

Abstract
Background & Aims
Liver fibrosis is the most worrisome feature of Nonalcoholic Steatohepatitis (NASH). Growing evidence supports a link between hepatocyte apoptosis and liver fibrogenesis. Our aim was to determine the therapeutic efficacy and safety of liver Bid, a key pro-apoptotic molecule, suppression using RNA interference (RNAi) for the treatment of fibrosis.
Methods
First, we optimized the delivery system for Bid siRNA in mice using ten different stealth RNAi siRNAs and two lipid formulations -Invivofectamine2.0 and a newly developed inviofectamine3.0 - that have been designed for high efficacy accumulation in the liver, assessed via real time PCR of Bid mRNA. Next, C57BL/6 mice were placed on a choline-deficient L-amino acid defined (CDAA) diet. After 19 wks of CDAA diet, a time-point that results in severe fibrotic-NASH, mice were injected with the selected Bid siRNA-Invivofectamine3.0 biweekly for three weeks. Additionally hepatocyte-specific Bid-deficient (BidΔhep) mice were placed on CDAA diet for 20 wks.
Results
A maximum Bid knockdown was achieved at 1.5 mg/kg siRNA with inviofectamine3.0, whereas it was at 7 mg/kg with Invivofectamine2.0. In NASH mice, after 3 wks of treatment, BID protein was reduced to 10% and this was associated with an improvement in liver fibrosis and inflammation associated with a marked reduction in TUNEL-positive cells, caspase 3 activation, and a reduction in mitochondrial BAX and BAK. BidΔhep mice showed similar protection from fibrotic changes.
Conclusion
Our data demonstrate that liver Bid suppression by RNAi technology, as well as hepatocyte-specific Bid-deficiency, improves liver fibrosis coupled with a reduction of inflammation in experimental NASH. These findings are consistent with existing evidence that hepatocyte apoptosis triggers HSC activation and liver fibrosis and suggest that Bid inhibition may be useful as an antifibrotic NASH therapy.
Keywords: Bid, liver inflammation, liver fibrosis, apoptosis, mitochondrial dysfunction
Introduction
Metabolic nonalcoholic fatty liver disease (NAFLD) has become one of most common forms of chronic liver disease worldwide. Growing evidence demonstrates that patients within the NAFLD spectrum who have progressed to nonalcoholic steatohepatitis (NASH), in particular NASH and fibrosis, are at a higher risk for disease-related morbidity and mortality [1-3]. The development of novel, effective therapies for patients with more advanced forms of the disease are urgently needed [4]. Hepatocellular apoptosis is emerging as an important, if not critical, mechanism contributing to the progression of fibrotic NASH [5]. In hepatocytes, certain lipids, such as free fatty acids (FFAs), can upregulate the expression of cell death receptors, as well as induce organelle stress, in particular mitochondrial dysfunction (commonly referred to as lipotoxicity), which may lead to apoptosis [5]. Fibrosis is based on the activation of hepatic stellate cells (HSCs) and experimental studies suggest that hepatocyte apoptosis and the resulting apoptotic bodies are important activators of HSCs [6]. Indeed, apoptotic bodies from hepatocytes are engulfed by HSCs, stimulating the fibrogenic activity of these cells; DNA fragments from apoptotic hepatocytes can also activate HSCs [6]. Notably, attenuation of hepatocyte apoptosis by inhibition of caspases, in particular caspase 3 and 8, reduces fibrogenesis in animal models of NASH [7, 8] thus establishing the proof of concept for anti-apoptotic NASH therapy.
BID is a BH3-only BCL-2 family member that is cleaved by caspase-8 into its active form, truncated BID (tBID), which links the extrinsic and intrinsic apoptosis pathways. tBID formation is crucial for the amplification of apoptotic death signals in cells like hepatocytes (called type 2 cells), where activation of the mitochondrial pathway is essential for cell death to occur. BID, however, is dispensable for apoptosis in most other cell types (called type 1 cells). We recently demonstrated that hepatocyte-specific Bid-deficient mice are resistant to the lethal effects of Fas activation in vivo [9]. Here we tested the hypothesis that selective ablation of BID in hepatocytes can effectively reduce liver injury and fibrosis associated with NASH. To test this hypothesis in this study, we used two different approaches: Bid knockdown in wild type (WT) mice via RNAi technology, and hepatocyte-specific Bid-deficient (BidΔhep) mice, both animal group were fed a CDAA diet.
Materials and Methods
siRNA screening
10 different stealth RNAi™ siRNAs were synthesized from Life Technologies (Life Technologies, Carlsbad, USA). Bid target sequences; Bid1: 5′-AGCACAUCACAGACCUGCUGGUGUU-3′, Bid2: 5′-CCGCUCCUUCAACCAAGGAAGAAUA-3′, Bid3: 5′-AGGAAGAAUAGAGCCAGAUUCU GAA-3′, Bid4:5′-CAGAUUCUGAAAGUCAGGAAGAAAU-3′, Bid5:5′-GAAAGUCAGGAAGAAAU CAUCCACA-3′, Bid6: 5′- CAGCUAGCCGCACAGUUCAUGAAUG-3′, Bid7: 5′-GAGAACGACAA GGCCAUGCUGAUAA-3′, Bid8: 5′- GCCAUGCUGAUAAUGACCAUGCUGU-3′, Bid9:5′-CACCA UCUUUGCUCCGUGAUGUCUU-3′, Bid10:5′-CCUAUGUGAGGAACUUGGUUAGAAA-3′. To determine the best Bid target sequence, stealth RNAi ™ siRNAs were combined with invivofectamine2.0 (Life Technologies, Carlsbad, CA, USA) for making complexes according to the manufacturer's instruction and complexes were injected into BALB/C mice at 4 mg/kg. After 2 days of injection, liver Bid mRNA expression level was detected by qPCR. The three most effective stealth RNAi ™ siRNA complexes (Bid3, Bid4, and Bid10) with invivofectamine2.0 were further injected into BALB/C at 7 mg/kg and Bid mRNA expression level was checked by qPCR at 14 days post-injection. The selected Bid siRNA, Bid3, was combined with a new lipid-based delivery reagent, inviofectamine3.0 according to the manufacturer's instruction and the complex was injected into the BALB/C mice at 1.5 mg/kg. After 2 days of injection, liver Bid mRNA expression level was checked by qPCR.
