

Abstract
Background
Colorectal cancer is an abnormal tissue development in the colon or rectum. Most of CRCs develop due to somatic mutations, while only a small proportion is caused by inherited mutations. Familial adenomatous polyposis is an inherited genetic disease, which is characterized by colorectal polyps. It is caused by inactivating mutations in the Adenomatous polyposis coli gene. Mice carrying and non-sense mutation in Adenomatous polyposis coli gene at site R850, which designated ApcR850X/+ (Min), develop intestinal adenomas, while the bulk of the disease is in the small intestine. A number of genetic modifier loci of Min have been mapped, but so far most of the underlying genes have not been identified. In our previous studies, we have shown that Collaborative Cross mice are a powerful tool for mapping loci responsible for phenotypic variation. As a first step towards identification of novel modifiers of Min, we assessed the phenotypic variation between 27 F1 crosses between different Collaborative cross mice and C57BL/6-Min lines.
Results
Here, C57BL/6-Min male mice were mated with females from 27 Collaborative cross lines. F1 offspring were terminated at 23 weeks old and multiple phenotypes were collected: polyp counts, intestine length, intestine weight, packed cell volume and spleen weight. Additionally, in eight selected F1 Collaborative cross-C57BL/6-Min lines, body weight was monitored and compared to control mice carry wildtype Adenomatous polyposis coli gene. We found significant (p < 0.05) phenotypic variation between the 27 F1 Collaborative cross-C57BL/6-Min lines for all the tested phenotypes, and sex differences with traits; Colon, body weight and intestine length phenotypes, only. Heritability calculation showed that these phenotypes are mainly controlled by genetic factors.
Conclusions
Variation in polyp development is controlled, an appreciable extent, by genetic factors segregating in the Collaborative cross population and suggests that it is suited for identifying modifier genes associated with ApcMin/+ mutation, after assessing sufficient number of lines for quantitative trait loci analysis.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-016-0349-6) contains supplementary material, which is available to authorized users.
Keywords: ApcMin/+, Colorectal cancer, Collaborative cross, Familial adenomatous polyposis, Genetic modifier, Moms, Phenotyping, Recombinant Inbred lines
Background
Genome-wide association studies (GWAS) have identified numerous common alleles that affect cancer susceptibility [1–4], yet the combined effects of the alleles currently discovered are too small to explain the bulk of heritable disease risk. This suggests, that the genetic influence on cancer susceptibility might not be entirely due the combined additive effects of individual alleles. Experimental mouse models of cancer are ideal for examining the effects of genetic modifiers that act epistatically with known susceptibility loci. Epistasis is difficult to detect in human GWAS due to the very large sample size required. However, it is straightforward to engineer mutant mice in which a known susceptibility locus is altered to change the risk of disease. By crossing the mutant onto a population of mice with different genetic backgrounds of naturally occurring variation, it is theoretically possible to map modifier loci.
The great majority of colorectal cancers (CRCs) develop from adenomas [5, 6]. Apc mutations are found in the germ line of patients with Familial Adenomatous Polyposis (FAP) and in over 80 % of sporadic colorectal adenomas [7–11]. Apc is a tumor suppressor gene [7], that encodes a large protein [8] that regulates Wnt pathway activity through its interaction with the transcription factor β-catenin [12–17]. Apc also interacts with numerous actin and microtubule-associated proteins [17–19]. Roles for Apc in cell migration have been demonstrated in vitro and in mouse models [13, 20].
Several mouse models with heterozygous germline mutations of Apc exist, and all of these develop adenomatous intestinal polyps [21, 22]. Different mutations are associated with different disease severities. For example, ApcR850X/+ (Min) mice typically develop ~100 intestinal polyps at 3–4 months of age, whereas the Apc1322T/+ mice develop polyps earlier in life, and the Apc1638N/+ model produces only a handful of polyps at over a year of age [21–24]. Like humans with germline mutations in Apc, Min mice are predisposed to intestinal adenoma formation, which is influenced by modifier loci carried by different inbred strains. Assessing Apc modifiers in the mouse is usually quantitative by counting numbers of intestinal polyps. The Mom1 (modifier of Min 1) locus was the first Apc modifier to be identified [21, 25]. It encodes secretory phospholipase A2 (Pla2), a Paneth cell-specific marker with effects on tumorgenesis that remain unclear [24, 26]. Another modifier, Mom2, is an Atp5a1 mutation that acts by suppressing loss of heterozygosity through cell lethality [14, 27, 28]. Other Mom loci have been mapped, but no other genes been cloned [29]. The most comprehensive modifier screen to date in a population of female BXH recombinant inbred mice backcrossed to C57BL/6-Min males [29] identified five modifier loci. Given that the genetic backgrounds of inbred strains have profound effects on polyp numbers, further modifiers almost certainly exist. These might affect not only polyp numbers, but other traits such as progression to cancer and location in the bowel [30].
The Collaborative Cross (CC) is a panel of recombinant inbred (RI) strains derived from a genetically diverse set of eight founder strains: A/J, C57BL/6 J, 129S1/SvImJ, NOD/LtJ, NZO/HiLtJ, CAST/EiJ, PWK/PhJ and WSB/EiJ. It was designed to provide complex trait analysis with greater power than previous approaches [31, 32]. A full description of the CC population under development at Tel-Aviv University is provided in recent publications [33–40]. The key features of the CC in relation to modifier mapping are the very large number of variants segregating in the population (there are over 36 million SNPs) [36], and the relatively high level of recombination present compared to other mouse RI sets (4.4 million SNPs segregate between the founders C57BL/6 J and C3H/HeJ of the BXH mice [41]). Three founders of the CC are wild-derived strains, representing the three different mice subspecies: Mus musculus castaneus (CAST/EiJ), Mus musculus musculus (PWK/PhJ) and Mus musculus domesticus (WSB/EiJ), which contribute many sequence variants not segregating among classical strains descended from Mus musculus domesticus [41–43], and account for the bulk of the variants segregating in the CC. Consequently mapping of quantitative trait loci (QTLs) using the CC often identifies QTLs involving contrasts between the wild-derived strains [37]. By incorporating polymorphisms data from the genome sequences of the CC founders [36], and restricting attention to variants whose differences across the founders are consistent with the pattern of action of the QTL, candidate genes under QTLs can be identified [36, 37, 44–47].
As a first step towards mapping modifiers of ApcMin/+ in CC mice, we assessed polyp-related phenotypes dissection in F1 hybrids of females from 27 CC lines to C57BL/6-Min male mice. In this report, we analyze phenotypic variation and the broad sense heritability (H2) of these traits.
