Asano K, et al. (2003). Association between hyperglycemia and the no‐reflow phenomenon in patients with acute myocardial infarction. J Am Coll Cardiol 41: 1–7. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT (1995). Regulation of membrane potential and diameter by voltage‐dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol 269: H348–H355. [DOI] [PubMed] [Google Scholar]
- Ko EA, Han J, Jung ID, Park WS (2008). Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res 44: 65–81. [DOI] [PubMed] [Google Scholar]
- Kosiborod M, Inzucchi SE, Krumholz HM, Xiao L, Jones PG, Fiske S, et al. (2008). Glucometrics in patients hospitalized with acute myocardial infarction: defining the optimal outcomes‐based measure of risk. Circulation 117: 1018–1027. [DOI] [PubMed] [Google Scholar]
- Lee TS, Saltsman KA, Ohashi H, King GL (1989). Activation of protein kinase C by elevation of glucose concentration: proposal for a mechanism in the development of diabetic vascular complications. Proc Natl Acad Sci U S A 86: 5141–5145. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Li H, Chai Q, Gutterman DD, Liu Y (2003). Elevated glucose impairs cAMP‐mediated dilation by reducing Kv channel activity in rat small coronary smooth muscle cells. Am J Physiol Heart Circ Physiol 285: H1213–H1219. [DOI] [PubMed] [Google Scholar]
- Liu Y, Terata K, Rusch NJ, Gutterman DD (2001). High glucose impairs voltage‐gated K+ channel current in rat small coronary arteries. Circ Res 89: 146–152. [DOI] [PubMed] [Google Scholar]
- Marfella R, Rossi F, Giugliano D (2001). Hyperglycemia and QT interval: time for re‐evaluation. Diabetes Nutr Metab 14: 63–65. [PubMed] [Google Scholar]
- Marzian S, Stansfeld PJ, Rapedius M, Rinne S, Nematian‐Ardestani E, Abbruzzese JL, et al. (2013). Side pockets provide the basis for a new mechanism of Kv channel‐specific inhibition. Nat Chem Biol 9: 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden EP, Clarke JG, Davies GJ, Kaski JC, Haider AW, Maseri A (1991). Effect of intracoronary serotonin on coronary vessels in patients with stable angina and patients with variant angina. N Engl J Med 324: 648–654. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele C, Paturzo F, Teperino R, Sakane F, Fiory F, Oriente F, et al. (2007). Glucose regulates diacylglycerol intracellular levels and protein kinase C activity by modulating diacylglycerol kinase subcellular localization. J Biol Chem 282: 31835–31843. [DOI] [PubMed] [Google Scholar]
- NICE Guidelines (2014). Type 2 diabetes: the management of type 2 diabetes. Available at: https://www.nice.org.uk/guidance/cg87/resources/guidance‐type‐2‐diabetes‐pdf (October 2015).
- Nobe K, Sakai Y, Nobe H, Takashima J, Paul RJ, Momose K (2003). Enhancement effect under high‐glucose conditions on U46619‐induced spontaneous phasic contraction in mouse portal vein. J Pharmacol Exp Ther 304: 1129–1142. [DOI] [PubMed] [Google Scholar]
- Nobe K, Miyatake M, Nobe H, Sakai Y, Takashima J, Momose K (2004). Novel diacylglycerol kinase inhibitor selectively suppressed an U46619‐induced enhancement of mouse portal vein contraction under high glucose conditions. Br J Pharmacol 143: 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobe K, Nezu Y, Tsumita N, Hashimoto T, Honda K (2008). Intra‐ and extrarenal arteries exhibit different profiles of contractile responses in high glucose conditions. Br J Pharmacol 155: 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MW, Barber PA, Desmond PM, Baird TA, Darby DG, Byrnes G, et al. (2002). Acute hyperglycemia adversely affects stroke outcome: a magnetic resonance imaging and spectroscopy study. Ann Neurol 52: 20–28. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al., NC‐IUPHAR (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbow RD, Hardy ME, Standen NB, Davies NW (2006). Glucose reduces endothelin inhibition of voltage‐gated potassium channels in rat arterial smooth muscle cells. J Physiol 575: 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbow RD, Norman RI, Everitt DE, Brignell JL, Davies NW, Standen NB (2009). Endothelin‐1 and angiotensin II inhibit arterial voltage‐gated K+ channels through different protein kinase C isoenzymes. Cardiovasc Res 83: 493–500. [DOI] [PubMed] [Google Scholar]
- Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ (2010). PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol 11: 103–112. [DOI] [PubMed] [Google Scholar]
- Schaffler A, Arndt H, Scholmerich J, Palitzsch K (1998). Acute hyperglycaemia causes severe disturbances of mesenteric microcirculation in an in vivo rat model. Eur J Clin Invest 28: 886–893. [DOI] [PubMed] [Google Scholar]
- Serova M, Ghoul A, Benhadji KA, Cvitkovic E, Faivre S, Calvo F, et al. (2006). Preclinical and clinical development of novel agents that target the protein kinase C family. Semin Oncol 33: 466–478. [DOI] [PubMed] [Google Scholar]
