

Abstract
Malignant melanoma is considered the most lethal skin cancer if not detected and treated at its early stages. About 10% of melanoma patients report a family history of melanoma; however, individuals with features of true hereditary melanoma (i.e. unilateral lineage, multi-generational, multiple primary lesions, and early onset of disease) are in fact quite rare. Although many new loci have been implicated in hereditary melanoma, CDKN2A mutations remain the most common. Familial melanoma in the presence of multiple atypical nevi should raise suspicion for a germline CDKN2A mutation. Such patients have a high risk of developing multiple primary melanomas and internal organ malignancies especially pancreatic cancer; thus, a multidisciplinary approach is necessary in many cases. The value of dermoscopy examination and total body photography performed at regular intervals has been suggested by a number of studies, and should therefore be considered for these patients and their first degree relatives. In addition, genetic counseling with the possibility of testing can be a valuable adjunct for familial melanoma patients. But, this must be performed with care and only by qualified individuals trained in cancer risk analysis.
Keywords: familial melanoma syndromes, mixed cancer syndromes, FAMMM, CDKN2A, CDK4, melanoma genetics
1.1. General Considerations for Hereditary Melanoma
Cutaneous malignant melanoma (CMM) can be highly lethal if it is not detected and treated at early stages. The incidence of melanoma has increased during the past several decades. In developed countries, CMM is the 6th most common cancer accounting for more than 47,000 deaths worldwide annually (45% occurring in Europe). The rise in incidence affects both young and older populations, while the global projected incidence of melanoma for the year 2025 is estimated to be 317,000 new cases compared to the 200,000 cases reported in 2008.1
About 7–15% of melanoma cases occur in patients with a family history of melanoma; however, this does not necessarily indicate that a single genetic mutation is being transmitted in those kindreds.2 Most cases of familial melanoma are due to shared sun exposure experiences among family members with susceptible skin types.2 In aggregate, about 45% of familial melanomas are actually associated with germline mutations in CDKN2A or CDK4. There does not appear to be another major locus beyond CDKN2A, as the prevalence of the new melanoma predisposition genes are quite rare (see Part 2). Although great strides have been made in identifying other novel co-segregating variants within melanoma kindreds, it is likely that many rare disease-causing mutations remain undiscovered.3 The term “mixed cancer syndromes” (MCS) can be applied to familial conditions for which there is a high incidence of various cancers in general, including melanoma. In the past few years, melanomas have also been found to arise in families that are generally prone to specific patterns of malignancies. Thus, the term “melanoma tumor syndromes” might be more appropriate to discriminate it from hereditary melanoma, where the dominant cancer phenotype is that of CMM.
1.2. Familial atypical multiple mole-melanoma (FAMMM; OMIM 155601) and FAMMM-Pancreatic Cancer (FAMMM-PC; OMIM 606719) syndromes
The first documented case of familial melanoma was reported by Norris in 1820. His patient was a 59-year-old man with melanoma, a high total body nevus count, and a family history of melanoma.4 Over a century after Norris made his observations, Lynch and Krush5 described the familial atypical multiple mole-melanoma (FAMMM syndrome) which comprised an association between pancreatic cancer, multiple nevi, and melanoma. Contemporaneously, Clark described a similar phenotype, the B-K mole syndrome, consisting of familial melanoma in the setting of numerous atypical nevi.6 In the early 1990′s several groups reported germline mutations in the cell cycle gene, p16 (now CDKN2A), among a subset of FAMMM kindreds.7, 8
1.2.a. Clinical features of FAMMM
FAMMM is a clinical phenotype comprised of numerous nevi (Figure 1a), some atypical, and a family history of melanoma; some diagnostic elements of the FAMMM phenotype are outlined in Table 1. Documenting a thorough family history of cancer, particularly melanoma, is of utmost importance as it is a critical element of the FAMMM syndrome. Particular attention should be paid to the age at which CMM and other cancers (Table 2) have been diagnosed in family members, as well as family skin phototype (i.e. red hair/fair skin) since these traits may be associated with higher disease penetrance.9 In those suspected of having FAMMM, careful examination of all nevi should be performed not only on the patient of interest, but also their first- and second-degree relatives.
Figure 1. The familial atypical multiple mole-melanoma phenotype.
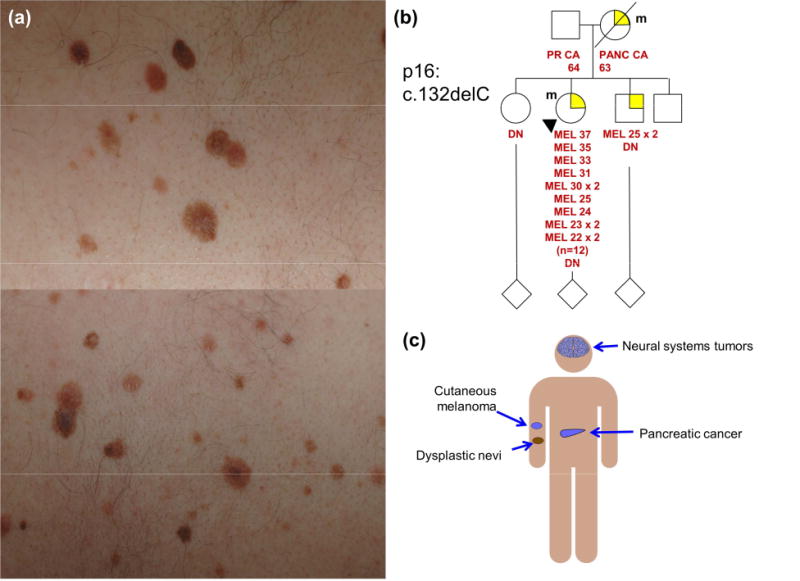
(a) Clinically atypical moles frequently associated with FAMMM syndrome. (b) Pedigree of a FAMMM kindred showing multiple early onset cutaneous melanomas (proband and brother) and pancreatic cancer (PANC CA; mother). The patient and mother are carriers of a p16 mutation (m). (c) Patients with FAMMM syndrome (in particular those with germline CDKN2A mutations) are at risk for cutaneous melanoma, pancreatic cancer and neural systems tumors (melanoma-astrocytoma syndrome).
Table 1.
Criteria for the diagnosis of FAMMM. All criteria should be present in order to make a diagnosis.22
|
Table 2.
