

Abstract
Molecular biology, genomics and proteomics methods have been utilized to reveal a non-annotated class of endogenous polypeptides (small proteins and peptides) encoded by short open reading frames (sORFs), or small open reading frames (smORFs). We refer to these polypeptides as s(m)ORF-encoded polypeptides or SEPs. The early SEPs were identified via genetic screens, and many of the RNAs that contain s(m)ORFs were originally considered to be non-coding; however, elegant work in bacteria and flies demonstrated that these s(m)ORFs code for functional polypeptides as small as 11-amino acids in length. The discovery of these initial SEPs led to searches for these molecules using methods such as ribosome profiling and proteomics, which have revealed the existence of many SEPs, including novel human SEPs. Unlike screens, –omics methods do not necessarily link a SEP to a cellular or biological function, but functional genomic and proteomic strategies have demonstrated that at least some of these newly discovered SEPs have biochemical and cellular functions. Here, we provide an overview of these results and discuss the future directions in this emerging field.
Introduction
Studies over the past few decades have revealed several unprecedented classes of biologically active molecules in the genome with regulatory roles in diverse physiological processes (Ambros, 2004, Vander Heiden, 2011, Fatica and Bozzoni, 2014, Yore et al., 2014). In particular, recent work has revealed a novel class of bioactive peptides in a variety of organisms that are derived from short open reading frames (sORFs) or small open reading frames (smORFs) (Hashimoto et al., 2008, Slavoff et al., 2013, Ma et al., 2014) (Figure 1). Unlike classical peptide hormones and neuropeptides, which are translated as larger precursor proteins followed by limited proteolytic processing (Holst, 2007, Steiner, 1998), these s(m)ORF-encoded polypeptides (SEPs) are short peptides encoded directly from s(m)ORFs (Galindo et al., 2007, Kondo et al., 2010, Magny et al., 2013, Pauli et al., 2014). A small number of well-studied SEPs have indicated that these polypeptides may act as important regulators in many fundamental biological processes, such as metabolism (Dong et al., 2013), development (Kondo et al., 2010, Pauli et al., 2014), and cell death (Hashimoto et al., 2001), but little is known about the biological activities, regulation, or even total number of SEPs. Therefore, discovery and functional characterization of SEPs will expand our knowledge of the composition of the genome and proteome, and provide fundamental insights into the molecular biology of cells.
Figure 1. Schematic of proteins and s(m)ORF-Encoded Polypeptides (SEPs) expression.
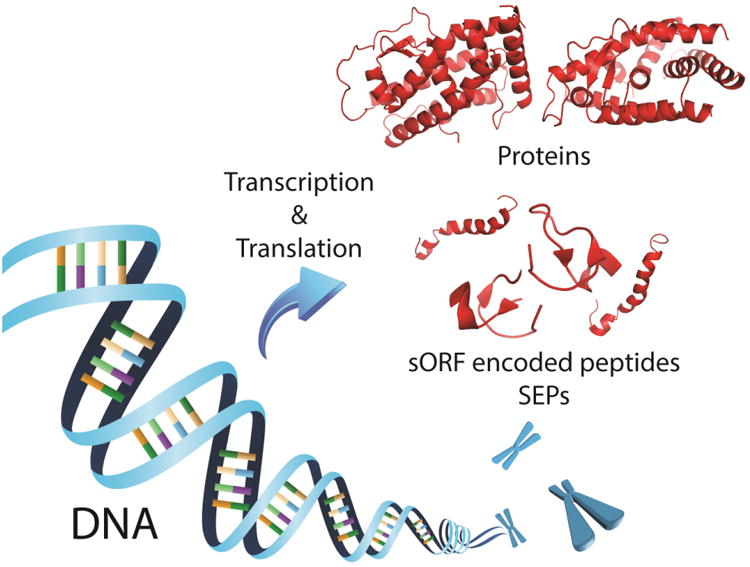
Although satisfactory classifications for these small peptides have not been clearly defined, we consider SEPs as generally less than 150 amino acids in length because we have found a number of non-annotated SEPs in this length range (Slavoff et al., 2013, Ma et al., 2014). The small size of SEPs hampers their discovery by both computational and experimental approaches (Dinger et al., 2008, Andrews and Rothnagel, 2014). On the one hand, it is challenging to apply bioinformatics methods to predict expression from s(m)ORFs by simply grafting widely-used gene prediction algorithms for large proteins. These programs usually assess coding potential of ORFs (i.e. protein-coding regions) by a number of stringent criteria, which recognize certain patterns in the transcripts, such as promoter sequences, polyadenylation signals, AUG-start codon usage, and sequence conservation (Zhang, 2002, Brent and Guigo, 2004, Lin et al., 2011). However, these features are not as rich in s(m)ORFs as in long protein coding genes, resulting in a high false positive rate to distinguish between coding and non-coding s(m)ORFs. On the other hand, non-annotated SEPs are not in standard proteomics databases and therefore cannot be discovered by direct detection. Even for SEPs that are in these databases, their small size and lower abundance make them more difficult to detect. In addition, many recently identified SEPs are derived from s(m)ORFs with non-AUG codons, which makes the identification process even more challenging (Ivanov et al., 2011, Menschaert et al., 2013). With improved strategies in computational approach, proteomics and next-generation sequencing, there have been great advances to address these challenges, and this has resulted in identification and validation of hundreds of new SEPs.
In this review, we describe various strategies to discover and identify SEPs, and overview several characteristics of SEPs based on recent proteomics results in the K562 human leukemia cell line. In addition, for SEPs that are discovered through these global identification strategies, we will discuss functional approaches to investigate the biology of these polypeptides.
