

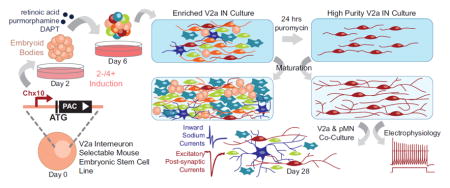
Abstract
Challenges in parsing specific contributions to spinal microcircuit architecture have limited our ability to model and manipulate those networks for improved functional regeneration after injury or disease. While spinal interneurons (INs) have been implicated in driving coordinated locomotor behaviors, they constitute only a small percentage of the spinal cord and are difficult to isolate from primary tissue. In this study, we employed a genetic strategy to obtain large quantities of highly enriched mouse embryonic stem cell (ESC)-derived V2a INs, an excitatory glutamatergic IN population that is defined by expression of the homeodomain protein Chx10 during development. Puromycin N-acetyltransferase expression was driven by the native gene regulatory elements of Chx10 in the transgenic ESC line, resulting in positive selection of V2a INs after induction and treatment with puromycin. Directly after selection, approximately 80% of cells are Chx10+, with 94% Lhx3+; after several weeks, cultures remain free of proliferative cell types and mature into normal glutamatergic neurons as assessed by molecular markers and electrophysiological methods. Functional synapses were observed between selected ESC-derived V2a INs and motor neurons when co-cultured, demonstrating the potential of these cells to form neural networks. While ESC-derived neurons obtained in vitro are not identical to those that develop in the spinal cord, the transgenic ESCs here provide a unique tool to begin studying V2a INs in isolation or for use in in vitro models of spinal microcircuits.
Keywords: puromycin selection, electrophysiology, transcription factor, neuronal differentiation, spinal cord injury
Graphical Abstract
Introduction
Neural networks in the form of central pattern generators (CPGs) are capable of generating rhythmic motor outputs that are essential to a range of sophisticated locomotor behaviors, but the microcircuit architecture involved has been much harder to characterize due to the diversity of cell types and lack of spatial organization in the spinal cord (Arber, 2012; Rybak et al., 2015). This is not only detrimental to our understanding of CPG circuitry but also hinders manipulations of those networks for improved functional outcomes after disease or trauma (Courtine et al., 2009; Harkema, 2008). Recent work using genetic ablation and ex vivo electrophysiological characterization of isolated spinal cord preparations has helped identify unique transcriptional markers to define the spinal interneuron (INs) populations that comprise these local spinal circuits (Arber, 2012; Azim et al., 2014; Crone et al., 2008; Gosgnach et al., 2006; Jessell, 2000; Kiehn, 2006; Lanuza et al., 2004; Zhang et al., 2008). However, the dependence on animal models precludes high-throughput pharmacological testing or in vitro modeling of spinal circuitry which may aid in the development of targeted therapeutics that promote neural regeneration and plasticity. Here we describe a method to generate large quantities of highly enriched INs from embryonic stem cells (ESCs), focusing on the acquisition of V2a INs.
V2a INs are defined by expression of the homeodomain protein Chx10 and are involved in CPG and propriospinal networks in the spinal cord and respiratory centers of the hindbrain (Al-Mosawie et al., 2007; Azim et al., 2014; Crone et al., 2008; Crone et al., 2012; Dougherty and Kiehn, 2010a, b; Lundfald et al., 2007; Peng et al., 2007). They are an ipsilaterally projecting glutamatergic premotor population with conserved locomotor functions in zebrafish and mice (Crone et al., 2008; Dougherty and Kiehn, 2010b; Kimura et al., 2006). V2a INs are distributed homogeneously along the rostrocaudal axis of the spinal cord in early mouse embryos but are localized to the ventral horn in the adult (Dougherty and Kiehn, 2010a; Francius et al., 2013). Genetic ablation studies have demonstrated their role in coordinating left-right alternation and skilled-reaching, as well as modulation of locomotor variability and rhythmic breathing (Azim et al., 2014; Crone et al., 2008; Crone et al., 2012; Crone et al., 2009; Dougherty and Kiehn, 2010a; Zhong et al., 2010; Zhong et al., 2011).
Robust “highly enriched” neuronal cultures are desirable because they can provide mechanistic insights otherwise confounded by mixed culture conditions. V2a INs, among others, are difficult to isolate from primary tissue in part because they make up a relatively small fraction of the total cells in the spinal cord (Crone et al., 2008). Self-renewing pluripotent cells, such as ESCs, are an attractive alternative to sorting primary tissue because they can be differentiated into a variety of cell types in large quantities for in vitro study or transplantation. By adapting established motor neuron (MN) differentiation protocols (Wichterle et al., 2002), we have previously shown that directed differentiation of ESCs into V2a INs is possible by exposing embryoid bodies (EBs) to retinoic acid (RA); a weak sonic hedgehog (Shh) agonist, purmorphamine; and a Notch-inhibitor, DAPT (Brown et al., 2014). However, despite our ability to derive V2a INs from ESCs, post-mitotic Chx10+ cells constitute only ~15% of the total cell population post-induction, which is further diluted as glial cells proliferate with time (Brown et al., 2014). Methods including fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting have been used to isolate single cell populations, but they are limited by the availability of antibodies to lineage-specific surface antigens, which have not been identified for many ventral IN populations, and require dissociation processes that can be harmful to mature neurons. While FACS can be used with transgenic reporter or lineage tracing cells, it can significantly compromise the viability of mature neurons and retains the potential for contamination.
Transgenic selection of desired ESC-derived populations has proven to be an effective method to generate isolated populations of a variety of cell types, including progenitor motor neurons (pMNs) and MNs (Anderson et al., 2007; Li et al., 1998; Marchetti et al., 2002; McCreedy et al., 2014a; McCreedy et al., 2012; Soria et al., 2000). Using lineage-specific promoters to drive antibiotic resistance, differentiation of the transgenic ESC line and subsequent antibiotic treatment results in highly enriched cultures that persist through maturation in vitro and in vivo after transplantation (McCreedy et al., 2014a; McCreedy et al., 2012; McCreedy et al., 2014b). In this study, we generated a selectable “Chx10-Puro” ESC line and investigated whether it could be used to obtain V2a INs that were comparable to endogenous V2a IN populations.
Methods
ESC Culture
Transgenic and RW4 mouse ESCs were cultured on gelatin-coated T-25 flasks in complete media, consisting of Dulbecco’s Modified Eagle Medium (DMEM; Life Technologies #11965-092, Carlsbad, CA) containing 10% newborn calf serum (Life Technologies #16010-159), 10% fetal bovine serum (Life Technologies #26140-079), and 1x Embryomax Nucleosides (Millipore #ES-008-D, San Francisco, CA). ESCs were passaged every two days at a 1:5 ratio in fresh complete media containing 1000 U/mL leukemia inhibitory factor (LIF; Millipore # ESG1106) and 100μM β-mercaptoethanol (BME; Life Technologies #21985-023).
Chx10-Puro Selection Vector
The targeting vector was constructed from a Gateway-compatible plasmid (pStart-K; Addgene #20346, Cambridge, MA) using a 750 bp Sal1-Asc1 fragment containing 5′ untranslated sequences and a 750 bp Asc1-Not1 fragment containing 3′ genomic sequences of Chx10 exon 1. A PAC/pGKneo dual resistance cassette was inserted between the two arms as previously described (McCreedy et al., 2012). The dual resistance cassette contains from 5′ to 3′: Asc1 site, Kozak sequence, puromycin cassette with bgh polyA signal (PKO-Select Puro; Agilent Genomics, Santa Clara, CA), floxed phosphoglycerate kinase I promoter driving the neomycin phosphotransferase gene (PGK-neo) with bgh polyA signal, and AscI site (Kozak, 1986; Thomas and Capecchi, 1987; Wu et al., 2008). Gateway recombination with the LR Clonase II Kit (Life Technologies #11791) was used to transfer aTTL-flanked regions into the pWS-TK3 vector, which contains the thymidine kinase gene for negative selection (Supplementary Figure 1A) (Wu et al., 2008).