Animal Studies
The use and care of the animals was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Diego (UCSD). Male BALB/C or C57BL/6 mice, 20 – 25 grams of body weight, were purchased from Harlan Laboratories (CA, USA) and were aged between 6 and 8 weeks at the beginning of this study. BALB/C mice were used for siRNA screening. C57BL/6 mice were fed a choline deficient amino acid defined (CDAA) (Dyets, Inc., Bethlehem, PA, USA) diet for 22 weeks to induce NASH. During the last 3 weeks of the feeding course, mice fed with a CDAA diet received weekly administration of the Bid siRNA complex, Negative siRNA complex, or Control (PBS) via intravenous injection (1.5 mg/kg at 1st week, and 0.5 mg/kg at 2nd and 3rd weeks). BidΔhep mice were fed a CDAA diet for 20 weeks to induce NASH.
Liver and blood sample preparation
All mice were sacrificed at the termination of treatment (22 weeks of CDAA diet) under anesthesia via i.p. injection using a 21G needle and a mixture of 100 mg/kg of Ketamine and 10 mg/kg of Xylazine dissolved in a 0.9% saline solution with euthanasia carried out by carbon dioxide exposure. Whole mouse blood was collected by cardiac puncture and disgorged into tubes with or without anticoagulant. Liver tissue was fixed in 10% formalin for 24 h and embedded in paraffin, quickly frozen in OCT (Sakura Finetek, Torrance, CA, USA), and incubated with RNAlater Solution (Life Technologies) for RNA extraction. The remaining liver tissue was quickly frozen in liquid nitrogen and stored at -80°C. Serum was used for ALT measurement via Infinity ALT (Thermo Scientific, Waltham, MA, USA) or insulin level using mouse ultrasensitive insulin ELISA (ALPCO, Salem, NH, USA).
Measurement of extracellular vesicles
Blood was centrifuged at 1,200 g for 15 min and 12,000 g for 12 min at 22°C to obtain platelet free plasma (PFP). PFP was incubated with Calcein-AM (Life Technologies) for 30 minutes at room temperature. EV count was performed using 2.5-μm Alignflow alignment beads (Life Technologies) as the size standards for flow cytometry, BD LSRII Flow Cytometer System, (BD Biosciences, San Jose, CA). The data were analyzed using FlowJo software (TreeStar Inc., Ashland, OR).
Liver histology and Immunostaining
Tissue sections were prepared and stained for hematoxylin and eosin. Steatosis and liver fibrosis were assessed via Sirius Red staining - liver sections were incubated for 2 h at room temperature with Fast Green FCF (Fisher scientific, Pittsburgh, PA, USA) and Direct Red (Sigma-Aldrich, St. Louis, MO, USA) in saturated picric acid (Sigma-Aldrich). Immunohistochemistry staining for myeloperoxidase (Myeloperoxidase Ab-1, Thermo Scientific) or Ly6C (Abcam, Cambridge, MA, USA) was performed in paraffin embedded or frozen liver sections according to the manufacturer's instruction. All pictures were taken by NanoZoomer 2.0HT Slide Scanning System (Hamamatsu, Japan) and quantitated on Image J software. Frozen liver sections were stained for active BAX with anti-BAX (6A7) (Abcam) antibody followed by the Alexa 488 anti-mouse 2nd antibody (Life Technologies) according to the manufacturer's instruction, Oil Red O staining using Oil red O (Sigma-Aldrich) in 60% 2-Propanol (Sigma-Aldrich), or for terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay (Roche, Drive Pleasanton, CA, USA). Oil Red O, active BAX, or TUNEL staining was observed using immunofluorescence microscopy (Olympus, USA).
Liver cell isolation
Liver cells were collected as previously described [10]. Briefly, C57BL/6 or BidΔhep mouse liver was digested with collagenase perfusion through portal vein and isolated parenchymal cells with centrifugation at 50 g for 1 min following centrifugation with Nycodenz gradient at 2000 g for 20 min for non-parenchymal cells.
In-vitro cell culture studies
HepG2 cells were grown and maintained in Dulbecco's Modified Eagle Medium (Gibco, Camarillo, CA) supplemented with 10% fetal bovine serum (Cellgro, Manassas, VA), Sodium pyruvate (Gibco), Penicillin and Streptomycin (growth medium) at 37°C in a 5% CO2 incubator. HepG2 were reverse transfected with Bid or Negative Silencer Select siRNA (Life Technologies) with Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's instruction. At 36 h post-transfection, cells were incubated with 50 ng/ml anti-human CD95 (Jo2) (BD Biosciences) for 12 h and were collected for RNA extraction or caspase 3 activity assay (Promega, Madison, WI).
Immunoblot Analysis
For immunoblot analysis 50μg of whole-liver lysate, as well as mitochondria or cytosolic fraction with mitochondria isolation kit (Thermo scientific, Rockford, IL, USA), was resolved by a 4–20% gradient gel, transferred to a nitrocellulose membrane, and blotted with the appropriate primary antibodies. Membranes were incubated with peroxidase-conjugated secondary antibody (Cell signaling, Danvers, MA, USA). Protein bands were visualized using an enhanced chemiluminescence reagent and digitized using a CCD camera (ChemiDoc®, BioRad, Hercules, CA, USA). Expression intensity was quantified by ImageLab (BioRad). A rabbit anti-Bid, anti-BAX, anti-BAK, anti-cytochrome C, anti-cleaved caspase 3, anti-caspase 3, anti-cleaved caspase 8, or anti-caspase 8 antibody was purchased from Cell Signaling and anti-α-SMA and anti-GAPDH were purchased from GeneTex (Irvine, CA, USA). Protein load was verified using GAPDH (GeneTex), or PORIN (GeneTex) antibody.
Real-time PCR
Total RNA was isolated from liver tissue using Trizol (Life Technologies) followed by an RNA purification column (Life Technologies) from cultured cells using RNA purification column according to the manufacturer's instruction. The cDNA was synthesized from 1μg of total RNA using the SuperScript VILO cDNA Synthesis kit (Life Technologies). Real-time PCR quantification for liver mRNA expression was performed using a TaqMan gene expression assay from Life Technologies, or SYBR-Green, and the CFX96 Thermal Cycler from BioRad. The sequences of the primers used for quantitative PCR are listed in supplemental Table 1. Mean values were normalized to β2 microglobulin for mRNA.
Statistical analyses
All data are expressed as mean ± SEM unless otherwise noted. Data were analyzed using Oneway Anova in siRNA screening and experimental NASH model or t-tests in BidΔhep mice using Graph Pad (Graph Pad Software Inc., CA, USA) for comparison of continuous variables. Differences were considered to be significant at p ≤ 0.05.