Discovery of SEPs
As mentioned, SEPs have been found in several different organisms, including bacteria (Wadler and Vanderpool, 2007), plants (Rohrig et al., 2002, Yang et al., 2011, De Coninck et al., 2013), yeast (Kastenmayer et al., 2006), worms (Gleason et al., 2008), flies (Galindo et al., 2007, Magny et al., 2013) and humans (Hashimoto et al., 2001). Screening studies looking for key regulators of certain phenotypes resulted in serendipitous discovery of a few short bioactive peptides encoded directly by s(m)ORFs, which revealed the existence of this class of non-annotated genes.
The discovery of a SEP in E. coli, for example, began with research aimed at understanding the role of the sugar transport small RNA (SgrS), a non-coding RNA. The expression of SgrS is inversely correlated with glucose flux into the cell and SgrS was shown to operate through an RNA-dependent mechanism. The 3′ end of the SgrS RNA sequence is able to bind ptsG mRNA that encodes the major E. coli glucose transporter and inhibit translation of this protein, but the role of the 5′ end of the SgrS sequence, upstream of the nucleotides involved in base pairing with the ptsG mRNA, remained a mystery (Maki et al., 2010, Rice and Vanderpool, 2011). Recent work revealed that the 5′ part of SgrS encodes a SEP, a 43-amino acid peptide referred to as SgrT, which inhibits glucose influx by directly binding and inhibiting the glucose transporter and therefore plays a central role in cellular metabolism (Wadler and Vanderpool, 2007).
Work in flies has also revealed important SEPs. The discovery of the shortest known SEP, the 11-amino acid peptide encoded by the polished rice (pri) gene, also began with studies that looked for regulators responsible for developmental defects in Drosophila legs. As a result, the pri gene was identified from a transcript that had been previously classified as a putative non-coding RNA (Galindo et al., 2007, Kondo et al., 2007) (Figure 2). The Pri SEP has been demonstrated to trigger N-terminal truncation of a transcription factor, Shavenbaby (Svb). Upon cleavage, Svb converts from a repressor to an activator, and in turn contributes to epidermal differentiation in Drosophila (Kondo et al., 2010). Moreover, subsequent phylogenetic analysis revealed that pri belongs to an ancient gene family that can be traced back at least 440 million years. This nucleotide-level conservation indicates that the functional role of this SEP has probably been conserved through insect evolution as well (Galindo et al., 2007).
Figure 2. Discovery and characterization of Pri/Tal SEP.
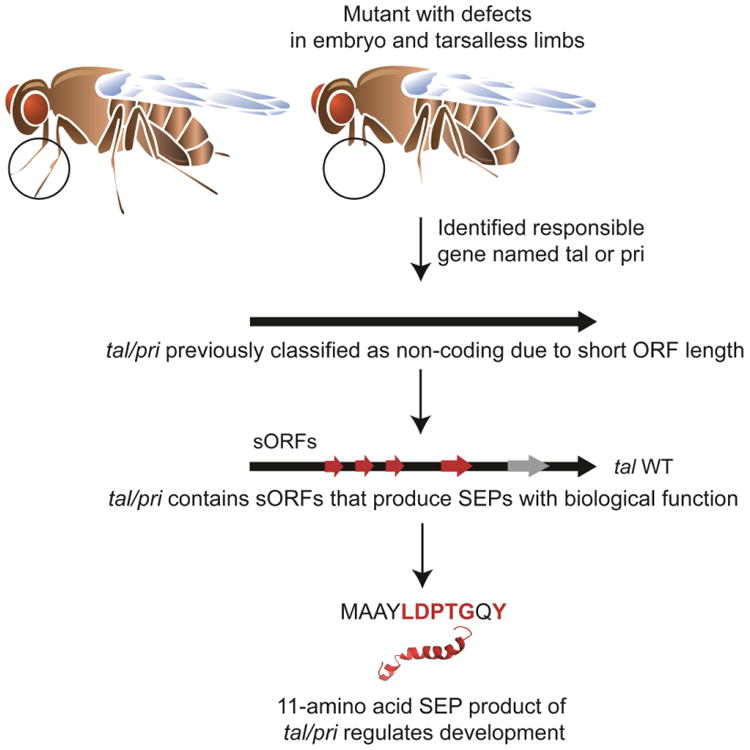
Polished rice (pri)/Tarsal-less (tal) transcript was initially classified as non-coding RNA. It contains several open reading frames (ORFs) smaller than 50 amino acids in length. An ORF coding for an 11 amino acid-long peptide has a highly conserved motif (amino acids labeled in red) and mediates the function of the gene. Deletion of this ORF leads to abnormal differentiation of Drosorphila legs. This is one example that demonstrates: 1) SEPs can arise from a polycistronic messenger or a putative non-coding RNA; 2) SEPs can have crucial biological function such as epidermal differentiation in this case.
Functional SEPs are also present in humans. Humanin was discovered during a screen for cDNAs from the nervous system that prevented cell death by the amyloid precursor protein (APP) in an effort to discover new genes that could prevent Alzheimer's disease. In this work, a plasmid containing a cDNA library was introduced into mammalian cells and these cells were then induced to produce APP, which leads to the death of most cells. Plasmids from surviving cells were then isolated, amplified, and the screen repeated an additional four times. One cDNA was particularly effective at preventing apoptosis in this screen, and further analysis revealed that the functional element of this gene was a 75-bp open reading frame (ORF) encoding a 24-amino acid peptide, which was named humanin. The RNA that encodes humanin is the mitochondrial 16s ribosomal RNA, which was thought to be a non-coding RNA (Hashimoto et al., 2001). Subsequent work reported that humanin prevents cell death via a protein-protein interaction (PPI) with Bcl-2–associated X protein (Bax) that prevents Bax activation (Guo et al., 2003, Zhai et al., 2005).