Electroporation and Clonal Analysis
A CRISPR/Cas9 system was used to insert our resistance cassette into the Chx10 locus with high efficiency. The vector containing the Chx10 guide RNA (gChx10) was generated by the Genome Engineering Core at Washington University in St. Louis. The Chx10 gRNA sequences target the ATG start site of Chx10 exon 1 and were placed into a derivative of Addgene plasmid #43860; no common SNPs were found and off target profiles were excellent with at least 3 bp of mismatch between the target and any other site in the genome, which dramatically reduces the probability of an off target cut (Veres et al., 2014). The p3s-Cas9HC vector contains the Cas9 open reading frame (Addgene plasmid #43945) (Cho et al., 2013). 1x107 RW4 ESCs were resuspended in electroporation buffer (20 mM HEPES pH 7.5, 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, and 6 mM dextrose) with 8 μg Chx10-Puro selection vector, 1 μg Cas9 vector, and 1 μg gChx10 vector. Cells were electroporated at 0.23 kV and 960 μF in a 0.4 cm cuvette (Bio-Rad #165-2081, Hercules, CA) then plated on a 100 mm gelatin-coated petri dishes in complete media with LIF and BME for the cells to recover overnight. For the remainder of the expansion, cells were exposed to media containing LIF, BME, 150 nM fialuridine (FIAU, Moravek #M251, Brea, CA) and 40 μg/mL geneticin (G418, Life Technologies #10131) which was replaced every 2 days. After 10 days, single colonies were picked and plated into individual wells of a gelatin-coated 96-well plate. Clones were screened for correct insertion by junction PCR (Chx10Forward: GCCAACCGAGGAGAGCTAGAAGGTT; PACReverse: GCGCCAGGAGGCCTTCCATCTGTT-GCT) and expanded accordingly. The amplification region spanned from the endogenous 5′ UTR (outside of the Chx10-Puro selection vector homology arm) into the PAC cassette to confirm targeted insertion (Supplemental Figure 1B). Fifty-six transgenic clones were screened for puromycin resistance in Chx10+ interneurons following V2a induction (see V2a Induction and V2a Selection).
Transgenic clones with puromycin-resistant Chx10+ INs were analyzed for PAC copy number by quantitative real-time polymerase chain reaction (qRT-PCR) using a customized TaqMan Copy Number Assay; mouse Tert was used as the endogenous control and the assay was prepared according to the manufacturer’s instructions. Calculations were completed using CopyCaller Software (v2.0, Applied Biosystems). The calculated copy number of each clone was normalized to the calculated copy number for the Hb9-Puro ESC cell line, which was previously determined to contain a single PAC insertion (McCreedy et al., 2014a).
Cre Excision
The PGK promoter and neomycin phosphotransferase genes were removed from an individual Chx10-Puro clone using the pTurboCre plasmid (gift from Timothy Ley). The pTurboCre plasmid (10ug) and an mRFP-expressing plasmid (1ug) were prepared for transfection using Lipofectamine 3000 (Life Technologies #L3000-001) according to the manufacturer’s instructions; cells were transfected for 24 hours. ESC cultures were then dissociated and plated sparsely (3 x 104 cells) in a 100 mm gelatin-coated petri dish with complete media, LIF, and BME. Single colonies were picked after 10 days and plated in 96-well gelatin coated plates. When confluent, the ESCs were split and tested for sensitivity to neomycin by exposure to 40 μg/mL geneticin for 5 days; those not sensitive were discarded. A single transgenic clone was used as the Chx10-Puro ESC line used for the remaining studies.
V2a Induction
V2a INs were obtained from ESCs using a “2−/4+” induction protocol. EBs were formed by transferring 1 x 106 ESCs into 10 mL of DFK5 media on an agar-coated 100 mm petri dish for two days (2−). DFK5 is a DMEM/F12 base media containing 5% Knockout Serum Replacement (Life Technologies #10828-028), 50 μM Nonessential Amino Acids (Life Technologies #11140-050), 100 μM BME, 1:100 100x Insulin-Transferrin-Selenium (Life Technologies #41400-045), 100 μM BME, 1:200 100x EmbryoMax Nucleosides. EB media was then replaced with 10 mL DFK5 containing 10nM retinoic acid (RA; Sigma #R2625) and 1μM purmorphamine (EMD Millipore #540223) for another two days (2+). For the final 2 days (4+), the media was replaced with DFK5 containing 10nM RA, 1μM purmorphamine and 5μM N-{N-(3,5-difluorophenacetyl-L-alanyl))-(S)-phenylglycine-t-butyl-ester (DAPT; Sigma #D5942). In induced V2a control cultures, the Shh antagonist cyclopamine (Cyc, 1 mM; Sigma #C4116) was used instead of purmorphamine.
V2a Selection and Culture
To determine cell viability after selection, transgenic ESCs induced using the 2−/4+ protocols were dissociated with 0.25% Trypsin-EDTA (Life Technologies #25200-056) for 10 minutes with agitation and quenched with complete media containing 0.001% DNAse (Sigma #DN25). Dissociated cells were counted and centrifuged at 1200 rpm for 5 minutes then re-suspended in a selection media of DFK5NB containing B-27 supplement (Life Technologies #17504-044), glutaMAX (Life Technologies #35050-061), 5 ng/mL glial-derived neurotrophic factor (GDNF; Peprotech #450-10, Rocky Hill, NJ), 5 ng/mL brain derived neurotrophic factor (BDNF; Peprotech #450-02, Rocky Hill, NJ), 5 ng/mL neurotrophin-3 (NT-3; Peprotech #450-03), and 2 μg/mL or 4 μg/mL puromycin in water (Sigma #P8833). DFK5NB is a combination of DFK5 media and Neurobasal Media (Life Technologies #21103-049) mixed at a 1:1 ratio. For selected and unselected cultures respectively, 5x106 cells/cm2 and 5x105 cells/cm2 were plated on individual laminin coated 24-well plates or 35mm dishes. After 24 hours of selection, the media was replaced with DFK5NB containing B27, Glutamax, NT-3, GDNF, and BDNF. The media was replenished as needed every two days.
Post-Selection Cell Viability
To determine the cell viability after 24 hours of selection with puromycin, cells induced using the 2-/4+ protocol were rinsed twice with DFK4 media to remove debris then incubated for 30 minutes in fresh DFK5 media containing calcein-AM (Life Technologies # C481). Fluorescent images were captured using a MICROfire camera attached to an Olympus IX70 inverted microscope. Quantification was completed by flow cytometry (n=4 biological replicates, see Flow Cytometry).
qRT-PCR
qRT-PCR was performed and analyzed as previously described (Brown et al., 2014). Briefly, cDNA was synthesized using a High Capacity RNA-to cDNA Kit (Invitrogen). The cDNA was combined with TaqMan Gene Expression Assays (Applied Biosystems, Carlsbad, CA) and TaqMan Fast Advanced Master Mix (Applied Biosystems). qRT-PCR was performed using a Step One Plus Applied Biosystems thermocyler with the following protocol: 95°C for 20s; 40 cycles of 95°C for 1s and 60°C for 20s. There were n=3 technical replicates completed per run and n=4 biological replicates for each condition. For PAC expression, samples were treated with DNAse (Qiagen #7924, Valencia, CA) prior to analysis.
Immunocytochemistry
Immunocytochemistry staining was performed as previously described (McCreedy et al., 2014a). Antibodies used include mouse anti-Chx10 (1:1000, Santa Cruz # sc-374151, Santa Cruz, CA), mouse anti-Lhx3 (1:1000, Lim3, Developmental Studies Hybridoma Bank (DSHB) #67.4E12, Iowa City IA), mouse anti-Hb9 (1:20, DSHB #81.5C10), mouse anti-Lim1/2 (1:50, DSHB #4F2), mouse anti-Evx1 (1:50, DSHB #99.1-3A2), rabbit anti-beta-tubulin III (1:1000, βtubIII, Covance #811801, Princeton, NJ), rabbit anti-Ki67 (1:250, Abcam #ab15580), guinea pig anti-Vglut2 (1:2500, EMD Millipore #AB2251), mouse anti-MAP2 (1:250, EMD Millipore #AB5622), rabbit anti-mouse anti-SV2 (1:100, DSHB #SV2), and secondary Alexa Fluor conjugated goat antibodies (1:200, Life Technologies). Nuclei were counterstained with Hoescht (1:1000, Life Technologies #H3569). For cell counting experiments, live cells were identified by phase contrast and Hoechst; only cells with apparent processes were included. Cell counts were performed using ImageJ (NIH); there were n=4 biological replicates for each condition with >450 cells counted per replicate.