Materials and Methods
Breeding, genotyping and phenotyping F1 CC-C57BL/6-Min mice
The C57BL/6-Min and CC mice were provided by the Small Animal Facility at Sackler Faculty of Medicine, Tel Aviv University (TAU). The CC lines were between the 11th and 38th generation of inbreeding by full-sib mating [34, 48]. They were genotyped using the MEGA-MUGA (60 k SNPs) genotyping array, which showed that each line was at least 85 % homozygous. Full details of the development of the CC are given in Iraqi et al. 2008 [34]. All experimental mice and protocols were approved by the Institutional Animal Care and Use Committee of TAU (approval numbers: M-08-075; M-12-024). Institutional Animal Care and Use Committee of TAU adherers to the Israeli guidelines that follow NIH/USA animal care and use protocols. Mice were housed on hardwood chip bedding in open-top cages at the animal facility and were given tap water and rodent chow ad libitum. F1-CC-C57BL/6-Min mice were monitored daily for their overall health status. Mice which lost 10 % of their bodyweight between two measured points, or over 20 % of their initial bodyweight, or were observed to be suffering (less movement and activity), and based on the consultation with the Veterinarian at the small animal unit, were terminated.
At 4 weeks old, 0.5 cm tail biopsies were collected and DNA isolated by NaOH boiling method [49]. The F1 mice were genotyped by PCR for the ApcMin/+ mutant allele, using primers MAPC-min (TTCTGAGAAAGACAGAAGTTA), MAPC-15 (TTCCACTTTGGCATAAGG), and MAPC-9 (GCCATCCCTTCACGT). For Apc wild type alleles, we used primers MAPC-15 and MAPC-9, which amplify 618 bp PCR products, while for the mutant form we used MAPC-min and MAPC-15, which amplify 317 bp PCR products [50]. For later identification each mouse was ear-labelled.
After five months the F1-CC-C57BL/6-Min mice were terminated, their small intestine and colon extracted and washed with PBS. The small intestine were divided into three segments (SB1-proximal, SB2-middle and SB3-distal), and the colon was kept as whole and spread over 3 mm paper. The intestines were fixed in 10 % Neutral Buffered Formalin (NBF) for overnight, and stained by 0.02 % methylene blue. The samples were then examined by binuclear. The number and sizes of polyps in each of the four intestinal sub-regions were recorded as described in Rudling et al. 2006 [51]. The total body weight, weight of spleen, weight and length of intestine and the total packed cell volume (PCV) were also measured at the terminal point. Additionally presented number of polyps per centimeter of intestine, which was calculated using the follow formula: Number of polyp per cm = Total number of polyps/Total length of intestine. Spleen weight was normalized to total body weight using the follow formula: Spleen %Weight = (Spleen weight (g)/total body weight (g)) × 100. Intestine weight was normalized to total body weight using the follow formula: Intestine %Weight = (Intestine weight (g)/total body weight (g)) × 100. Blood was collected by heart puncture [52, 53] into hematocrit heparinized capillary by hematocrit centrifuge CM-70 (ELMI). The % packed cell volume (%PCV) was calculated based on formula: (Total plasma (uL) × 100)/total blood volume (uL).
For 40 selected males from 8 F1-CC-C57BL/6 lines (with representation of control (Apc+/+) and affected (ApcMin/+) mice) body weight was monitored along the experiment at 5 time points, at the weaning (8 weeks old), 12, 16, 19 and 23 weeks old.
Data analysis
Data analysis performed using a statistical software package SPSS version 19. Analysis of variance (ANOVA) was performed to test the differences of polyp’s number, intestinal %weight and length, spleen %weight, %PCV between 27 F1 CC-C57BL/6-Min lines, and body weight between 8 F1 CC-C57BL/6 lines. Clustering analysis of total polyps was performed by Duncan’s test. All the results represented as Mean ± Standard Error, if n = 1 represented only the value of the phenotype.
Correlation between traits (polyp number, intestinal %weight and length, polyps per cm, %PCV and spleen %weight) was measured by Pearson product-moment correlation coefficient.
Heritability (H2) was estimated as the proportion of phenotypic variation explained by differences between F1 CC-C57BL/6-Min lines in the ANOVA, i.e. H2 = Vg/(Vg + Ve).
Results
Breeding
C57BL/6-Min males were crossed to females from 27 CC lines, total progeny of 403 F1 CC-C57BL/6 mice were generated as detailed in Additional file 1: Table 1S. We found that 179/403 mice (44.42 %; 91 males and 88 females) were heterozygous for the knockout allele (carried the ApcMin/+ allele) and therefore were F1 CC-C57BL/6-Min. The other progeny were wild-type for the Apc allele, and are henceforth denoted as F1 CC-C57BL/6. A second set of 40 male mice including 25 carried ApcMin/+ allele and 15 non carrier (control) of 8 CC lines were also generated, separately to study body weigh differences between these two genotypes.
Body weight monitoring of F1 CC-C57BL/6 lines
A previous study showed that ApcMin/+ mice lose their body weight during polyp’s development [54, 55]. Here, body weight performances of males from 8 randomly selected F1 CC-C57BL/6-Min lines was monitored during the experiment at 5 time points, and compared to the corresponding F1 CC-C57BL/6 wild type lines, results presented in Fig. 1.
Fig. 1.
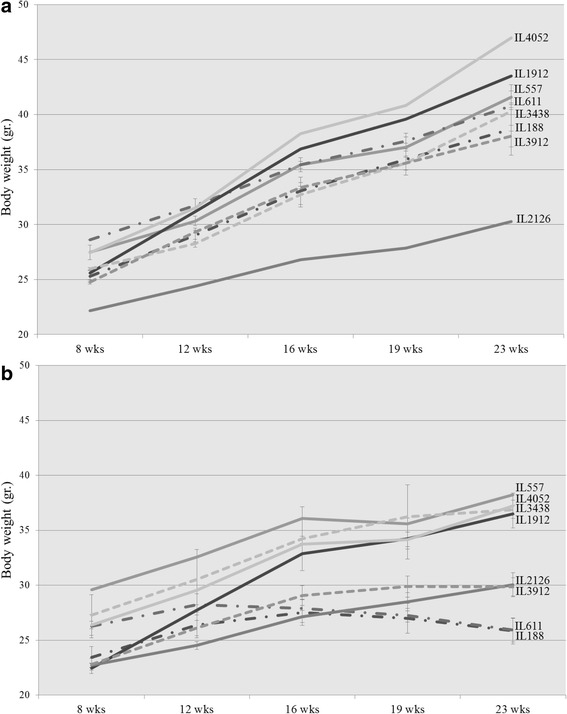
Body weight (±SE) of the a control and b Apc Min/+ groups at 5 time points (8, 12, 16, 19 and 23 weeks old) of 8 different set of CC lines. The X-axis represents the time points (in weeks) while the Y-axis represents values of body mass (g). One-way ANOVA performed for statistical analysis, p < 0.05
At the initial time point (8 weeks old) the body weight of control lines varied between 22.17 g (IL2126) to 28.6 g (IL611) (Fig. 1a). Body weight of ApcMin/+ carriers varied between 22.45 ± 0.46 g (Mean ± Standard Error) (IL1912) to 29.6 g (IL557) (Fig. 1b). One-way ANOVA showed that body weight significantly (p < 0.05) different between lines, within both controls and ApcMin/+ groups. In contrast no significant differences were found at the initial time point between the control and ApcMin/+ groups, within each examined line.