The serendipitous discovery of these SEPs suggests that translation of s(m)ORFs and production of small bioactive peptides in the proteome are much more pervasive than previously appreciated. s(m)ORFs are a common feature in the genome, located mostly within sequences of non-coding RNAs and 5′-UTR of protein coding genes (Calvo et al., 2009, Ladoukakis et al., 2011). The coding potential of s(m)ORFs has previously been neglected mainly due to the difficulty in distinguishing them from non-coding genes by prevalent bioinformatics methods, as well as the challenge to detect their translation products from a bulk of peptide sequences though current proteomics approaches (Falth et al., 2006, Crappe et al., 2013). With the purpose of looking for new SEPs that are biologically relevant, extensive efforts have been performed to re-evaluate the coding possibility of s(m)ORFs by using improved computational and experimental strategies. For example, Pauli and colleagues revisited the zebrafish transcripts by integrating ribosome profiling data, and identified 700 novel protein-coding transcripts from non-annotated translated ORFs, of which 81% were conserved in other vertebrates. Among these new ORFs, 28 secreted peptides were further isolated which contained putative signal sequences but lacked the predicted transmembrane domains. Follow-up studies with one of these s(m)ORFs, referred to as toddler, indicated that it is able to produce a 58-amino acid polypeptide. The toddler SEP was demonstrated to activate G-protein-coupled signaling by binding to the APJ/Apelin receptor and consequently promote cell movement during zebrafish gastrulation (Pauli et al., 2014).
The second example involves the discovery of two functional SEPs of 28 and 29 amino acids that are encoded by putative non-coding RNA pncr003:2L. Inspired from the evidence that the pri s(m)ORF was initially misannotated as a non-coding RNA, a pool of all polyadenylated, polysome-associated putative non-coding RNA in Drosophila was re-examined using an improved bioinformatics approach, which resulted in two s(m)ORFs, pncr003:2L, driven by strong Kozak sequences. Subsequent studies indicated that pncr003:2L peptides are expressed in somatic and cardiac muscles, where they regulate muscle contraction by modulating Ca2+ trafficking. More importantly, these two SEPs were conserved across evolution as they showed structural and functional homology with s(m)ORFs sarcolipin (sln) and phospholamban (pln) in vertebrates, where they play a role in regulating Ca2+ transport into ER following muscle contraction (Magny et al., 2013).
These examples along with recent computational analyses have suggested that many s(m)ORFs are translated, however, their coding potential in general has not been interrogated systematically and experimental evidence that these s(m)ORFs are able to generate stable polypeptides is still lacking (Vanderperre et al., 2013). One reasonable strategy to address this issue is to take advantage of ribosome profiling analyses (Bazzini et al., 2014, Smith et al., 2014) (Figure 3). Ribosome profiling is an emerging sequencing technique that can provide a global snapshot of all mRNAs being actively translated (Ingolia, 2014, Ingolia et al., 2009). Whereas RNA-seq sequences all of the transcripts present in a sample; ribosome profiling sequences ribosome-protected mRNA fragments that map back to the genome and de novo assemble into transcripts. The combination of these techniques enables the identification of translation start sites and alternative splicing forms to provide a more comprehensive coverage of the trascriptome. In addition, the amount of normalized mRNA reads in sequencing results is proportional to ribosome distribution and density on corresponding transcripts, which could provide quantitative information about translation as well (Ingolia et al., 2012). Therefore, ribosome profiling has become increasingly popular to assess coding potential of ORFs in the genome, especially those previously unannotated or considered as non-coding RNAs (Smith et al., 2014). As a result, plenty of new transcripts have been identified which consist of either novel exons, alternative initiation and splicing sites of annotated genes or entire new ORFs that had been classified as non-coding regions in the transcriptome, including 5′UTRs and long non-coding RNAs (lncRNAs) (Chew et al., 2013, Ingolia et al., 2014). Among them, quite a few are s(m)ORFs that encode for SEPs.
Figure 3. SEP discovery workflow applying both the ribosome profiling technique and mass spectrometry.
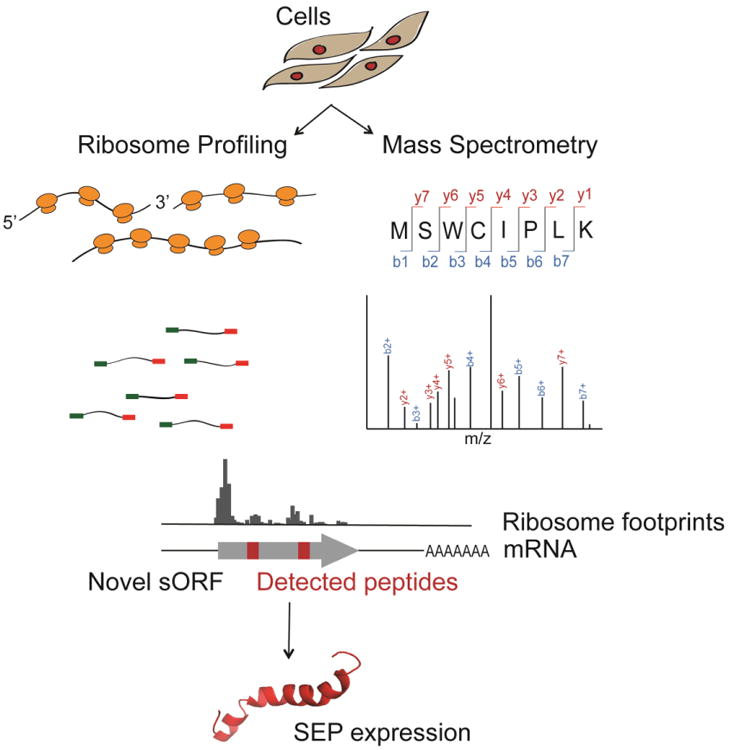
mRNA transcripts are isolated from cells and followed by a ribosome profiling experiment. The ribosome footprints are mapped to the genome and de novo assembled into transcripts, leading to the discovery of novel s(m)ORFs with coding potential. Simultaneously, the small proteome is extracted from the cell lysate and analyzed by mass spectrometry. Detected peptides are mapped to the transcriptome from which the peptides are translated. Ribosome profiling and mass spectrometry analysis together lead to the discovery of novel genes coding for SEPs.
