

SUMMARY
XPG is a structure-specific endonuclease required for nucleotide excision repair, and incision-defective Xpg mutations cause the skin cancer-prone syndrome xeroderma pigmentosum. Truncating mutations instead cause the neurodevelopmental progeroid disorder Cockayne syndrome, but little is known about how XPG loss results in this devastating disease. We identify XPG as a partner of BRCA1 and BRCA2 in maintaining genomic stability through homologous recombination (HRR). XPG depletion causes DNA double-strand breaks, chromosomal abnormalities, cell-cycle delays, defective HRR, inability to overcome replication fork stalling, and replication stress. XPG directly interacts with BRCA2, RAD51, and PALB2, and XPG depletion reduces their chromatin binding and subsequent RAD51 foci formation. Upstream in HRR, XPG interacts directly with BRCA1. Its depletion causes BRCA1 hyper-phosphorylation and persistent chromatin binding. These unexpected findings establish XPG as an HRR protein with important roles in genome stability and suggest how XPG defects produce severe clinical consequences including cancer and accelerated aging.
INTRODUCTION
Unrepaired DNA lesions from both environmental and endogenous sources impede replication fork progression and result in replication stress (Magdalou et al., 2014; Zeman and Cimprich, 2014). Repair intermediates, e.g. single-strand breaks, also can interfere with replication and be processed by it into more severe lesions, DNA double-strand breaks (DSBs). In addition, encounters with transcription pose a major problem for the replication machinery (Bermejo et al., 2012; Helmrich et al., 2013), which is likely a more severe problem when transcription itself is stalled. Thus, defects in coordination between DNA replication and DNA repair can lead to genomic instability, developmental and immunological abnormalities, and cancer and/or aging (Marteijn et al., 2014; Zeman and Cimprich, 2014).
A major mechanism for rescuing stalled replication forks involves homologous recombination repair (HRR) of the resulting DSB mediated by RAD51 (Prakash et al., 2015; San Filippo et al., 2008). HRR is essential for cell survival, and in its absence, lethal chromosome breaks occur during replication (Sonoda et al., 1998). HRR is initiated by nucleolytic processing of the broken DNA ends to give single-stranded (ss) DNA tails, which become coated by the major eukaryotic ssDNA binding protein, replication protein A (RPA). RAD51 is recruited to ssDNA and loaded onto it through displacement of RPA by the breast cancer-associated protein BRCA2 (Jensen et al., 2010; Liu et al., 2010; Thorslund et al., 2010) in partnership with PALB2 (Xia et al., 2006). In contrast (Roy et al., 2012), the breast cancer-associated protein BRCA1 functions early in HRR to regulate end resection (Coleman and Greenberg, 2011) and to recruit BRCA2 through interaction with PALB2 (Sy et al., 2009). The RAD51 nucleoprotein presynaptic filament catalyzes the search for homology between the ssDNA end and the intact sister chromatid, invading the duplex DNA to form a DNA joint called the D-loop in a process that also requires PALB2 and RAD51AP1 (Buisson et al., 2010; Dray et al., 2010; Wiese et al., 2007). Accessory proteins including RAD52 (Feng et al., 2011) and five RAD51 paralogs, XRCC2/3 and RAD51B/C/D (Chun et al., 2013), are important in the critical early step of RPA displacement and RAD51 filament formation, which manifests at the cellular level as RAD51 foci.
The DNA repair protein XPG was first identified as a structure-specific endonuclease required for nucleotide excision repair (NER). Initiation of NER requires binding of XPC to the helix distortion caused by a lesion, followed by further opening of the DNA by the transcription/repair factor TFIIH to form an NER bubble, then lesion verification by TFIIH together with XPA. Sequential cuts at the bubble junctions by ERCC1/XPF and XPG excise the lesion-containing strand (Marteijn et al, 2014). Transcription-coupled repair (TCR) is a specialized process that preferentially removes transcription-blocking lesions from transcribed DNA strands through recognition of stalled RNA polymerase by the CSB protein, rather than recognizing the lesion itself (Fousteri and Mullenders, 2008; Vermeulen and Fousteri, 2013). TCR therefore does not involve XPC, but requires other NER proteins including XPG.
Defects in NER result in the skin cancer-prone, sun-sensitive disorder xeroderma pigmentosum (XP), and point mutations in XPG that inactivate its endonuclease (XP-G patients) cause XP (Nouspikel et al., 1997; O’Donovan and Wood, 1993). In contrast, patients with rare truncating mutations in XPG have the combined diseases of XP with Cockayne syndrome (CS), the signature molecular defect of which is loss of TCR (Emmert et al., 2002; Lindenbaum et al., 2001; Nouspikel et al., 1997). Rather than cancer susceptibility, XP-G/CS presents as severe, primarily postnatal, progressive neurodevelopmental abnormalities with mental retardation, dramatic growth failure, greatly accelerated symptoms of aging, and death in early childhood (Scharer, 2008). Mouse models recapitulate the patient phenotypes. The original Xpg knockout resulted in death before weaning (Harada et al., 1999), but a recent conditional knockout mouse in a hybrid strain background survives to 15–18 weeks and displays many progressive progeroid features, including early cessation of growth, cachexia, kyphosis, and extensive neurodegeneration (Barnhoorn et al., 2014). In striking contrast, mice with point mutations inactivating XPG enzymatic activity are UV sensitive but otherwise normal (Shiomi et al., 2004; Tian et al., 2004), similar to XP-G patients. Thus, distinct from its NER incision function, XPG is essential for normal postnatal development in mammals. However, the nature of this requirement has been unclear.
XPG has multiple non-enzymatic functions outside its role in NER that might contribute to the fatal postnatal phenotype associated with its loss. It interacts directly with both RNA polymerase II and the CSB protein that is essential for initiation of TCR, and it has been implicated in early steps of TCR (Sarker et al., 2005) beyond its requirement for repair per se. In addition, XPG forms a complex with TFIIH and has been reported to be important for its stable association with the CAK kinase subunit (Ito et al., 2007). XPG also has a role in the early steps of base excision repair (BER) of oxidative DNA damage through direct interaction with, and stimulation of, the NTH1 glycosylase (Klungland et al., 1999; Weinfeld et al., 2001).
To better understand the critical postnatal function(s) of XPG, we undertook a series of biochemical and cell biological studies directed toward identifying other protein partners of XPG and elucidating the consequences of its absence. Our unexpected findings establish XPG as a member of the HRR pathway whose loss results in an inability to recover from replication stress, leading to DNA damage and genomic instability. These results suggest a basis for the hitherto unexplained, unusually severe phenotypes of Xpg knockout mice and, importantly, of XP-G/CS patients.
RESULTS
Loss of XPG causes spontaneous DNA damage independent from defective NER
We compared the growth rate of normal (wild type, WT) primary human fibroblasts to that of primary fibroblasts from two unrelated XP-G/CS patients, each with severely truncating XPG mutations (Nouspikel et al., 1997; Okinaka et al., 1997) and no detectable XPG protein (Figure 1B). The XP-G/CS cells had a much slower proliferation rate (Figure 1A), with a doubling time of 38 h compared to 22 h for WT, and they accumulated in G2/M (Figure S1A).
Figure 1. Loss of XPG causes spontaneous DNA damage independent from defective NER.
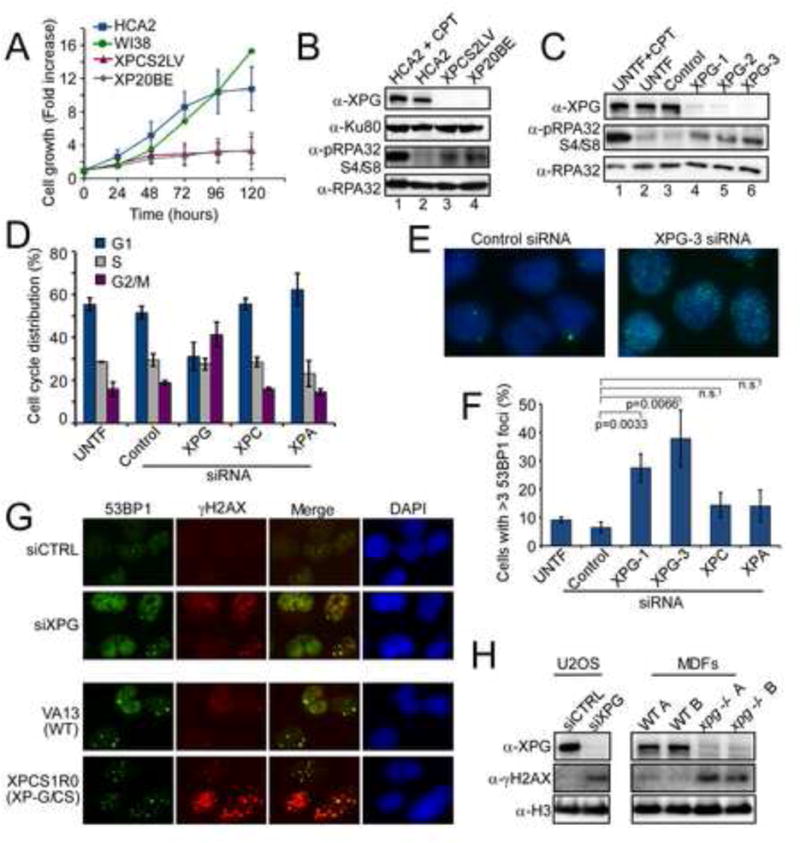
(A) Impaired cell growth of XP-G/CS patient primary fibroblasts, XPCS2LV and XP20BE, compared to normal human fibroblasts, HCA2 and WI38, of the same population doubling age. Data are mean ± SD (N=3); XP20BE (N=2); WI38 was a single experiment.