During the experiment ApcMin/+ carriers displayed three patterns of body weight changes: body weight loss (marked as - · - · - · - in Fig. 1b), Plateau (---- in Fig. 1b) and continues body weight accumulation ( in Fig. 1b). While all the control lines gained weight continuously during the experiment.
As shown in Fig. 1b, two lines – IL188 and IL611 lost body weight during the experiment. Table 1 shows delta body weight between 23 and 8 wks. old, was -0.34 g and 2.39 g, for IL611 and IL188, respectively. Line IL188 carries ApcMin/+ lost -1.15 g of total body weight between weeks 19 to 23. Over the entire experiment, IL188 mice carries ApcMin/+ gained weight, but started losing weight, only late in the study, whereas IL611 lost weight relative to initial weight. The control mice of same lines gained 12.18 g (IL611) and 13.4 g (IL188), which were significantly different from the ApcMin/+ carriers group. Interestingly, ApcMin/+ carriers from these lines also developed high number of polyps, and will be presented later on.
Table 1.
Summary of collected phenotypes at 23 weeks old: Total polyps, %PCV, Spleen %Weight and Delta body weight (Body weight 23 weeks old-Body weight 8 weeks old), for control and Apc Min/+ mice from 8 F1 CC-C57BL/6
| CC Line | Apc | N (mice) | Total Polyps | %PCV | Spleen %Weight | Delta Body weight (gr.) |
|---|---|---|---|---|---|---|
| IL611 | Min | 3 | 55.33 ± 10.91 | 25.96 ± 4.32 | 2.7637 ± 0.677 | -0.34 |
| WT | 2 | 0 | 54.62 ± 0.1 | 0.1962 ± 0.0007 | 12.18 | |
| IL188 | Min | 8 | 70.38 ± 10.78 | 29.45 ± 3.8 | 1.9698 ± 0.3419 | 2.39 |
| WT | 3 | 0 | 54.93 ± 2.53 | 0.1762 ± 0.0667 | 13.4 | |
| IL3438 | Min | 2 | 67.5 ± 24.5 | 38.93 ± 6.67 | 1.2124 ± 0.691 | 9.59 |
| WT | 2 | 0 | 53.75 ± 4.55 | 0.1729 ± 0.024 | 14.36 | |
| IL3912 | Min | 3 | 42.33 ± 5.36 | 35.2 ± 2.29 | 0.7768 ± 0.1209 | 7.03 |
| WT | 2 | 0 | 56.62 ± 4.06 | 0.1699 ± 0.0496 | 13.28 | |
| IL2126 | Min | 4 | 14.75 ± 4.05 | 51.75 ± 1.54 | 0.2966 ± 0.0602 | 7.33 |
| WT | 1 | 0 | 51.92 | 0.1651 | 8.11 | |
| IL557 | Min | 1 | 53 | 34.25 | 1.4379 | 8.65 |
| WT | 3 | 0 | 55.6 ± 5.92 | 0.2656 ± 0.0357 | 14.12 | |
| IL1912 | Min | 2 | 43.5 ± 1.5 | 41.84 ± 1.84 | 0.5044 ± 0.0786 | 14.04 |
| WT | 1 | 0 | 53.75 | 0.1609 | 17.9 | |
| IL4052 | Min | 2 | 9 | 48.96 ± 5.38 | 0.3218 ± 0.017 | 10.85 |
| WT | 1 | 0 | 60.26 | 0.1703 | 19.51 |
The ApcMin/+ carriers group of lines IL3438 and IL3912 (Fig. 1b) exhibited Plateau pattern during the final weeks of the experiment (after week 20). Table 1 shows that delta body weight for these lines was 9.59 g (IL3438) and 7.03 g (IL3912). Control mice from the same lines gained 14.36 g (IL3438), which wasn’t significantly different from ApcMin/+ carriers, and 13.28 g (IL3912) which was significantly different (p < 0.01) compared to ApcMin/+ carriers. Values for the rest tested phenotypes of these lines summarized in Table 1.
The remaining lines – IL2126, IL557, IL1912 and IL4052, which are also presented in Fig. 1b, have demonstrated continues increase in body weight throughout the experiment. Line IL2126 has exhibited no significant difference in weight accumulation tendency between the experimental (7.33 g) and control groups (8.11 g). Line IL557 showed significant difference (p < 0.039) in delta body weight accumulation between the experimental (8.65 g) and control groups (14.12 g). Line IL1912 showed no significant difference in delta body weight accumulation between the experimental (14.04 g) and control groups (17.9 g). Line IL4052 showed no significant difference in delta body weight accumulation between the experimental (10.85 g) and control groups (19.51 g). Values for the rest tested phenotypes summarized in Table 1.
At the final time point of the experiment (23 wks. old) lines from both groups, control and ApcMin/+ carriers, displayed significant differences in their body weight values. Additionally at this step of the experiment we found significant variation between control and ApcMin/+ carriers group within each line.
Polyp counts in F1 CC-C57BL/6-Min mice
Mice were terminated at age of 23 weeks and the gastrointestinal tract (small intestine and colon) was extracted for polyps count, results are presented in Fig. 2. Across the 27 F1 CC-C57BL/6-Min lines mean polyp count was 27.3 ± 1.49. The cohort exhibited a wide range of phenotypic variability, from 9 polyps (IL4052) to 69 ± 6.83 (IL3348). One-way ANOVA revealed significant variation between CC lines (p < 0.01). Polyp counts were approximately normally distributed, suggesting intervention of numerous genetic factors in this trait. Duncan’s analysis divided tested lines into 9 clusters, presented by continuous lines under the graph in Fig. 2. Interestingly, mean polyp count in the parental line C57BL/6-Min was 45.50 ± 5.98, so that the majority (20/27, 74.07 %) of F1 CC-C57BL/6-Min lines have fewer polyps than the parental line, despite the fact that the number of defective Apc alleles is the same in all cases. When we compared the polyp counts in the male mice of the 8 CC lines assessed in the second set of lines presented in Fig. 1a and Table 1 and Additional file 1: Table 7S, majority of lines were consistent on both sets. Notably, that SE is very high in many lines. Based on one way ANOVA, we have found that some lines are significantly different between male and female, mice. As for polyp counts, we have found that some lines are significantly different with this trait between both sexes as presented in Additional file 1: Table 7S, however, overall across all the tested lines sex was significant different with polyp counts.