Ribosome profiling has revealed that there could be pervasive translation of s(m)ORFs outside annotated protein coding genes (Ingolia et al., 2014). First, scanning 40S ribosomal subunits and other non-specific protein binders, as well as non-productive binding to single ribosomes could contribute to footprints but not translation (Wilson and Masel, 2011). Second, different interpretations of ribosome profiling data, especially for non-annotated transcripts, could result in completely opposite conclusions (Guttman et al., 2013). Third, improved ribosome profiling approaches need to be developed specifically for s(m)ORF analyses, since s(m)ORFs that encode SEPs are much shorter and smaller in size, potentially making traditional ribosome profiling less suitable (Aspden et al., 2014). Therefore, demonstrating protein-coding potential of s(m)ORFs by detecting experimentally their stable protein products becomes extremely necessary and important.
Recent advances in mass spectrometry (MS) proteomics provide a powerful tool to discover SEPs from cell and tissue lysates (Ferguson et al., 2009). These MS experiments differ from ribosome profiling because they are able to detect polypeptides translated from s(m)ORFs and thereby validate the protein-coding potential of the s(m)ORF (Figure 3). For example, Oyama and colleagues developed a proteomics approach in an attempt to discover novel (non-annotated) coding sequences (CDS) in mammalian cells. The key step in their approach was the generation of their own protein database through the ‘6-way translation’ of annotated RNA sequences in the RefSeq database. By using the entire RNA sequences instead of just those regions thought to be coding, this protein database included any proteins found in 5′UTRs or 3′UTRs, as well as identified frame shifted variants of known genes. This approach identifies peptides and proteins that would be missed by traditional proteomics experiments that rely on annotated protein genes. As a result, a total of four SEPs have been discovered in K562 human leukemia and HEK293 cell lines with a length distribution of 88-148 amino acids (Oyama et al., 2007). To improve on these results, we utilized next-generation RNA sequencing (RNA-Seq) to identify all possible protein-coding mRNA transcripts, including non-annotated transcripts (i.e. transcripts that exist but are not in the NCBI RefSeq database). The RNA-Seq data was translated into all possible reading frames to create a database that is expected to contain all of the polypeptide sequences that could theoretically be produced in the cell. Using this database, we identified more than 300 additional human SEPs from several mammalian cell lines (K562, MCF10A, MDAMB231 cells) as well as human tissue samples, demonstrating the prevalence of this class of polypeptides. In addition, a few SEPs were detected in every sample we analyzed, which indicates that SEPs might be ubiquitous and serve fundamental (i.e. housekeeping) roles (Slavoff et al., 2013, Ma et al., 2014).
SEP Characteristics
So far, we have identified 285 novel SEPs from the K562 human leukemia cell line through the peptidomic approach discussed above. In order to obtain a better understanding of SEPs, we examined several global properties of the molecules we discovered, such as length distribution, start codon usage as well as locations in the genome, and found that they possess many unique characteristics.
Our proteomics analysis using trypsin-digested samples detects small pieces of peptide fragments from a bulk of peptide samples, however, it is not able to obtain full protein-level SEP sequence coverage and in particular the N- and C-terminus of SEPs are missing in most cases. Therefore, we have to assign start and stop codons for each SEP in the corresponding s(m)ORF to determine its length. For example, we assigned the first downstream in-frame stop codons in SEP-encoding s(m)ORFs as stop sites. Likewise, the closest upstream in-frame AUG was assumed to be the start codon. If no upstream AUG was present, the initiation codon was considered to be an in-frame near-cognate non-AUG codon embedded within a Kozak-consensus sequence (Kozak, 1986). The near-cognate codon usually has a single nucleotide difference from AUG (e.g. CUG), which has been shown as a translation-initiating site in ribosome profiling experiments. In a few cases, neither of these conditions was met and the codon immediately following an upstream stop codon was defined as the start site. By doing this, we intended to determine the maximum SEP length, while not biasing the analysis toward shorter SEP lengths. Using this approach, we determined the SEPs to range between 8 and 149 amino acids in length, with the majority (>90%) being smaller than 100 amino acids long. In particular, we found that the SEP length could be fitted into a Gaussian distribution with a population centroid between 26-50 amino acids (Slavoff et al., 2013, Ma et al., 2014).