Flow Cytometry
A modified 2−/4+ protocol was used to obtain data on early expressing transcription factors in selected ESC-derived neuronal cultures. Briefly, EBs were dissociated at 2−/3+ and 5x106 cells were plated in individual wells of a laminin-coated 24-well plate in DFK5 media containing 10 nM RA, 1 μM purmorphamine, 5 μM DAPT, and 2 or 4 μg/mL puromycin for 24 hours. Surviving cells were dissociated with 0.25% Trypsin-EDTA for 5 minutes and quenched with complete media. Staining was conducted using the Transcription Buffer Set (Becton Dickinson #562725, Franklin Lakes, NJ) according to the manufacturer’s protocols with antibodies as described in “Immunocytochemistry.” Data was collected using a BD Canto II Flow Cytometer (Becton Dickinson). Between 10,000–100,000 events were recorded per condition (n≥4 biological replicates per condition) and analyzed using FloJo software (FlowJo, Ashland, OR); debris was removed from analysis using forward scatter versus side scatter and Hoechst versus forward scatter plots. Gating parameters were set using control groups stained only with secondary antibodies.
Single-Cell Electrophysiology
Cells were induced using the 2−/4+ protocol, dissociated onto laminin coated 35 mm dishes at 2 x 106 and selected for 24 hours with puromycin. Cultures were grown for up to 10 days post-selection in DFK5NB media with growth factors prior to recording. Cultures were perfused with Tyrode’s solution (150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM glucose, and 10 mM HEPES, at pH 7.4). Whole-cell electrodes were filled with internal solution (140 mM K-glucuronate, 10mM NaCl, 5 mM MgCl2, 0.2 mM EGTA, and 10 mM HEPES, at pH 7.4, supplemented with 5 mM Na-ATP and 1 mM Na-GTP). The open tip resistance was 2–5 MOhm. External solutions were delivered by local perfusion from an 8-barrelled pipette. Voltage-gated channel antagonists tetrodotoxin (TTX, 500 nM), tetraethylammonium chloride (TEA, 30 mM) and 4-aminopyridine (4-AP, 5 mM) were dissolved in Tyrode’s solution. Ligand-gated channel agonists were dissolved at 100 μM in 160 mM NaCl, 2 mM CaCl2, 10 mM HEPES. Currents were recorded under voltage clamp with an Axopatch 200A amplifier, filtered at 1 kHz and digitized at 10 kHz using pClamp software (Molecular Devices, Sunnyvale, CA). Evoked synaptic currents were detected by simultaneous whole-cell recordings from adjacent pairs of cells in Chx10-Puro/Olig2-Puro mixed cultures. Brief steps to 0 mV from a holding potential of −80 mV elicited fast inward sodium currents that escaped voltage control in the axon of presynaptic cells and evoked excitatory postsynaptic currents in the adjacent cell, which was held at −80 mV.
V2a and pMN Co-Culture
Mixed Chx10-Puro and Olig2-Puro cultures were established to assess synapse formation between V2a INs and MNs. A constitutively active fluorescent reporter was knocked into the Rosa26 domain of the Chx10-Puro line as described above using the Ai9 plasmid (gifted by Hongkui Zeng, Addgene plasmid #22799)(Madisen et al., 2010), pTurboCre, the Cas9 plasmid, and Rosa26 guide RNAs (gR26) generated by the Genome Engineering Core at Washington University in St. Louis. Clones were screened for correct insertion by junction PCR (R26Forward: TCCCAAAGTCGCTCTGAGTT; CAGReverse: CCATCGCTGCACAAAATAAT) and expanded accordingly. The previously established Olig2-Puro ESC line contains the PAC gene under the expression of Olig2, which marks the pMN domain (McCreedy et al., 2012). The Olig2-Puro and Chx10-Puro ESCs were induced according to their respective 2-/4+ protocols (McCreedy et al., 2012), dissociated onto laminin (Life Technologies #23017-015) coated 35mm dishes at 2 x 106 at a 1:1 ratio and selected for 24 hours in DFK5NB with 4 μg/mL puromycin. The media was replaced with DFK5NB containing B27, Glutamax, NT-3, GDNF, and BDNF and was replenished as needed every two days for up to 7 days. Neurobasal media containing the supplements and growth factors was used for the remaining duration of culture up to 4 weeks.
Statistical Analysis
Statistical analyses were performed using Statistica software (v5.5, StatSoft, Tulsa, OK). Significance was determined using Scheffe’s post hoc test for analysis of variance (ANOVA) with confidence as indicated. Average values reported with error bars are the standard deviation or standard error of the mean (SEM) as indicated.
Results
Puromycin resistance coincides with Chx10 expression in transgenic ESC line
In order to isolate V2a INs from a heterogeneous population of differentiated ESCs, we developed a genetic strategy to positively select for cells expressing the defining transcription factor Chx10 (Figure 1). CRISPR-assisted homologous recombination was used to target the first exon of the mouse Chx10 locus for puromycin N-acetyltransferase (PAC) insertion, thereby placing antibiotic resistance under the control of the native Chx10 gene regulatory elements. A single clone with targeted genomic insertion (Supplementary Figure 1B) and one copy of the PAC gene (Supplementary Figure 1C) was treated with Cre recombinase to remove the neomycin selection cassette. The resulting Chx10-Puro ESC line was used for all subsequent analyses in this study.
Figure 1. Schematic showing PAC insertion into the Chx10 gene locus.

(1) gChx10 guide RNAs mediate double stranded break at the ATG start codon. (2) Homologous recombination incorporated targeting vector cassette into the first exon. PAC expression was driven by the native Chx10 upstream promotors. The ubiquitously expressed PGK promoter drove neomycin phosphotransferase to screen cells with the incorporated dual cassette. (3) Cre recombinase was used to remove the PGK-neo cassette. (4) The final Chx10-Puro construct expresses PAC when Chx10 is expressed.
To determine whether Chx10 expression resulted in the expression of PAC in the Chx10-Puro line, Chx10-Puro ESCs were differentiated using a 2−/4+ V2a induction protocol - cells were aggregated for two days to form EBs, then exposed to RA, purmorphamine, and DAPT for four days. Unmodified RW4 ESCs and cultures induced using the Shh antagonist Cyc were used as controls. qRT-PCR demonstrated that when induced with the 2−/4+ protocol, RW4 and Chx10-Puro cultures express comparable levels of Chx10 that are significantly higher than uninduced ESC and Cyc control cultures (p<0.0001) (Figure 2A). However, only induced Chx10-Puro cultures expressed PAC, which disappeared when Shh signaling was suppressed with Cyc. This suggests that PAC expression is activated with Chx10 expression in the Chx10-Puro line.
Figure 2. PAC expression in Chx10-Puro ESC line allows for enrichment of neuronal cells.

(A) Chx10 and PAC mRNA expression in ESCs, ESCs induced with RA and puromycin (Pur), and ESCs induced with RA and cyclopamine (Cyc). (B) Schematic of 2−/4+induction protocol followed by 24 hours of puromycin (Puro) selection. (C) Live cell viability assay (Scale bars = 100 μm) and (D) flow cytometry in ESCs, ESCs induced with RA and Pur, and ESCs induced with RA and Cyc following puromycin selection with 2 μg/mL or 4 μg/mL; control cultures without puromycin were run in parallel. *p<0.0001 compared to control group. (E) Phase contrast with tdTomato overlay demonstrating neuronal morphology in selected cultures (Scale bar = 50 μm).
To confirm cell viability post-selection, cultures were dissociated following the induction and plated on laminin-coated wells in DFK5NB media containing puromycin for 24 hours (Figure 2B). In the absence of puromycin, cells were viable in all conditions as evidenced by calcein AM staining (Figure 2C). Following puromycin selection, viable cells were only apparent in induced Chx10-Puro cultures (Figure 2G,H). The addition of puromycin killed all RW4 cultures as well as Chx10-Puro cultures induced with Cyc. Analysis by flow cytometry indicated 12.0 ± 3.6% and 11.0 ± 3.6% of cells were viable after selection with 2 μg/mL and 4 μg/mL puromycin, respectively, compared to 85.81 ± 3.79% viable cells in unselected cultures (p<0.0001) (Figure 2D). Surviving cells demonstrated neuronal morphology, including phase bright cell bodies and neurite extensions that are consistent with V2a induction (Figure 2E).
V2a IN markers are enriched in selected Chx10-Puro cells
Immunocytochemistry and flow cytometry were used to confirm V2a IN identity in selected cultures using antibodies for the pan-neuronal marker (beta tubulin III (β-tubIII)) and defining transcription factors for V2a INs (Chx10, Lhx3), motor neurons (Hb9, Lhx3), V1 INs (Lim1/2), and V0 INs (Evx1, Lim1/2). The p2 progenitor marker Lhx3 is expressed in both V2 INs and immature MNs (Sharma et al., 1998), but expression of Lhx3 in post-mitotic cells has been used as a marker for V2a INs as it drives expression of Chx10 in certain contexts (Crone et al., 2008; Tanabe et al., 1998; Thaler et al., 1999). To capture early, transient expression of these transcription factors, a shorter, modified 2−/3+ induction protocol was used to ensure high protein expression post-selection for detection and quantification (Figure 3A).