| CDKN2A | CDK4 |
|---|---|
| Uveal melanoma | – |
| Breast cancer | Breast cancer (Phyllodes tumor) |
| Ovarian tumors | Ovarian tumors |
| Cervical cancer | Cervical cancer |
| Endometrial cancer | – |
| Pancreatic cancer | Pancreatic cancer |
| Stomach cancer | Stomach cancer |
| Esophageal cancer | – |
| Colon cancer | Colon cancer |
| Lung cancer | Lung cancer |
| Leukemia | – |
| Lymphoma (Hodgkin) | Lymphoma |
| Brain/CNS tumors | – |
| Renal cell carcinoma | – |
| Urinary bladder carcinoma | – |
| Prostate cancer | Prostate cancer |
| Hepatic cancer | – |
| Sarcomas | – |
| Parotic gland tumors | – |
| Tonsillar tumors | – |
| Nasopharyngeal/laryngeal tumors | – |
| Tongue cancer | – |
Nevi in patients with FAMMM are phenotypically diverse (Figure 1a). It is not unusual to observe multiple nevi with marked atypia, some bearing a striking resemblance to melanoma, interspersed between numerous benign-looking nevi. While, atypical nevi are more likely to undergo malignant transformation when compared to banal nevi; melanomas in FAMMM patients, however, often develop on normal skin.10, 11
While it is clear that patients with FAMMM syndrome have a dramatically increased risk of melanoma, it is less clear whether there are inherent differences between FAMMM-associated and sporadic melanomas. Patients with FAMMM seem to be more prone to developing superficial spreading and nodular melanomas,12 which is interesting in light of other findings that suggest CDKN2A-mutant CMMs are significantly less invasive (lower Clark levels) than CDKN2A-wild-type CMMs.13 No statistically significant differences in location and Breslow thickness have been reported between sporadic melanoma controls and FAMMM patients. Sargen et al. have recently reported that CDKN2A mutation positive CMMs tend to possess histologic features compatible with superficial spreading melanomas, including higher pigmentation (Ptrend = 0.02), increased pagetoid scatter (Ptrend = 0.07) and a non-spindle cell morphology in the vertical growth phase.14 However, more information is required in order to establish specific histopathologic features indicative of a CMM from a CDKN2A mutation positive patient. Gillgren et al. found that familial melanomas have a tendency to occur on the trunk more so than on the head and neck.15 Recent studies have shown similar rates of somatic BRAF and NRAS mutations in patients with or without germline CDKN2A mutations. Zebary et al. reported that BRAF and NRAS mutations occurred in 43% and 11% of CMMs, respectively, in CDKN2A mutation carriers, compared to 39% and 14% of CMMs in non-CDKN2A mutation controls16; similar findings are echoed by others.17 The pattern of metastasis between familial and sporadic melanoma patients does not appear to differ so a distinct post-melanoma follow-up program is probably not necessary for FAMMM patients.18 However, as will be discussed below, the risk of pancreatic cancer among some patients with FAMMM does warrant special consideration.
An example of a typical FAMMM pedigree is shown in Figure 1b. The proband presented with multiple atypical nevi (>200) and has had more than 10 histologically-confirmed CMMs. Her mother developed pancreatic cancer at age 63 and succumbed to the disease. Genetic testing revealed a single base pair deletion (c.132delC) which was shared by both the proband and the mother. The key elements of FAMMM are embodied in this pedigree- early age of onset (age 22), multiplicity of CMMs (n=12), family history of melanoma, dysplastic nevi and pancreatic cancer and a documented deleterious co-segregating mutation on one side of the family.
1.2.b. The Genetics of FAMMM
The dominant molecular pathway involved in FAMMM is shown in Figure 2. Xeroderma pigmentosum is an extremely rare condition and as such will only be discussed briefly. The CDKN2A gene is located on chromosome 9p21.3 and its alterations are most commonly associated with FAMMM syndrome. Typically, germline mutations of CDKN2A seen in CMM and pancreatic cancer (PC)-prone kindreds are missense or nonsense mutations that impair the inhibitory functions of p16 and/or p14ARF. CDKN2A is comprised of 4 exons (1α, 1β, 2 and 3) which are used to encode for two proteins- p16 (1α, 2 and 3) and p14ARF (exons 1β, 2 and 3). p16 inhibits cyclin-dependent kinase (CDK) 4 and CDK6, thereby preventing the phosphorylation of retinoblastoma protein (RB1). A hypophosphorylated RB1 molecule sequesters and prevents the transcription factor E2F1 from inducing S-phase genes and triggering G1-to-S transition. On the other hand, p14ARF antagonizes HDM2, which ubiquitinates the tumor suppressor, p53, thereby condemning p53 for proteasomal degradation.19 Accelerated destruction of p53 abolishes the normal DNA damage and G2 checkpoint responses.19 Therefore, inactivation of the CDKN2A locus enhances proliferation and reduces apoptosis. The prevalence of germline CDKN2A mutations has been found to vary with geography and the family context.3, 20, 21 In a meta-analysis by Goldstein and colleagues, 39% of families (with >3 affected) had germline CDKN2A mutations, ranging from 20% (32/162) in Australia to 45% (29/65) in North America to 57% (89/157) in Europe.22 Similarly, in a study of Greek families, Nikolaou et al. reported that 22% of familial melanoma cases and 57% of individuals with multiple primary melanomas carried a CDKN2A mutation.23 When melanoma cases were ascertained independent of family history, there was a much lower rate of mutation. The frequency of CDKN2A mutations in patients with a single primary melanoma or multiple primary melanomas were 1.2% and 2.9%, respectively.24 The likely explanation is that other co-inherited modifiers (e.g. additional risk conferring variants mutations) exist in a pedigree or that select members of some families share extremely high levels of sun exposure histories.
Figure 2. Pathways linked to FAMMM predisposition.
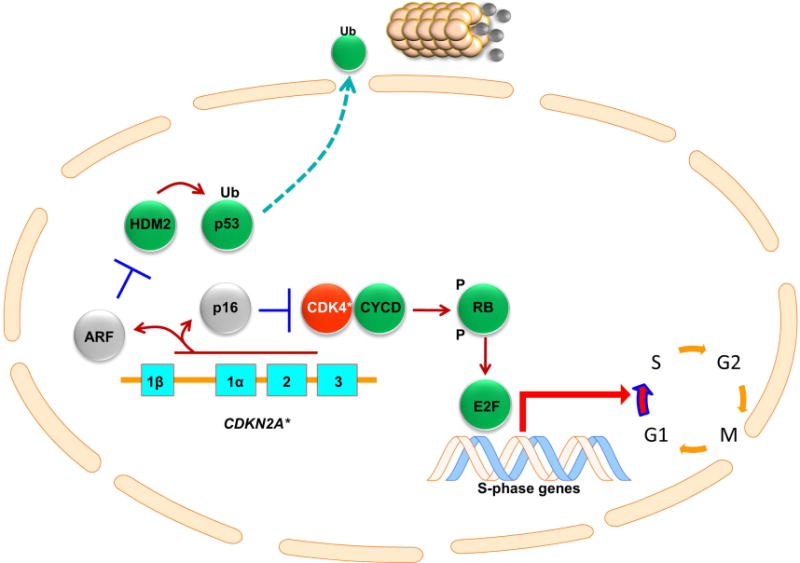
CDKN2A is comprised of 4 exons- 1α, 1β, 2 and 3. Exons 1α, 2 and 3 encode for p16 while exons 1β, 2 and 3 encode for p14ARF (ARF). p16 inhibits CDK4, which, without p16, binds cyclin D (CYCD) and phosphorylates (P) the retinoblastoma protein (RB). This in turn releases E2F transcription factors, which induces G1 phase genes and triggers G1-S cell cycle transition. p14ARF inhibits HDM2, which normally ubiquitinates (Ub) p53 condemning it to destruction by the proteasome. Mutations in CDKN2A (CDKN2A*) leads to loss of p14ARF and p16 function (gray color) while mutations in CDK4 (CDK4*) renders CDK4 resistant to p16 inhibition thereby activating CDK4 activity (red color); non-mutated genes are shown in green.
CDKN2A mutation penetrance (or the likelihood of developing melanoma over time) also varies by geography. The estimated penetrance rates are 30–91%, 50–76%, and 13–58% among patients ages 50–80 in Australia, the United States, and Europe, respectively. These broad risk differences could also be attributed to different sun exposure patterns and the presence of other genetic risk factors in the families.25, 26 For instance, co-inheritance of melanocortin 1 receptor (MC1R) variants and specific interleukin-9 and glutathione S-transferase theta 1 variants have been described as risk modifiers for CDKN2A-mutation penetrance.9, 27, 28 In a population-based study, Begg et al. found that the estimated risks of CMM among CDKN2A mutation carriers were 14%, 24% and 28% by ages 50, 70 and 80 years, respectively;29 the lower risk estimates may reflect the lack of other melanoma-risk variants in these sporadic cases.