Another interesting feature of SEPs involves the preponderance of non-canonical translation start sites. Two thirds of the detected SEPs (190/285) do not initiate at AUG codons, among which 56 SEPs start translation from a near-cognate non-AUG codon. These near-cognate codons differ from AUG by a single base and in most cases are embedded within a Kozak sequence, which increases translation initiation. Several lines of evidence indicate that SEPs can be translated using non-AUG start codons. First, ribosome profiling has revealed liberal use of non-AUG initiation (Van Damme et al., 2014). Second, transient expression of non-AUG containing SEPs in HEK293 cells produced full-length SEPs, which verifies that s(m)ORFs with non-AUG start codons are translated. Third, we validated the start codon for a single SEP as well as the requirement of a Kozak sequence by site-directed mutagenesis. In this experiment, we found that an ACG start codon was used instead of an AUG. Mutation of the ACG to an AUG resulted in increased translation, while mutation of the Kozak sequence inhibited translation, demonstrating the requirement for the Kozak sequence to be present for the initiation at ACG (Slavoff et al., 2013). Together these data provided strong evidence that SEPs can be produced from non-AUG, near cognate, initiation codons.
Next, we analyzed locations of these SEP-encoding s(m)ORFs in the genome and found that more than 70% (201/285) SEP producing RNA transcripts are not annotated in the RefSeq database, which highlights the importance of the custom protein database derived from RNA-Seq data in our approach and indicates that our strategy is able to provide superior coverage of small SEPs in the entire genome. Also the remaining 84 SEPs that are encoded from annotated RefSeq transcripts fall into five major categories: (i) those located in the 5′-UTR, (ii) those located in the 3′-UTR, (iii) those located in a different reading frame inside an annotated protein coding sequence (CDS), (iv) those located on non-coding RNAs (ncRNAs) and (v) those located on antisense transcripts (Figure 4). The locations of these s(m)ORFs mirror the distribution obtained from ribosome profiling indicating that our proteomics coverage achieves the necessary breadth and depth to reveal global properties of s(m)ORFs (Ingolia et al., 2011, Smith et al., 2014).
Figure 4. SEPs detected in K562 cells that are derived from RefSeq transcripts and their locations.
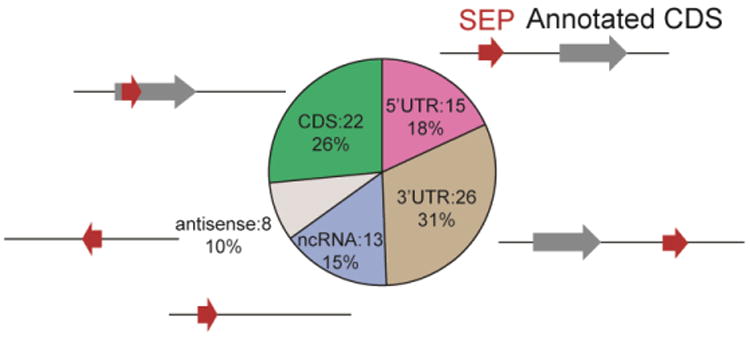
The s(m)ORFs are found on coding RNAs at the 5′UTR, 3′UTR and CDS and on non-coding RNAs. Gray arrows represent annotated protein coding ORFs, and red arrows represent s(m)ORF encoding SEPs and their relative locations to the annotated ORFs.
Furthermore, by carefully examining SEP producing transcripts, we noticed that several SEPs are translated from alternative splicing of annotated protein coding genes. For example, the DEDD2-SEP encoding s(m)ORF is a frameshifted sequence within the main CDS of the DEDD2 transcript, which normally couldn't be translated according to a traditional ribosome scanning mechanism. It turns out that the DEDD2 RNA has two splicing variants. One is for full-length DEDD2 protein expression and the other is a truncated mRNA sequence wherein the first start codon is that of the DEDD2-SEP s(m)ORF (Slavoff et al., 2013). Therefore, alternative splicing of annotated protein coding genes is one of the SEP producing mechanisms, which might be functionally relevant as well since higher organisms are able to create multiple functions from single genes for genetic efficiency.
Recent studies on aminoacyl tRNA synthetases (AARSs) indicated that alternative splicing events occur in all human AARSs, which retain only noncatalytic domains by splicing out catalytic domains at the mRNA level. Some of these catalytically inactive splice variants fall into the SEP regime in terms of their small size. Interestingly, each of the AARS variants demonstrated novel regulatory activities spanning a variety of physiological processes, including cell differentiation and proliferation, transcriptional regulation, and inflammatory response, which are distinct from the original aminoacylation function (Lo et al., 2014). In addition to the fact that alternative splicing is able to produce functional SEPs, SEPs can also be generated via polycistronic translation of a given RNA transcript. For example, the aforementioned pri (tal) mRNA is a polycistronic transcript with four individual s(m)ORFs, which produces three 11-aa peptides and one 32-aa peptide with a conserved LDPTGXY motif in all of them. The expression of four SEP isoforms has been verified in vitro and in vivo and all of them exert the same biological functions (Galindo et al., 2007) (Figure 2).
Mechanistic Investigation
There are some SEPs with demonstrated biological activities. The majority of bioactive SEPs were discovered as a consequence of phenotypic screens. These unbiased methods were able to identify SEPs that could affect a phenotype. A number of approaches have been taken to understand the molecular mechanisms underlying the biological functions of these SEPs. The functional characterization and mechanistic investigation of SEPs will provide valuable information about SEP biology. If the SEP is known to contribute to a certain phenotype, it may be connected to other known regulators that lead to the same phenotype. For example, the 11 amino acid-long Pri SEP has been shown to play a role in Drosophila embryogenesis, as embryos that lack pri gene display prominent defects in trichome formation, though the molecular mechanism was not clear at that time. It is known that a transcription factor Shavenbaby (Svb) is responsible for trichome formation as well as direct regulation of downstream gene expression. So it is likely that Pri SEP works together with Svb in Drosophila embryo development. Indeed, studies on a pri loss-of-function mutant indicated that pri is specifically required for the expression of Svb regulating genes, and subsequent work revealed that Pri SEP exerts its function by cleavage of the N-terminus of the Svb protein, which converts it from a transcription activator to a repressor (Kondo et al., 2010). Therefore, study of potential links between SEPs and other regulators that share the same phenotype is an efficient strategy to investigate SEP functions at the molecular level.