Figure 3. Characterization of Chx10-Puro ESC-derived subpopulations.

(A) Schematic of modified 2-/4+ induction protocol. (B) Counting analysis of β-tubulin III (β-tubIII) distribution in control and puromycin-selected cultures. *p<0.05 compared to control group. (C) Flow cytometry analysis of ventral IN subpopulations in control and puromycin-selected cultures. *p<0.0001 compared to control group. #p<0.05 compared to control group. (D) Chx10 and (E) immunoflourescence analysis in control and puromycin-selected cultures. Scale bars = 200 μm.
Neuronal enrichment was quantified by counting the number of cell nuclei that co-localize with β-tubIII. Approximately 86.81 ± 5.40% of cells were β-tubIII+ in unselected control cultures; the percentage increased significantly to 98.27 ± 0.25% or 100% (p<0.05) following selection with 2 μg/mL or 4 μg/mL puromycin respectively. (Figure 3B). Analysis of the population distribution by flow cytometry confirmed that selection constitutes an enrichment of V2a cell identity (Figure 3C, Table 1, Supplementary Figure 2). While unselected cultures contained cells from all the ventral IN subtypes (Figure C, Supplementary Figure 3), a significant majority of cells were Chx10+ (Figure 3D) and/or Lhx3+ (Figure 3E) after treatment with puromycin (p<0.0001). The combination of staining and quantification suggest that there are negligible quantities of V0, V1 (p<0.0001) or MNs (p<0.05) that survive selection (Figure 3C, Supplementary Figure 3).
Table 1. Quantification of IN subtypes by flow cytometry.
Flow cytometry analysis of ventral IN subpopulations in control and puromycin-selected cultures.
| Chx10 | Lhx3 | Hb9 | Evx1 | Lim1/2 | |
|---|---|---|---|---|---|
| Control | 27.50 ± 9.21 | 53.93 ± 1.30 | 8.60 ± 4.52 | 23.80 ± 4.17 | 15.16 ± 1.53 |
| 2 μg/mL | 80.37 ± 3.27* | 93.96 ± 0.62* | 1.17 ± 1.06# | 0.61 ± 0.66* | 3.21 ± 0.40* |
| 4 μg/mL | 70.25 ±12.90* | 86.97 ± 5.83* | 1.97 ± 1.42# | 2.26 ± 1.16* | 0.76 ± 0.64* |
Values presented as mean ± standard deviation.
denotes p<0.0001 compared to control group.
denotes p<0.05 compared to control group.
While the intent of this study was to generate spinal V2a INs using Chx10 as the definitive transcription factor for identification, Chx10 is also expressed in the developing retina (Dhomen et al., 2006; Rowan and Cepko, 2004). The expression of two retinal developmental markers, Rax and Six3 (Loosli et al., 1999; Muranishi et al., 2012), was assessed and they were not significantly upregulated in selected cultures compared to ESC controls, but Chx10 expression was significantly upregulated, as expected (Supplementary Figure 4A–B). Cultures were stained for Isl1, which is a developmental marker for MNs in the spinal cord, but in combination with Chx10 and Lhx3 is also a marker for retinal bipolar cells (Elshatory et al., 2007). In selected cultures, 3.09 ± 0.93% and 0.47 ± 0.55% of cells were Isl1+ in cultures treated with 2 μg/mL and 4 μg/mL puromycin respectively, which were significantly lower (p<0.05) compared to 8.10 ± 1.78% of cells in unselected induced cultures (Supplementary Figure 4C–D). Interestingly, using a higher concentration of puromycin resulted in a lower percentage of cells expressing V2a markers (Figure 3C), but also significantly reduces the number cells that express Isl1 compared to using 2 μg/mL puromycin (p<0.05). These more highly enriched (4 μg/mL puromycin) cultures were used for all maturation and electrophysiology experiments.
Selected Chx10-Puro neurons are post-mitotic and achieve functional maturity
Because neuronal differentiation of ESCs can give rise to proliferative cell types that reduce the purity of the culture, selected Chx10-Puro cultures were evaluated for mitotic activity. An antibody for the proliferation marker Ki67 was used to detect and quantify mitotic cells via immunocytochemistry and flow cytometry (Figure 4A,B Supplementary Figure 2). Ki67+ cells comprised15.13 ± 2.58% of cells observed in unselected cultures, which was significantly decreased when treated with 2 ug/mL (1.80 ± 0.10%) or 4ug/mL (0.83 ± 0.29%) puromycin (p<0.0001). Unselected cultures also visibly showed a rapid increase in the number of cell nuclei over 2 weeks (Figure 4D).
Figure 4. Maturation of post-mitotic V2a INs.

(A) Immunofluorescence analysis of the proliferation marker Ki67 in unselected and selected cultures directly after selection. (B) Flow cytometry analysis of Ki67 in control and puromycin-selected cultures. *p<0.0001 compared to control group. (C) Schematic of 2-/4+ induction protocol, selection, and extended-culture. Media was replaced with DFK5NB with growth factors directly after selection for up to two weeks post-selection. (D) Mature Vglut2, NF, and MAP2 staining in unselected and selected cultures after two weeks in culture.
Most V2a INs in vivo express excitatory vesicular glutamate transporter 2 (Vglut2) (Lundfald et al., 2007). To confirm functional maturation of selected Chx10-Puro cultures, cells were induced using the 2−/4+ protocol, dissociated onto laminin-coated plates and selected for 24 hours with puromycin, then grown for 14 days post-selection in DFK5NB media with growth factors (Figure 4C). Selected cells abundantly express Vglut2 and the mature dendritic marker, microtubule associated protein 2 (MAP2) (Figure 4D). Vglut2+ neurons appear to be the majority of cells in selected cultures compared to only a small fraction in unselected cultures. Taken together, these data suggest that we have generated highly enriched, post-mitotic V2a INs.
Selected V2a INs demonstrate mature electrophysiological profiles
Whole cell current and voltage clamp recordings were performed to evaluate the functional maturity of cells in puromycin-selected cultures up to 12 days post-selection (d12). Cell capacitance, which is proportional to surface area, was low on d2 (17.1±1.95 pF, n=5) but increased to approximately 30 pF by d3 and remained relatively constant through d12 (Table 2). Input resistance was highest on d2 (2.22±0.66 GΩ, n=5) and declined as cells matured (415±80 MΩ, d12, n=7). Cell resting potentials were relatively depolarized at early time points after selection, but by d9-d12 the mean Vrest was approximately −50 mV. When membrane potential was maintained near −60 mV by steady current injection under current clamp nearly all cells with neuronal morphology were capable of firing action potentials upon stimulation with a depolarizing current pulse (Figure 5A). Of these, 35.7% fired single action potentials, 35.7% fired several action potentials near the beginning of an 800 msec pulse but then adapted, while the remaining 28.6% exhibited tonic firing throughout the depolarizing pulse. Under whole-cell voltage clamp, depolarizing steps from −80 mV elicited fast inward currents that were sensitive to blocking by the voltage-gated sodium channel antagonist tetrodotoxin (TTX, 0.5 μM). Traces in Figure 5B show TTX-sensitive current obtained by subtracting current recorded in TTX at each test potential from current recorded in control solution. In addition, all cells displayed transient and sustained outward currents mediated by voltage-gated potassium channels (Figure 5C, D). Consistent with previous work in other cell types (Bean, 2007), the organic cation tetraethylammonium (TEA) blocked sustained currents, whereas transient currents were selectively inactivated at a holding potential (Vh) of −40 mV (Figure 5C) or by application of 4-aminopyridine (data not shown). The transient outward currents required less depolarization for activation as shown in the plots of peak and steady-state (SS) outward currents as a function of test potential (Fig. 5D).
Table 2.