Various studies have also demonstrated a much lower median age of onset of CMM in patients from germline CDKN2A mutation families (33–45 years) compared to patients without a CDKN2A mutation (53–61 years); this trend remains largely consistent irrespective of a geographic region.3, 30, 31 There are reports of CDKN2A kindreds where CMM has occurred in the early teens and twenties.20 As would be expected, the increased risk of CMM in these patients does not diminish with their first diagnosis as they also have a much higher 5-year cumulative incidence of a second melanoma compared to mutation-negative controls (23.4% and 2.3%, respectively).32
Germline CDK4 mutations have also been described in the FAMMM syndrome, albeit rarely.33–35 As alluded to above, CDK4, which is the target for p16 inhibition, plays an important role in normal cell cycle progression (Figure 2). The oncogenic CDK4 mutations described in affected families translates into a substitution of arginine-24, which disrupts p16 binding.35 Puntervoll et al. have reported an increased CMM risk in 17 families from 8 different countries that harbor CDK4 mutations. Out of 103 patients with one CMM, 41.7% developed a second primary CMM, and 21.1% developed CMM before the age of 30 (median age 39 years). Furthermore, 70–75% of patients exhibited multiple atypical nevi, which was considered to be a modifier for CMM risk, since these patients developed CMMs at a younger age.36 This study investigated the clinical phenotype of these CDK4-mutant melanoma families and determined that it is indistinguishable from the more well characterized CDKN2A-mutant melanoma phenotype, i.e. high burden of atypical nevi, early age of disease onset, and predilection for multiple primary melanoma.36 Since p16 directly interacts with CDK4, it is not surprising that the two phenotypes overlap significantly; in essence, the same biochemical event (increased RB phosphorylation) occurs with either mutation (Figure 2).
1.3. Melanoma-Astrocytoma Syndrome (OMIM 155755)
Melanoma-Astrocytoma Syndrome (MAS) is a variant of FAMMM that may be more linked to the loss of p14ARF function.37 Larger scale chromosome 9p21 alterations (including deletions involving the CDKN2A/CDKN2B/CDKN2BAS gene cluster up to the MLLT3 gene) have been described in some isolated cases.38–41 Kaufman et al. described this syndrome in 1993 when they reported concurrent CMMs and multiple types of nervous system tumors (NSTs) in eight members of a family over three generations.42 Later, Azizi et al. reported that 17 individuals with CMM, among 15 families, had one or more additional relatives with tumors of the nervous system.43 Conflicting data exist on this very rare syndrome. Generally patients are young (age <30) and can develop CMM either before and after the NSTs.38–40, 44 A positive association between radiotherapy for the NSTs and the incidence of CMM in these patients has been proposed, but remains unsubstantiated.41
1.4. Management of FAMMM patients including CDKN2A carriers
The significant risk of melanoma inherent to the FAMMM syndrome means that these patients need heightened dermatologic surveillance. Some general management considerations for patients with hereditary syndromes is presented in Table 3. It must be mentioned that given the rarity of melanoma syndromes, most data regarding follow-up recommendations are mainly based on small studies or expert opinion. Given the plethora of clinically atypical moles, dermoscopy is an important tool in approaching patients with FAMMM. It is also prudent for children from FAMMM kindreds to undergo routine skin examinations from late adolescence (Level of evidence: IV). This recommendation is supported by observational studies showing that FAMMM patients tend to develop melanomas at much younger ages. Surveillance of FAMMM patients should entail an extensive baseline total body skin examination (TBSE), including the scalp, oral mucosa, genital area, and nails. Most authors suggest that screening performed at 6 month intervals are adequate10, 20, 45, 46 although formal prospective trials of outcome do not exist (Level of evidence: IV). Haenssle et al. have reported that patients with FAMMM may develop up to 1 new melanoma every 3 years of follow-up and suggest that 3 month interval examinations may be more appropriate.47 At this point, though, there are no data supporting that 3 month interval examinations may be superior to 6 month interval examinations regarding patient outcomes. (Level of evidence: IV). Nevi should be checked for any changes in morphology (e.g. color or symmetry) and size. Since these patients may have many atypical nevi, lesions that stand out, exhibiting the so called “ugly duckling sign,” may warrant special attention. Beyond a thorough TBSE, the utility of more advanced techniques, such as total body photography (TBP) and sequential digital dermoscopy imaging (SDDI), for patients at extreme risk for melanoma has also been suggested48 although the adoption of these procedures may be limited by practice logistics. Moloney et al. reported that in 311 high-risk patients evaluated at 6 month intervals, 38% of post-baseline melanomas were detected using TBP and 39% with SDDI. Importantly, these tools allowed for earlier detection and treatment, which are known to impact melanoma outcome.49 In the Moloney study, most of the excised melanomas were categorized as in situ tumors, and the ratio of benign to malignant excised lesions was reported to be 1.6:1.49 In addition, Rademaker et al. have reported that the melanomas diagnosed in patients after TBP and SDDI examination where thinner compared to those diagnosed with clinical inspection. (69% <0.75 mm Breslow thickness compared to 52%, p=0.0216).50 Previous studies have also advocated the benefit of TBP in earlier diagnosis of melanoma.51, 52 An interesting point, though, is that these studies do not report patient outcomes, and thus their impact on patient survival is unknown. Also, the recommendations supported by those studies depend on the notion that earlier recognition of melanomas may lead to an overall better patient outcome. A small number of studies have reported evidence to support this.53 However, the exact frequency of follow up (follow up at 3 month, 6 month or 1 year intervals) is not clear. When planning follow up visits for high risk patients, two factors must be weighted: one is the psychological burden of having to be examined at specific intervals and the other is the cost-effectiveness of this process. Risser et al reported that the number of biopsies performed for patients undergoing TBP and clinical inspection was the same. Therefore, the cost-effectiveness of TBP use is questionable.54 It must be mentioned, though, that patients selected for TBP belong to high risk groups. In addition, the decision to perform a biopsy of a suspicious lesion after TBSE, is taken mostly due to nevus morphology at the time of examination, while TBP relates to morphologic changes over time (i.e. morphology changing from previous examination).55 Therefore, in theory, melanomas could be diagnosed earlier if TBP is used. In a recent study by Watts et al. a cost analysis of the surveillance of high risk melanoma patients was done. It was concluded that although these patients are indeed more costly to follow-up, it is overall cheaper to screen than having to later treat a stage IV melanoma. It is important though to find a golden rule where cost and patient benefit are perfectly balanced.56 Patients must be encouraged and taught to perform self-examination at regular intervals either alone, or with the assistance of a spouse or relative. Furthermore, routine sun protective behaviors must be reinforced at every visit. Screening, of all family members of FAMMM kindreds should be encouraged. Preemptive removal of observable stable or benign-appearing nevi is not recommended since the practice has not been shown to reduce melanoma risk meaningfully and is associated with increased morbidity and costs. (Level of evidence: IV) Isolated lesions which are visually inaccessible to the patient (such as mid lower back or scalp) may be removed prophylactically.
Table 3.