(B) pRPA32 (Ser4/8) amount in undamaged XP-G/CS patient cells, XPCS2LV and XP20BE, compared to normal human fibroblasts, HCA2. HCA2 cells treated with CPT (2 μM for 1 h, harvested after 4 h) were a positive control. Ku80 was a loading control.
(C) pRPA32 amounts in U2OS cells untransfected (UTF) with or without CPT treatment (2 μM, 1 h), or transfected with control siRNA or 3 different siRNAs targeting XPG.
(D) Cell-cycle progression of siRNA transfected U2OS cells. Knockdown of XPG was with a mixture of two siRNAs, XPG-1 and XPG-3. Data are the mean ± SD for N=3. While depletion of XPG, but not of other NER proteins, resulted in G2/M accumulation, the S-phase fraction did not change (28.5%, 29.5%, and 27.5% for untransfected, control KD, and XPG siRNA, respectively).
(E and F) Immunostaining for 53BP1 in U2OS cells transfected with siRNAs, fixed after 72 h and quantified for cells with >5 53BP1 foci. Data are the mean ± SEM from N=3. XPC and XPA are N=2.
(G) Immunostaining for 53BP1 (green) and γH2AX (red) in U2OS cells transfected with siRNAs. Merged images (foci overlap = yellow) and DAPI (blue) stained nuclei are shown. VA13 (wild-type) and XP-G/CS patient cell line XPCS1R0 were stained as noted for U2OS cells.
(H) γH2AX amounts in U2OS cells depleted of XPG (left) and in primary mouse dermal fibroblasts (MDFs) from two different wt or xpg−/− mice (right). Histone H3 was a loading control.
See also Figure S1.
We examined XP-G/CS cells for increased pRPA32 as a sign of replication stress (Zeman and Cimprich, 2014). Stalled replication forks lead to formation of stretches of single-stranded DNA (ssDNA) and hence to loading of RPA, which is rapidly phosphorylated in the RPA32 subunit (Binz et al., 2004). Consistent with high replication stress, undamaged patient cells lacking XPG had a dramatic increase in pRPA32 compared to undamaged WT (Figure 1B, lanes 3,4 vs. 2), similar to WT after 1 h treatment with 2 μM camptothecin (CPT; lane 1), which causes ssDNA breaks that are converted to DSBs by replication.
Use of patient cells has the disadvantage of absence of isogenic WT controls. In addition there is a possibility that compensatory secondary mutations or epigenetic changes may have occurred during cell culture. Therefore, we used three independent siRNAs to transiently reduce XPG protein to barely detectable levels in U2OS cells (Figure 1C) and compared them to a non-specific siRNA (Control) and untransfected cells (UNTF). XPG depletion caused reduced growth rate and accumulation in G2/M but no change in the fraction of S-phase cells (Figures 1D, S1B,C). Depletion of XPG strongly induced pRPA32 (Figure 1C, lanes 4–6). A similar outcome was observed in XPG-depleted HeLa cells, where the increase was approximately equal to that caused by the addition of 1 μM mitomycin C (MMC) to controls (Figure S1D).
We also measured the impact of XPG loss on foci formation for common markers of DNA damage. XPG-depleted U2OS cells had both significantly elevated 53BP1 foci (Figure 1E,F) and γH2AX foci (Figure 1G), as well as increased γH2AX by western analysis (Figure 1H). Large numbers of γH2AX foci and increased γH2AX were also observed in SV40-transformed XPCS1RO cells from an XP-G/CS patient (Ellison et al., 1998), but not in SV40-transformed WT (VA13) cells (Figures 1G, S1F, lane 2 vs. 1). Importantly, we similarly observed elevated γH2AX in primary dermal fibroblasts derived from xpg−/−mice (Barnhoorn et al., 2014) compared to WT littermate controls (Figures 1H, S1G).
Together these results strongly suggest that loss of XPG, either genetically in human or mouse cells or by siRNA depletion from two different human cell lines, leads to large numbers of DSBs. To exclude that the effects were due to loss of NER, we similarly depleted U2OS cells of either XPC or XPA, since both are essential for global NER and the latter is also essential for TCR. Neither depletion increased pRPA32 (Figure S1E), 53BP1 foci formation (Figure 1F), or G2/M accumulation (Figures 1D, S1C). We conclude that the DNA damage responses engendered by XPG depletion are due to loss of a function distinct from its NER role.
XPG depletion leads to genomic instability
Consistent with induction of DSBs, XPG depletion significantly increased micronuclei in U2OS in the absence of any damaging treatment (Figure 2A). In contrast, depletion of XPC or XPA had no effect, thus ruling out a role for NER. We next examined metaphase spreads of HeLa cells and found a significant ~5-fold elevation in chromatid breaks after depletion of XPG (Figure 2B). This result suggests a role for XPG in HRR, since chromatid breaks are a hallmark of defects in this BRCA-mediated pathway (Roy et al., 2012). Supporting this idea, XPG-depleted fibroblasts were sensitive to MMC (Figure S2), which causes DNA crosslinks that are repaired by HRR (Moynahan et al., 2001), and MMC treatment of XPG-depleted HeLa cells further elevated the level of chromatid breaks (Figure 2B,D). XPG-depleted cells also had acentric and double-minute chromosomal fragments as well as complex aberrations, not observed in control cells (Figure 2C,D). Collectively, these results establish that XPG functions to maintain genomic integrity in undamaged cells and show that its requirement is even more critical after DNA damaging treatments.
Figure 2. XPG is required for genome stability and cell survival.
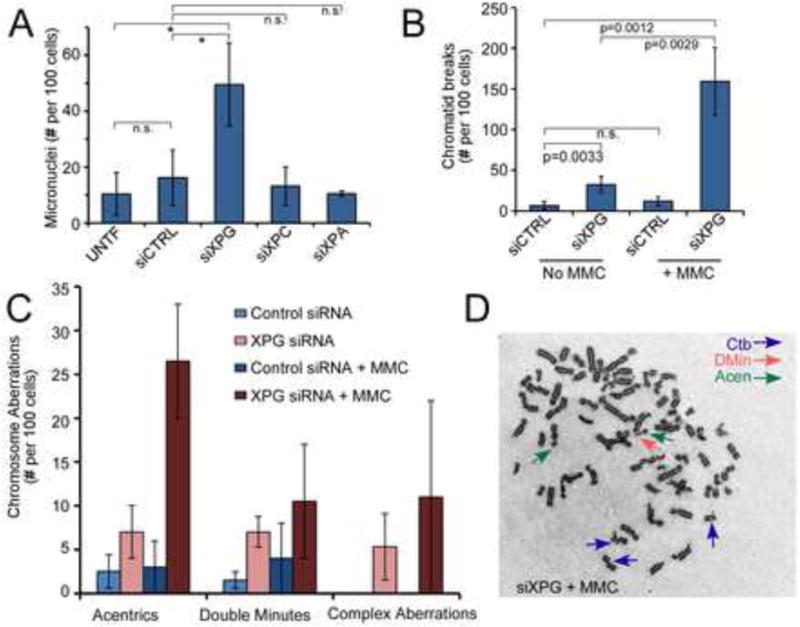
(A) Micronuclei in siRNA transfected U2OS cells. Data represent the mean ± SD for N=2, with XPG depletion significantly higher than control (*, <0.05; n.s., not significant).