Electrophysiological properties of selected Chx10-Puro cells.
| Parameter | d2–4 | d9–12 |
|---|---|---|
| Capacitance (pF) | 33.1 ± 3.2 (19) | 29.8 ± 2.1 (38) |
| Input Resistance (GOhm) | 1.7 ± 0.24 (19) | 0.57 ± 0.12 (38) * |
| Tau (msec) | 52.1 ± 7.1 (19) | 22.8 ± 9.1 (38) * |
| V rest (mV) | −37.6 ± 1.5 (18) | −51.2 ± 1.8 (28) * |
| % V rest < −50 mV | 5.60% | 53.5% * |
| Rheobase (pA) | 13.1 ± 6.3 (7) | 49.7 ± 12.0 (21) * |
| 1st spike latency (msec) | 91.1 ± 22.4 (7) | 76.1 ± 15.0 (21) |
| 1st spike amplitude (mV) | 70.4 ± 7.1 (7) | 92.5 ± 4.1 (21) * |
| 1st spike overshoot (mV) | 11.9 ± 6.8 (7) | 31.4 ± 4.1 (21) * |
| 1st spike threshold (mV) | −38.9 ± 1.9 (7) | −37.4 ± 1.4 (21) |
| 1st spike width (msec) | 6.5 ± 0.9 (7) | 2.7 ± 0.3 (21) * |
| 10 Hz 1st latency (msec) | 41.9 ± 5.9 (4) | 31.4 ± 3.1 (14) |
| 10 Hz 1st frequency (Hz) | 11.3 ± 0.5 (4) | 13.9 ± 1.3 (14) |
| 10 Hz frequency adaptation | 1.3 ± 0.03 (4) | 1.4 ± 0.14 (14) |
| 10 Hz after potential (mv) | −2.4 ± 2.7 (4) | −0.15 ± 0.62 (14) |
| Sag at −90 mV (mV) | 2.4 ± 1.0 (4) | 2.4 ± 0.72 (14) |
| Peak I Na (nA) | −1.87 ± 0.27 (16) | −2.29 ± 0.22 (29) |
| Vm for peak I Na (mV) | −26.6 ± 2.0 (16) | −32.6 ± 1.6 (29) * |
Values presented as mean ± SEM (number of cells) (* denotes significantly different from d2–4 by t-test, Mann-Whitney rank sum test, or Z-test). Cell capacitance and input resistance were determined from 10 mV voltage clamp steps from a holding potential of −80 mV. First spike latency, amplitude, absolute amplitude, threshold and half-width were determined for the first spike recorded at threshold depolarization. In addition, 800 msec depolarizations that elicited spiking with an average frequency of 10 Hz were used to measure 1st latency, instantaneous frequency from the first inter-spike interval (ISI), and frequency adaptation (ratio of last to first ISI). Sag in voltage responses was determined for 800 msec hyperpolarizing current injections from −60 mV.
Figure 5. Action potentials and voltage-gated currents in selected Chx10-Puro cells.

(A) Action potential firing patterns recorded on d9 in 3 different selected cells stimulated with 800 msec square pulse current injections. (B) Currents mediated by voltage-gated sodium channels sensitive to tetrodotoxin (TTX) on d2 evoked by steps from a holding potential of −80 mV to test potentials ranging from −60 to +25 mV. Traces show the difference between currents recorded in the absence and presence of 0.5 μM TTX. Peak inward current plotted as a function of test potential. (C) Transient and sustained outward potassium currents on d4 evoked by 130 msec voltage steps from holding potentials of −80 mV (above) or −40 mV (below) to test potentials ranging from −100 mV to +60 mV in the presence of 0.5 μM TTX. Exposure to 30 mM tetraethylammonium (TEA) blocked the sustained current, whereas holding at −40mV inactivated the transient current. (D) Current-voltage relations for transient (peak TEA) and sustained (SS) outward currents.
To test for functional neurotransmitter receptors, cells were exposed to 100 μM selective agonists for AMPA/kainite, NMDA, glycine and GABA. Agonist activated inward currents were observed while holding cells at a fixed potential of −80 mV (Figure 6A). As the cells matured from d3 to d20 or more, the amplitude of these agonist-gated currents increased (Figure 6B). Agonist-evoked currents were also recorded as the membrane potential was slowly ramped between −110 and +110 mV (Figure 6C). Currents elicited by the inhibitory transmitters GABA and glycine reversed polarity near the estimated equilibrium potential for chloride ions (−54 mV) given the composition of our internal and external solutions. In contrast, the currents evoked by NMDA and kainate reversed much closer to 0 mV, consistent with monovalent cation permeability.
Figure 6. Currents activated by transmitter receptor agonists in selected Chx10-Puro cells.

(A) Whole-cell currents evoked by brief application of 100 μM kainate, GABA, glycine or NMDA (plus 1 μM glycine) as indicated by the filled bars. Holding potential, −80 mV; 9 days after puromycin selection (* denotes significant difference from d3 and d9, # denotes significant difference from d3). (B) Peak agonist-gated current at −80 mV (mean ± SEM) recorded in cells 3, 9 or more than 20 days after puromycin selection. (C) Agonist-evoked currents recorded during voltage ramps from −110 to +110 mV at 1.2 mV/msec, 28 days after puromycin selection. GABA and glycine evoked currents that reversed polarity at −53.8 ± 1.7 mV (n=4) and −55.3 ± 1.7 mV (n=4), respectively, consistent with activation of chloride-selective channels. Currents evoked by kainate and NMDA reversed polarity at 5.9 ± 2.6 mV (n=4) and 19.8 ± 4.1 mV (n=4), respectively, consistent with monovalent cation permeability.
Overall, the membrane properties of our selected Chx10-Puro cells are broadly consistent with previous electrophysiological characterization of V2a interneurons in acute tissue slice and isolated spinal cord preparations (Dougherty and Kiehn, 2010a; Zhong et al., 2010). Recordings from native neurons in these preparations have revealed a variety of action potential patterns including tonic, adapting and single spike firing, as are also seen in our cultures.
Synapses observed in mixed V2a and pMN culture
The ability of ESC-derived neuronal populations to form synapses is critical for their use in cell therapy or for in vitro modeling of neural circuitry. Endogenous V2a INs primarily synapse onto MNs, but have also been observed to synapse onto other V2a INs. To model these interactions, selected ESC-derived V2a INs and pMNs were co-cultured for up to 4 weeks and examined for functional connectivity (Figure 7A–D). A previously established Olig2-Puro ESC line was used to obtain a highly enriched pMN culture, which gives rise to a variety of cell types including MNs, oligodendrocytes, and astrocytes. The presence of glia has been shown to be beneficial to spontaneous electrical activity and for improved synapse formation in culture (Boehler et al., 2007; Pfrieger and Barres, 1997). A constitutively-active red fluorescent reporter was knocked into the Chx10-Puro line to identify V2a INs in co-culture conditions (Figure 7E–G); MNs were identified by their distinct morphology (Figure 7B). Robust staining for synaptic vesicles (SV2) was observed in both V2a INs and MNs after 3 weeks; 98.67 ± 1.03% of Vglut2+ cells in these cultures co-stained with SV2 (Figure 7H–J).
Figure 7. Chx10-Puro and Olig2-Puro co-culture.

(A) Schematic of 2-/4+ induction protocol fpr Chx10-Puro and Olig2-Puro cells, followed by selection and up to four weeks of co-culture. Media is switched from DFK5NB to Neurobasal after one week. (B) Phase contrast and (C,D) Hoescht staining of mixed culture demonstrates variety of cell morphologies and cell debris. (E) TdTomato allows for identification of Chx10-Puro cells. (F,G, H) Vglut2 staining appears almost exclusively in V2a INs. (I,J) SV2 staining is prevalent in most neurons and indicative of synaptic activity. Scale bars = 200 μm.
To test for functional synaptic connections, we performed simultaneous whole-cell recordings from adjacent pairs of cells on d21 to d31 (Finley et al., 1996). Both cells were held at −80 mV and brief (5–10 msec) voltage steps to 0 mV were delivered at 0.2 Hz, first to one cell and then the other. As shown in Figure 8, the voltage steps in a labeled Chx10-Puro cell elicited inward sodium currents that escaped from voltage control in the presynaptic axon and evoked excitatory postsynaptic currents (EPSCs) in the adjacent Chx10-Puro cell. In all, we obtained successful simultaneous recordings from 9 cell pairs with 18 potential presynaptic cells, including 13 labeled Chx10-Puro cells and 5 unlabeled cells. Upon stimulation, most of the cells evoked EPSCs (11 of 13 labeled Chx10-Puro cells and 3 of 5 unlabeled cells) with an average amplitude of 520 ± 170 pA and a mean synaptic delay of 2.0 ± 0.2 msec from the peak of presynaptic inward current to the initial rise of the EPSC. The remaining 2 labeled Chx10-Puro cells and 2 unlabeled Olig2-Puro cells failed to evoke any postsynaptic response, suggesting either they did not make functional contact with the adjacent cell or they did not release a transmitter capable of eliciting fast postsynaptic currents. Importantly, the AMPA/kainate receptor antagonist NBQX (30 μM) blocked synaptic transmission mediated by all of the presynaptic Chx10-Puro cells, as well as the 3 unlabeled presynaptic cells, confirming that they exhibit an excitatory glutamatergic phenotype. Superfusion with the GABAA receptor antagonist bicuculline methiodide (200 μM) did not affect evoked synaptic transmission in any of our paired recordings, but did block spontaneous IPSCs that were observed in several of the recorded cells in mature Chx10-Puro/Olig2-Puro mixed cultures. Similarly, NBQX also blocked spontaneous EPSCs observed while recording from individual cells in mixed cultures.