Results
Bid suppression in NASH mice using RNAi technology
In order to achieve efficient gene knockdown using RNAi technology, we initially concentrated our efforts on identifying and selecting a target sequence. For this study we synthesized 10 different target sequences and checked the liver Bid mRNA expression level using a low dose RNAi treatment (4 mg/kg) for the short time point at day 2 or a high dose RNAi treatment (7 mg/kg) for the long time point at day 14. We selected three siRNAs - Bid3, Bid4, and Bid10 -from the short time point, and then decided that Bid3 was the best target sequence to produce an efficient Bid knockdown (p<0.001) (Fig 1A, B). We next compared knockdown efficiency using Invivofectamine2.0, which was used for the initial siRNA screenings, to the next generation siRNA delivery reagent, Invivofectamine3.0, that has been designed for high efficacy accumulation in the liver [11]. We observed significant Bid knockdown at 1.5 mg/kg with Invivofectamine3.0 (p<0.001) as compared to 7 mg/kg of Invivofectamine2.0 (Fig. 1C). We next used our most efficient construct, Bid3 siRNA, for the treatment protocol. For this we placed C57BL/6 mice on a CDAA diet for 19 wks, which causes severe steatohepatitis and liver fibrosis, we then injected buffer (Control), Neg siRNA complex, or Bid siRNA complex weekly for three weeks, 1.5 mg/kg initially and 0.5 mg/kg on week 2 and week 3 to provide a booster effect (Fig 1D). After 22 wks of CDAA diet and the aforementioned three weekly siRNA injections, mice were sacrificed. We confirmed via qPCR and Western blotting that liver Bid mRNA (p<0.001), as well as BID protein (p<0.001), was significantly reduced by our specific Bid siRNA complex therapy (Fig. 1E-1G).
Fig. 1. Hepatic Bid suppression in mice via siRNA.
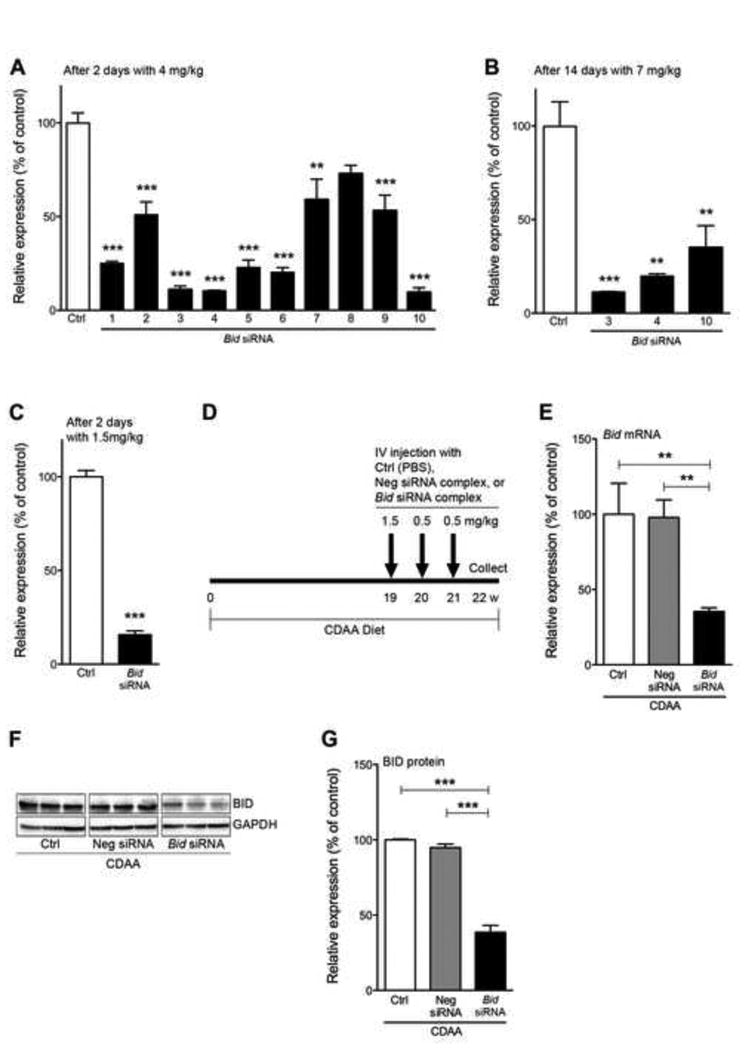
Optimization of the siRNA delivery system in wild type mice (A-C) and Bid suppression in NASH mice (D-G). (A) Relative expression of liver Bid mRNA after 2 days with 4 mg/kg Bid siRNA (10 different target sequences, no.1-10) -Invivofectamine2.0 complex. **P<0.01, ***P<0.001. (B) Relative expression of liver Bid mRNA after 14 days with 7 mg/kg Bid siRNA (no.3, 4, or 10) -Invivofectamine2.0 complex. **P<0.01, ***P<0.001. (C) Relative expression of liver Bid mRNA 2 days with 1.5 mg/kg Bid siRNA (no.3) -Invivofectamine3.0 complex. (D) Experimental design. (E) Relative expression of liver Bid mRNA at termination of treatment (22 wks) with control, Neg siRNA, or Bid siRNA. **P<0.01, ***P<0.001. (F) Protein expression of BID in whole liver by immunoblotting from mice fed a CDAA diet administered with control, Neg siRNA, or Bid siRNA. (G) Bar graph shows quantification of BID protein expression from immunoblotting. ***P<0.001. Values are mean ± SEM. Ctrl: Control, Neg siRNA: negative siRNA
Bid knockdown reduces circulating levels of extracellular vesicles and improves inflammation in mice fed a CDAA diet
The Bid siRNA treatment in NASH mice did not affect mouse body weight (Fig. 2A), ratio of liver weight / body weight (Fig. 2B), the serum levels of the enzyme ALT (Supplementary Fig. 1A), or serum levels of insulin (Supplementary Fig. 1B). The number of circulating extracellular vesicles (EVs), a novel non-invasive biomarker of liver damage in NASH [12], showed a trend towards decrease in CDAA-fed mice treated with Bid siRNA but this was not statistically significant (Fig. 2C). Histological examination showed that mice fed a CDAA diet for 22 weeks showed severe inflammatory activity and significant lipid accumulation in the liver (Fig. 2D-E). Notably, liver damage was significantly reduced in CDAA fed mice treated with Bid siRNA (Fig. 2D), although the degree of liver steatosis was not changed (Fig. 2E). The reduction of liver damage with concomitant reduction in number of circulating EVs led us to further investigate liver inflammation. The degree of neutrophilic infiltration, assessed via MPO staining, was significantly reduced in NASH mice treated with Bid siRNA when compared to control (p<0.01) or Neg siRNA (p<0.01) treated NASH mice (Fig. 2F and 2H). Moreover, infiltrated Ly6C positive inflammatory monocytes were reduced in the Bid siRNA treatment animal, whereas aggregates of Ly6C positive cells were observed in control or Neg siRNA treated NASH mice (Fig. 2G). The expression of liver inflammatory genes, such as IL-6 (p<0.05), MIP-1α (p<0.05), and KC (p<0.05) was reduced in Bid siRNA treated NASH mice when compared to control animals (Fig. 2I).