Fig. 2.
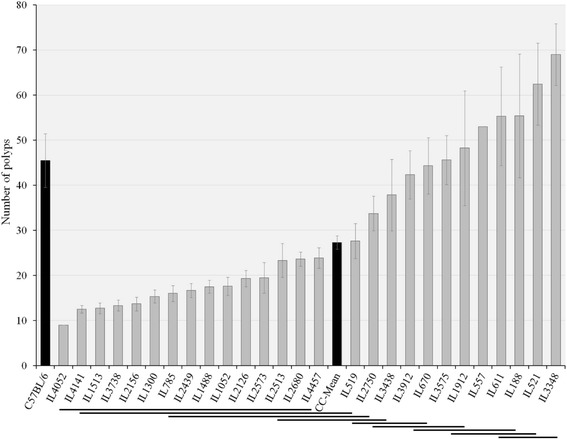
Average number of polyps (±SE) in 27 F1 CC-C57BL/6-Min lines (n = 1-16 mice/line). The X-axis represents CC lines, C57BL/6 strain carrying the Apc Min/+ mutation (first black column) and CC-Mean (black column ~ in the middle), while the Y-axis number of polyps. Continuous lines under the graph represent 9 clusters based on Duncan’s Least Significant Range (LSR) test. One-way ANOVA performed for statistical analysis, p < 0.05
Next we asked if different segments of the intestine exhibit differential polyp distribution. The small intestine was subdivided into 3 parts (small intestine proximal-SB1, middle-SB2 and distal-SB3), and the colon was related separately. Polyp counts were recorded in each region. Figure 3 shows the distribution of polyps in the four components of gastrointestinal tract in the 27 F1 CC-C57BL/6-Min lines and C57BL/6-Min. Overall, polyps were distributed approximately equally between 4 segments: SB1 with 5.96 ± 0.4 polyps (21.58 %), SB2 with 7.73 ± 8.17 (26.98 %), SB3 with 7.97 ± 7.69 (28.97 %) and colon with 6.2 ± 3.9 (22.47 %). The percentages of polyp counts in C57BL/6 J-Min strain were 23.63 % (SB1), 37.91 % (SB2), 26.92 % (SB3) and 11.54 % (colon).
Fig. 3.
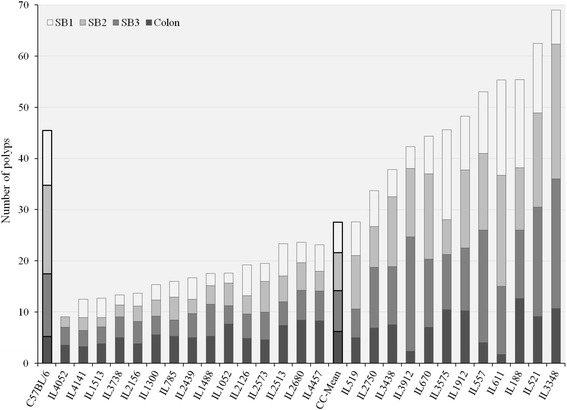
Distribution of all counted polyp in three parts of small intestine: SB1-proximal, SB2-middle, SB3-distal and colon. The X-axis represents CC lines, C57BL/6 strain carrying the Apc Min/+ mutation (first black column) and CC-Mean (black column ~ in the middle), while the Y-axis number of polyps. One-way ANOVA performed for statistical analysis, p < 0.05
Interestingly some lines showed approximately the same number of polyps, which were differentially distributed across the intestine. For example, line IL611 had 55.33 ± 10.91 and line IL188 55.4 ± 13.71 polyps. However, in IL611 only 3.01 % of polyps were in the colon; while IL188 had 22.74 % of polyps in the same segment. Similarly in SB2, IL611 showed 39.16 % polyps and IL188 22.02 %. This suggests that different genetic factors might be involved in polyp development throughout the gastrointestinal tract.
Intestine Length
Since tumor development might affect the length and weight of the intestine, we asked if these phenotypes also differed across the population. Additional file 1: Figure 1S (presented in supplementary matherials) shows the distribution of intestine length (cm) across the F1 CC-C57BL/6-Min lines and the C57BL/6-Min parental strain. The mean intestine length of the entire population was 47.92 ± 0.28 cm, ranging from 43.52 ± 0.85 cm (IL2126) to 51.83 ± 1.21 cm (IL3348). One-way ANOVA showed that intestine length varied significantly (p < 0.01) between 27 F1 CC-C57BL/6-Min lines. The mean intestine length for the parental strain, C57BL/6-Min, was 48.55 ± 1.55 cm.
Number of polyps per centimeter
Next, number of polyps per cm of intestine was calculated (as described in Matirials and Methods section), results presented in Additional file 1: Figure 2S. As for entire population, avarge number of polyps per cm was 0.5676 ± 0.0291. Lowest value of 0.1877 ± 0.0356 polyps per cm was recorded for line IL4052, which had overall only 9 polyps. Highest value of 1.3267 ± 0.0421 polyps per cm was recorded for line IL3348, which had overall 69 ± 6.836 polyps. The parental line C57BL/6 showed 1.142 ± 0.0297 polyps per cm.
Additionally, histogram plot in Additional file 1: Figure 3S shows frequency of number of lines within different ranges of polyps per cm values. It was found that majority of tested CC lines, 9 lines, developed between 0.2721 ± 0.1815 to 0.4143 ± 0.0283 polyps per cm of intestine. As for extreme cases: one line (IL4052) developed 0.1877 ± 0.0356 polyps per cm, and 2 lines (IL521 and IL3348) developed more than 1.2209 ± 0.0177 polyps per cm.
Intestine Weight
Additional file 1: Figure 4S shows the distribution of intestine %weight. The mean across the entire population was 2.63 ± 0.71 %, ranging from 1.63 ± 0.16 % (IL2750) to 4.82 ± 0.71 % (IL611). One-way ANOVA showed that the intestine %weight varied significantly (p < 0.01) between tested lines. The mean intestine %weight in C57BL/6-Min was 4.5 ± 0.08.