Obtain thorough medical history from patient:
|
Physical examination
|
Clinical recommendations
|
The association between pancreatic cancer (PC) and FAMMM syndrome is well documented, with an estimated risk 13–22 times higher than that of the average population; this risk increases to 38 fold in CDKN2A-mutant FAMMM patients.20, 57, 58 Pancreatic cancer seems to be the second most commonly observed malignancy in FAMMM patients harboring a CDKN2A mutation.3, 59 In a study by Goldstein et al. it was reported that PC was observed in 28% of CDKN2A-mutant families compared to only 6% of CDKN2A-wild-type families. Conversely, 74% of families with PC harbored a CDKN2A mutation compared to 33% of “melanoma only” families.3 Another study estimated that 17% of CDKN2A-mutant patients would develop PC by 75 years of age.60 In general, the mean age of onset for PC ranges from 65–71 years.20, 45, 61 It is unclear whether the age of onset for PC is lower for patients with FAMMM compared to sporadic cases. Various studies on this topic have reported mixed data, with only one study by James et al. showing a statistically significant difference in age of PC diagnosis between the two groups. Of note, smoking was a strong confounding factor in this study.61, 62 Evidence regarding the association of FAMMM and other cancers is more equivocal. Associations with digestive tract, breast, and respiratory tract cancer, among others, have been described; however, CDKN2A mutation status does not seem to influence the age of onset in these cancers.20, 59, 60, 63
The role of genetic testing for hereditary melanoma has been somewhat controversial since dermatologic management of individuals with familial melanoma (i.e. surveillance, sun protection education) rarely requires knowledge of the patient’s CDKN2A status. However, as alluded to above, the melanoma phenotype may be a window to a latent PC risk. Thus, a basic understanding of genetic risk assessment and counseling is worthwhile though referral to a genetic counselor for more formal evaluation is preferred especially given the time constraints of a busy dermatologic practice. The following are just a sampling of some fundamental discussion points (also outlined in Figure 3).
Figure 3. Genetic counseling algorithm for FAMMM patients.
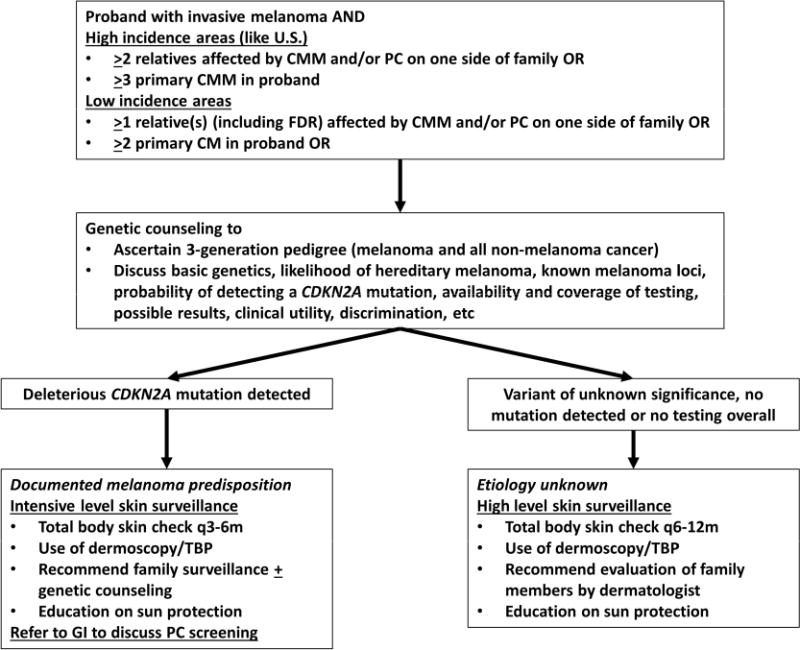
Patients with a personal history of melanoma may be considered for genetic counseling if certain criteria from high and low incidence area are met. The United States and Australia would be considered high incidence areas while England and Greece would be considered low incidence areas. A genetic counselor would ascertain a 3-generation pedigree and discuss the likelihood of hereditary melanoma, the molecular genetics related to familial melanoma risk, testing options, costs, risks of discrimination and possible test results. If patient undergoes CDKN2A testing and a deleterious mutation is detected, intensive skin surveillance is recommended along with a referral to GI for discussion of pancreatic cancer screening. If testing is not pursued or if a normal or variant of unknown significance result is returned, the etiology of the familial pattern remains unknown. Given the family history, the patient is considered high risk and should undergo high level skin surveillance. Abbreviations, PC, pancreatic cancer; TBP, total body photography, CMM, cutaneous malignant melanoma.
[1]. Does my patient have hereditary melanoma?
The individual seeking counseling is known as the “proband.” Currently, there are no firm criteria that would allow easy diagnosis of a proband with “hereditary melanoma.” Some have adopted the “rule of 3′s,”64 i.e. one individual with invasive CMM along with 2 additional members with either CMM and/or PC on one side of the family OR one individual with 3 primary CMMs. One caveat is that severely photodamaged patients may develop 3 melanomas especially later in life as sun damage accumulates. One perhaps slightly more stringent practical criterion would be a “3 by 40” modification where individuals with 3 CMMs diagnosed before age 40 may be more likely to be under genetic influences.
The benefit of formal genetic counseling is the analysis of an in-depth ≥3-generation pedigree. Although dermatologic charts may document a family history of melanoma, other critical information may not be ascertained. For instance, 3 melanoma cases in a small pedigree is different than 3 melanoma cases in a large extensive pedigree. The age of onset, current age and other concurrent non-melanoma cancers all contribute to the final interpretation of hereditary melanoma or mixed cancer syndrome. The presence of pancreatic cancer in a kindred is also important in assessing genetic risk. Formal training is typically required for accurate pedigree acquisition, and a full family history essential to accurate risk assessment.
[2]. What are the possible genes to be tested?
To understand genetic testing, fundamental principles of genetics must first be reviewed. Hereditary melanoma, like nearly all cancer syndromes, with the exception of xeroderma pigmentosum, is autosomal dominant.19 Thus, there is a 50% chance of sharing a mutation among 1st degree relatives. The major locus to be considered in a patient with FAMMM syndrome is CDKN2A although a small percentage (<1%) of FAMMM patients also harbor CDK4 mutations. In addition, there are likely many other unknown predisposing loci. Therefore, the first important message is that CDKN2A will be normal in a majority of individuals suspected of having FAMMM especially in high melanoma incidence areas like the United States. This is because there are environmental factors and other genetic factors (whether they be dominant genes or polygenetic factors) which have yet to be discovered.
[3]. Who is likely to be a carrier?
Phenotypically, the presence of multiple atypical nevi is not enough for the diagnosis of FAMMM though some it has been published that their presence in family members correlates with a three-fold higher likelihood of carrying a genetic mutation.33 It is important to note, however, that the presence of atypical nevi is not a carrier signature as non-carriers even in CDKN2A-mutated families, can exhibit multiple atypical moles. Thus, there is a complex relationship between melanomas and atypical, or dysplastic, nevi. Two statistical models have been developed in order to assist in identifying CDKN2A-mutation bearing individuals or families. MELPREDICT is based on logistic regression while MelaPRO (which can be obtained as part of CancerGene-https://www4.utsouthwestern.edu/breasthealth/cagene/) incorporates three different penetrance models (Bayes-Mendel algorithm).31, 32 These models provide a probability of mutation carriage for any given proband (MELPREDICT) or family member (MelaPRO) based on the family cancer pattern. Cancer risk counselors usually have access to MelaPRO as other similar algorithms, such as BRCAPRO for determining BRCA1/2 carrier risk, have been part of the counseling practice.
[4]. What are the possible results?
The identification of a deleterious CDKN2A mutation (“positive result”) establishes a disease-causing mutation in the kindred. First degree relatives (parents, siblings, children) will have a 50% chance of harboring the same mutation and risk. Since the penetrance is never 100%, there will be carriers in family who may not develop melanoma although the risk will be substantially higher than population rates. If unaffected relatives undergo subsequent testing and are found to have a normal CDKN2A, their risks may still elevated because of other risk factors, such as an MC1R variant or excessive sun exposure. However, a non-carrier will have a substantially lower risk of malignancy than a carrier though it may not return to general population risk levels.