Fig. 2. Bid knockdown reduces circulating levels of extracellular vesicles and improves inflammation independent of steatosis in mice fed a CDAA diet.
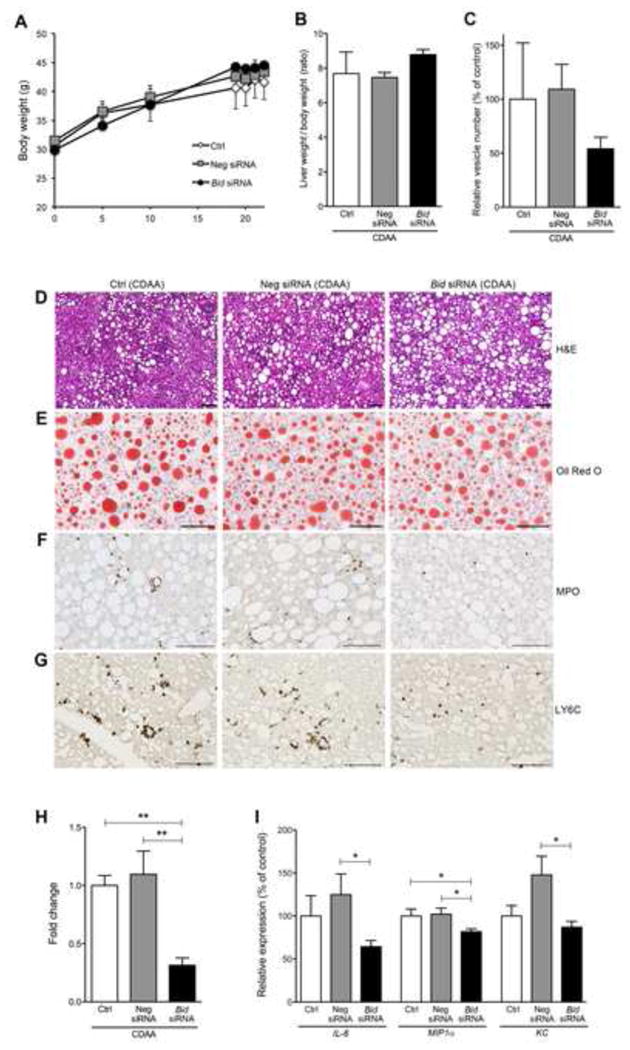
The effect of Bid suppression in (A) body weight (B) ratio of liver weight / body weight, and (C) circulating extracellular vesicles in mice fed a CDAA diet administered with control, Neg siRNA, or Bid siRNA. Liver histology of Bid suppression revealed no change in steatosis, but did show reduced liver inflammation in comparison to control (D-G). (D) Haematoxylin-eosin staining of liver sections in mice fed a CDAA diet administered with control, Neg siRNA, or Bid siRNA. Scale bar, 100 μm. (E) Oil Red O staining of liver sections in mice fed a CDAA diet administered with control, Neg siRNA, or Bid siRNA. Scale bar, 100 μm. (F) Immunohistochemical staining specific for MPO (neutrophils) (F) or Ly6C (G) of liver sections in mice fed a CDAA diet administered with control, Neg siRNA, or Bid siRNA. Scale bar, 100 μm. (H) Bar graph shows quantification of MPO positive cells. ** p<0.01. (I) Gene expression of inflammatory genes as measured by qPCR. All gene expression levels were normalized to housekeeping control, β2 microglobulin, and shown relative to the expression levels of mice fed a CDAA diet administered with control. *p<0.05. Values are mean ± SEM. Ctrl: Control, Neg siRNA: negative siRNA
Liver fibrosis is reversed in CDAA mice treated with Bid siRNA
A reduction of liver damage in NASH mice treated with Bid siRNA led us to examine specific markers linked to liver fibrogenesis and fibrosis, namely hepatic stellate cell (HSCs) activation and collagen deposition. The mice fed a CDAA diet for 22 wks, coupled with the Neg siRNA injection protocol, showed an increase in collagen deposition, as assessed via morphometric quantitation of Sirius Red stained livers, whereas the collagen deposition was significantly reduced in NASH mice treated with Bid siRNA (p<0.05) (Fig. 3A and 3B). Furthermore, α-SMA protein expression in the liver was significantly reduced in Bid siRNA treated NASH mice when compared to control treated (p<0.05) or Neg siRNA treated (p<0.01) NASH mice (Fig. 3C and 3D). Moreover, the expression of liver fibrogenic genes, such as TIMP-1 (p<0.05), CTGF (p<0.05), was significantly decreased in Bid siRNA treated NASH mice when compared to control animals, while α-SMA expression showed a similar trend but the reduction was not significant (Fig. 3E).
Fig. 3. Bid siRNA treatment reversed NASH fibrosis through a reduction in HSC activation.