- Shaifta Y, Snetkov V, Prieto‐Lloret J, Knock G, Smirnov S, Aaronson P, et al. (2015). Sphingosylphosphorylcholine potentiates vasoreactivity and voltage‐gated Ca2 + entry via NOX1 and reactive oxygen species. Cardiovasc Res 106: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims MW, Winter J, Brennan S, Norman RI, Ng GA, Squire IB, et al. (2014). PKC‐mediated toxicity of elevated glucose concentration on cardiomyocyte function. Am J Physiol Heart Circ Physiol 307: H587–H597. [DOI] [PubMed] [Google Scholar]
- Squire IB, Nelson CP, Ng LL, Jones DR, Woods KL, Lambert PC (2010). Prognostic value of admission blood glucose concentration and diabetes diagnosis on survival after acute myocardial infarction: results from 4702 index cases in routine practice. Clin Sci (Lond) 118: 527–535. [DOI] [PubMed] [Google Scholar]
- Stott JB, Jepps TA, Greenwood IA (2014). Kv7 potassium channels: a new therapeutic target in smooth muscle disorders. Drug Discov Today 19: 413–424. [DOI] [PubMed] [Google Scholar]
- Straub SV, Girouard H, Doetsch PE, Hannah RM, Wilkerson MK, Nelson MT (2009). Regulation of intracerebral arteriolar tone by Kv channels: effects of glucose and PKC. Am J Physiol Cell Physiol 297: C788–C796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamareille S, Terwelp M, Amirian J, Felli P, Zhang XQ, Barry WH, et al. (2013). Endothelin‐1 release during the early phase of reperfusion is a mediator of myocardial reperfusion injury. Cardiology 125: 242–249. [DOI] [PubMed] [Google Scholar]
- Tesfamariam B, Cohen RA (1992). Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol 263: H321–H326. [DOI] [PubMed] [Google Scholar]
- Velmurugan GV, White C (2012). Calcium homeostasis in vascular smooth muscle cells is altered in type 2 diabetes by Bcl‐2 protein modulation of InsP3R calcium‐release channels. Am J Physiol Heart Circ Physiol 302: H124–H134. [DOI] [PubMed] [Google Scholar]
- Wen HC, Huang WC, Ali A, Woodgett JR, Lin WW (2003). Negative regulation of phosphatidylinositol 3‐kinase and Akt signalling pathway by PKC. Cell Signal 15: 37–45. [DOI] [PubMed] [Google Scholar]
- Wulff H, Yarov‐Yarovoy V (2013). Channels: sticking to nooks and crannies. Nat Chem Biol 9: 473–474. [DOI] [PubMed] [Google Scholar]
- Zamami Y, Takatori S, Yamawaki K, Miyashita S, Mio M, Kitamura Y, et al. (2008). Acute hyperglycemia and hyperinsulinemia enhance adrenergic vasoconstriction and decrease calcitonin gene‐related peptide‐containing nerve‐mediated vasodilation in pithed rats. Hypertens Res 31: 1033–1044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 K+‐induced increases in [Ca2+]i and vasoconstriction are increased in the presence of high glucose and reversed by Gö6976. Isolated rat mesenteric arteries were stimulated to contract by two applications of 60 mM KCl (60K) separated by a 30 min washout period. Example traces showing 60K(1) and a second response to 60K (60K(2)) after 30 min incubation in (A) 5 mM glucose, (B) 20 mM glucose and (C) 20 mM glucose in the presence of the cPKC inhibitor Gö6976 (300 nM) immediately after washout of the first 60K addition. (D) Mean data showing increases in tension (mN/mm) and Fura‐2 ratio for 60K(2) relative to 60K(1) (%) and the reversal of the high glucose potentiation in the presence of Gö6976 (**P<0.01; ***P<0.001, repeated measures ANOVA with Bonferroni's post‐hoc test, or unpaired t‐test).
Figure S2 Glucose‐mediated increases in high extracellular [K+]‐induced vasoconstriction of mesenteric smooth muscle are mimicked by Kv1 channel blockade. (A) Example I/V relationships from MASMCs in 5 mM glucose before (i) and after (ii) perfusion for 9 min with the Kv1 family channel inhibitor Psora‐4 (100 nM). (B) Mean I/V relationship from MASMCs before and after Psora‐4 (n=6 cells). (C) Mean data for membrane potential in the absence and presence of Psora‐4 (***P<0.001, paired t‐test: 6 cells from ≥3 animals). (D) Example trace showing 60K(1) and second 60K response (60K(2)) after 30 min incubation with Psora‐4 and 5 mM glucose with a third 60K addition in the presence of Psora‐4 and 20 mM glucose. (E) Mean data showing increases in 60K(2) relative to 60K(1) (%) in the presence of Psora‐4 (***P<0.001, repeated measures ANOVA with Bonferroni's post‐hoc test, ≥ 6 vessels from ≥ 3 animals). (F) Example trace showing the response of mesenteric arteries to 125K‐induced contraction in 5 and 20 mM glucose. (G) Mean data showing a lack of potentiation of the 125K‐response in 20 mM glucose suggesting that the small hyperpolarizing effect from K+ currents seen in 60K is abolished by further decreasing the K+ gradient (≥ 8 vessels from ≥ 3 animals for each dataset).