Figure 8. Evoked excitatory synaptic transmission between selected Chx10-Puro cells.

Simultaneous whole-cell recordings of pre-and postsynaptic currents from two adjacent Chx10-Puro cells 31 days after puromycin selection. Voltage steps to 0 mV from a holding potential of −80 mV elicited inward sodium current, and uncompensated capacity transients, in the presynaptic cell. Excitatory synaptic currents (EPSCs) were evoked in the postsynaptic cell with a delay of 3.2 msec, measured from the peak of presynaptic sodium current to the initial rise of the EPSC. Exposure to the AMPA/kainate receptor antagonist NBQX (30 μM) completely blocked the EPSC, while the GABAA receptor antagonist bicuculline methiodide (0.2 mM) had no effect. Each panel shows 3 superimposed traces in each condition. Presynaptic stimuli were delivered at 0.2 Hz; postsynaptic holding potential, −80 mV.
Discussion
Chx10-Puro mouse ESC line yields highly enriched V2a INs
While optimized induction protocols have been developed to obtain several ventral spinal populations from ESCs (Brown et al., 2014; McCreedy et al., 2014a; McCreedy et al., 2012; Wichterle et al., 2002; Xu and Sakiyama-Elbert, 2015), the heterogeneity of resulting cultures make the study or transplantation of single cell types unfeasible. By using defining transcription factor promoters to drive puromycin resistance in ESC-derived cell populations, our lab and others have been able to positively select for enriched populations of beta cells, cardiomyocytes, endothelial cells, neurepithelial progenitors, pMNs, and MNs (Anderson et al., 2007; Li et al., 1998; Marchetti et al., 2002; McCreedy et al., 2014a; McCreedy et al., 2012; Soria et al., 2000). Here we generated a Chx10-Puro ESC line and confirmed that, after induction and puromycin treatment, the vast majority of viable cells were Chx10+ and Lhx3+. By modulating the concentration of puromycin, contamination due to other ventral cell types and Chx10+/ Lhx3+/Isl1+ retinal bipolar cells could be reduced. Within two weeks, selected Chx10-Puro cells mature into Vglut2+ glutamatergic neurons; Vglut2 presents as punctate staining along the neurite surface, however strong cytosolic staining was also observed at early time points. This may be caused by an upregulation of protein expression during early stages of maturation, or, in selected cultures, due to a lack of supportive cells that aid in maturation. Within four weeks of culture (Figure 7E–G), the cytosolic component is reduced. We also report a higher yield of Chx10+ cells in unselected cultures than previously published (Brown et al., 2014), likely because the cells are maintained as EBs for the duration of the induction.
Our data suggests 80–90% purity in selected Chx10-Puro cultures, but it is probable that quantification by flow cytometry is an underestimation of true population counts and the purity of the selected cultures is likely to be higher than we observed due to transcription factor inactivation. While increasing the concentration of puromycin was expected to improve the yield of cells positive for V2a markers, instead we saw a downward trend despite the elimination of potential retinal cells marked by Isl1 expression and other ventral markers. A rapid decrease in the number of Chx10 and Lhx3 expressing cells was also observed with time (data not shown). Given that selected cultures are not overtaken by proliferative cell types and remain primarily glutamatergic neuronal cells when mature, it is conceivable that when selected with 4 μg/mL puromycin, early V2a INs and retinal cells die before PAC expression is sufficient to counter puromycin activity, and that the surviving cells analyzed begin to lose V2a marker expression but represent a more mature V2a population. This is at odds with endogenous cells in animal models, where immunohistochemistry has been used to identify Chx10+ and Lhx3+ cells in spinal cord slices into maturity (Al-Mosawie et al., 2007; Crone et al., 2008; Crone et al., 2009; Dougherty and Kiehn, 2010a, b; Lundfald et al., 2007). Lineage tracing using cre-recombinase would enable a clearer delineation of total subtype yields in ESC-derived cultures. Selected Chx10-Puro V2a INs also exhibited morphological and electrophysiological heterogeneity consistent with observations ex vivo, which seems to imply at least some degree of diversity (Al-Mosawie et al., 2007; Dougherty and Kiehn, 2010a, b; Kimura et al., 2006). Several dozen transcription factors have been detected to identify discrete ventral IN subpopulations in the last few years alone (Francius et al., 2013), including at least one functionally distinct subpopulation of Chx10+ cells, a Shox2+/Chx10+ population, termed V2d (Dougherty et al., 2013). The availability of markers to classify V2 IN subpopulations is of major interest and would help to further characterize the purified population.
Functional activity in purified ESC-derived V2a populations mimics ex vivo data
Most native V2a interneurons in acute spinal cord preparations appear capable of spiking repeatedly throughout the duration of a prolonged depolarizing pulse, although a substantial minority exhibit other firing patterns including burst firing that adapts and single spiking (Dougherty and Kiehn, 2010a; Zhong et al., 2010). Our selected Chx10-Puro cells also exhibit a range of action potential firing, as well as other electrophysiological properties that are generally consistent with work on native V2a cells. Maturing selected Chx10-Puro cells expressed receptors for the major fast excitatory and inhibitory spinal neurotransmitters, suggesting that they should be capable of functional integration into spinal circuits.
V2a INs in vivo play a critical role in CPGs, networking with other cell types in order to achieve coordinated locomotion (Azim et al., 2014; Crone et al., 2008; Crone et al., 2012; Crone et al., 2009; Zhong et al., 2010; Zhong et al., 2011). In the present study, we demonstrate the ability of selected Chx10-Puro V2a INs to make functional excitatory synapses onto each other, as well as onto presumptive MNs. In the instances where there was no apparent connection between Olig2-Puro cells and adjacent labeled Chx10-Puro cells, it is possible that the unlabeled Olig2-Puro cells were MNs releasing acetylcholine, which did not acutely activate channels and thus did not produce a fast post synaptic current. The connections observed during paired recordings were likely monosynaptic because of their large amplitudes and short synaptic delays. In all cases tested, the AMPA/kainate receptor antagonist, NBQX, produced complete block of evoked transmission, demonstrating that the transmitter released by presynaptic Chx10-Puro cells activates postsynaptic glutamate receptors. Thus, our paired recordings provide conclusive evidence that selected Chx10-Puro cells are glutamatergic.
Previous work by Zhong et al. (2010) provided evidence for selective electrical coupling between V2a INs with similar action potential firing characteristics, a feature that might help to coordinate rhythmic activity. We saw no examples of electrical coupling in our paired recordings either from 9 cell pairs in mature Chx10-Puro/Olig2-Puro co-cultures (d20 to d31), or in preliminary recordings from 6 pairs in d10 Chx10-Puro cultures; however, we were not able to characterize the spiking properties of these cells to determine whether or not both cells in a pair displayed similar firing patterns.
Chx10-Puro cells for in vitro modeling
While most in vitro studies that use neuronal cell cultures have investigated functional properties, advances in culture methods, geometric and spatial patterning, instrumentation, and signal processing make possible “lab on a chip” technologies for brain and spinal cord (Wheeler and Brewer, 2010). V2a INs occupy a significant role in the CPG and, compared to other IN types, have well characterized network properties that make them an attractive population with which to design an experimental platform (Dougherty and Kiehn, 2010a). A minimalist approach could be taken, whereby V2a INs are observed in isolation, but transcription-factor driven selectable ESC lines could also be used modularly to generate complex cultures that remain well defined. We demonstrate the feasibility of such an approach here using both the Chx10-Puro and Olig2-Puro ESC lines to create a simple model of V2a-MN interactions. Because ESC induction protocols can be manipulated to enrich from distinct anatomical regions (Lippmann et al., 2015; Okada et al., 2004), there is a significant degree of flexibility possible using selectable lines.