(B, C) Chromatid breaks (B) and an array of chromosome aberrations (C) in siCTRL or siXPG depleted HeLa cells either mock or MMC-treated (100 nM, 1 h), scored 24 h following MMC. Data represent the mean ± SD for N=4 (no MMC) or N=2 (with MMC).
(D) Giemsa-stained metaphase spread of MMC-treated XPG-depleted HeLa cells. Arrows show examples of chromatid breaks, double-minute or acentric fragments.
See also Figure S2.
XPG mediates recovery from replication stress
Next, we examined the ability of XPG to mitigate replication stress induced by CPT. Compared to siRNA control, CPT treatment of XPG depleted cells led to a significantly greater induction of both ssDNA as marked by pRPA32 and of DSBs as marked by γH2AX. Consistent with defective repair of DSBs at replication forks, γH2AX persisted and accumulated for at least 72 h (Figure 3A,B). XPG depletion also led to a major delay in cell cycle re-entry after CPT (Figure 3C,D) and to CPT hypersensitivity (Figure S3A).
Figure 3. XPG mediates recovery from replication stress.
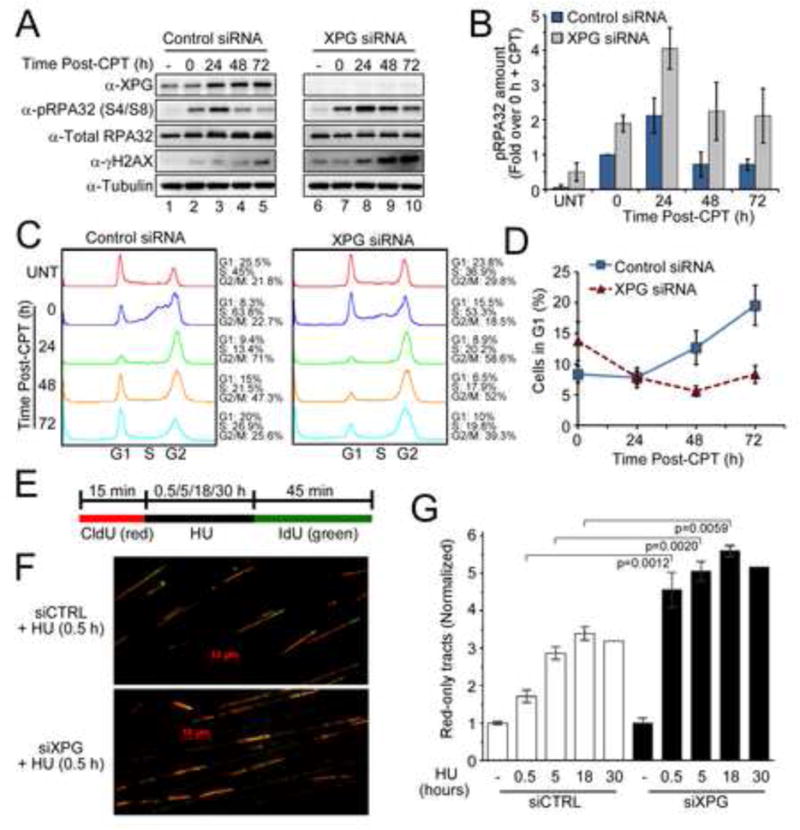
(A) pRPA32 (S4/S8) and γH2AX level in U2OS cells transfected with siRNAs (48 h), treated with or without CPT (20 nM, 24 h), followed by CPT removal and harvest at the indicated times.
(B) Quantification of pRPA32 (S4/S8) protein from (A). Data represent the mean ± SD for N=3.
(C) Cell cycle progression of U2OS cells treated with CPT (A).
(D) G1 cells after CPT treatment (A). Data represent the mean ± SD for N=3.
(E) Diagram of the DNA fiber assay showing the addition of nucleotide analog, CldU (red), for 15 min, addition of HU for 0.5, 5, 18 or 30 h, then IdU (green) for 45 min.
(F) DNA fibers from U2OS cells transfected with siRNAs and treated with HU for 30 min.
(G) Relative fraction of red-only DNA fibers in U2OS cells transfected with siRNAs as indicated and treated with HU for various times. Values were normalized to those in the same cells without HU. The percentages of red-only tracts among all structures assessed were similar and not significantly different (p=0.105) between untreated siCTRL cells (18.55% ± 0.7621) and untreated XPG-depleted cells (14.31% ± 1.877). Data represent the mean ± SEM for N=3 (18 h, N=2; 30 h, N=1).
See also Figure S3.
To directly test the possibility that XPG facilitates replication restart after replication stress, we performed DNA fiber labeling to examine replication fork progression in the presence or absence of hydroxyurea (HU). U2OS cells were pulse-labeled with CldU (red), incubated without or with HU to arrest replication forks, then labeled with IdU (green) (Figures 3E,F, S3B, 3C). Tracts labeled only in red indicate either forks that terminated during the labeling period or forks that stalled or collapsed and were unable to restart. Without HU, the frequency of red-only tracts was not significantly different between control and XPG-depleted cells (Figures 3G, S3C). However, after 30 min of HU treatment the frequency of stalled forks was significantly elevated in XPG-depleted cells (Figure 3F,G). Longer HU treatment further increased the frequency of stalled forks in both control and XPG-depleted cells, but the frequency remained significantly higher at every time point when XPG was depleted (Figure 3G). These results establish a previously unsuspected requirement for XPG in recovery from replication stress.
XPG protein is induced in response to replication stress and accumulates at sites of DSBs
During recovery from CPT damage, XPG protein increased approximately 2-fold and remained elevated for at least 72 h (Figures 3A, 4A). Treatment with the MG132 inhibitor showed that XPG is not regulated by proteasomal degradation (Figure S4A). Rather, qRT-PCR analysis revealed that XPG mRNA levels increased following CPT (Figures 4B, S4B). This up-regulation of XPG is surprising, since it does not occur after UV damage (Christmann et al., 2006). It is consistent with the idea that XPG plays an important role in the cellular response to replication stress.
Figure 4. XPG increases after replication stress and accumulates at sites of DSBs.
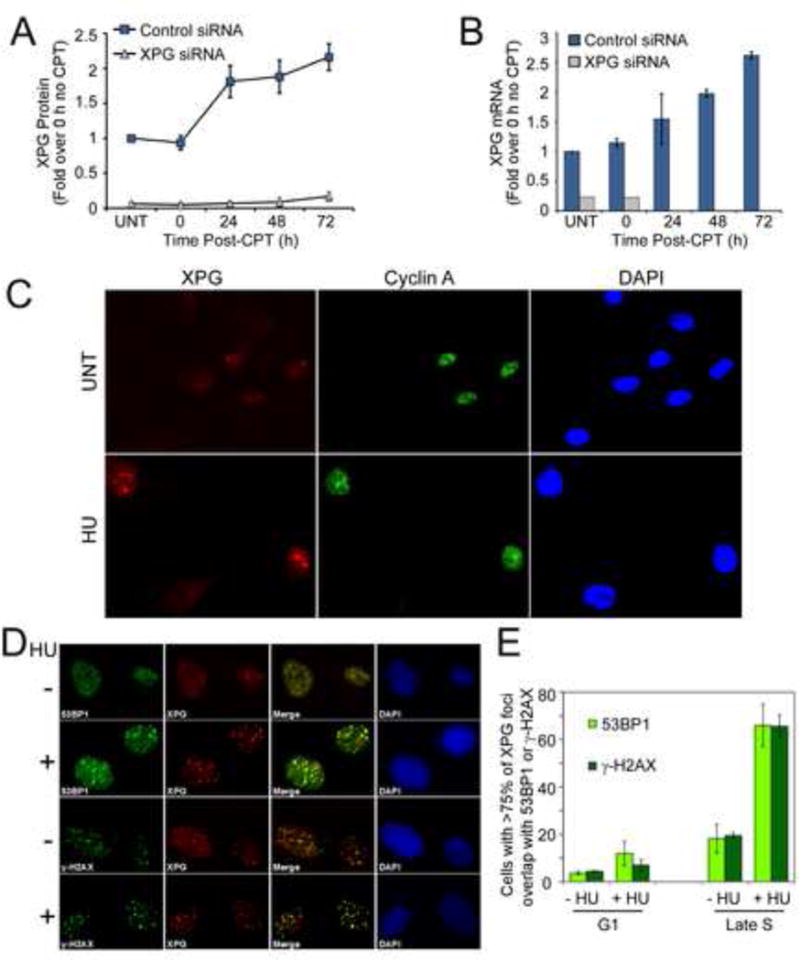
(A) XPG protein amount (Figure 3A) quantified from U2OS cells after CPT treatment with or without siRNA knockdown, shown with the mean ± SD for N=3.