In an affected patient with FAMMM who returns a normal CDKN2A result (“negative result”) or a “variant of unknown significance (VUS),” little advice can be offered. The patient may harbor a high-risk mutation in an undiscovered gene and therefore the risk is incalculable. These patients should continue to undergo the same dermatologic surveillance. For a non-affected member of a FAMMM kindred, there is no role for genetic testing without concomitant evaluation of at least one if not two other affected relatives from the same family. In short, the designation of “carrier” vs “non-carrier” can only be made if the familial mutation can be identified.
[5]. How can I use the results?
CDKN2A mutation carriers should be referred to a healthcare provider familiar with PC screening (Level of evidence: IV)65 in addition to ongoing intensive dermatologic surveillance at q3–6 month intervals with the possible use of TBP. Relatives of CDKN2Amutation carriers, regardless of genetic test results, should continue to be under careful dermatologic surveillance and strict sun protection.
No low-cost, gold standard screening approach exists for PC though studies in high-risk cohorts have demonstrated that early, pre-invasive pancreatic lesions can be detected with screening programs and then treated preemptively.66 Although PC screening lies outside the purview of dermatologists, familiarity with the available screening modalities is useful. Currently, these include endoscopic retrograde cholangiopancreatography (ERCP), which is able to detect small tumors but has associated complications due to its invasive nature, CT and MRI, which are less sensitive but also less invasive, and endoscopic ultrasound (EUS), which is the most sensitive and safe option at this time.67 (Level of evidence: IV) Some authors suggest that screening should start at the age of 50 years, or 10 years earlier than the PC age of onset in the family, but no specific consensus exists for a specific protocol in cancer screening of CDKN2A mutation carriers.13 Patients with FAMMM who forego testing, test negative for CDKN2A or who have a VUS should remain under careful dermatologic surveillance but PC screening is probably not necessary.
[6]. How will the patient use the results?
Families in general share exposure risks (e.g. sunny vacations together), risk conferring traits (e.g. sun sensitive skin, blue eyes) and disease-causing variants (e.g. CDKN2A mutation). The lack of a high-risk mutation in CDKN2A should not empower patients to abandon sun protective practices. Parents should also recognize that their children will continue to need strict sun protection even in the face of a normal CDKN2A. “True Negatives”, however, would not need to undergo PC screening however. There are various psychological benefits from undergoing genetic testing in CDKN2A families, including decreased anxiety.68
[7]. Will my patient experience genetic discrimination?
In 2008, the U. S. Government passed the Genetic Information Non-discrimination Act (GINA; http://www.ginahelp.org) which protects all individuals from health and employment discrimination based on genetic information. The Genetic Information Non-discrimination Act (GINA) went into effect in 2009 and provides comprehensive protection against genetic discrimination for all Americans. Under GINA, it is against the law for most health insurers to use genetic test results or family history information as a pre-existing condition. Additionally, under the GINA law, most health insurers cannot use genetic information to make decisions regarding eligibility, premiums, underwriting or coverage. It is also against the law for employers with 15 or more employees to use genetic information in hiring, firing, promotion, or other employment decisions. GINA does not protect against discrimination from life insurance, disability insurance or long-term care insurance companies. GINA’s protections do not apply to the US military or employees of the federal government who get care through the Federal Employees Health Benefits Plans, however, these groups have their own policies in place that may protect their members from insurance discrimination. For more information about the protections offered by GINA visit www.ginahelp.org.
1.5 Part 1 Conclusion
Malignant melanoma pathogenesis is multifactorial and complicated. However, hereditary cancer syndromes, such as FAMMM, provide excellent genetic models for studies that may increase early detection rates and improve existing prevention, and management protocols. Patients with a high nevus count and multiple atypical nevi should always be asked about personal or family history of melanoma or other internal organ cancer. Considering the fact that up to 10% of melanomas may be familial, increased physician awareness can lead to faster diagnosis of melanomas and, through patient education, to improved preventive behaviors.
KEY POINTS.
Hereditary melanomas can appear as part of a Familial Melanoma Syndrome or a Mixed Cancer Syndrome
A positive association between melanoma, multiple nevi, pancreatic cancer, and CDKN2A mutations is now well established
Patients suspected to have FAMMM present with multiple atypical nevi (>50) and have a positive personal and/ or family history for melanoma
FAMMM patients present with melanomas at a younger age and are at a higher risk to develop a second primary melanoma compared to the general population
Patients with FAMMM may also develop cutaneous melanomas on normal skin, despite the large number of atypical nevi that they may present with
The CDKN2A locus is the major recurrent source of germline mutagenesis in hereditary melanoma
The prevalence of germline CDKN2A mutations in melanoma families and CDKN2A mutation penetrance vary with geographic location
Patients that harbor the mutation have a higher risk of developing melanoma, however some evidence suggests that these CMMs may be less invasive than CDKN2A-wild-type CMMs
Patients with FAMMM syndrome should undergo total body skin examination, dermoscopic examination of clinically atypical nevi with possible total body photography every three to six months
Children from families with FAMMM may begin screening in late adolescence.
Due to the reported association between CDKN2A mutations and internal organ malignancies (specifically pancreatic cancer), all patients suspected to harbor the mutation should be referred to a specialist for appropriate screening
Acknowledgments
We acknowledge the contribution of many other authors whose work was not cited in this review due to space constraints. Mentorship and supervision during the writing of this manuscript was supported by a grant from the NIH (to H.T. K24 CA149202) and by the generous donors to the MGH on behalf of melanoma research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Editors
The editors involved with this CME activity and all content validation/peer reviewers of the journal-based CME activity have reported no relevant financial relationships with commercial interest(s).
Authors
The authors involved with this journal-based CME activity have reported no relevant financial relationships with commercial interest(s).
Planners
The planners involved with this journal-based CME activity have reported no relevant financial relationships with commercial interest(s). The editorial and education staff involved with this journal-based CME activity have reported no relevant financial relationships with commercial interest(s).