Another strategy is based on the hypothesis that SEPs exert their biological function through interacting with other proteins or biomolecules. Therefore, if the binding protein(s) could be identified, we could use this information to develop and test SEP functions according to the known roles of the interaction partners. For example, co-immunoprecipitation of MRI-2-SEP from HEK293 cells has yielded two proteins, Ku70 and Ku80, which are heterodimeric proteins involving in the non-homologous end joining (NHEJ) pathway of DNA double-strand break (DSB) repair. Therefore, we hypothesized that MRI-2-SEP may also play a role in NHEJ pathway through protein-protein interactions with the Ku70/Ku80 heterodimer. Subsequent studies confirmed this assumption and showed that MRI-2 accumulates in the nucleus upon Ku overexpression and induction of DSBs, and enhances the rate of NHEJ in vitro (Slavoff et al., 2014) (Figure 5). Last but not least, gene expression profiling is also a useful tool to analyze and elucidate SEP functions. This strategy provides global gene expression patterns for control cells and cells with overexpression and/or knockdown of a given SEP. Any gene or gene sets that exhibit different expression levels between the two conditions might be regulated by the SEP. In particular, if the same genes change in overexpression and knockdown studies, this would indicate that SEP regulation of the genes is specific and proteins encoded by these genes may have potential roles relevant to SEP activity.
Figure 5. SEPs subcellular localization and their involvement in protein-protein interaction.
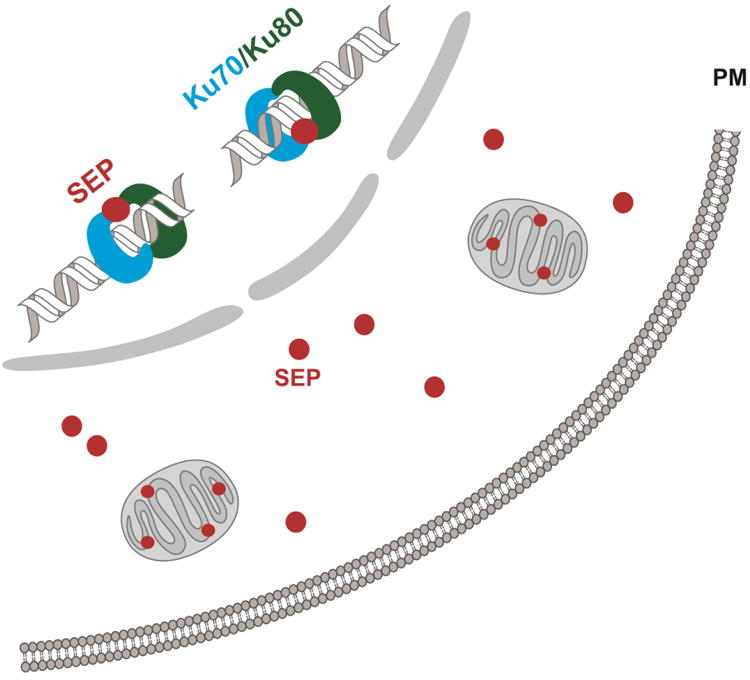
SEPs are expressed at various subcellular locations such as in the nucleus, mitochondria, as well as the cytosol, suggesting that SEPs can have molecular activities in the cell. In one example (Slavoff et al.), a SEP participates in protein-protein interaction with Ku70/Ku80 complex and plays a regulatory role in the DNA non-homologous end joining (NHEJ) repair process.
Future Directions
The discovery of biological active SEPs indicates that the human proteome is significantly more complex than previously appreciated. As an emerging field of research, we believe that we have only begun to explore the breadth and diversity of this exciting new family of polypeptides. Future efforts will move towards greater SEP detection and better understanding of their roles in a variety of biological processes. In particular, several questions need to be addressed.
How many SEPs in total are in the proteome? Current computational and experimental approaches have identified hundreds of SEPs, however, due to the limitation of each approach, it is entirely possible that there are many more as-yet-undiscovered polypeptides encoded from s(m)ORFs. Therefore, improved strategies are required to detect more SEPs. For example, can more accurate algorithms for s(m)ORF identification and coding potential prediction be developed? And can proteomics be improved for better peptide detection? In addition, most of the previous studies focused on intracellular SEPs. However, secreted SEPs may be equally important in terms of their regulatory roles as many bioactive peptides are secreted and act as signaling molecules to trigger a variety of biological activities (Saltiel and Kahn, 2001, van den Pol, 2012).
In addition to improve detection, quantitative methods for SEP levels also need to be optimized. Can different SEP levels among cells and tissues in different biological states be measured? One challenge with SEPs is that they are shorter and less abundant than average proteins so fewer peptides are detected by proteomics and their detection can be stochastic (detection in 25-50% of all runs). After improvement of detection sensitivity, the next step will be to start to quantify SEPs between biological samples (e.g. disease versus normal tissue) to determine which SEPs are important in different biological processes. In addition, quantitative analysis can be coupled to anatomical SEP profiling to help identify SEPs with tissue-specific functions (Margolis et al., 2005, Regard et al., 2008). In particular, the distinct expression of SEPs in disease cells/tissues may eventually impact medicine and human health by revealing novel pathways for pathogenesis.