A general caveat in the use of pluripotent stem cells for modeling or therapy is the gap between neurons obtained in the dish and those that develop normally in vivo. As evidenced by the selected Chx10-Puro cultures generated in this study, there remain some phenotypic and functional differences between ESC-derived V2a INs and endogenous cells. Some variation is expected, especially given the abnormal isolation of these neurons during a critical period of maturation, and it is possible that a longer duration of maturation or optimization of culture conditions might improve comparability. However, it is similarly possible that the significant differences in development cannot be overcome and the cells here merely mimic V2a IN properties while incapable of the complex functional activity expected of endogenous V2a INs. Compounding the issue of characterization is a lack of comprehensive genetic profiles to identify functionally distinct IN subtypes. Creating tools to investigate these potential differences is beneficial to our understanding of spinal cord development and for improving stem cell therapies.
Conclusions
By knocking PAC into the Chx10 locus of mouse ESCs, the addition of puromycin to differentiated ESC cultures killed Chx10 negative cells. V2a IN enrichment was evident directly after selection of differentiated “Chx10-Puro” cells and persisted through maturation into functional glutamatergic neurons. Electrophysiology demonstrated that selected V2a INs are capable of spontaneously firing action potentials and respond to a range of agonists. Finally, our selected V2a INs form synapses with each other and with MNs in co-cultures. Together our findings suggest that selected Chx10-Puro V2a INs, while not identical to native V2a INs, are broadly comparable and can be a powerful resource for in vitro modeling of neural networks, investigation of neuronal development and diversification, and targeted cell therapies.
Supplementary Material
(A) Schematic of Chx10-PAC targeting vector. (B) jPCR validation of PAC insertion into first exon of Chx10; the 2.4 kb fragment is an amplification from the endogenous 5′ UTR into the PAC cassette. (C) Copy number analysis of PAC in parental RW4 cells, Hb9-Puro ESCs (McCreedy et al., 2014a), and Chx-10 Puro ESCs. GAPDH was used as a control.
Representative flow cytometry analysis of IN subtypes and proliferation marker Ki67 in unselected (gray) and selected (black) differentiated Chx1-Puro cultures.
(A) Hb9, (B) Evx1, and (C) Lim1/2 immunofluorescence analysis in control and puromycin-selected cultures. Scale bars = 200 μm
(A) Chx10, (B) Rax and Six3 mRNA expression in unselected and selected Chx10-Puro ESCs after modified 2-/3+ protocol (C) Quantification of Isl1 in control and puromycin-selected cultures. *p<0.05 compared to control group, **p<0.05 compared to control group and 2 μg/mL puromycin selection. (D) Immunoflourescence analysis in control and puromycin-selected cultures for Isl1. Scale bars = 200 μm
Highlights.
Antibiotic selection of transgenic mouse ESCs yields highly enriched V2a interneurons
Purity of ESC-derived V2a IN culture persists through glutamatergic maturation
Electrophysiological properties of selected V2a INs are similar to endogenous cells
Selected ESC-derived V2a INs form synapses with motor neurons when co-cultured
Acknowledgments
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under grants R01 NS090617 (S.E.S), R01NS051454 (S.E.S) and NS30888 (J.E.H.). N.I. was supported by an Individual Predoctoral National Research Service Award (F31NS090760).
Abbreviations
- CPG
central pattern generator
- Cyc
cyclopamine
- DAPT
N-{N-(3,5-difluorophenacetyl-L-alanyl))-(S)-phenylglycine-t-butyl-ester
- EB
embryoid body
- EPSC
excitatory post synaptic current
- ESC
embryonic stem cell
- FACS
Fluorescence-activated cell sorting
- IN
interneuron
- MAP2
microtubule associated protein 2
- MN
motor neuron
- PAC
puromycin N-acetyltransferase
- pMN
progenitor motor neuron
- Pur
purmorphamine
- Puro
puromycin
- RA
retinoic acid
- SCI
spinal cord injury
- Shh
sonic hedgehog
- TEA
tetraethylammonium chloride
- TTX
tetrodotoxin
- Vglut2
vesicular glutamate transporter 2
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Mosawie A, Wilson JM, Brownstone RM. Heterogeneity of V2-derived interneurons in the adult mouse spinal cord. The European journal of neuroscience. 2007;26:3003–3015. doi: 10.1111/j.1460-9568.2007.05907.x. [DOI] [PubMed] [Google Scholar]
- Anderson D, Self T, Mellor IR, Goh G, Hill SJ, Denning C. Transgenic enrichment of cardiomyocytes from human embryonic stem cells. Molecular therapy : the journal of the American Society of Gene Therapy. 2007;15:2027–2036. doi: 10.1038/sj.mt.6300303. [DOI] [PubMed] [Google Scholar]
- Arber S. Motor circuits in action: specification, connectivity, and function. Neuron. 2012;74:975–989. doi: 10.1016/j.neuron.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Azim E, Jiang J, Alstermark B, Jessell TM. Skilled reaching relies on a V2a propriospinal internal copy circuit. Nature. 2014 doi: 10.1038/nature13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nature reviews Neuroscience. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Boehler MD, Wheeler BC, Brewer GJ. Added astroglia promote greater synapse density and higher activity in neuronal networks. Neuron glia biology. 2007;3:127–140. doi: 10.1017/S1740925X07000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Butts JC, McCreedy DA, Sakiyama-Elbert S. Generation of V2a interneurons from mouse embryonic stem cells. Stem Cells and Development. 2014 doi: 10.1089/scd.2013.0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature biotechnology. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Courtine G, Gerasimenko Y, van den Brand R, Yew A, Musienko P, Zhong H, Song B, Ao Y, Ichiyama RM, Lavrov I, Roy RR, Sofroniew MV, Edgerton VR. Transformation of nonfunctional spinal circuits into functional states after the loss of brain input. Nature neuroscience. 2009;12:1333–1342. doi: 10.1038/nn.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone SA, Quinlan KA, Zagoraiou L, Droho S, Restrepo CE, Lundfald L, Endo T, Setlak J, Jessell TM, Kiehn O, Sharma K. Genetic ablation of V2a ipsilateral interneurons disrupts left-right locomotor coordination in mammalian spinal cord. Neuron. 2008;60:70–83. doi: 10.1016/j.neuron.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Crone SA, Viemari JC, Droho S, Mrejeru A, Ramirez JM, Sharma K. Irregular Breathing in Mice following Genetic Ablation of V2a Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:7895–7906. doi: 10.1523/JNEUROSCI.0445-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone SA, Zhong G, Harris-Warrick R, Sharma K. In mice lacking V2a interneurons, gait depends on speed of locomotion. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:7098–7109. doi: 10.1523/JNEUROSCI.1206-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhomen NS, Balaggan KS, Pearson RA, Bainbridge JW, Levine EM, Ali RR, Sowden JC. Absence of chx10 causes neural progenitors to persist in the adult retina. Investigative ophthalmology & visual science. 2006;47:386–396. doi: 10.1167/iovs.05-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KJ, Kiehn O. Firing and cellular properties of V2a interneurons in the rodent spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010a;30:24–37. doi: 10.1523/JNEUROSCI.4821-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KJ, Kiehn O. Functional organization of V2a-related locomotor circuits in the rodent spinal cord. Annals of the New York Academy of Sciences. 2010b;1198:85–93. doi: 10.1111/j.1749-6632.2010.05502.x. [DOI] [PubMed] [Google Scholar]
- Dougherty KJ, Zagoraiou L, Satoh D, Rozani I, Doobar S, Arber S, Jessell TM, Kiehn O. Locomotor rhythm generation linked to the output of spinal shox2 excitatory interneurons. Neuron. 2013;80:920–933. doi: 10.1016/j.neuron.2013.08.015. [DOI] [PubMed] [Google Scholar]
- Eiraku M, Sasai Y. Mouse embryonic stem cell culture for generation of three-dimensional retinal and cortical tissues. Nature protocols. 2012;7:69–79. doi: 10.1038/nprot.2011.429. [DOI] [PubMed] [Google Scholar]
- Elshatory Y, Deng M, Xie X, Gan L. Expression of the LIM-homeodomain protein Isl1 in the developing and mature mouse retina. The Journal of comparative neurology. 2007;503:182–197. doi: 10.1002/cne.21390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley MF, Kulkarni N, Huettner JE. Synapse formation and establishment of neuronal polarity by P19 embryonic carcinoma cells and embryonic stem cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:1056–1065. doi: 10.1523/JNEUROSCI.16-03-01056.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francius C, Harris A, Rucchin V, Hendricks TJ, Stam FJ, Barber M, Kurek D, Grosveld FG, Pierani A, Goulding M, Clotman F. Identification of multiple subsets of ventral interneurons and differential distribution along the rostrocaudal axis of the developing spinal cord. PloS one. 2013;8:e70325. doi: 10.1371/journal.pone.0070325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosgnach S, Lanuza GM, Butt SJ, Saueressig H, Zhang Y, Velasquez T, Riethmacher D, Callaway EM, Kiehn O, Goulding M. V1 spinal neurons regulate the speed of vertebrate locomotor outputs. Nature. 2006;440:215–219. doi: 10.1038/nature04545. [DOI] [PubMed] [Google Scholar]