(B) XPG mRNA quantified by qRT-PCR in U2OS cells treated with CPT (20 nM, 24 h). Result in cells transfected with XPG siRNA is shown for undamaged cells (UNT) and 0 h after CPT. Data represent the mean ± SD for N=2 independent experiments run in triplicate.
(C) Immunostaining of XPG (red) and Cyclin A (green) in asynchronous normal human fibroblasts (HCA2) untreated or treated with HU (30 mM, 60 min).
(D) Immunostaining of XPG (red) and either 53BP1 (green) or γH2AX (green) in hTERT-immortalized HCA2 cells synchronized into late S-phase, and then mock (−) or HU (+) treated (30 mM, 1 h). Merged images (foci overlap = yellow) and DAPI (blue) stained nuclei are shown.
(E) Quantification of XPG foci overlap with 53BP1 and γH2AX in G1 or late S-phase. Data represents the mean ± SEM for N=2.
See also Figure S4.
We used immunofluorescence to investigate localization of XPG in normal human fibroblasts and observed distinct nuclear XPG foci in S-phase cells marked by staining for Cyclin A (Figure 4C), whereas G1 cells exhibited very few XPG foci (Figure S4C). The XPG foci became noticeably larger and brighter after HU treatment (Figure 4C). We confirmed that the foci reflect localized XPG by their absence in XP-G/CS cells, by the same pattern of foci formation using a second XPG antibody (Trego et al., 2011) (Figure S4D), and by their reduction after siRNA depletion (Figure S4D).
Since stalled replication forks cause DSBs after collapse, we investigated whether XPG foci overlap with 53BP1 and γH2AX foci in cells synchronized into S-phase with or without HU treatment (Figure 4D). In contrast to G1 cells, approximately 20% of undamaged S-phase cells contained XPG foci that overlapped with 53BP1 and γH2AX, and this fraction increased to ~70% after HU (Figure 4E). We also observed overlapping XPG and 53BP1 foci in asynchronously growing cells after ionizing radiation treatment (Figure S4E).
XPG interacts with HRR proteins
Consistent with recruitment of XPG to HRR at DSBs caused by collapsed replication forks, XPG foci strongly overlapped with RAD51 foci in mid-S phase cells (Figure 5A). To test the possibility that XPG interacts with HRR proteins, we performed reciprocal co-immunoprecipitations (co-IPs) from nuclear extracts of U2OS cells. Pull-down of XPG, BRCA2, or RAD51 in each case resulted in co-IP of the other two, and also of PALB2 (Figure 5B). Moreover, XPG foci overlapped with BRCA2 foci in normal human fibroblasts treated with HU (Figure 5C).
Figure 5. XPG interacts with HRR proteins.
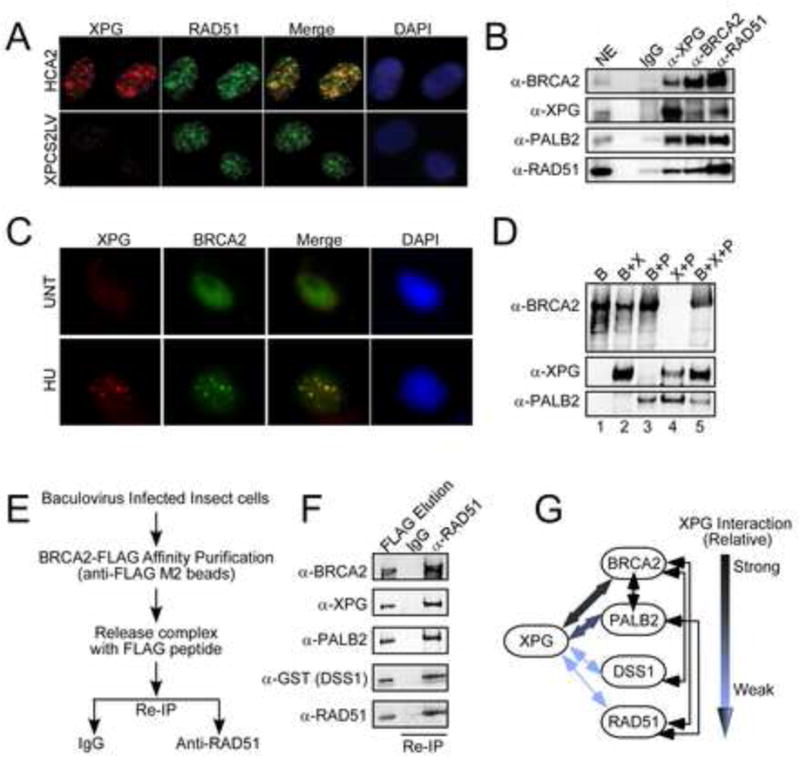
(A) Immunostaining of XPG (red) and RAD51 (green) in HCA2-hTERT (upper panel) and XPG null cells (XPCS2LV, lower panel) synchronized in mid-S phase. The merged images (foci overlap = yellow), and DAPI (blue) stained nuclei are shown.
(B) Co-IP of XPG, BRCA2, RAD51, and PALB2 from U2OS cell nuclear extracts.
(C) Immunostaining of XPG (red) and BRCA2 (green) foci in asynchronous HCA2-hTERT fibroblasts either untreated, or treated with HU (10 mM, 18 h). The merged image (foci overlap = yellow) and DAPI (blue) stained nuclei are shown.
(D) Affinity purification of FLAG-BRCA2 (B) with either XPG (X) and/or PALB2 (P) proteins from coninfected insect cell extracts. In the X+P co-expression (no BRCA2), XPG was FLAG-tagged.
(E) Schematic of FLAG-affinity purification, followed by elution and IP.
(F) Affinity purification of FLAG-BRCA2, then IP with α-RAD51 or control IgG from co-infected insect cell extracts reveals stable complex of five HRR proteins.
(G) Schematic of XPG interactions with HRR proteins.
See also Figure S5.
We next co-expressed HRR proteins in insect cells to identify direct interactions among them. XPG and RAD51 interacted weakly (Figure S5A), but XPG formed a tight complex with either BRCA2 or PALB2 (Figure 5D, lanes 2,4), and the three proteins formed a stable trimeric complex (lane 5). For other co-expression combinations, we first carried out affinity purification of FLAG-tagged BRCA2, elution with FLAG peptide, and IP of another of the co-expressed proteins (Figure 5E) to reveal stable trimeric and tetrameric complexes (Figures S5B,D). Since interaction of BRCA2 with the small highly acidic protein DSS1 facilitates HRR (Kristensen et al., 2010), with DSS1 acting as a DNA mimetic to displace RPA and allow RAD51 loading (Zhao et al., 2015), we wondered whether it might also participate in the HRR complex with XPG. Indeed, XPG interacted weakly with DSS1, although much less strongly than the interaction between BRCA2 and DSS1 (Figure S5C). Importantly, XPG formed a stable, five-membered HRR complex with BRCA2, PALB2, RAD51 and DSS1 upon co-expression from five separate baculoviruses. The complex robustly survived affinity purification, release, and re-IP (Figure 5E,F). Together, these protein-protein interactions (summarized with relative affinities, Figure 5G) substantiate a role for XPG in HRR and strongly suggest that it functions with BRCA2 to promote presynaptic filament formation.
XPG promotes HRR and loads RAD51, BRCA2 and PALB2 following replication stress
We examined whether XPG promotes HRR by measuring gene conversion using a DR-GFP reporter construct (Pierce et al., 1999) integrated into a U2OS cell line (Xia et al., 2006). Depletion of XPG with either of two siRNAs significantly reduced gene conversion to about 50% of control (Figures 6A,S6A). This reduction is comparable to that from loss of RAD51AP1 (Figure 6A) or XRCC3 (Wiese et al., 2007). In contrast, depletion of the NER protein XPC had no effect (Figure 6A), and reduced gene conversion was not due to reduced I-SceI endonuclease expression (Figure S6B). Based on these results, we tested whether XPG depletion sensitizes cells to PARP inhibition, as reported for other HRR proteins (McCabe et al., 2006). We found strong sensitization, comparable to that caused by depletion of BRCA2 itself (Figure 6B).
Figure 6. XPG promotes HRR and chromatin binding of BRCA2, PALB2, and RAD51.