Current studies have revealed a few unique features of SEPs, such as pervasive expression starting from non-AUG codons, but a more comprehensive knowledge of SEP functions— biochemical, cellular and physiological— will help reveal any general roles for these polypeptides. For example, the combination of ribosome profiling with proteomics can help define the exact boundaries and start codons for the s(m)ORFs that produce SEPs. If certain SEPs that are co-regulated, for example, share a non-AUG start codon that might suggest a more global method for their regulation. Moreover, it is unlikely that all SEPs are biologically active as some may represent translational noise. Conservation analysis is a useful tool to predict active SEPs but it may not be the only way (Figure 6). Data on the half-life and abundance of SEPs might reveal those SEPs that are long-lived and abundant, which might have the best chance to partake in cellular or physiological functions. Therefore, methods to measure the half-lives of SEPs will be of great value in studying their function. In addition, though SEPs are essentially small proteins, the understanding of their regulation is nowhere near as far along as other proteins. The Sgrs gene for example regulates its SEP in the presence of excess glucose by transcriptional regulation, but it is possible, even likely, that other SEPs are regulated at post-translational level. If so, how can these post-translational modifications be discovered and what are their roles in SEP biology?
Figure 6. SEP conservation.
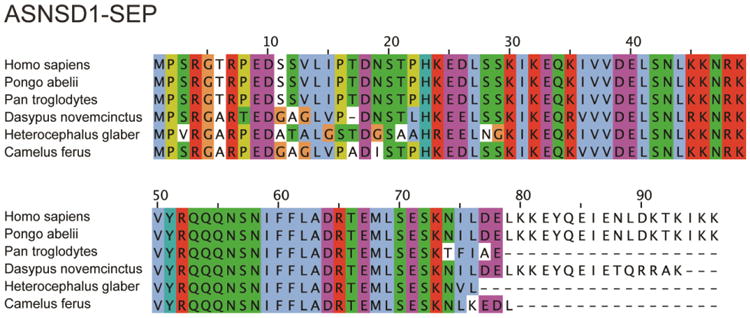
ASNSD1-SEP is a 96 amino acid-long SEP detected in K562, MCF10A and MDAMB231 cell lines. It is highly conserved across mammals, indicating its potential biological function.
Several studies revealed that SEPs play pivotal roles in a variety of cellular processes, and aberrant regulation of these bioactive polypeptides leads to developmental defects as well as pathogenesis. As more and more SEPs have been identified, functional and mechanistic exploration becomes increasingly necessary. Knowledge of their modes of action and roles in biology will make tremendous contributions to a new layer of regulation in our genome and proteome and could potentially offer novel therapeutic interventions to human diseases.
Acknowledgments
We thank Cynthia Donaldson and Joan Vaughan for insightful discussions and helpful comments on the manuscript.
Footnotes
Declaration of Interest: The authors would like to thank the NIH for support (R01 GM102491). The authors have no conflict of interest to declare.
References
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet. 2014;15:193–204. doi: 10.1038/nrg3520. [DOI] [PubMed] [Google Scholar]
- Aspden JL, Eyre-Walker YC, Phillips RJ, et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 2014;3:e03528. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Johnstone TG, Christiano R, et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. Embo J. 2014;33:981–993. doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent MR, Guigo R. Recent advances in gene structure prediction. Curr Opin Struct Biol. 2004;14:264–272. doi: 10.1016/j.sbi.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci USA. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew GL, Pauli A, Rinn JL, et al. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140:2828–2834. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crappe J, Van Criekinge W, Trooskens G, et al. Combining in silico prediction and ribosome profiling in a genome-wide search for novel putatively coding sORFs. BMC Genomics. 2013;14:648. doi: 10.1186/1471-2164-14-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coninck B, Carron D, Tavormina P, et al. Mining the genome of Arabidopsis thaliana as a basis for the identification of novel bioactive peptides involved in oxidative stress tolerance. J Exp Bot. 2013;64:5297–5307. doi: 10.1093/jxb/ert295. [DOI] [PubMed] [Google Scholar]
- Dinger ME, Pang KC, Mercer TR, et al. Differentiating Protein-Coding and Noncoding RNA: Challenges and Ambiguities. PLoS Comput Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Wang DX, Liu P, et al. Zm908p11, encoded by a short open reading frame (sORF) gene, functions in pollen tube growth as a profilin ligand in maize. J Exp Bot. 2013;64:2359–2372. doi: 10.1093/jxb/ert093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falth M, Skold K, Norrman M, et al. SwePep, a database designed for endogenous peptides and mass spectrometry. Mol Cell Proteomics. 2006;5:998–1005. doi: 10.1074/mcp.M500401-MCP200. [DOI] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- Ferguson JT, Wenger CD, Metcalf WW, et al. Top-down proteomics reveals novel protein forms expressed in Methanosarcina acetivorans. J Am Soc Mass Spectrom. 2009;20:1743–1750. doi: 10.1016/j.jasms.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo MI, Pueyo JI, Fouix S, et al. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5:e106. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason CA, Liu QL, Williamson VM. Silencing a candidate nematode effector gene corresponding to the tomato resistance gene Mi-1 leads to acquisition of virulence. Mol Plant Microbe Interact. 2008;21:576–585. doi: 10.1094/MPMI-21-5-0576. [DOI] [PubMed] [Google Scholar]