Packed Cell Volume
ApcMin/+ mice suffer from severe anemia [56], and severe anemia is the main cause of death in these mice [57]. In order to investigate this phenmenon in the F1 CC-C57BL/6-Min population, the packed cell volume (PCV) was measured at the terminal point of experiment, results are summarized in Additional file 1: Figure 5S. The overall mean of entire population was 44.6 ± 0.7 %, and individual lines ranged from 25.96 ± 4.32 % (IL611) to 51.94 ± 1.33 % (IL4457). Normal PCV values are in the range 45-55 % [56], so 51.85 % (14/27) of the CC-C57BL/6-Min lines were below the normal range. One-way ANOVA revealed that %PCV varied significantly (p < 0.01) between F1 CC-C57BL/6-Min lines. The parental C57BL/6-Min line showed the lowest %PCV value of 24.35 ± 2.13 %. When we compared the %PCV in the male mice of the 8 CC lines assessed in the second set of lines presented in Fig. 1a and Table 1, majority of lines were consistent on both sets. Notably, that SE is very high in many lines.
Spleen Weight
It has been reported that ApcMin/+ mice suffer from splenomegaly, (enlargement of the spleen) [58]. Additional file 1: Figure 6S shows the distribution of spleen %weights. The mean of the tested 27 F1 CC-C57BL/6-Min lines was 0.53 ± 0.05 %, ranging from 0.18 ± 0.02 % (IL3738) to 2.76 ± 0.67 % (IL611). One-way ANOVA showed that spleen %weight varied significantly (p < 0.01) between lines. The parental line, C57BL/6-Min, had relatively high value of 1.52 ± 0.22 %. When we compared the spleen weight in the male mice of the 8 CC lines assessed in the second set of lines presented in Fig. 1a and Table 1, majority of lines were consistent on both sets. Notably, that SE is very high in many lines.
Correlation between phenotypes
Next we were interested to investigate the relationship between polyp’s development and related phenotypes. Table 2 shows Pearson correlations between traits across the 27 F1 CC-C57BL/6-Min lines.
Table 2.
Pearson correlation values between traits: total polyps, intestine length, intestine %weight, %PCV, spleen %weight and polyps per cm
| Total Polyps | Intestine Length | Intestine %Weight | %PCV | Spleen %Weight | Polyps per cm | |
|---|---|---|---|---|---|---|
| Total Polyps | 1 | 0.589** | 0.695** | -0.841** | 0.743** | 0.998** |
| Intestine Length | 0.589** | 1 | 0.266 | -0.457* | 0.317 | 0.539** |
| Intestine %Weight | 0.695** | 0.266 | 1 | -0.769** | 0.815** | 0.714** |
| %PCV | -0.841** | -0.457* | -0.769** | 1 | -0.846** | -0.852** |
| Spleen %Weight | 0.743** | 0.317 | 0.815** | -0.846** | 1 | 0.764** |
| Polyps per cm | 0.998** | 0.539** | 0.714** | -0.852** | 0.764** | 1 |
*Correlation is significant at p < 0.05 **Correlation is significant at p < 0.01
The length of intestine and total number of polyps is positively correlated (Pearson correlation coefficient (r) = 0.589, p < 0.01). This suggests that a high number of polyps are associated with longer intestinal track. The scatter plot in Fig. 4a (R2 = 0.346) shows data distribution of these two traits.
Fig. 4.
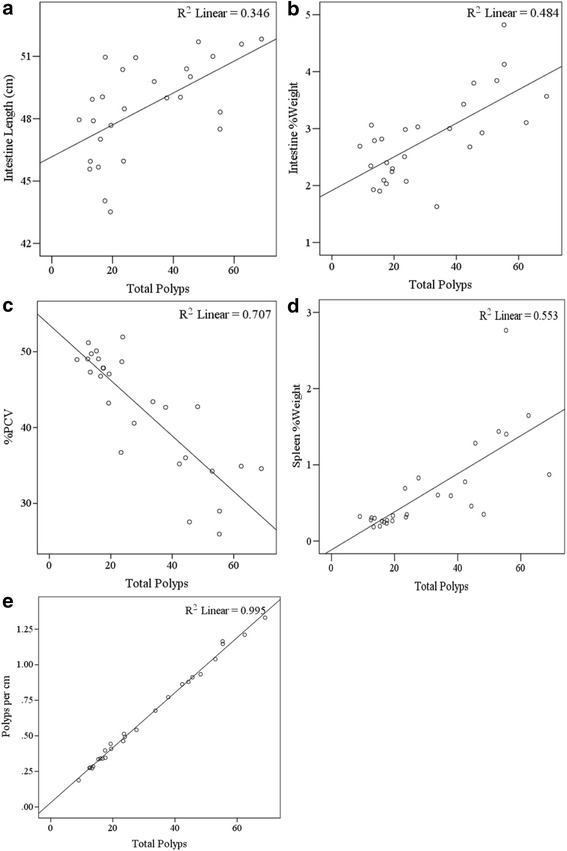
Scatter plot with tread lines of total polyps vs. a. Intestine length (R2 = 0.346), b. Intestine %Weight (R2 = 0.484), c. % PCV (R2 = 0.707), d. Spleen %Weight (R2 = 0.553) and e. Polyps per cm (R2 = 0.995)
Similarly intestine %weight is positively correlated with total polyps count (r = 0.695, p < 0.01). Suggesting that a high number of polyps are associated with heavier intestinal track. The scatter plot in Fig. 4b (R2 = 0.484) shows data distribution of these two traits.
In contrast, a negative correlation (r = -0.841, p < 0.01) was observed between %PCV and total number of polyps, i.e. high number of polyps are associated with low values of %PCV. The scatter plot in Fig. 4c (R2 = 0.707) shows data distribution of these two traits.
The spleen %weight and total number of polyps were positively correlated (r = 0.743, p < 0.01). This suggests that high numbers of polyps are associated with heavier spleen, indicating splenomegaly. The scatter plot in Fig. 4d (R2 = 0.553) shows data distribution of these two traits.
As was expected there is strong positive correlation was found between %PCV and spleen %weight. Pearson test showed negative correlation (r = -0.846, p < 0.01) between these traits, suggesting that anemia accompanied by splenomegaly.
As was expected there is strong positive correlation between Total polyps and Polyps per cm (r = 0.998, p < 0.01), corresponding scatter plot in Fig. 4e (R2 = 0.995). Additionally significant positive correlations were found between Polyps per cm and Intestine length (r = 0.539, p < 0.01), Intestine %Weight (r = 0.714, p < 0.01) and Spleen %Weight (r = 0.764, p < 0.01), significant negative correlation was found with %PCV (r = -0.852, p < 0.01).