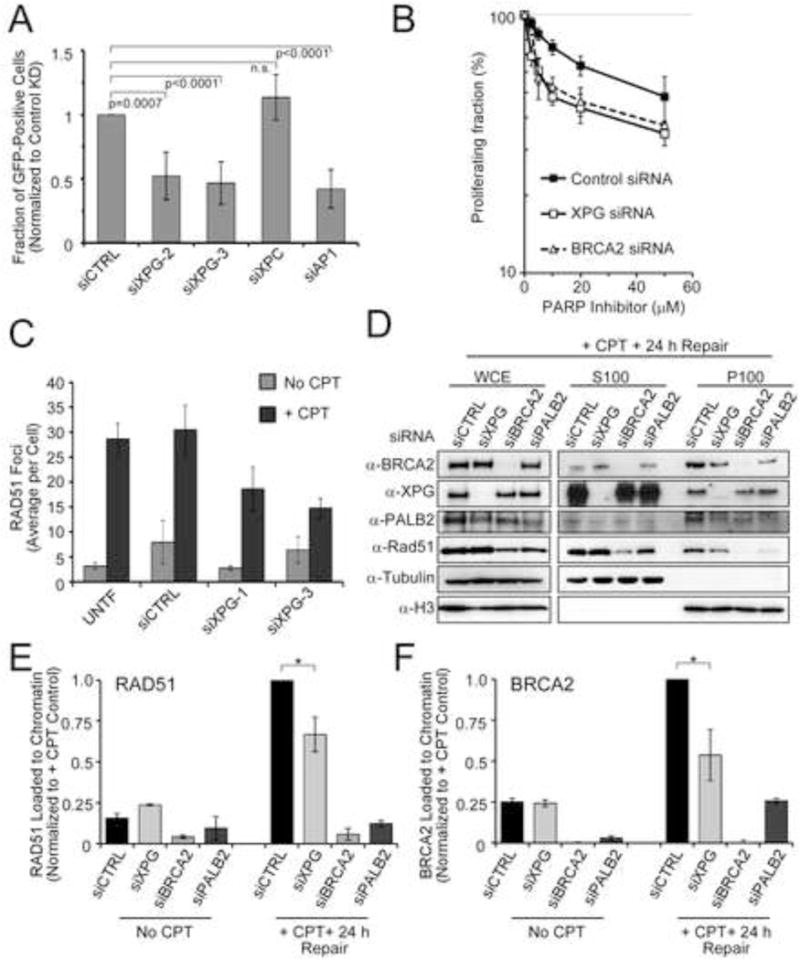
(A) DR-U2OS cells were transfected with siRNAs, followed by transfection with an I-SceI expression plasmid. Data represent the mean ± SD for N=5–13.
(B) Normal human fibroblasts (HCA2-hTERT) transfected with siRNAs were assayed for survival by BrdU incorporation after PARP inhibition by ABT-888. Data represent the mean ± SEM for N=3 (siCTRL, siXPG) and N=2 (siBRCA2).
(C) RAD51 foci formation revealed by immunostaining was quantified in U2OS cells transfected with siRNAs and then treated with or without CPT (20 nM, 24 h). Data represent the mean ± SEM for N=3.
(D) U2OS cells were transfected with siRNAs, treated with CPT (20 nM, 24 h), and harvested for whole cell extracts or fractionated into soluble (S100) or chromatin-bound (P100) proteins.
(E, F) Quantification of RAD51 (E) and BRCA2 (F) chromatin loading from (D). Data represent the mean ± SEM for N=2, with XPG depletion significantly lower than control (*, <0.05).
See also Figure S6.
To understand the mechanism by which XPG promotes HRR, we examined whether loss of XPG affects RAD51 foci formation after CPT. Consistent with slow resolution of pRPA32 (Figure 3), RAD51 foci formation was attenuated (Figures 6C,S6C). This reduction could be due either to overall destabilization of RAD51 protein or its reduced chromatin localization. To distinguish between these, we used biochemical fractionation to examine whole cell extracts (WCE) vs. the soluble (S100) or chromatin-bound (P100) fractions from cells treated or not with CPT. Depletion of XPG did not affect the amount of RAD51 in WCE (Figures 6D,S6D) but significantly reduced RAD51 chromatin loading after CPT (Figure 6E). Furthermore, XPG depletion also significantly reduced BRCA2 chromatin loading (Figure 6F) and reduced PALB2 on chromatin (Figure S6E). Conversely, BRCA2 or PALB2 depletion had no effect on XPG loading (Figure S6F).
XPG modulates BRCA1 phosphorylation and chromatin loading
If XPG exclusively acts with BRCA2 and PALB2 to promote HRR, then the competing single-strand annealing (SSA) pathway, which is mediated by RAD52 and repairs DSBs through annealing short homologous sequences on either side of the break (Lok et al., 2013), should increase after XPG depletion, as it does when either BRCA2 or RAD51 is depleted (Stark et al., 2004). However, in contrast to the dramatic increase in SSA observed upon BRCA2 depletion, depletion of XPG led to a significant decrease in SSA (Figure 7A) as measured in U2OS SA-GFP cells (schematic in Figure S7A) (Gunn and Stark, 2012), while as expected from this pathway’s requirement for extensive resection, depletion of the CtIP nuclease virtually eliminated SSA (Figure 7A). We also tested for an effect of XPG on non-homologous end joining (NHEJ), which predominates outside of S/G2 (Escribano-Diaz et al., 2013). Using U2OS EJ5-GFP cells (schematic in Figure S7B) (Gunn and Stark, 2012), we found that depletion of XPG did not alter NHEJ (Figure S7C). We then asked whether XPG was epistatic with RAD52 in SSA but instead found that simultaneous knockdown of both XPG and RAD52 reduced SSA to a level significantly below that from depletion of RAD52 alone (Figure 7A). Since XPG and RAD52 thus are not epistatic in SSA, we hypothesized that XPG may act upstream of both HRR and SSA, perhaps through interaction with BRCA1, which is required for both.
Figure 7. XPG promotes SSA and affects BRCA1 function.
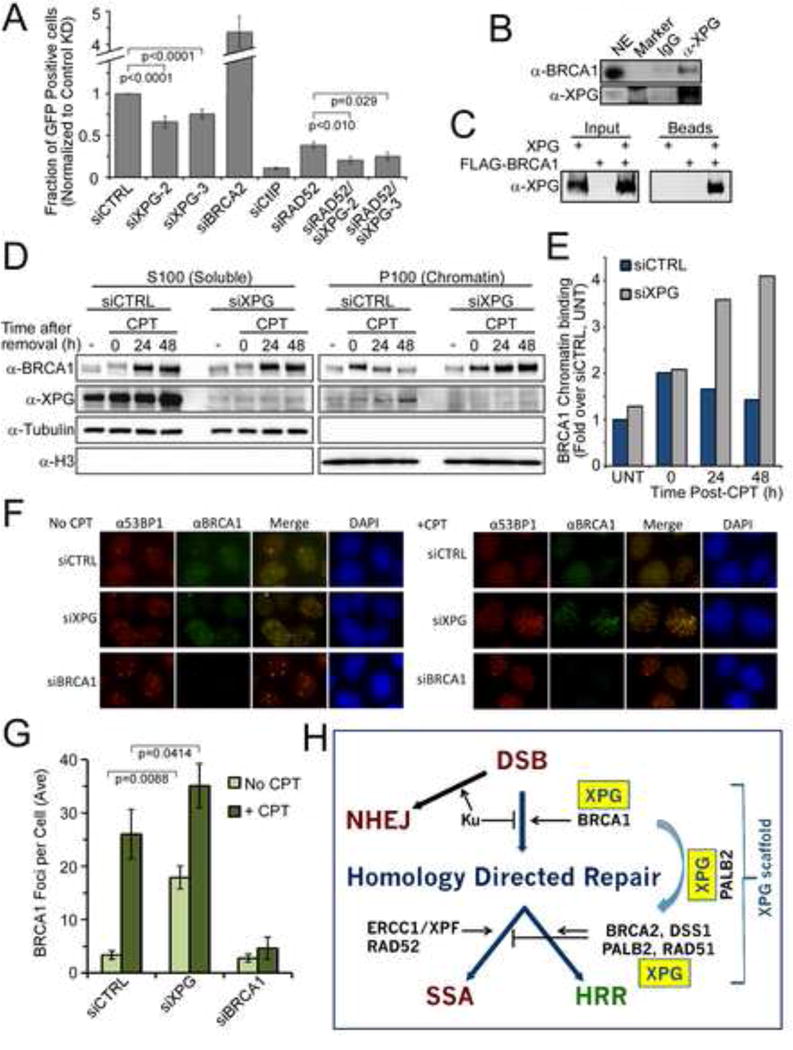
(A) SA-U2OS cells (diagrammed in Figure S7A), were transfected with siRNAs, followed by transfection with an I-SceI expression plasmid. Data represent the mean ± SEM for N=6–15.