- Harkema SJ. Plasticity of interneuronal networks of the functionally isolated human spinal cord. Brain research reviews. 2008;57:255–264. doi: 10.1016/j.brainresrev.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nature reviews Genetics. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- Kiehn O. Locomotor circuits in the mammalian spinal cord. Annual review of neuroscience. 2006;29:279–306. doi: 10.1146/annurev.neuro.29.051605.112910. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Okamura Y, Higashijima S. alx, a zebrafish homolog of Chx10, marks ipsilateral descending excitatory interneurons that participate in the regulation of spinal locomotor circuits. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:5684–5697. doi: 10.1523/JNEUROSCI.4993-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Lanuza GM, Gosgnach S, Pierani A, Jessell TM, Goulding M. Genetic identification of spinal interneurons that coordinate left-right locomotor activity necessary for walking movements. Neuron. 2004;42:375–386. doi: 10.1016/s0896-6273(04)00249-1. [DOI] [PubMed] [Google Scholar]
- Li M, Pevny L, Lovell-Badge R, Smith A. Generation of purified neural precursors from embryonic stem cells by lineage selection. Current biology : CB. 1998;8:971–974. doi: 10.1016/s0960-9822(98)70399-9. [DOI] [PubMed] [Google Scholar]
- Lippmann ES, Williams CE, Ruhl DA, Estevez-Silva MC, Chapman ER, Coon JJ, Ashton RS. Deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem cell reports. 2015;4:632–644. doi: 10.1016/j.stemcr.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loosli F, Winkler S, Wittbrodt J. Six3 overexpression initiates the formation of ectopic retina. Genes & development. 1999;13:649–654. doi: 10.1101/gad.13.6.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundfald L, Restrepo CE, Butt SJ, Peng CY, Droho S, Endo T, Zeilhofer HU, Sharma K, Kiehn O. Phenotype of V2-derived interneurons and their relationship to the axon guidance molecule EphA4 in the developing mouse spinal cord. The European journal of neuroscience. 2007;26:2989–3002. doi: 10.1111/j.1460-9568.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti S, Gimond C, Iljin K, Bourcier C, Alitalo K, Pouyssegur J, Pages G. Endothelial cells genetically selected from differentiating mouse embryonic stem cells incorporate at sites of neovascularization in vivo. Journal of cell science. 2002;115:2075–2085. doi: 10.1242/jcs.115.10.2075. [DOI] [PubMed] [Google Scholar]
- McCreedy DA, Brown CR, Butts JC, Xu H, Huettner JE, Sakiyama-Elbert S. Generation of High Purity Cholinergic Motoneurons by Hb9-Enhancer Driven Antibiotic Resistance in Genetically Engineered Mouse Embryonic Stem Cells. Biotechnoogy and Bioengineering. 2014a doi: 10.1002/bit.25260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreedy DA, Rieger CR, Gottlieb DI, Sakiyama-Elbert SE. Transgenic enrichment of mouse embryonic stem cell-derived progenitor motor neurons. Stem cell research. 2012;8:368–378. doi: 10.1016/j.scr.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreedy DA, Wilems TS, Xu H, Butts JC, Brown CR, Smith AW, Sakiyama-Elbert SE. Survival, Differentiation, and Migration of High-Purity Mouse Embryonic Stem Cell-derived Progenitor Motor Neurons in Fibrin Scaffolds after Sub-Acute Spinal Cord Injury. Biomaterials science. 2014b;2:1672–1682. doi: 10.1039/c4bm00106k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranishi Y, Terada K, Furukawa T. An essential role for Rax in retina and neuroendocrine system development. Development, growth & differentiation. 2012;54:341–348. doi: 10.1111/j.1440-169X.2012.01337.x. [DOI] [PubMed] [Google Scholar]
- Okada Y, Shimazaki T, Sobue G, Okano H. Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Developmental biology. 2004;275:124–142. doi: 10.1016/j.ydbio.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Peng CY, Yajima H, Burns CE, Zon LI, Sisodia SS, Pfaff SL, Sharma K. Notch and MAML signaling drives Scl-dependent interneuron diversity in the spinal cord. Neuron. 2007;53:813–827. doi: 10.1016/j.neuron.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Rowan S, Cepko CL. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Developmental biology. 2004;271:388–402. doi: 10.1016/j.ydbio.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Rybak IA, Dougherty KJ, Shevtsova NA. Organization of the Mammalian Locomotor CPG: Review of Computational Model and Circuit Architectures Based on Genetically Identified Spinal Interneurons(1,2,3) eNeuro. 2015;2 doi: 10.1523/ENEURO.0069-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Sheng HZ, Lettieri K, Li H, Karavanov A, Potter S, Westphal H, Pfaff SL. LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell. 1998;95:817–828. doi: 10.1016/s0092-8674(00)81704-3. [DOI] [PubMed] [Google Scholar]
- Soria B, Roche E, Berna G, Leon-Quinto T, Reig JA, Martin F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes. 2000;49:157–162. doi: 10.2337/diabetes.49.2.157. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Ito M, Hosaka Y, Sato T, Ito E, Suzuki K, Takahashi M. Effect of percutaneous transvenous mitral commissurotomy on postexercise breathlessness as determined by ventilation during recovery from constant workload exercise. The American journal of cardiology. 1998;82:1132–1135. A1139. doi: 10.1016/s0002-9149(98)00573-6. [DOI] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron. 1999;23:675–687. doi: 10.1016/s0896-6273(01)80027-1. [DOI] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell stem cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler BC, Brewer GJ. Designing Neural Networks in Culture: Experiments are described for controlled growth, of nerve cells taken from rats, in predesigned geometrical patterns on laboratory culture dishes. Proceedings of the IEEE Institute of Electrical and Electronics Engineers. 2010;98:398–406. doi: 10.1109/JPROC.2009.2039029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Wu S, Ying G, Wu Q, Capecchi MR. A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nature protocols. 2008;3:1056–1076. doi: 10.1038/nprot.2008.70. [DOI] [PubMed] [Google Scholar]
- Xu H, Sakiyama-Elbert SE. Directed Differentiation of V3 Interneurons from Mouse Embryonic Stem Cells. Stem cells and development. 2015 doi: 10.1089/scd.2015.0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Narayan S, Geiman E, Lanuza GM, Velasquez T, Shanks B, Akay T, Dyck J, Pearson K, Gosgnach S, Fan CM, Goulding M. V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron. 2008;60:84–96. doi: 10.1016/j.neuron.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Droho S, Crone SA, Dietz S, Kwan AC, Webb WW, Sharma K, Harris-Warrick RM. Electrophysiological characterization of V2a interneurons and their locomotor-related activity in the neonatal mouse spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:170–182. doi: 10.1523/JNEUROSCI.4849-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Sharma K, Harris-Warrick RM. Frequency-dependent recruitment of V2a interneurons during fictive locomotion in the mouse spinal cord. Nature communications. 2011;2:274. doi: 10.1038/ncomms1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic of Chx10-PAC targeting vector. (B) jPCR validation of PAC insertion into first exon of Chx10; the 2.4 kb fragment is an amplification from the endogenous 5′ UTR into the PAC cassette. (C) Copy number analysis of PAC in parental RW4 cells, Hb9-Puro ESCs (McCreedy et al., 2014a), and Chx-10 Puro ESCs. GAPDH was used as a control.
Representative flow cytometry analysis of IN subtypes and proliferation marker Ki67 in unselected (gray) and selected (black) differentiated Chx1-Puro cultures.
(A) Hb9, (B) Evx1, and (C) Lim1/2 immunofluorescence analysis in control and puromycin-selected cultures. Scale bars = 200 μm
(A) Chx10, (B) Rax and Six3 mRNA expression in unselected and selected Chx10-Puro ESCs after modified 2-/3+ protocol (C) Quantification of Isl1 in control and puromycin-selected cultures. *p<0.05 compared to control group, **p<0.05 compared to control group and 2 μg/mL puromycin selection. (D) Immunoflourescence analysis in control and puromycin-selected cultures for Isl1. Scale bars = 200 μm