Figure S3 Enhanced K+‐induced contraction by monosaccharides is dependent of glycolytic metabolism. Isolated rat mesenteric arteries were stimulated to contract by two applications of 60 mM KCl (60K(1) and 60K(2)) separated by a period of 30 min. (A) 60K(1) contraction was in solution containing the non‐metabolized monosaccharide L‐glucose (5 mM) with 60K(2) added after 30 min incubation with the L‐glucose (20 mM). (B) Mean data showing the % of 60K(2) in L‐ and D‐glucose for comparison. (C) 60K(1) contraction was in solution containing fructose (5 mM) with 60K(2) added after 30 min incubation with 20 mM fructose. (D) Mean data showing the % of 60K(2) in 20 mM fructose to 20 mM D‐glucose (≥ 6 vessels from ≥ 3 animals for each dataset).
Figure S4 Additive and non‐additive effects of high glucose for ATII and ET1 on Kv current inhibition in rat mesenteric artery smooth muscle cells. Representative whole‐cell current recordings of Kv current from MASMC recorded in 0 mM glucose, after 9 min of perfusion with 20 mM glucose, and after a further 9 min of perfusion with ATII (100 nM) or ET1 (10 nM). Mean current (normalized to cell capacitance) in 0 mM glucose, after 9 min of perfusion with 20 mM glucose and after a further 9 min of perfusion with either ATII or ET1. Kv current was reduced on switching to 20 mM glucose (P<0.001, repeated measures ANOVA with Bonferroni's post hoc test) with a further inhibition on co‐addition of ATII (***P<0.001), but not ET1 (P>0.05, repeated measures ANOVA with Bonferroni's post hoc test, n=3 animals (6 cells) for each dataset).
Figure S5 U46619‐ and UTP‐induced constrictions of rat mesenteric artery are enhanced by high extracellular glucose in a conventional PKC isoenzyme‐dependent manner. (A) Example traces of U46619 (3‐1000 nM)‐induced constrictor responses from rat mesenteric arterial rings in 5 (i) or 20 (ii) mM glucose. (B) U46619 concentration‐response curves in the presence of 5 and 20 mM glucose. (C) Mean contractile responses to U46619 (100 nM) in 5 and 20 mM glucose in the absence or presence of Gö6976 or Tat‐PKC α&β. (D) Example traces of UTP (3‐1000 nM)‐induced constrictor responses from rat mesenteric arterial rings in 5 (i) or 20 (ii) mM glucose. Addition of 100 nM AngII to rat mesenteric arterial rings caused contraction in both 5 mM (i) and 20 mM (ii) glucose but was substantially enhanced in 20 mM as shown in Figure 5. (E) UTP concentration‐response curves in the presence of 5 and 20 mM glucose. (F) Mean contractile responses to UTP (100 μM) in 5 and 20 mM glucose in the absence or presence of Gö6976 or Tat‐PKC α&β.
Figure S6 Vasodilator responses to a β‐adrenoceptor agonist are unaffected by elevated extracellular glucose concentrations in isolated rat mesenteric artery. (A) Representative traces showing vasodilator responses of agonist (100 μM UTP) pre‐constricted small rat mesenteric arteries by cumulative additions of isoprenaline in either 5 mM (i) and 20 mM (ii) glucose. (B) Mean contractile response to 60K and UTP in 5 and 20 mM glucose. Consistent with previous figures both vasoconstrictor activities are potentiated in elevated glucose (**P<0.01, ***P<0.001, unpaired Student's t‐test). (C) Isoprenaline concentration‐response curves normalized to the peak contraction induced by UTP in 5 or 20 mM glucose (mean IC50 values 11.5 ± 2.6 and 12.3 ± 1.9 nM at 5 and 20 mM glucose, respectively; P>0.05, n ≥ 3 animals (8 vessels) for each data‐point). (D) Representative traces of vasodilator responses of agonist (U46619, 100 nM) pre‐constricted porcine coronary arterioles by cumulative additions of isoprenaline in 5 (i) and 20 mM (ii) glucose. (E) Mean contractile response to 60K and 100 nM U46619 in 5 and 20 mM glucose demonstrating an enhanced vasoconstrictor response in elevated glucose (***P<0.001, unpaired Student's t‐test). (F) Isoprenaline concentration‐response curves normalized to the peak contraction induced by U46619 in 5 or 20 mM glucose (mean IC50 values 6.3 ± 2.1 and 6.1 ± 1.9 nM in 5 and 20 mM glucose respectively; P>0.05, n≥4 animals (16 vessels) for each data‐point).
Supporting info item