(B) Co-IP of BRCA1 with XPG from U2OS cell nuclear extracts.
(C) Affinity purification of FLAG-BRCA1 with XPG from co-infected insect cell extracts.
(D) BRCA1 chromatin loading in U2OS cells transfected with siRNAs, treated with CPT (20 nM, 24 h), harvested at the indicated times after CPT removal, and fractionated into soluble (S100) or chromatin-bound (P100) proteins.
(E) Quantification of BRCA1 chromatin loading.
(F) Immunofluorescence of BRCA1 (green) and 53BP1 (red) foci in U2OS cells transfected with siRNAs, and 48 h later treated with CPT (20 nM, 24 h) (right panel) or mock treated (left panels). The merged image (overlap = yellow), and DAPI (blue) stained nuclei are shown.
(G) BRCA1 foci (Figure 7F) were quantified and plotted. Data represent the mean ± SEM for N=3.
(H) Model for XPG participation in HRR. XPG regulates BRCA1 action in initiation of homology-directed repair, and depletion of XPG reduces both HRR and SSA. XPG additionally acts in a complex with BRCA2, PALB2, DSS1, and RAD51 as a recombination mediator to promote RAD51 presynaptic filament formation in HRR.
See also Figure S7.
We therefore asked whether XPG depletion affects BRCA1 function. By co-IP, XPG and BRCA1 are indeed associated in human cell extracts (Figure 7B). We further verified that they interact directly by co-IP from co-infected insect cells (Figures 7C, S7D). Chromatin fractionation experiments revealed a surprise. In striking contrast to the decreased loading of BRCA2, PALB2, and RAD51, depletion of XPG led to increased and persistent BRCA1 chromatin binding after CPT (Figures 7D,E). In control cells the chromatin bound fraction of BRCA1 was highest immediately after CPT removal and then slowly declined. However, chromatin-bound BRCA1 increased dramatically with time in cells depleted for XPG. These results suggest that XPG is required for BRCA1 release. A possible mechanism is suggested by the fact that, while mobility of BRCA1 is reduced in control cells immediately after removal of CPT and returned to normal with time, the reduced mobility of BRCA1 in XPG-depleted cells was both more pronounced and in fact increased with time (Figures 7D, S7E). The altered mobility was entirely due to hyper-phosphorylation, as demonstrated by phosphatase treatment of the modified form (Figure S7G). Inhibiting ATM or ATR did not reduce the interaction between XPG and BRCA1 (Figure S7H).
XPG depleted cells had significantly increased BRCA1 foci both in untreated cells and after CPT (Figures 7F,G), consistent with increased chromatin binding. As expected, depletion of BRCA1 resulted in increased DNA damage as marked by 53BP1 foci (Figure S7I). Notably, BRCA1 depletion led to an increase in total XPG protein (Figures S7F), similar to that observed as a result of replication stress (Figure 4A), whereas XPG amounts did not change with PALB2 or BRCA2 depletion (Figure 6D).
Taken together, our findings establish that XPG has an important, complex role in responding to replication stress and maintaining genome stability (Figure 7H) through direct protein-protein interactions with key factors both for initiation of HRR and SSA (BRCA1) and for presynaptic filament formation (BRCA2, PALB2, DSS1, and RAD51).
DISCUSSION
Our work reveals an unexpected, multi-faceted function for XPG in HRR that is clearly distinct from its role in NER. Specifically, we show that XPG forms a higher-order complex with BRCA2, PALB2, RAD51 and DSS1, that it also interacts with BRCA1, and that loss of XPG leads to a significant reduction of HRR. The reduced HRR reflects decreased RAD51 foci formation caused by decreased chromatin binding of RAD51, BRCA2, and PALB2. The deleterious cellular consequences observed upon XPG depletion are consistent with its requirement in HRR. These include increased pRPA32 signaling, DSB formation, G2/M accumulation, and sensitivity to the crosslinking agent MMC, the Topoisomerase I inhibitor CPT, and PARP inhibition. Furthermore, stalled replication forks accumulate dramatically upon XPG depletion, correlating with induction of genomic instability as marked by chromatid breaks and micronuclei even in otherwise undamaged cells.
The observed effects of XPG loss on RAD51, BRCA2, and PALB2, together with its direct interactions with each of these proteins, suggest that XPG functions in HRR as a recombination mediator to facilitate presynaptic filament formation. It is perhaps important in this context that XPG interacts directly with RPA through an acidic region in the XPG spacer domain (He et al., 1995), and that the spacer also contains a ubiquitin binding motif (UBM) (Hofmann, 2009). The biological relevance of the XPG interaction with RPA has primarily been viewed in the context of NER, where the interaction may assist in coordinating DNA re-synthesis with XPG incision (Fagbemi et al., 2011). However, since ubiquitylated RPA is involved in regulating repair at stalled replication forks (Elia et al., 2015b), it is possible that the interaction is also functionally important for localizing XPG to stalled replication forks and/or for coordinating RPA removal in complex with BRCA2/PALB2/DSS1.
The interpretation that XPG functions as an additional recombination mediator, while consistent with most of our observations, would not predict its effects on BRCA1. BRCA1 is phosphorylated by ATM and ATR in response to damage or replication stress respectively, and the phosphorylation is important for BRCA1 focal localization (Cortez et al., 1999; Tibbetts et al., 2000). However, little is known about subsequent regulatory steps, including the mechanism of BRCA1 dephosphorylation or consequences of its failure. XPG involvement in regulating BRCA1 activity, perhaps by promoting phosphatase action, is strongly suggested by the striking hyperphosphorylation, increased foci formation, and persistent chromatin binding of BRCA1 when XPG is lost. It is even possible that the downstream effects of XPG loss on RAD51 foci formation and HRR are simply an indirect consequence of its requirement for regulating BRCA1 function. However, the multiple downstream protein-protein interactions of XPG would be difficult to reconcile with this view. We therefore favor the possibility that XPG has two distinct functions in HRR, one in the initiation step through regulatory effects on BRCA1 and another through direct participation in RAD51 presynaptic filament formation with BRCA2 and PALB2.
The most well-understood function of XPG is its required role as the 3′ endonuclease in NER and transcription-coupled NER (TC-NER). However, several lines of evidence strongly suggest that the dramatic BRCA-like phenotype upon loss of XPG as reported here is separable from this role. In support of this idea, depletion of XPC or XPA, which also block NER or both NER and TC-NER respectively, did not increase DSBs or genomic instability. Furthermore, there are strong biological arguments supporting a critical non-enzymatic role for XPG. Data from both mouse models and human patients establish that while XPG is required for normal postnatal development, its endonuclease activity is not. In both cases, inactivating point mutations cause only UV sensitivity, whereas knockouts (mouse) or truncations (patients) cause the severe CS phenotype and very early death (Barnhoorn et al., 2014).
We suggest that, independently of its catalytic activity, XPG functions as a scaffold protein at multiple steps in HRR. However, we have been unable to rescue the HRR defect in either XP-G/CS patient cells or siRNA knockdowns by ectopic expression of WT XPG cDNA (e.g., Figure S1F, lanes 3,4), despite the fact that the NER defect is complemented (Ellison et al., 1998; Staresincic et al., 2009). There are several possible explanations for this observation. Notably, native XPG expression is apparently tightly regulated and responds to replication stress (Fig. 4A,B), and it is possible that this regulation is critical for its HRR function. Furthermore, XPG protein is heavily post-translationally modified, including multiple phosphorylations, ubiquitination, and likely sumoylation (Elia et al., 2015a). A lack of UTR sequences in ectopic expression may result in the dysregulation of RNA-mediated cotranslational protein modifications (Kramer et al., 2009). In addition, there are at least three protein coding isoforms of XPG/ERCC5 (GENCODE build 19 and later, http://gencodegenes.org) plus a gene-merge transcript, BIVM-ERCC5, of unknown function. Thus, ectopically expressed XPG, although active as an endonuclease, may lack some properties that are critical for its homeostasis and scaffolding functions. Of note, inability to rescue knockdowns by ectopic expression is not without precedent and has recently been observed by us in HRR for the NUCKS1 protein (Parplys et al., 2015). Notwithstanding the failure of ectopically expressed XPG to complement phenotypes other than those related to NER incision, the biology of different classes of XPG mutations (inactivating point mutations vs. truncations or deletions) in human patients and in mouse models argues very strongly that XPG has critical functions distinct from its endonuclease activity. Our results as reported here strongly suggest that the XPG role(s) in HRR and in replication fork restart are one such function.