Heritability
Our last aim of this repost was to determine whether variation in polyp development and other tested phenotypes has a genetic basis in F1 CC-C57BL/6-Min population. In order to answer this question, heritiabilities (H2) values were calculated, results summarized in Table 3. All the phenotypes have high heritability values, meaning that tested traits have strong genetic basis. The highest value of 0.641 was found for the phenotype of all counted polyps in along the gastrointestinal tract (Total Polyps), while the lowest value of 0.305 was found for the number of polyps in the colon.
Table 3.
Mean square between and within groups, and H2 calculations for traits of: total number of polyps, polyp’s distribution in different parts of intestine (SB1, SB2, SB3) and Colon, intestine length, polyps per cm, intestine %weight, %PCV and spleen %weight
| Total polyps | SB1 | SB2 | SB3 | Colon | Intestine Length | Polyps per cm | Intestine %Weight | %PCV | Spleen %Weight | |
|---|---|---|---|---|---|---|---|---|---|---|
| MS between | 1874.734 | 124.804 | 265.143 | 241.036 | 40.197 | 37.665 | 0.678 | 2.992 | 334.972 | 1.654 |
| MS within | 150.509 | 12.580 | 32.091 | 26.832 | 10.510 | 8.471 | 0.059 | 0.521 | 43.910 | 0.202 |
| H2 | 0.641 | 0.581 | 0.530 | 0.553 | 0.305 | 0.349 | 0.621 | 0.424 | 0.508 | 0.528 |
Discussion
This is the first analysis of intestinal polyp’s development and related traits in 27 F1 CC-C57BL/6 lines carrying the ApcMin/+ mutation. All the tested phenotypes showed wide variability, suggesting that the diverse genetic background of the CC lines is contributes to this variation. Analysis of heritability confirmed this statement by showing that the majority of phenotypic variation is due to genetics.
Previous studies have shown that ApcMin/+ mice lose body weight during disease progression [59], which mimics the loss of weight observed in cancer patients, especially of gastrointestinal cancers [60]. In our study, in eight randomly selected lines, we found that body weight at the start of experiment showed significant variation between lines, but there were no significant differences between knockout and control mice within each line. This suggests that initially the presence of ApcMin/+ mutation has no influence on body weight. However, throughout the experiment we identified 3 different patterns of body weight performances in ApcMin/+: loss of body weight, plateau and continuous accumulation of body weight. Resistant lines accumulate their body weight continuously and do not develop multiple polyps, while susceptible lines lose body weight, presumably because of the polyp burden. These results emphasize the importance of genetic background in the response to ApcMin/+ mutation.
Previous studies have shown that genetically diverse mouse strains, carrying ApcMin/+ mutation, vary in intestinal tumor development [29]. Our results support this finding. Additionally, we found that polyp counts in F1 CC-C57BL/6-Min lines are distributed below and above the parental (C57BL/6-Min) mean value. We conclude that the CC population contains suppressors and enhancers genetic elements of the ApcMin/+ mutation.
The different four parts (SB1, SB2, SB3 and colon) of the gastrointestinal tract have specific physiological function, due to different gene expression profiles, pH and microbiota [61]. In a previous study with ApcMin/+ mice it was reported that the density of polyps varies across the gastrointestinal tract [23]. Our results confirm this observation and suggest that tumorgenesis in different parts of intestine is controlled by different genes. Additionally current results suggest that high levels of polyp proliferation in ApcMin/+ mice affect the length and weight of the intestine. The much polyps mouse have the longer and heavier the gastrointestinal tract.
Our data also support the findings that ApcMin/+ mice suffer from anemia [56] as we observed a negative correlation between the number of polyps and %PCV. Susceptible lines with high total polyp counts had low %PCV values, while resistant lines for polyp development displayed normal %PCV values.
ApcMin/+ mice also develop splenomegaly [59]. Possible causes include viral or bacterial infections, cirrhosis, venous pressure in spleen or liver, metastasis of solid tumors or hemolytic anemia [60]. Our results confirm that ApcMin/+ mice develop splenomegaly concurrently with polyp development. Additionally strong negative correlation was found between the size of spleen and %PCV values. Therefore, we suggest that the main cause for splenomegaly is the hemolytic anemia. Several studies have already showed that erythrocytes trapped in the spleen cause the enlargement of this organ and increase anemia [59].
Finally we conclude that modifiers for ApcMin/+ which affect intestinal tumor development segregate in the CC population, and that these loci are likely to be mappable by future QTL analysis. Unfortunately, the number assessed CC lines in this study (27 lines) were not sufficient and to provide enough statistical power for mapping QTL, and it is believed with more lines (additional 20 or so lines) to be assessed in the future, strong and significant QTL and suggested candidate genes will be achieved.
Conclusions
Variation in polyp development is controlled, an appreciable extent, by genetic factors segregating in the Collaborative cross population and suggests that it is suited for identifying modifier genes associated with ApcMin/+ mutation, after assessing sufficient number of lines for quantitative trait loci analysis. The expected findings may be used for early prediction of potential intestine cancer development in host carrying susceptible genetic factors, thus can be applied for better control and sufficient apply therapy tools and approaches.
Availability of Data
All raw collected data which used in this manuscript will be, freely available from http://phenome.jax.org/.
Acknowledgements
This work was supported by the Wellcome Trust grants 090532/Z/09/Z, 085906/Z/08/Z, 083573/Z/07/Z, and 075491/Z/04, Cancer Research Counsel-UK, and the Cancer Biology Research Center of Tel Aviv University. We thank Tel-Aviv University for their core funding and technical support.