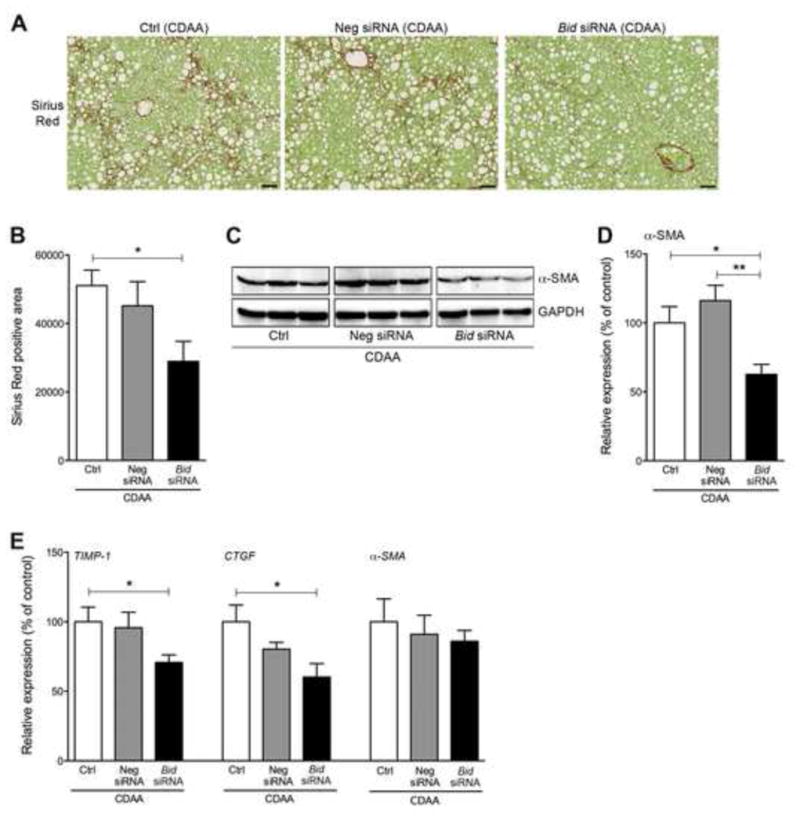
(A) Sirius Red staining of liver sections in mice fed a CDAA diet administered with control, Neg siRNA, or Bid siRNA. Scale bar, 250 μm. (B) Bar graph shows quantification of Sirius Red positive area from liver sections. * p<0.05. (C) Protein expression of a-SMA in whole liver using immunoblotting. (D) Bar graph shows quantification of positive area of Sirius Red from liver sections stained with Sirius Red. **p<0.01, * p<0.05. (E) Gene expression of fibrogenic genes as measured by qPCR. All gene expression levels were normalized to housekeeping control, β2 microglobulin, and shown relative to the expression levels of mice fed a CDAA diet treated with control. * p<0.05. Values are mean ± SEM. Ctrl: Control, Neg siRNA: negative siRNA
Bid knockdown reduces liver cell apoptosis via protection of mitochondrial function
BID protein, a BH3-only subgroup of the BCL-2 family, triggers cell death in hepatocytes through the translocation of its activated cleaved form to the outer mitochondrial membrane resulting in mitochondrial permeabilization and dysfunction, a process that is key during lipotoxicity associated with NASH [13]. Therefore, we hypothesized that the beneficial effect observed in the Bid siRNA treated mice is due, at least in part, to mitochondrial protection and a reduction in cell death. TUNEL positive cells were significantly reduced in Bid siRNA treated NASH mice when compared to Neg siRNA (p<0.001) or control (p<0.001) treated NASH mice (Fig. 4A and 4B). The reduction of cell death via Bid suppression led us to further investigate mitochondrial dysfunction. Mitochondrial permeabilization was determined by assessing cytochrome c release from the mitochondria into the cytosol in the livers of the different groups of mice. Untreated mice and those treated with Neg siRNA showed a significant reduction of cytochrome c in the mitochondrial fraction, while cytochrome c was kept within the mitochondrial fraction in the livers of mice treated with Bid siRNA (P<0.05) (Fig. 4C and 4D). In addition, the recruitment of BAX and BAK proteins to the mitochondria was reduced in NASH mice treated with Bid siRNA when compared to Neg siRNA (BAX: P<0.01) or control mice (BAX: P<0.05, BAK: P<0.001) (Fig. 4C-4E). Active BAK expression, visualized via immunofluorescence, was also reduced in NASH mice treated with Bid siRNA when compared to Neg siRNA or control mice (Supplementary figure 2A). Furthermore, the inhibition of mitochondrial dysfunction resulted in a reduction of cleaved caspase 3 (downstream of mitochondrial dysfunction) in the Bid siRNA treatment group as compared to the Neg siRNA or control (p<0.05) groups (Fig. 4G and 4H), whereas no difference was detected in cleaved caspase 8 expression (upstream of BID pathway) (Fig. 4G and 4H), as well as full caspase 8 and full caspase 3 (Fig. 4G). To confirm the link between the inhibition of apoptotic cell death and BID reduction in vitro, we established BID reduced HepG2 cells uisng Bid siRNA following stimulation with Jo2 and assessed caspase 3 activity. The activity of caspase 3 activity was significantly inhibited in both, 50% or 80% of Bid mRNA reduction (Supplementary Fig. 2B and 2C).
Fig. 4. Bid suppression reduced cell death through inhibition of mitochondrial dysfunction.
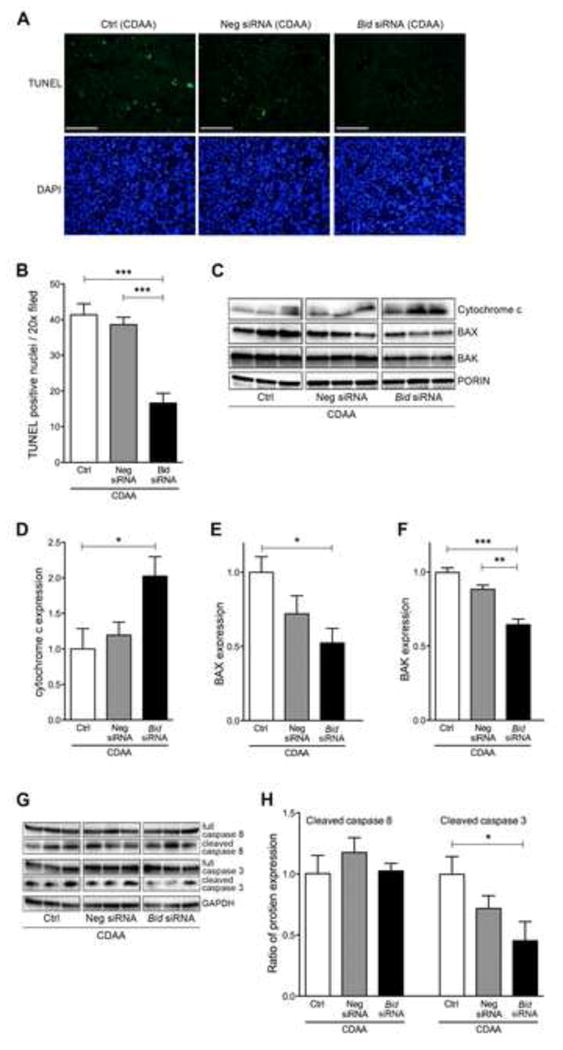
(A) TUNEL staining of liver sections in mice fed a CDAA diet administered with control, Neg siRNA, or Bid siRNA. Scale bar, 100 μm. (B) Bar graph shows quantification of TUNEL positive cells. *** p<0.001. (C) Protein expression of cytochrome c, BAX, BAK, and PORIN in mitochondrial fraction of liver using immunoblotting. Bar graph shows quantification of (D) cytochrome c, (E) BAX, and (F) BAK. ***p<0.001, **p<0.01, * p<0.05. (G) Protein expression of cleaved caspase 3, full caspase 3, cleaved caspase 8, full caspase 8, and GAPDH in whole liver using immunoblotting. (H) Bar graph shows quantification of cleaved caspase 3 or cleaved capase 8. * p<0.05. Values are mean ± SEM. Ctrl: Control, Neg siRNA: negative siRNA