References (Part I)
- 1.Bray F, Jemal A, Grey N, Ferlay J, Forman D. Global cancer transitions according to the Human Development Index (2008–2030): a population-based study. Lancet Oncol. 2012;13(8):790–801. doi: 10.1016/S1470-2045(12)70211-5. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein AM, Tucker MA. Genetic epidemiology of cutaneous melanoma: a global perspective. Arch Dermatol. 2001;137(11):1493–1496. doi: 10.1001/archderm.137.11.1493. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, Azizi E, Bianchi-Scarra G, Bishop DT, Bressac-de Paillerets B, Bruno W, Calista D, Cannon Albright LA, Demenais F, Elder DE, Ghiorzo P, Gruis NA, Hansson J, Hogg D, Holland EA, Kanetsky PA, Kefford RF, Landi MT, Lang J, Leachman SA, Mackie RM, Magnusson V, Mann GJ, Niendorf K, Newton Bishop J, Palmer JM, Puig S, Puig-Butille JA, de Snoo FA, Stark M, Tsao H, Tucker MA, Whitaker L, Yakobson E, Melanoma Genetics C High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66(20):9818–9828. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- 4.Norris W. Case of fungoid disease. Edinb Med Surg J. 1820;16:562–565. [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch HT, Krush AJ. Hereditary and malignant melanoma: implications for early cancer detection. Can Med Assoc J. 1968;99:789–792. [PMC free article] [PubMed] [Google Scholar]
- 6.Clark WH, Jr, Reimer RR, Greene M, Ainsworth AM, Mastrangelo MJ. Origin of familial malignant melanomas from heritable melanocytic lesions. ‘The B-K mole syndrome’. Arch Dermatol. 1978;114(5):732–738. [PubMed] [Google Scholar]
- 7.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH, Jr, Tucker MA, Dracopoli NC. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8(1):15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 8.Gruis NA, van der Velden PA, Sandkuijl LA, Prins DE, Weaver-Feldhaus J, Kamb A, Bergman W, Frants RR. Homozygotes for CDKN2 (p16) germline mutation in Dutch familial melanoma kindreds. Nat Genet. 1995;10(3):351–353. doi: 10.1038/ng0795-351. [DOI] [PubMed] [Google Scholar]
- 9.Bergman W, Gruis NA. Management of melanoma families. Cancers (Basel) 2010;2(2):549–566. doi: 10.3390/cancers2020549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czajkowski R, Placek W, Drewa G, Czajkowska A, Uchanska G. FAMMM syndrome: pathogenesis and management. Dermatol Surg. 2004;30(2 Pt 2):291–296. doi: 10.1111/j.1524-4725.2004.30088.x. [DOI] [PubMed] [Google Scholar]
- 11.Kelly JW, Yeatman JM, Regalia C. A high incidence of melanoma found in patients with multiple dysplastic naevi by photographic surveillance. Med J Aust. 1997;167(4):191–194. doi: 10.5694/j.1326-5377.1997.tb138843.x. [DOI] [PubMed] [Google Scholar]
- 12.Ford D, Bliss JM, Swerdlow AJ, Armstrong BK, Franceschi S, Green A, Holly EA, Mack T, MacKie RM, Osterlind A, et al. Risk of cutaneous melanoma associated with a family history of the disease. The International Melanoma Analysis Group (IMAGE) Int J Cancer. 1995;62(4):377–381. doi: 10.1002/ijc.2910620403. [DOI] [PubMed] [Google Scholar]
- 13.Eckerle Mize D, Bishop M, Resse E. Familial Atypical Multiple Mole Melanoma Syndrome. Bethesda (MD): National Center for Biotechnology Information (US); 2009. [PubMed] [Google Scholar]
- 14.Sargen MR, Kanetsky PA, Newton-Bishop J, Hayward NK, Mann GJ, Gruis NA, Tucker MA, Goldstein AM, Bianchi-Scarra G, Puig S, Elder DE. Histologic features of melanoma associated with CDKN2A genotype. J Am Acad Dermatol. 2015;72(3):496–507. e497. doi: 10.1016/j.jaad.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillgren P, Brattstrom G, Frisell J, Palmgren J, Ringborg U, Hansson J. Body site of cutaneous malignant melanoma–a study on patients with hereditary and multiple sporadic tumours. Melanoma Res. 2003;13(3):279–286. doi: 10.1097/00008390-200306000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Zebary A, Omholt K, van Doorn R, Ghiorzo P, Harbst K, Hertzman Johansson C, Hoiom V, Jonsson G, Pjanova D, Puig S, Scarra GB, Harland M, Olsson H, Egyhazi Brage S, Palmer J, Kanter-Lewensohn L, Vassilaki I, Hayward NK, Newton-Bishop J, Gruis NA, Hansson J, Melanoma Genetics C Somatic BRAF and NRAS mutations in familial melanomas with known germline CDKN2A status: a GenoMEL study. J Invest Dermatol. 2014;134(1):287–290. doi: 10.1038/jid.2013.270. [DOI] [PubMed] [Google Scholar]
- 17.Lee JH, Choi JW, Kim YS. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br J Dermatol. 2011;164(4):776–784. doi: 10.1111/j.1365-2133.2010.10185.x. [DOI] [PubMed] [Google Scholar]
- 18.Hornbuckle J, Culjak G, Jarvis E, Gebski V, Coates A, Mann G, Kefford R. Patterns of metastases in familial and non-familial melanoma. Melanoma Res. 2003;13(1):105–109. doi: 10.1097/00008390-200302000-00017. [DOI] [PubMed] [Google Scholar]
- 19.Marzuka-Alcala A, Gabree MJ, Tsao H. Melanoma susceptibility genes and risk assessment. Methods Mol Biol. 2014;1102:381–393. doi: 10.1007/978-1-62703-727-3_20. [DOI] [PubMed] [Google Scholar]
- 20.Lynch HT, Brand RE, Hogg D, Deters CA, Fusaro RM, Lynch JF, Liu L, Knezetic J, Lassam NJ, Goggins M, Kern S. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families: the familial atypical mole melanoma-pancreatic carcinoma syndrome. Cancer. 2002;94(1):84–96. doi: 10.1002/cncr.10159. [DOI] [PubMed] [Google Scholar]
- 21.Rulyak SJ, Brentnall TA, Lynch HT, Austin MA. Characterization of the neoplastic phenotype in the familial atypical multiple-mole melanoma-pancreatic carcinoma syndrome. Cancer. 2003;98(4):798–804. doi: 10.1002/cncr.11562. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein AM, Chan M, Harland M, Hayward NK, Demenais F, Bishop DT, Azizi E, Bergman W, Bianchi-Scarra G, Bruno W, Calista D, Albright LA, Chaudru V, Chompret A, Cuellar F, Elder DE, Ghiorzo P, Gillanders EM, Gruis NA, Hansson J, Hogg D, Holland EA, Kanetsky PA, Kefford RF, Landi MT, Lang J, Leachman SA, MacKie RM, Magnusson V, Mann GJ, Bishop JN, Palmer JM, Puig S, Puig-Butille JA, Stark M, Tsao H, Tucker MA, Whitaker L, Yakobson E, Lund Melanoma Study G, Melanoma Genetics C Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44(2):99–106. doi: 10.1136/jmg.2006.043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikolaou V, Kang X, Stratigos A, Gogas H, Latorre MC, Gabree M, Plaka M, Njauw CN, Kypreou K, Mirmigi I, Stefanaki I, Tsao H. Comprehensive mutational analysis of CDKN2A and CDK4 in Greek patients with cutaneous melanoma. Br J Dermatol. 2011;165(6):1219–1222. doi: 10.1111/j.1365-2133.2011.10551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berwick M, Orlow I, Hummer AJ, Armstrong BK, Kricker A, Marrett LD, Millikan RC, Gruber SB, Anton-Culver H, Zanetti R, Gallagher RP, Dwyer T, Rebbeck TR, Kanetsky PA, Busam K, From L, Mujumdar U, Wilcox H, Begg CB, Group GEMS The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol Biomarkers Prev. 2006;15(8):1520–1525. doi: 10.1158/1055-9965.EPI-06-0270. [DOI] [PubMed] [Google Scholar]