The result of XPG depletion is an approximate 50% reduction in HRR that was consistently observed for several different end points. However, the induction of DSBs, genomic instability, and stalled replication forks when XPG is depleted greatly exceeds that expected from a two-fold decrease in HRR. These discrepancies suggest that other functions of XPG, in addition to the new HRR role(s) described here, likely contribute to genomic stability and replication fork maintenance.
One possibility is that XPG plays an additional role either in replication fork protection or reversal. It is now clear that elaborate mechanisms exist for protecting replication forks that have encountered blocks and for restoring their progress. These mechanisms operate prior to the crisis state of collapse and DSB induction, after which HRR is clearly required. They include protection from excessive resection/degradation, which involves non-HRR functions of both BRCA1 and BRCA2 (Schlacher et al., 2011; Schlacher et al., 2012). Replication fork reversal to form a “chicken-foot” structure is also central to avoiding fork breakage and allowing replication restart, and it is mediated by RAD51 in a role outside of its recombinase function (Zellweger et al., 2015). Processing of reversed forks to allow replication restart requires WRN protein (Thangavel et al., 2015), and a non-enzymatic function of WRN has been implicated in protecting nascent DNA upon fork stalling (Su et al., 2014). Since we show that XPG interacts directly with BRCA1, BRCA2, and RAD51, and since it also has a direct, functional interaction with WRN (Trego et al., 2011), one possibility to explain the extreme accumulation of stalled forks when XPG is lost is that it additionally serves as an important co-factor in one or more of these processes.
Another possibility that must be considered relates to the recent demonstration that the presence of XPG endonuclease activity in cells lacking various RNA processing factors causes extensive DSB formation and genomic instability from cleavage of R-loops (Sollier et al., 2014), which are RNA-DNA hybrids with displaced ssDNA that can form behind transcription. However, we show the opposite: DSBs – presumably at stalled replication forks – and genomic instability are caused by loss of XPG. It is presently unclear whether XPG processes R-loops under normal conditions (i.e. in the presence of RNA processing factors), and if so whether such cleavage is beneficial or deleterious. It is possible that, in the absence of XPG, unprocessed R-loops could lead to DSBs when encountered by replication forks. In this context it is interesting that R-loop cleavage by XPG also requires the TCR protein CSB (Sollier et al., 2014), whose activity is increased by interaction with XPG (Sarker et al., 2005). Loss of either XPG or CSB would therefore both prevent TCR, leading to a dramatic increase in stalled transcription, and increase R-loop formation or persistence. The combined effect could significantly increase replication fork stalling – which would be poorly resolved in the absence of XPG, leading to high levels of replication stress. Thus the relationship between TCR, R-loops, replication fork stalling and collapse, and HRR is evidently complex and certainly poorly understood at present. What is now clear, however, is that XPG occupies a central position in these intertwined processes. Interestingly, CSB has recently been implicated both in recruitment of HRR proteins to oxidative damage at sites of active transcription (Wei et al., 2015) and in the mitotic checkpoint and pathway choice for DSB repair (Batenburg et al., 2015). Although the detailed connection between these findings for CSB and the HRR role of XPG reported here remains to be elucidated, it is increasingly evident that TCR defects in general are likely to have an impact that extends well beyond maintenance of transcription to include effects on genome stability.
In summary, we have demonstrated an unexpected, critical role for XPG in replication fork maintenance and preservation of genomic stability through participation in HRR after replication stress via direct interactions with key HRR proteins. Cell cycle defects, large numbers of DSBs, HRR defects, and elevated DNA damage response signaling ensue when cells are deprived of XPG. The severe disease presentation that manifests in patients with truncating mutations in XPG or in mouse models with deletion of Xpg has never been adequately explained and cannot be reconciled with loss of NER alone. Loss of the HRR role for XPG that is described here, and possibly of additional XPG functions in processing of stalled forks, may represent a major contributing factor in the CS phenotype. The assignment of XPG as an important player in BRCA-mediated HRR and the greatly elevated genomic instability that occurs in its absence also raise the possibility that it functions as a previously unrecognized tumor suppressor gene for breast, ovarian, and other BRCA-associated cancers.
EXPERIMENTAL PROCEDURES
Cell culture and siRNA transfection
Primary and immortalized human cell lines were cultured under ambient oxygen and 10% CO2 in DMEM. Primary mouse dermal fibroblasts were cultured at 3% oxygen, 5% CO2 in DMEM/Hams-F12 media. Both were supplemented with fetal calf serum and 1% antibiotic/antimycotic. siRNAs (40 nM) were transiently transfected with Lipofectamine™RNAiMAX (Invitrogen) on two consecutive days, followed by re-plating and incubation for 24–72 h prior to experimentation.
Whole cell extracts, cellular fractionation, and phosphatase treatment
Cells were lysed in SDS sample buffer (3% SDS, 10% glycerol, 100 mM Tris-HCl, pH 6.8) and heated at 95° C. For experiments with BRCA2, cell lysates were not heated but were needle sheared in the same buffer. Protein concentrations were determined by the BCA assay (Pierce). Fractionation into an S100 fraction containing cytoplasmic and nuclear proteins and P100 fraction containing chromatin, nuclear matrix, and insoluble proteins was performed as previously described (Xia et al., 2006). For phosphatase treatment, the P100 pellets were mock or lambda phosphatase treated according to the manufacturer’s recommendation (NEB), then needle sheared in SDS buffer.
Drug treatments and cellular proliferation
Unless otherwise noted, cells were exposed to 20 nM camptothecin (CPT) or DMSO for 24 h in growth medium. After treatment, cultures were washed with PBS and returned to growth medium or harvested. Mitomycin C (MMC) exposure was in growth medium for 1 h, followed by PBS wash and incubation for 24 h prior to analysis. PARP inhibitor ABT-888 treatment was for 1 h prior to addition of BrdU. Hydroxyurea (HU) treatment was for 1 h at 30 mM or for 16 h at 5 or 10 mM. To measure proliferation after damage, cells were incubated in the presence of BrdU (20 μM) for 72 h, harvested, and analyzed by FACS to detect BrdU-positive cells.
DNA fiber assay, micronuclei and chromosome analysis
The DNA fiber assay, micronuclei assay and chromosome analysis were performed as described (Parplys et al., 2014) (Groesser et al., 2007; Wiese et al., 2007).
Statistics
Statistical significances were determined using the Student’s T test.
Supplemental experimental procedures
Details of cells used, insect cell expression and purification methodologies, reporter assays for gene conversion and SSA, antibodies and protocols used for immunofluorescence and westerns, and other detailed experimental procedures and associated references are found in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Maria Jasin, Jeremy Stark, and Orlando Schärer for cell lines, Ben Brown (ENCODE Consortium) for helpful discussions of ERCC5 coding potential, David Schild for discussions and comments on the manuscript, Gerald Fontenay for assistance with BioSig3D, Caroline Ho, Stanley Leung, Iris Lee and Aya Iwamoto for assistance with western blots, and Cliff Ng for major laboratory support. This work was supported by NIH grants R01 ES019935 (P.K.C.; J.C.), P01 CA092584 (P.K.C.; P.S.), R21 CA187765 (P.K.C.; K.S.T.), P01 AG017242 (J.H.J.H.; J.C.; P.K.C.), R01 ES015252 (P.S.), and R01 ES021454 (C.W.), and by the DOE Low Dose Radiation Research Program, Contract DEAC02-05CH11231.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
K.S.T., T.G., A.C.P., M.S.T., C.W., and P.K.C. designed the studies and analyzed the data. K.S.T., T.G., A.R.D., A.C.P., B.R., A.H., B.S., W.Z., M.R.N., and C.W. performed the experiments and analyzed the data. J.M.P. generated preliminary data that motivated this study. P.S., C.W., J.H.J.H., and J.C. edited the manuscript. K.S.T. and P.K.C. wrote the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and Supplemental Experimental Procedures.