Additional file
Table 1S: Summary of all generated progeny, divided by gender & Min carriers. Figure 1S. Intestinal length (cm) (±SE). The X-axis represents CC lines, C57BL/6 strain carrying the Apc Min/+ mutation (first black column) and CC-Mean (black column ~ in the middle), while the Y-axis centimeter. One-way ANOVA performed for statistical analysis, p < 0.05. Figure 2S. Number of polyps per cm (±SE). The X-axis represents CC lines, C57BL/6 strain carrying the Apc Min/+ mutation (first black column) and CC-Mean (black column ~ in the middle), while the Y-axis is number of polyps. One-way ANOVA performed for statistical analysis, p < 0.05. Figure 3S. Frequency of lines as for the number of polyps per cm. The X-axis represents number of polyps per cm, while the Y-axis is the frequency. Number inside each column is the number of CC lines within each range of polyps. Figure 4S. Intestine %Weight (±SE). The X-axis represents CC lines, C57BL/6 strain carrying the Apc Min/+ mutation (first black column) and CC-Mean (black column ~ in the middle), while the Y-axis %Weight. One-way ANOVA performed for statistical analysis, p < 0.05. Figure 5S. %PCV (±SE). The X-axis represents CC lines, C57BL/6 strain carrying the Apc Min/+ mutation (first black column) and CC-Mean (black column ~ in the middle), while the Y-axis %. One-way ANOVA performed for statistical analysis, p < 0.05. Figure 6S. Spleen %Weight (±SE). The X-axis represents CC lines, C57BL/6 strain carrying the Apc Min/+ mutation (first black column) and CC-Mean (black column ~ in the middle), while the Y-axis %Weight. One-way ANOVA performed for statistical analysis, p < 0.05. Figure 7S. Average number of polyps (±SE) in 22 F1 CC-C57BL/6-Min lines (n = 1-16 mice/line) based on male and female. The X-axis represents male and female of each studied CC line, while the Y-axis number of polyps. (PDF 998 kb)
Footnotes
Competing of interests
The authors declare no potential conflict of interest with respect to financial or Non-financial competing interests, the authorship and/or publication of this article.
Authors’ contributions
AD and DB performed the experiments on the mice and collected the data and helped to draft the manuscript. IT, RM and FAI conceived of and designed the study, helped with collection, analysis and interpretation of the data and drafted the manuscript. All authors approved the final version for publication.
Contributor Information
Alexandra Dorman, Email: alexandra.dorm@gmail.com.
Daria Baer, Email: dariabaer@gmail.com.
Ian Tomlinson, Email: iant@well.ox.ac.uk.
Richard Mott, Email: rmott@well.ox.ac.uk.
Fuad A. Iraqi, Email: fuadi@post.tau.ac.il
References
- 1.Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 2010;42:973–979. doi: 10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy MI, Abecasis RG, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, et al. Genome-wide association studies complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 4.Zanke BW, Greenwood CMT, Rangrej J, Kustra R, Tenesa A, Farrington SM, et al. Genome-wide scan indentifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007;39:989–994. doi: 10.1038/ng2089. [DOI] [PubMed] [Google Scholar]
- 5.Barrett J, Jiwa M, Rose P, Hamilton W. Pathways to the diagnosis of colorectal cancer: an observational study in three UK cities. Fam Pract. 2005;14:9–15. doi: 10.1093/fampra/cmi093. [DOI] [PubMed] [Google Scholar]
- 6.Auwera I, Laere SJ, Bosch SM, Eynden GG, Trinh BX, Dam PA, et al. Aberrant methylation of the Adenomatous Polyposis Coli (APC) gene promoter is associated with the inflammatory breast cancer phenotype. Br J Cancer. 2008;99:1735–1742. doi: 10.1038/sj.bjc.6604705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, et al. Identification of deletion mutations and three new genes at the familial polyposis. Cell. 1991;66:601–613. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- 8.Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- 9.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 10.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 11.Fostira F, Thodi G, Sandaltzopoulos R, Fountzilas G, Yannoukakos D. Mutational spectrum of APC and genotype-phenotype correlations in Greek FAP patients. BMC Cancer. 2010;10:389–396. doi: 10.1186/1471-2407-10-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubinfeld B, Souza B, Albert I. Association of the APC gene product with betacatenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- 13.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK-3β to the APC–β-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 14.Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 15.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 16.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhurinsky J, Shtutman M, Ben-Ze’ev A. Plakoglobin and b-catenin: protein interactions, regulation and biological roles. J Cell Sci. 2000;113:3127–3139. doi: 10.1242/jcs.113.18.3127. [DOI] [PubMed] [Google Scholar]
- 18.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- 19.Silverman KA, Koratkar RA, Siracusa LD, Buchberg AM. Exclusion of Madh2, Madh4, and Madh7 as candidates for the modifier of Min 2 (Mom2) locus. Mamm Genome. 2003;14:119–129. doi: 10.1007/s00335-002-2214-3. [DOI] [PubMed] [Google Scholar]
- 20.Hirohashi S, Kanai Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci. 2003;94:575–581. doi: 10.1111/j.1349-7006.2003.tb01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moser AR, Dove WF, Roth KA, Gordon JIL. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. Journal Cell Biology. 1992;716:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moser AR, Mattes EM, Dove WF, Lindstrom MJ, Haag JD, Gould MN. Min, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc Natl Acad Sci U S A. 1993;90:8977–8981. doi: 10.1073/pnas.90.19.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cormier RT, Dove WF. Dnmt1N/+ reduces the net growth rate and multiplicity of intestinal adenomas in C57BL/ 6-Multiple intestinal neoplasia (Min)/+ mice independently of p53 but demonstrates strong synergy with the Modifier of Min 1AKR resistance allele. Cancer Res. 2000;60:3965–3970. [PubMed] [Google Scholar]
- 24.Cormier RT, Bilger AJ, Lillich RB, Halberg KH. The Mom1AKR intestinal tumor resistance region consists of Pla2g2a and a locus distal to D4Mit64. Oncogene. 2000;19:3182–3192. doi: 10.1038/sj.onc.1203646. [DOI] [PubMed] [Google Scholar]
- 25.Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 26.Cormier RT, Hong KH, Halberg RB, Hawkins TL, Richardson P. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet. 1997;17:88–91. doi: 10.1038/ng0997-88. [DOI] [PubMed] [Google Scholar]
- 27.Silverman KA, Koratkar RA, Siracusa LD, Buchberg AM. Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Res. 2002;12:88–97. doi: 10.1101/gr.206002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suraweera N, Haines J, McCart A, Rogers P, Latchford A, Coster M, et al. Genetic determinants modulate susceptibility to pregnancy-associated tumourigenesis in a recombinant line of Min mice. Hum Mol Genet. 2006;15:3429–3435. doi: 10.1093/hmg/ddl419. [DOI] [PubMed] [Google Scholar]