Germline Bid suppression in hepatocytes mimics the protection observed by RNAi therapy
Our findings showing the reduction of several features of disease severity in NASH mice treated with Bid siRNA led us to further explore cell specificity within these data. Since the IV injected siRNA-lipid complex mainly targets the liver, particularly hepatocytes [14], we hypothesized that BID knockdown in hepatocytes, rather than non-parenchymal cells in the liver, is crucial for the protective effect induced by this treatment. To test this hypothesis we used hepatocyte specific Bid deficient mice (BidΔhep) recently developed in our lab [9]. To confirm BID depletion in hepatocytes, we isolated hepatocytes and nonparenchymal cells (NPC). α-SMA was detected in NPC from WT and BidΔhep, but not in hepatocytes from WT and BidΔhep, as an indicator of appropriate isolation (Supplementary Fig. 3A). BID depletion was observed only in hepatocytes from BidΔhep mice, whereas BID was detected in hepatocytes from WT, as well as NPC from WT and BidΔhep mice (Supplementary Fig. 3A). We placed WT or BidΔhep mice on a CDAA diet for 20 weeks and at the completion of this period mice were sacrificed and tissues were harvested. Body weight, ratio of liver weight / body weight, and epididymal adipose tissue weight were similar between WT and BidΔhep mice fed a CDAA diet (Fig 5A, 5B, and Supplementary Fig. 4A). Furthermore, the serum levels of the enzyme ALT, or serum levels of insulin were similar between WT and BidΔhep mice fed a CDAA diet (Supplementary Fig. 4B and 4C). The degree of collagen deposition within the liver, as visualized by Sirius Red staining (p<0.05) (Fig. 5C and 5D) and qPCR for fibrogenic genes, was reduced in CDAA fed BidΔhep mice when compared to CDAA fed control animals (Supplementary Fig. 4D). The degree of neutrophilic infiltration, assessed via MPO staining, was significantly reduced in BidΔhep mice when compared to CDAA fed control animals (p<0.05) (Fig. 5E and 5F). Markers of liver inflammation – F4/80, Ly6c, IL-6, IL-1β , and MIP1α – were non-significantly reduced in BidΔhep mice as determined by qPCR (Fig 5G). These changes were associated with a significant reduction in TUNEL positive cells in BidΔhep mice when compared to WT animals (p<0.05) (Fig 5H).
Fig. 5. Hepatocyte BID is the key to control liver damage.
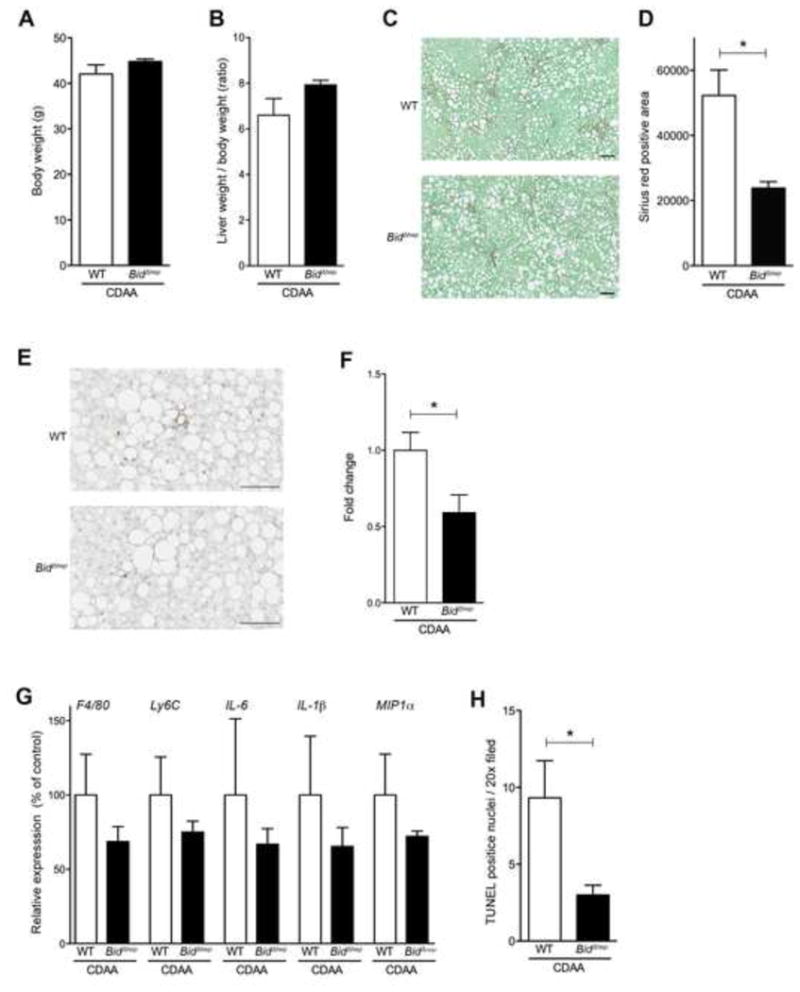
hepatocyte-specific BID knockout (BidΔhep) mice fed a CDAA diet for 20 weeks had reduced liver damage compared to wild type mice fed the same diet. (A) The difference between wild type (WT) and hepatocyte-specific BID knockout mice fed a CDAA diet in (A) body weight and (B) ratio of liver weight / body weight. (C) Sirius-red staining of liver sections from WT or BidΔhep mice fed a CDAA diet. Scale bar, 100 μm. (D) Bar graph shows quantification of Sirius Red positive area.. * p<0.05. (E) Immunohistochemical staining specific for MPO (neutrophils) in liver sections from WT or BidΔhep mice fed a CDAA diet. Scale bar, 100 μm. (F) Bar graph shows the quantification of MPO positive cells. * p<0.05. (G) Gene expression of inflammatory genes as measured by qPCR. All gene expression levels were normalized to housekeeping control, β2 microglobulin, and shown relative to the expression levels of WT mice fed a CDAA diet. * p<0.05. (H) Bar graph shows quantification of TUNEL positive cells. * p<0.05. Values are mean ± SEM.