- 25.Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B, Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland EA, Mann GJ, Mantelli M, Nancarrow D, Platz A, Tucker MA, Melanoma Genetics C Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94(12):894–903. doi: 10.1093/jnci/94.12.894. [DOI] [PubMed] [Google Scholar]
- 26.Cust AE, Harland M, Makalic E, Schmidt D, Dowty JG, Aitken JF, Agha-Hamilton C, Armstrong BK, Barrett JH, Chan M, Chang YM, Gascoyne J, Giles GG, Holland EA, Kefford RF, Kukalizch K, Lowery J, Randerson-Moor JA, Schmid H, Taylor CF, Whitaker L, Hopper JL, Newton-Bishop JA, Mann GJ, Bishop DT, Jenkins MA. Melanoma risk for CDKN2A mutation carriers who are relatives of population-based case carriers in Australia and the UK. J Med Genet. 2011;48(4):266–272. doi: 10.1136/jmg.2010.086538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang XR, Pfeiffer RM, Wheeler W, Yeager M, Chanock S, Tucker MA, Goldstein AM. Identification of modifier genes for cutaneous malignant melanoma in melanoma-prone families with and without CDKN2A mutations. Int J Cancer. 2009;125(12):2912–2917. doi: 10.1002/ijc.24622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaudru V, Lo MT, Lesueur F, Marian C, Mohamdi H, Laud K, Barrois M, Chompret A, Avril MF, Demenais F, Bressac-de Paillerets B. Protective effect of copy number polymorphism of glutathione S-transferase T1 gene on melanoma risk in presence of CDKN2A mutations, MC1R variants and host-related phenotypes. Fam Cancer. 2009;8(4):371–377. doi: 10.1007/s10689-009-9249-5. [DOI] [PubMed] [Google Scholar]
- 29.Begg CB, Orlow I, Hummer AJ, Armstrong BK, Kricker A, Marrett LD, Millikan RC, Gruber SB, Anton-Culver H, Zanetti R, Gallagher RP, Dwyer T, Rebbeck TR, Mitra N, Busam K, From L, Berwick M, Genes E, Melanoma Study G Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J Natl Cancer Inst. 2005;97(20):1507–1515. doi: 10.1093/jnci/dji312. [DOI] [PubMed] [Google Scholar]
- 30.Masback A, Olsson H, Westerdahl J, Sandberg T, Borg A, Jonsson N, Ingvar C. Clinical and histopathological features of malignant melanoma in germline CDKN2A mutation families. Melanoma Res. 2002;12(6):549–557. doi: 10.1097/00008390-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 31.FitzGerald MG, Harkin DP, Silva-Arrieta S, MacDonald DJ, Lucchina LC, Unsal H, O’Neill E, Koh J, Finkelstein DM, Isselbacher KJ, Sober AJ, Haber DA. Prevalence of germ-line mutations in p16, p19ARF, and CDK4 in familial melanoma: analysis of a clinic-based population. Proc Natl Acad Sci U S A. 1996;93(16):8541–8545. doi: 10.1073/pnas.93.16.8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Rhee JI, Krijnen P, Gruis NA, de Snoo FA, Vasen HF, Putter H, Kukutsch NA, Bergman W. Clinical and histologic characteristics of malignant melanoma in families with a germline mutation in CDKN2A. J Am Acad Dermatol. 2011;65(2):281–288. doi: 10.1016/j.jaad.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 33.Zuo L, Weger J, Yang Q, Goldstein AM, Tucker MA, Walker GJ, Hayward N, Dracopoli NC. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12(1):97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 34.Soufir N, Avril MF, Chompret A, Demenais F, Bombled J, Spatz A, Stoppa-Lyonnet D, Benard J, Bressac-de Paillerets B. Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. The French Familial Melanoma Study Group. Hum Mol Genet. 1998;7(2):209–216. doi: 10.1093/hmg/7.2.209. [DOI] [PubMed] [Google Scholar]
- 35.Molven A, Grimstvedt MB, Steine SJ, Harland M, Avril MF, Hayward NK, Akslen LA. A large Norwegian family with inherited malignant melanoma, multiple atypical nevi, and CDK4 mutation. Genes Chromosomes Cancer. 2005;44(1):10–18. doi: 10.1002/gcc.20202. [DOI] [PubMed] [Google Scholar]
- 36.Puntervoll HE, Yang XR, Vetti HH, Bachmann IM, Avril MF, Benfodda M, Catricala C, Dalle S, Duval-Modeste AB, Ghiorzo P, Grammatico P, Harland M, Hayward NK, Hu HH, Jouary T, Martin-Denavit T, Ozola A, Palmer JM, Pastorino L, Pjanova D, Soufir N, Steine SJ, Stratigos AJ, Thomas L, Tinat J, Tsao H, Veinalde R, Tucker MA, Bressac-de Paillerets B, Newton-Bishop JA, Goldstein AM, Akslen LA, Molven A. Melanoma prone families with CDK4 germline mutation: phenotypic profile and associations with MC1R variants. J Med Genet. 2013;50(4):264–270. doi: 10.1136/jmedgenet-2012-101455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Randerson-Moor JA, Harland M, Williams S, Cuthbert-Heavens D, Sheridan E, Aveyard J, Sibley K, Whitaker L, Knowles M, Bishop JN, Bishop DT. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet. 2001;10(1):55–62. doi: 10.1093/hmg/10.1.55. [DOI] [PubMed] [Google Scholar]
- 38.Bahuau M, Vidaud D, Jenkins RB, Bieche I, Kimmel DW, Assouline B, Smith JS, Alderete B, Cayuela JM, Harpey JP, Caille B, Vidaud M. Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res. 1998;58(11):2298–2303. [PubMed] [Google Scholar]
- 39.Randerson-Moor J, Kukalizch K, Bishop DT. Melanoma-Astrocytoma syndrome. Atlas Genet Cytogenet Oncol Haematol. 2006;10(4):288–291. [Google Scholar]
- 40.Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007;67(8):3963–3969. doi: 10.1158/0008-5472.CAN-06-2004. [DOI] [PubMed] [Google Scholar]
- 41.Frigerio S, Disciglio V, Manoukian S, Peissel B, Della Torre G, Maurichi A, Collini P, Pasini B, Gotti G, Ferrari A, Rivoltini L, Massimino M, Rodolfo M. A large de novo 9p21.3 deletion in a girl affected by astrocytoma and multiple melanoma. BMC Med Genet. 2014;15:59. doi: 10.1186/1471-2350-15-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufman DK, Kimmel DW, Parisi JE, Michels VV. A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology. 1993;43(9):1728–1731. doi: 10.1212/wnl.43.9.1728. [DOI] [PubMed] [Google Scholar]
- 43.Azizi E, Friedman J, Pavlotsky F, Iscovich J, Bornstein A, Shafir R, Trau H, Brenner H, Nass D. Familial cutaneous malignant melanoma and tumors of the nervous system. A hereditary cancer syndrome. Cancer. 1995;76(9):1571–1578. doi: 10.1002/1097-0142(19951101)76:9<1571::aid-cncr2820760912>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen K, Ingvar C, Masback A, Westerdahl J, Borg A, Sandberg T, Jonsson N, Nagel J, Olsson H. Melanoma and nonmelanoma skin cancer in patients with multiple tumours–evidence for new syndromes in a population-based study. Br J Dermatol. 2004;150(3):531–536. doi: 10.1111/j.1365-2133.2003.05852.x. [DOI] [PubMed] [Google Scholar]
- 45.Goldstein AM, Struewing JP, Chidambaram A, Fraser MC, Tucker MA. Genotype-phenotype relationships in U.S. melanoma-prone families with CDKN2A and CDK4 mutations. J Natl Cancer Inst. 2000;92(12):1006–1010. doi: 10.1093/jnci/92.12.1006. [DOI] [PubMed] [Google Scholar]