References
- Barnhoorn S, Uittenboogaard LM, Jaarsma D, Vermeij WP, Tresini M, Weymaere M, Menoni H, Brandt RM, de Waard MC, Botter SM, et al. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet. 2014;10:e1004686. doi: 10.1371/journal.pgen.1004686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batenburg NL, Thompson EL, Hendrickson EA, Zhu XD. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015;34:1399–1416. doi: 10.15252/embj.201490041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45:710–718. doi: 10.1016/j.molcel.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Binz SK, Sheehan AM, Wold MS. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst) 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17:1247–1254. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann M, Tomicic MT, Origer J, Aasland D, Kaina B. c-Fos is required for excision repair of UV-light induced DNA lesions by triggering the re-synthesis of XPF. Nucleic Acids Res. 2006;34:6530–6539. doi: 10.1093/nar/gkl895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Buechelmaier ES, Powell SN. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol Cell Biol. 2013;33:387–395. doi: 10.1128/MCB.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- Dray E, Etchin J, Wiese C, Saro D, Williams GJ, Hammel M, Yu X, Galkin VE, Liu D, Tsai MS, et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol. 2010;17:1255–1259. doi: 10.1038/nsmb.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia AE, Boardman AP, Wang DC, Huttlin EL, Everley RA, Dephoure N, Zhou C, Koren I, Gygi SP, Elledge SJ. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol Cell. 2015a;59:867–881. doi: 10.1016/j.molcel.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia AE, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, Lowry E, Gygi SP, Scully R, Elledge SJ. RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol Cell. 2015b;60:280–293. doi: 10.1016/j.molcel.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison AR, Nouspikel T, Jaspers NG, Clarkson SG, Gruenert DC. Complementation of transformed fibroblasts from patients with combined xeroderma pigmentosum-Cockayne syndrome. Exp Cell Res. 1998;243:22–28. doi: 10.1006/excr.1998.4147. [DOI] [PubMed] [Google Scholar]
- Emmert S, Slor H, Busch DB, Batko S, Albert RB, Coleman D, Khan SG, Abu-Libdeh B, DiGiovanna JJ, Cunningham BB, et al. Relationship of neurologic degeneration to genotype in three xeroderma pigmentosum group G patients. J Invest Dermatol. 2002;118:972–982. doi: 10.1046/j.1523-1747.2002.01782.x. [DOI] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Fagbemi AF, Orelli B, Scharer OD. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair. 2011;10:722–729. doi: 10.1016/j.dnarep.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. 2011;108:686–691. doi: 10.1073/pnas.1010959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18:73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- Groesser T, Chun E, Rydberg B. Relative biological effectiveness of high-energy iron ions for micronucleus formation at low doses. Radiat Res. 2007;168:675–682. doi: 10.1667/RR0967.1. [DOI] [PubMed] [Google Scholar]
- Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol. 2012;920:379–391. doi: 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- Harada YN, Shiomi N, Koike M, Ikawa M, Okabe M, Hirota S, Kitamura Y, Kitagawa M, Matsunaga T, Nikaido O, et al. Postnatal growth failure, short life span, and early onset of cellular senescence and subsequent immortalization in mice lacking the xeroderma pigmentosum group G gene. Mol Cell Biol. 1999;19:2366–2372. doi: 10.1128/mcb.19.3.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Henricksen LA, Wold MS, Ingles CJ. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature. 1995;374:566–569. doi: 10.1038/374566a0. [DOI] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Nudler E, Tora L. Transcription-replication encounters, consequences and genomic instability. Nat Struct Mol Biol. 2013;20:412–418. doi: 10.1038/nsmb.2543. [DOI] [PubMed] [Google Scholar]
- Hofmann K. Ubiquitin-binding domains and their role in the DNA damage response. DNA Repair. 2009;8:544–556. doi: 10.1016/j.dnarep.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol Cell. 2007;26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer G, Boehringer D, Ban N, Bukau B. The ribosome as a platform for cotranslational processing, folding and targeting of newly synthesized proteins. Nat Struct Mol Biol. 2009;16:589–597. doi: 10.1038/nsmb.1614. [DOI] [PubMed] [Google Scholar]
- Kristensen CN, Bystol KM, Li B, Serrano L, Brenneman MA. Depletion of DSS1 protein disables homologous recombinational repair in human cells. Mutat Res. 2010;694:60–64. doi: 10.1016/j.mrfmmm.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Lindenbaum Y, Dickson D, Rosenbaum P, Kraemer K, Robbins I, Rapin I. Xeroderma pigmentosum/cockayne syndrome complex: first neuropathological study and review of eight other cases. Eur J Paediatr Neurol. 2001;5:225–242. doi: 10.1053/ejpn.2001.0523. [DOI] [PubMed] [Google Scholar]
- Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17:1260–1262. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdalou I, Lopez BS, Pasero P, Lambert SA. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol. 2014;30:154–164. doi: 10.1016/j.semcdb.2014.04.035. [DOI] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–481. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. [PubMed] [Google Scholar]
- Nouspikel T, Lalle P, Leadon SA, Cooper PK, Clarkson SG. A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Proc Natl Acad Sci USA. 1997;94:3116–3121. doi: 10.1073/pnas.94.7.3116. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- O’Donovan A, Wood RD. Identical defects in DNA repair in xeroderma pigmentosum group G and rodent ERCC group 5. Nature. 1993;363:185–188. doi: 10.1038/363185a0. [DOI] [PubMed] [Google Scholar]
- Okinaka RT, Perez-Castro AV, Sena A, Laubscher K, Strniste GF, Park MS, Hernandez R, MacInnes MA, Kraemer KH. Heritable genetic alterations in a xeroderma pigmentosum group G/Cockayne syndrome pedigree. Mutat Res. 1997;385:107–114. doi: 10.1016/s0921-8777(97)00031-1. [DOI] [PubMed] [Google Scholar]
- Parplys AC, Kratz K, Speed MC, Leung SG, Schild D, Wiese C. RAD51AP1-deficiency in vertebrate cells impairs DNA replication. DNA Repair. 2014;24:87–97. doi: 10.1016/j.dnarep.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parplys AC, Zhao W, Sharma N, Groesser T, Liang F, Maranon DG, Leung SG, Grundt K, Dray E, Idate R, et al. NUCKS1 is a novel RAD51AP1 paralog important for homologous recombination and genome stability. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, Jasin M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell. 2005;20:187–198. doi: 10.1016/j.molcel.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Scharer OD. Hot topics in DNA repair: the molecular basis for different disease states caused by mutations in TFIIH and XPG. DNA Repair. 2008;7:339–344. doi: 10.1016/j.dnarep.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi N, Kito S, Oyama M, Matsunaga T, Harada YN, Ikawa M, Okabe M, Shiomi T. Identification of the XPG region that causes the onset of Cockayne syndrome by using Xpg mutant mice generated by the cDNA-mediated knock-in method. Mol Cell Biol. 2004;24:3712–3719. doi: 10.1128/MCB.24.9.3712-3719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell. 2014;56:777–785. doi: 10.1016/j.molcel.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–1120. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Mukherjee S, Yang Y, Mori E, Bhattacharya S, Kobayashi J, Yannone SM, Chen DJ, Asaithamby A. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 2014;9:1387–1401. doi: 10.1016/j.celrep.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol. 2015;208:545–562. doi: 10.1083/jcb.201406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat Struct Mol Biol. 2010;17:1263–1265. doi: 10.1038/nsmb.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Jones DA, Smith M, Shinkura R, Alt FW. Deficiency in the nuclease activity of xeroderma pigmentosum G in mice leads to hypersensitivity to UV irradiation. Mol Cell Biol. 2004;24:2237–2242. doi: 10.1128/MCB.24.6.2237-2242.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, Abraham RT. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. doi: 10.1101/gad.851000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trego KS, Chernikova SB, Davalos AR, Perry JJ, Finger LD, Ng C, Tsai MS, Yannone SM, Tainer JA, Campisi J, et al. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle. 2011;10:1998–2007. doi: 10.4161/cc.10.12.15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen W, Fousteri M. Mammalian transcription-coupled excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012625. doi: 10.1101/cshperspect.a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Nakajima S, Bohm S, Bernstein KA, Shen Z, Tsang M, Levine AS, Lan L. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc Natl Acad Sci USA. 2015;112:E3495–3504. doi: 10.1073/pnas.1507105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinfeld M, Xing JZ, Lee J, Leadon SA, Cooper PK, Le XC. Factors influencing the removal of thymine glycol from DNA in gamma-irradiated human cells. Prog Nucl Acid Res Mol Biol. 2001;68:139–149. doi: 10.1016/s0079-6603(01)68096-6. [DOI] [PubMed] [Google Scholar]
- Wiese C, Dray E, Groesser T, San Filippo J, Shi I, Collins DW, Tsai MS, Williams GJ, Rydberg B, Sung P, et al. Promotion of homologous recombination and genomic stability by RAD51AP1 via RAD51 recombinase enhancement. Mol Cell. 2007;28:482–490. doi: 10.1016/j.molcel.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Vaithiyalingam S, San Filippo J, Maranon DG, Jimenez-Sainz J, Fontenay GV, Kwon Y, Leung SG, Lu L, Jensen RB, et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol Cell. 2015;59:176–187. doi: 10.1016/j.molcel.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.