Discussion
The main findings of the present study relate to the role of RNA-based therapy to modulate hepatic Bid, a key pro-apoptotic protein that triggers mitochondrial dysfunction during lipotoxicity, as a potential novel therapeutic strategy for NASH. Bid siRNA suppression via next generation siRNA technology lead to a reduction of fibrosis associated with a reduction in liver inflammation and apoptotic cell death. Bid knockdown in hepatocytes, rather than non-parenchymal cells in the liver, is crucial for the protective effect induced by this treatment as mice with hepatocyte-specific Bid deficiency showed a similarly protective phenotype.
Targeting hepatocyte cell death has evolved as an attractive, mechanism based treatment strategy for NASH [5, 15, 16]. However, the complexity of targeting cell death pathways relevant to NASH development comes from the recognition that, in many instances, hepatic cell death represents a highly heterogeneous process with frequent overlap and crosstalk between involved pathways. As a result, inhibiting a particular pathway may induce molecular transitions between different modalities triggering cell death by other mechanisms.
Indeed, studies blocking caspases, in particular caspase 8 have suggested a potential switch into a death receptor-induced receptor protein kinases 1 and 3 (RIP1 and RIP3)-dependent necroptotic cell death [17, 18]. Conversely, the use of selective RIP3 inhibitors have been recently shown to trigger apoptotic cell death [19]. We have recently demonstrated that hepatocyte-specific deletion of BID, which does not interfere with death receptor-induced caspase 8 activation, did not induce programmed necrosis and resulted in significant protection against Fas-induced liver injury [9] and hepatocarcinogenesis [20]. In addition, inhibiting cell death in extrahepatic tissues may result in unwanted side effects. Most non-hepatocytes are so-called type I cells, where death receptor-induced cell death is not dependent on BID signaling and mitochondrial amplification [21], thus inhibiting Bid in these cells might not play a crucial anti-apoptotic role. Hepatocytes, on the other hand, are classified as type II cells (where BID and mitochondria are necessary to amplify the apoptotic signal) [13]. Taken together, these data provide a strong rationale to target BID activation, as opposed to its upstream (caspase 8) or downstream (caspase 3) counterparts, as an ideal therapeutic strategy to reduce hepatocyte lipotoxicity, cell death and subsequent sterile inflammation. Indeed, in this study, we showed a reversal of liver damage via Bid siRNA treatment in a NASH mouse model even after being fed a CDAA diet for 19 weeks, a time point that results in significant liver fibrosis and inflammation.
We did not find any changes in the degree of steatosis as well as serum ALT levels induced by CDAA diet in the Bid siRNA treated group. In contrast, we found that this treatment had mainly an effect on cell death, inflammation, and fibrosis. The mechanisms of steatosis induced by the CDAA diet are complex an involved the presence of choline deficiency with decrease VLDL (very low-density lipoprotein) formation as well as an increase delivery of free fatty acids to the liver, and de novo lipogenesis [22]. These pathways are not dependent on hepatocyte Bid expression and the lack of protection by the siRNA therapy is in line with this concept. The lack of effect on serum ALT levels is more intriguing but there is growing evidence in the literature that in the context of metabolic changes in the liver related to fatty liver induction, serum ALT may reflect more these metabolic disruption than actual inflammation and liver injury. Indeed various clinical studies in patients with NAFLD have demonstrated that patients with elevated serum ALT levels may have only changes of steatosis without inflammation or fibrosis on liver biopsy while patients with normal serum ALT may present with the entire spectrum of disease including NASH with advanced fibrosis [23-25]. Another explanation is that the relatively short term treatment of 3 weeks did not give enough time for the ALT levels to decrease in serum.
Only a small fraction (∼5%) of our genes are targetable by small molecule therapeutics or antibodies, the so-called “druggable” genome. In contrast, due to the inherent selectivity of all expressed mRNA targets, including the vast “undruggable” genome, RNAi therapeutics [26-29] have great potential to revolutionize the treatment of NASH. RNAi has an EC50 ∼10-12 M (1 pM), and exquisite target selectivity for all mRNAs. Moreover, the liver is a particularly attractive organ for RNA-based therapy because siRNA penetrates the liver with high efficacy when administered via intravenous injection [30]. Furthermore, several carriers - lipid based or synthesized short interfering ribonucleic neutrals - have been developed for liver/hepatocyte specific delivery [31, 32]. Invivofectamine2.0, a lipid-based carrier, was developed by LifeTechnologies and is widely used for in vivo experiments. Recently, LifeTechnologies has developed a next generation lipid-based carrier for in vivo work, called Invivofectamine3.0, which increases delivery efficacy while minimizing potentially unwanted cytotoxicity. As a result, the siRNA dose that lead to the maximum Bid knockdown was 1.5 mg/kg with Invivofectamine3.0, instead of 7 mg/kg with Invivofectamine2.0. Invivofectamine3.0 may also have the benefit of minimizing off-target effects. Although we used BALB/C mice for our siRNA screening due to its easily visualized tail vein, the target sequence of siRNAs has the same effect in multiple mouse strains.
Our results indicate that Bid suppression via siRNA technology holds the potential to be a therapeutic target candidate for severe NASH. Weekly administration of the Bid siRNA complex for a total of three weeks to mice fed a CDAA diet for 19 weeks effectively reduced BID expression in the liver and was associated with a marked reduction in hepatocellular death, release of extracellular vesicles and sterile inflammation - changes that were associated with a significant anti-fibrotic effect. The protection was at least in part mediated through a decrease in mitochondrial permeabilization, subsequent release of cytochrome c into the cytosol, and caspase 3 activation. Furthermore, in order to test the hypothesis that the effect observed with the Bid siRNA therapy was mainly due to its effect on hepatocytes, we used hepatocyte specific BID deficient mice. The results demonstrated that BID deficient mice showed a similar level of protection from NASH induced by the CDAA diet as the one observed using the RNA-based therapy. These results point to the importance of Bid suppression in hepatocytes versus non-parenchymal cells for the therapeutic effect of BID inhibition. Since we observed a significant inhibition of caspase 3 activity in Jo2 stimulated cells in vitro that possess a 50-80% reduction in Bid mRNA level, the complete inhibition of BID may not be required to protect the liver from injury, thus pointing to siRNA as a viable therapy.
In summary, the present study shows that liver Bid suppression by RNAi technology, as well as hepatocyte-specific BID-deficiency, improves liver fibrosis combined with a reduction in cell death and sterile inflammation in experimental NASH. These findings are consistent with evidence that hepatocyte apoptosis is a key feature of lipotoxicity involved in NASH development and triggers HSC activation and liver fibrosis and suggest that BID inhibition may be useful as an antifibrotic NASH therapy.