- 46.Kefford RF, Newton Bishop JA, Bergman W, Tucker MA. Counseling and DNA testing for individuals perceived to be genetically predisposed to melanoma: A consensus statement of the Melanoma Genetics Consortium. J Clin Oncol. 1999;17(10):3245–3251. doi: 10.1200/JCO.1999.17.10.3245. [DOI] [PubMed] [Google Scholar]
- 47.Haenssle HA, Korpas B, Hansen-Hagge C, Buhl T, Kaune KM, Johnsen S, Rosenberger A, Schon MP, Emmert S. Selection of patients for long-term surveillance with digital dermoscopy by assessment of melanoma risk factors. Arch Dermatol. 2010;146(3):257–264. doi: 10.1001/archdermatol.2009.370. [DOI] [PubMed] [Google Scholar]
- 48.Salerni G, Carrera C, Lovatto L, Puig-Butille JA, Badenas C, Plana E, Puig S, Malvehy J. Benefits of total body photography and digital dermatoscopy (“two-step method of digital follow-up”) in the early diagnosis of melanoma in patients at high risk for melanoma. J Am Acad Dermatol. 2012;67(1):e17–27. doi: 10.1016/j.jaad.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moloney FJ, Guitera P, Coates E, Haass NK, Ho K, Khoury R, O’Connell RL, Raudonikis L, Schmid H, Mann GJ, Menzies SW. Detection of primary melanoma in individuals at extreme high risk: a prospective 5-year follow-up study. JAMA Dermatol. 2014;150(8):819–827. doi: 10.1001/jamadermatol.2014.514. [DOI] [PubMed] [Google Scholar]
- 50.Rademaker M, Oakley A. Digital monitoring by whole body photography and sequential digital dermoscopy detects thinner melanomas. J Prim Health Care. 2010;2(4):268–272. [PubMed] [Google Scholar]
- 51.Feit NE, Dusza SW, Marghoob AA. Melanomas detected with the aid of total cutaneous photography. Br J Dermatol. 2004;150(4):706–714. doi: 10.1111/j.0007-0963.2004.05892.x. [DOI] [PubMed] [Google Scholar]
- 52.Oliveria SA, Chau D, Christos PJ, Charles CA, Mushlin AI, Halpern AC. Diagnostic accuracy of patients in performing skin self-examination and the impact of photography. Arch Dermatol. 2004;140(1):57–62. doi: 10.1001/archderm.140.1.57. [DOI] [PubMed] [Google Scholar]
- 53.Curiel-Lewandrowski C, Kim CC, Swetter SM, Chen SC, Halpern AC, Kirkwood JM, Leachman SA, Marghoob AA, Ming ME, Grichnik JM. Survival is not the only valuable end point in melanoma screening. J Invest Dermatol. 2012;132(5):1332–1337. doi: 10.1038/jid.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Risser J, Pressley Z, Veledar E, Washington C, Chen SC. The impact of total body photography on biopsy rate in patients from a pigmented lesion clinic. J Am Acad Dermatol. 2007;57(3):428–434. doi: 10.1016/j.jaad.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 55.Marghoob AA, Halpern AC. Biopsy rates in patients with and without total body photography. J Am Acad Dermatol. 2008;58(5):894–895. doi: 10.1016/j.jaad.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 56.Watts CG, Cust AE, Menzies SW, Coates E, Mann GJ, Morton RL. Specialized surveillance for individuals at high risk for melanoma: a cost analysis of a high-risk clinic. JAMA Dermatol. 2015;151(2):178–186. doi: 10.1001/jamadermatol.2014.1952. [DOI] [PubMed] [Google Scholar]
- 57.Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, Fontaine LS, Organic SM, Dracopoli NC, Clark WH, Jr, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med. 1995;333(15):970–974. doi: 10.1056/NEJM199510123331504. [DOI] [PubMed] [Google Scholar]
- 58.Lynch HT, Fusaro RM, Lynch JF, Brand R. Pancreatic cancer and the FAMMM syndrome. Fam Cancer. 2008;7(1):103–112. doi: 10.1007/s10689-007-9166-4. [DOI] [PubMed] [Google Scholar]
- 59.Bergman W, Watson P, de Jong J, Lynch HT, Fusaro RM. Systemic cancer and the FAMMM syndrome. Br J Cancer. 1990;61(6):932–936. doi: 10.1038/bjc.1990.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden) Int J Cancer. 2000;87(6):809–811. [PubMed] [Google Scholar]
- 61.Hruban RH, Petersen GM, Ha PK, Kern SE. Genetics of pancreatic cancer. From genes to families. Surg Oncol Clin N Am. 1998;7(1):1–23. [PubMed] [Google Scholar]
- 62.James TA, Sheldon DG, Rajput A, Kuvshinoff BW, Javle MM, Nava HR, Smith JL, Gibbs JF. Risk factors associated with earlier age of onset in familial pancreatic carcinoma. Cancer. 2004;101(12):2722–2726. doi: 10.1002/cncr.20700. [DOI] [PubMed] [Google Scholar]
- 63.Debniak T, van de Wetering T, Scott R, Nagay L, Cybulski C, Gorski B, Jakubowska A, Gronwald J, Masojc B, Huzarski T, Byrski T, Nej-Wolosiak K, Kladny J, Maleszka R, Lubinski J. Low prevalence of CDKN2A/ARF mutations among early-onset cancers of breast, pancreas and malignant melanoma in Poland. Eur J Cancer Prev. 2008;17(5):389–391. doi: 10.1097/CEJ.0b013e3282f75eb1. [DOI] [PubMed] [Google Scholar]
- 64.Leachman SA, Carucci J, Kohlmann W, Banks KC, Asgari MM, Bergman W, Bianchi-Scarra G, Brentnall T, Bressac-de Paillerets B, Bruno W, Curiel-Lewandrowski C, de Snoo FA, Debniak T, Demierre MF, Elder D, Goldstein AM, Grant-Kels J, Halpern AC, Ingvar C, Kefford RF, Lang J, MacKie RM, Mann GJ, Mueller K, Newton-Bishop J, Olsson H, Petersen GM, Puig S, Rigel D, Swetter SM, Tucker MA, Yakobson E, Zitelli JA, Tsao H. Selection criteria for genetic assessment of patients with familial melanoma. J Am Acad Dermatol. 2009;61(4):677, e671–614. doi: 10.1016/j.jaad.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, Nio Y, Schulick RS, Bassi C, Kluijt I, Levy MJ, Chak A, Fockens P, Goggins M, Bruno M, International Cancer of Pancreas Screening C International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62(3):339–347. doi: 10.1136/gutjnl-2012-303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brentnall TA, Bronner MP, Byrd DR, Haggitt RC, Kimmey MB. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med. 1999;131(4):247–255. doi: 10.7326/0003-4819-131-4-199908170-00003. [DOI] [PubMed] [Google Scholar]
- 67.Grover S, Syngal S. Hereditary pancreatic cancer. Gastroenterology. 2010;139(4):1076–1080. 1080 e1071–1072. doi: 10.1053/j.gastro.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aspinwall LG, Taber JM, Leaf SL, Kohlmann W, Leachman SA. Genetic testing for hereditary melanoma and pancreatic cancer: a longitudinal study of psychological outcome. Psychooncology. 2013;22(2):276–289. doi: 10.1002/pon.2080. [DOI] [PubMed] [Google Scholar]
- 69.Borg A, Sandberg T, Nilsson K, Johannsson O, Klinker M, Masback A, Westerdahl J, Olsson H, Ingvar C. High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutationpositive melanoma families. J Natl Cancer Inst. 2000;92(15):1260–1266. doi: 10.1093/jnci/92.15.1260. [DOI] [PubMed] [Google Scholar]
- 70.Lynch HT, Fusaro RM, Kimberling WJ, Lynch JF, Danes BS. Familial atypical multiple mole-melanoma (FAMMM) syndrome: segregation analysis. J Med Genet. 1983;20(5):342–344. doi: 10.1136/jmg.20.5.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lynch HT, Fusaro RM, Pester J, Oosterhuis JA, Went LN, Rumke P, Neering H, Lynch JF. Tumour spectrum in the FAMMM syndrome. Br J Cancer. 1981;44(4):553–560. doi: 10.1038/bjc.1981.225. [DOI] [PMC free article] [PubMed] [Google Scholar]