- 29.Nnadi SC, Watson R, Innocent J, Gonye GE, Buchberg AM, Siracusa LD. Identification of five novel modifier loci of Apc (Min) harbored in the BXH14 recombinant inbred strain. Carcinogenesis. 2012;33(8):1589–1597. doi: 10.1093/carcin/bgs185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang EH, Park JC, Appelman H, Weinberg AD, Banerjee M, Logsdom CD, et al. Induction of inflammatory bowel disease accelerates adenoma formation in Min+/- mice. Surgery. 2005;139:782–788. doi: 10.1016/j.surg.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Chesler EJ, Miller DM, Branstetter LR, Galloway LD, Jackson BL. The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm Genome. 2008;19:382–389. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iraqi FA. Fine mapping of quantitative trait loci using advanced intercross lines of mice and positional cloning of the corresponding genes. Journal of Experimental Lung Research. 2000;26:641–649. doi: 10.1080/01902140150216729. [DOI] [PubMed] [Google Scholar]
- 33.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 34.Iraqi FA, Churchill G, Mott R. The Collaborative Cross, developing a resource for mammalian systems genetics: A status report of the Wellcome Trust cohort. Mamm Genome. 2008;19:379–381. doi: 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 35.Aylor DL, Valdar W, Foulds-Mathes W, Buus RJ, Verdugo RA, Baric RS, et al. Genetic analysis of complex traits in the emerging collaborative cross. Genome Res. 2011;21:1213–1222. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collaborative Cross Consortium The Genome Architecture of the Collaborative Cross Mouse Genetic Reference Population. Genetics. 2012;190(2):389–402. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durrant C, Tayem H, Yalcin B, Cleak J, Goodstadt L, de Villena PMF, et al. Collaborative Cross mice and their power to map host susceptibility to Aspergillus fumigates infection. Genome Res. 2011;21:1239–1248. doi: 10.1101/gr.118786.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shusterman A, Salyma Y, Nashef A, Soller M, Wilensky A, Mott R, et al. Genotype is an important determinant factor of host susceptibility to periodontitis in the Collaborative Cross and inbred mouse populations. BMC Genet. 2013;14:68. doi: 10.1186/1471-2156-14-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iraqi FA, Athamni H, Dorman A, Salymah Y, Tomlinson I, Nashif A, et al. Heritability and coefficient of genetic variation analyses of phenotypic traits provide strong basis for high-resolution QTL mapping in the CollaborativeCross mouse genetic reference population. Mamm Genome. 2014;25:109–119. doi: 10.1007/s00335-014-9503-5. [DOI] [PubMed] [Google Scholar]
- 40.Vered K, Durrant C, Mott R, Iraqi FA. Susceptibility to klebsiella pneumonaie infection in collaborative cross mice is a complex trait controlled by at least three loci acting at different time points. BMC Genomics. 2014;15:865. doi: 10.1186/1471-2164-15-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts A, Pardo-Manuel De Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome. 2007;18(6–7):473–481. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MFW, et al. Genealogies of mouse inbred strains. Nat Genet. 2000;24:23–25. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- 43.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477(7364):289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang M, Lemon WJ, Liu G, Wang Y, Iraqi FA, Malkinson AM, et al. Fine mapping and identification of candidate pulmonary adenoma susceptibility 1 genes using advanced intercross lines. Cancer Research Journal. 2003;63:3317–3324. [PubMed] [Google Scholar]
- 45.Broman KW. The genomes of recombinant inbred lines. Genetics. 2005;169(2):1133–1146. doi: 10.1534/genetics.104.035212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yalcin B, Flint J, Mott R. Using Progenitor Strain Information to Identify Quantitative Trait Nucleotides in Outbreed Mice. Genetics. 2005;171:673–681. doi: 10.1534/genetics.104.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mathes WF, Aylor DL, Miller DR, Churchill GA, Chesler EJ, de Villena FP, et al. Architecture of energy balance traits in emerging lines of the Collaborative Cross. Am J Physiol. 2010;300:1124–1134. doi: 10.1152/ajpendo.00707.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Welsh CE, Miller RD, Manly KF, Wang J, McMillan L, Morahan G, et al. Status and access to the Collaborative Cross population. Mamm Genome. 2012;23:706–712. doi: 10.1007/s00335-012-9410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29(1):52–54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- 50.Ju J, Hong J, Zhou JN, Pan Z, Bose M, Liao J, et al. Inhibition of intestinal tumorigenesis in Apcmin/+ mice by (-)-epigallocatechin-3-gallate, the major catechin in green tea. Cancer Res. 2005;65:10623–10631. doi: 10.1158/0008-5472.CAN-05-1949. [DOI] [PubMed] [Google Scholar]
- 51.Rudling R, Hassan AB, Kitau J, Mandir N, Goodlad RA. A simple device to rapidly prepare whole mounts of murine intestine. Cell Prolif. 2006;39:415–420. doi: 10.1111/j.1365-2184.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janakiram NB, Mohammed A, Qian L, Choi CI, Steele VE, Rao CV. Chemopreventive Effects of RXR-Selective Rexinoid Bexarotene on Intestinal Neoplasia of ApcMin/+ Mice. Neoplasia. 2012;14:159–168. doi: 10.1593/neo.111440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walker HK, Hall WD, Hurst JW. Clinical Methods: The History, Physical, and Laboratory Examinations 1990, 3rd edition. [PubMed]
- 54.Mehl KA, Davis JM, Clements JM, Berger FG, Pena MM, Carson JA. Decreased intestinal polyp multiplicity is related to exercise mode and gender in ApcMin/+ mice. J Appl Physiol. 2005;98(6):2219–2225. doi: 10.1152/japplphysiol.00975.2004. [DOI] [PubMed] [Google Scholar]
- 55.Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA. Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol. 2008;294:393–401. doi: 10.1152/ajpregu.00716.2007. [DOI] [PubMed] [Google Scholar]
- 56.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 57.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, et al. Mouse Model of Colonic Adenoma-Carcinoma Progression Based on Somatic Apc Inactivation. Cancer Res. 2007;67:9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- 58.Qadri SM, Mahmud H, Lang E, Gu S, Bobbala D, Zelenak C, et al. Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J Cell Mol Med. 2012;16(5):1085–1093. doi: 10.1111/j.1582-4934.2011.01387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mori K, Fujimoto-Ouchi K, Onuma E, Noguchi M, Shimonaka Y, Yasuno H, et al. Novel models of cancer-related anemia in mice inoculated with IL-6-producing tumor cells. Biomed Res. 2000;30(1):47–51. doi: 10.2220/biomedres.30.47. [DOI] [PubMed] [Google Scholar]
- 60.Lane SW, Sykes SM, Al-Shahrour F, Shterental S, Paktinat M, Lo Celso C, et al. The ApcMin mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood. 2010;115(17):3489–3497. doi: 10.1182/blood-2009-11-251728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. ApcMin: A mouse model for intestinal and mammary tumorigenesis. Eur J Cancer. 1995;31:1061–1064. doi: 10.1016/0959-8049(95)00181-H. [DOI] [PubMed] [Google Scholar]