Supplementary Material
Acknowledgments
Financial Disclosure: This work was supported by NIH grants U01AA022489 and DK082451 to AEF. UCSD Neuroscience Core for microscopy is supported by a grant P30 CA23100.
We thank the UCSD Neuroscience Core, especially Jennifer Santini for microscopy assistance.
Funding: The work was funded by NIH (U01AA022489 and DK082451 to AEF)
Abbreviations
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- CDAA
chorine-deficient L-amino acid defined
- BidΔhep
Hepatocyte-specific Bid-deficient
- EV
extracellular vesicle
Footnotes
Conflict of Interest: The authors state no conflict of interest, except Xavier De Mollerat Du Jeu and Andronikou Nektaria are employees of Life technologies
Author names in bold designate shared co-first authorship.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. Journal of gastroenterology and hepatology. 2013;28(Suppl 1):68–76. doi: 10.1111/jgh.12212. [DOI] [PubMed] [Google Scholar]
- 2.Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. Journal of hepatology. 2012;56:1384–1391. doi: 10.1016/j.jhep.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 3.Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U. S Hepatology. 2014;59:2188–2195. doi: 10.1002/hep.26986. [DOI] [PubMed] [Google Scholar]
- 4.Nascimbeni F, Pais R, Bellentani S, Day CP, Ratziu V, Loria P, et al. From NAFLD in clinical practice to answers from guidelines. Journal of hepatology. 2013;59:859–871. doi: 10.1016/j.jhep.2013.05.044. [DOI] [PubMed] [Google Scholar]
- 5.Wang K. Molecular mechanisms of hepatic apoptosis. Cell death & disease. 2014;5:e996. doi: 10.1038/cddis.2013.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guicciardi ME, Gores GJ. Apoptosis as a mechanism for liver disease progression. Seminars in liver disease. 2010;30:402–410. doi: 10.1055/s-0030-1267540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thapaliya S, Wree A, Povero D, Inzaugarat ME, Berk M, Dixon L, et al. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Digestive diseases and sciences. 2014;59:1197–1206. doi: 10.1007/s10620-014-3167-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatting M, Zhao G, Schumacher F, Sellge G, Al Masaoudi M, Gabetaler N, et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology. 2013;57:2189–2201. doi: 10.1002/hep.26271. [DOI] [PubMed] [Google Scholar]
- 9.Lazic M, Eguchi A, Berk MP, Povero D, Papouchado B, Mulya A, et al. Differential regulation of inflammation and apoptosis in Fas-resistant hepatocyte-specific Bid-deficient mice. Journal of hepatology. 2014;61:107–115. doi: 10.1016/j.jhep.2014.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Mollerat du Jeu X, Eguchi A, Nektaria A, Feldstein AE. Novel therapeutic nanoparticles for in vivo delivery of low dose siRNA in liver cells and for the treatment of liver fibrosis associated nonalcoholic steatohepatitis. Mol Ther. 2013;21:e18. [Google Scholar]
- 12.Povero D, Eguchi A, Li H, Johnson CD, Papouchado BG, Wree A, et al. Circulating Extracellular Vesicles with Specific Proteome and Liver MicroRNAs Are Potential Biomarkers for Liver Injury in Experimental Fatty Liver Disease. PloS one. 2014;9:e113651. doi: 10.1371/journal.pone.0113651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert review of gastroenterology & hepatology. 2011;5:201–212. doi: 10.1586/egh.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Disterer P, Al-Shawi R, Ellmerich S, Waddington SN, Owen JS, Simons JP, et al. Exon skipping of hepatic APOB pre-mRNA with splice-switching oligonucleotides reduces LDL cholesterol in vivo. Mol Ther. 2013;21:602–609. doi: 10.1038/mt.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eguchi A, Povero D, Alkhouri N, Feldstein AE. Novel therapeutic targets for nonalcoholic fatty liver disease. Expert opinion on therapeutic targets. 2013;17:773–779. doi: 10.1517/14728222.2013.789502. [DOI] [PubMed] [Google Scholar]
- 16.Hirsova P, Gores GJ. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cellular and molecular gastroenterology and hepatology. 2015;1:17–27. doi: 10.1016/j.jcmgh.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 19.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Molecular cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wree A, Johnson CD, Font-Burgada J, Eguchi A, Povero D, Karin M, et al. Hepatocyte-specific Bid depletion reduces tumor development by suppressing inflammation-related compensatory proliferation. Cell death and differentiation. 2015 doi: 10.1038/cdd.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loguercio C, Andreone P, Brisc C, Brisc MC, Bugianesi E, Chiaramonte M, et al. Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: a randomized controlled trial. Free radical biology & medicine. 2012;52:1658–1665. doi: 10.1016/j.freeradbiomed.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Kohli R, Feldstein AE. NASH animal models: are we there yet? Journal of hepatology. 2011;55:941–943. doi: 10.1016/j.jhep.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Molleston JP, Schwimmer JB, Yates KP, Murray KF, Cummings OW, Lavine JE, et al. Histological abnormalities in children with nonalcoholic fatty liver disease and normal or mildly elevated alanine aminotransferase levels. The Journal of pediatrics. 2014;164:707–713 e703. doi: 10.1016/j.jpeds.2013.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavine JE, Schwimmer JB, Nonalcoholic Steatohepatitis-Clinical Research N. Pediatric initiatives within the Nonalcoholic Steatohepatitis-Clinical Research Network (NASH CNR) Journal of pediatric gastroenterology and nutrition. 2003;37:220–221. doi: 10.1097/00005176-200309000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Adams LA, Feldstein AE. Nonalcoholic steatohepatitis: risk factors and diagnosis. Expert review of gastroenterology & hepatology. 2010;4:623–635. doi: 10.1586/egh.10.56. [DOI] [PubMed] [Google Scholar]
- 26.Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chemistry & biology. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G. Preclinical and clinical development of siRNA-based therapeutics. Advanced drug delivery reviews. 2015 doi: 10.1016/j.addr.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973–985. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer discovery. 2013;3:406–417. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
- 30.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nature reviews Drug discovery. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mishra N, Yadav NP, Rai VK, Sinha P, Yadav KS, Jain S, et al. Efficient hepatic delivery of drugs: novel strategies and their significance. BioMed research international. 2013;2013:382184. doi: 10.1155/2013/382184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meade BR, Gogoi K, Hamil AS, Palm-Apergi C, van den Berg A, Hagopian JC, et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nature biotechnology. 2014;32:1256–1261. doi: 10.1038/nbt.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.