- Guo B, Zhai D, Cabezas E, et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- Guttman M, Russell P, Ingolia NT, et al. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–251. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Kondo T, Kageyama Y. Lilliputians get into the limelight: Novel class of small peptide genes in morphogenesis. Dev Growth Differ. 2008;50:S269–S276. doi: 10.1111/j.1440-169X.2008.00994.x. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Tajima H, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci USA. 2001;98:6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15:205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, et al. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Stern-Ginossar N, et al. Ribosome Profiling Reveals Pervasive Translation Outside of Annotated Protein-Coding Genes. Cell Rep. 2014;8:1365–1379. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, et al. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov IP, Firth AE, Michel AM, et al. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res. 2011;39:4220–4234. doi: 10.1093/nar/gkr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenmayer JP, Ni L, Chu A, et al. Functional genomics of genes with small open reading frames (sORFs) in S-cerevisiae. Genome Res. 2006;16:365–373. doi: 10.1101/gr.4355406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Hashimoto Y, Kato K, et al. Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol. 2007;9:660–665. doi: 10.1038/ncb1595. [DOI] [PubMed] [Google Scholar]
- Kondo T, Plaza S, Zanet J, et al. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–339. doi: 10.1126/science.1188158. [DOI] [PubMed] [Google Scholar]
- Kozak M. Point Mutations Define a Sequence Flanking the Aug Initiator Codon That Modulates Translation by Eukaryotic Ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Ladoukakis E, Pereira V, Magny EG, et al. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 2011;12:R118. doi: 10.1186/gb-2011-12-11-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Gardiner E, Xu Z, et al. Human tRNA synthetase catalytic nulls with diverse functions. Science. 2014;345:328–332. doi: 10.1126/science.1252943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Ward CC, Jungreis I, et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 2014;13:1757–1765. doi: 10.1021/pr401280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magny EG, Pueyo JI, Pearl FM, et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;341:1116–1120. doi: 10.1126/science.1238802. [DOI] [PubMed] [Google Scholar]
- Maki K, Morita T, Otaka H, et al. A minimal base-pairing region of a bacterial small RNA SgrS required for translational repression of ptsG mRNA. Mol Microbiol. 2010;76:782–792. doi: 10.1111/j.1365-2958.2010.07141.x. [DOI] [PubMed] [Google Scholar]
- Margolis RN, Evans RM, O'Malley BW, et al. The Nuclear Receptor Signaling Atlas: development of a functional atlas of nuclear receptors. Mol Endocrinol. 2005;19:2433–2436. doi: 10.1210/me.2004-0461. [DOI] [PubMed] [Google Scholar]
- Menschaert G, Van Criekinge W, Notelaers T, et al. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol Cell Proteomics. 2013;12:1780–1790. doi: 10.1074/mcp.M113.027540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama M, Kozuka-Hata H, Suzuki Y, et al. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol Cell Proteomics. 2007;6:1000–1006. doi: 10.1074/mcp.M600297-MCP200. [DOI] [PubMed] [Google Scholar]
- Pauli A, Norris ML, Valen E, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343:1248636. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135:561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JB, Vanderpool CK. The small RNA SgrS controls sugar-phosphate accumulation by regulating multiple PTS genes. Nucleic Acids Res. 2011;39:3806–3819. doi: 10.1093/nar/gkq1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrig H, Schmidt J, Miklashevichs E, et al. Soybean ENOD40 encodes two peptides that bind to sucrose synthase. Proc Natl Acad Sci USA. 2002;99:1915–1920. doi: 10.1073/pnas.022664799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Slavoff SA, Heo J, Budnik BA, et al. A human short open reading frame (sORF)-encoded polypeptide that stimulates DNA end joining. J Biol Chem. 2014;289:10950–10957. doi: 10.1074/jbc.C113.533968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff SA, Mitchell AJ, Schwaid AG, et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, Alvarez-Dominguez JR, Kline N, et al. Translation of Small Open Reading Frames within Unannotated RNA Transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7:1858–1866. doi: 10.1016/j.celrep.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Gawron D, Van Criekinge W, et al. N-terminal Proteomics and Ribosome Profiling Provide a Comprehensive View of the Alternative Translation Initiation Landscape in Mice and Men. Mol Cell Proteomics. 2014;13:1245–1261. doi: 10.1074/mcp.M113.036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN. Neuropeptide transmission in brain circuits. Neuron. 2012;76:98–115. doi: 10.1016/j.neuron.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- Vanderperre B, Lucier JF, Bissonnette C, et al. Direct Detection of Alternative Open Reading Frames Translation Products in Human Significantly Expands the Proteome. PLoS One. 2013;8:e70698. doi: 10.1371/journal.pone.0070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadler CS, Vanderpool CK. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci USA. 2007;104:20454–20459. doi: 10.1073/pnas.0708102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Masel J. Putatively noncoding transcripts show extensive association with ribosomes. Genome Biol Evol. 2011;3:1245–1252. doi: 10.1093/gbe/evr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XH, Tschaplinski TJ, Hurst GB, et al. Discovery and annotation of small proteins using genomics, proteomics, and computational approaches. Genome Res. 2011;21:634–641. doi: 10.1101/gr.109280.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yore MM, Syed I, Moraes-Vieira PM, et al. Discovery of a Class of Endogenous Mammalian Lipids with Anti-Diabetic and Anti-inflammatory Effects. Cell. 2014;159:318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai D, Luciano F, Zhu X, et al. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- Zhang MQ. Computational prediction of eukaryotic protein-coding genes. Nat Rev Genet. 2002;3:698–709. doi: 10.1038/nrg890. [DOI] [PubMed] [Google Scholar]