

Abstract
Each year, approximately 3.8 million people suffer mild to moderate traumatic brain injuries (mTBI) that result in an array of neuropsychological symptoms and disorders. Despite these alarming statistics, the neurological bases of these persistent, debilitating neuropsychological symptoms are currently poorly understood. In this study we examined the effects of mTBI on the amygdala, a brain structure known to be critically involved in the processing of emotional stimuli. Seven days after lateral fluid percussion injury (LFPI), mice underwent a series of physiological and behavioral experiments to assess amygdala function. Brain injured mice exhibited a decreased threat response in a cued fear conditioning paradigm, congruent with a decrease in amygdala excitability determined with basolateral amygdala (BLA) field excitatory post synaptic potentials together with voltage sensitive dye imaging (VSD). Furthermore, beyond exposing a general decrease in the excitability the primary input of the amygdala, the lateral amygdala (LA), VSD also revealed a decrease in the relative strength or activation of internuclear amygdala circuit projections after LFPI. Thus, not only does activation of the LA require increased stimulation, but the proportion of this activation that is propagated to the primary output of the amygdala, the central amygdala, is also diminished following LFPI. Intracellular recordings revealed no changes in the intrinsic properties of BLA pyramidal neurons after LFPI. This data suggests that mild to moderate TBI has prominent effects on amygdala function and provides a potential neurological substrate for many of the neuropsychological symptoms suffered by TBI patients.
Keywords: traumatic brain injury, mTBI, amygdala, cued fear, threat response, voltage-sensitive dye, lateral fluid percussion, LFPI
Introduction
Traumatic brain injury (TBI) afflicts approximately 3.8 million people annually in the United States alone, with at least 5.3 million or 2% of the US population suffering persistent TBI-related disability (M. Faul and Coronado, 2015; Langlois et al., 2006; Rutland-Brown et al., 2006). Even mild to moderate traumatic brain injury (mTBI i.e. concussion), the most common form of TBI, results in debilitating symptoms and cognitive dysfunction. Whereas much of the animal research performed on mTBI has focused on cognition and memory (Xiong et al., 2013), cognitive deficits and memory impairment represent only a few of the debilitating neuropsychological symptoms suffered by mTBI patients. Many other prominent symptoms including, anxiety, aggression/irritability/rage, depression/anhedonia, apathy, and poor impulse control indicate emotional destabilization following mTBI (Bazarian et al., 2009; Jorge et al., 2004; Malkesman et al., 2013; Rao et al., 2009; Riggio, 2010).
Functional alterations in the amygdala, a brain region critically involved in the conscious and autonomic response to emotional stimuli, are associated with many if not all of the aforementioned symptoms (Cardinal et al., 2002; Duvarci and Pare, 2014; Ledoux, 2000; Phelps and LeDoux, 2005). Furthermore, if concussion induces acute physiological alterations in the amygdala, this could represent a potential substrate for the substantial comorbidity of traumatic brain injury and post-traumatic stress disorder (PTSD) (Riggio, 2010). While a limited number of human studies linking changes in amygdala size and diffusivity (diffusion tensor imaging) to TBI have been performed (Depue et al., 2014; Juranek et al., 2011), there exists a dearth of studies concerning amygdala physiology and dysfunction following mTBI. Understanding the effects of mTBI on amygdala function is critical for the development of therapeutic interventions to treat mild to moderate TBI and further our understanding how mTBI relates to PTSD.
Using a well-established mouse model of mTBI, lateral fluid percussion injury (LFPI) (Thompson et al., 2005; Xiong et al., 2013), physiological and behavioral experiments were performed to determine the effects of mTBI on amygdala function. As the amygdala is a critical component of threat response (freezing behavior), our studies began by testing animals for the acquisition and expression of threat response in a cued fear conditioning paradigm. Next, extracellular recording together with voltage sensitive dye (VSD) imaging was conducted to examine network activity and circuit function across multiple amygdala sub-regions, in brain slices from sham and brain-injured animals. VSD imaging allowed us to determine how brain injury effects local responses in the stimulated amygdala sub-regions, and further determine how this activation propagated to other amygdala nuclei; thus revealing potential changes in amygdala circuit dynamics. Finally, intracellular recordings were performed to assess intrinsic properties of amygdala neurons following LFPI. Through the combined use of physiological and behavioral techniques we have identified circuit level dysfunction in the amygdala corresponding to deficits in threat response in brain-injured animals.
Materials and Methods
Animals
All experiments were performed on 7 to 12 week-old, male C57BL/J6 mice (The Jackson Laboratory). All animals were group housed with free access to food and water. All procedures were performed in accordance with the guidelines published in the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Children's Hospital of Philadelphia Institutional Animal Care and Use Committee. Behavioral and physiological experiments were performed on separate cohorts of animals.
Lateral Fluid Percussion Injury
LFPI is a well-established model of mild to moderate traumatic brain injury that mimics many aspects of human TBI pathology and symptomatology (Carbonell et al., 1998; Thompson et al., 2005; Xiong et al., 2013). Animals were randomly assigned into three groups; Naïve animals that received no surgery or injury, but were treated otherwise identically; LFPI (surgery+injury); or sham (surgery only). Surgery and injury were performed on consecutive days, for full details see (Cole et al., 2010a; Witgen et al., 2005). Briefly, on day one mice were anesthetized with a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg) and placed in a stereotaxic frame (Stoelting). A 3 mm outer diameter trephine was then used to perform a craniectomy of the right parietal bone lateral of the sagittal suture between lambda and bregma, without disrupting the intact dura. A Luer-loc needle hub (3 mm inner diameter) was secured to the skull surrounding the craniectomy with adhesive and dental cement. The needle hub was filled with sterile saline solution and sealed overnight. On the following day, LFPI animals were anesthetized with isoflurane and connected to the LFPI device (Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA). The LFPI device was then triggered, delivering a 10–15 ms fluid pulse (peak pressure 1.5–1.8 atm) onto the intact dura of the brain to generate a mild to moderate TBI. The needle hub was removed and the incision sutured. Any animals with injuries resulting in dural breach or herniation were excluded from the study. Sham animals underwent an identical procedure without receiving the fluid pulse injury. Since no significant differences between sham and naïve animals were evident, these two groups were pooled for analysis and referred to as the sham group.
Cued Fear Conditioning
It is well established that acquisition and expression of a threat response, that is freezing, to a light and/or tone cue, paired with an aversive stimuli, is dependent upon physiological processes within the basolateral and central amygdalae (Dębiec et al., 2010; Duvarci and Pare, 2014; Phillips and Ledoux, 1992; Tronson et al., 2012). Fear conditioning paradigms pair an emotionally neutral stimulus or context (conditioned stimulus or CS) with an aversive stimulus (unconditioned stimulus or US), leading to the expression of a threat response to presentation of the neutral CS alone. As noted, both the context and cue become predictive of the US and elicit a threat response. For these experiments the context was altered between training (context A) and testing (context B) to isolate the light/tone (CS) cued response from the hippocampal dependent contextual response.
The cued fear conditioning paradigm used in this study was modified from experiments described in (Newton et al., 2004; Wolff et al., 2014). The CS consisted of simultaneous auditory (75dB, white noise, 20s) and light stimuli (yellow light pulses, 20s, flickering at 4 Hz) generated by built in audio and light stimuli generators (Med Associates, St. Albans, VT, USA). The US consisted of a footshock (1.05mA, 1.5s) delivered through the metal grid floor. During CS-US pairings, the US was delivered immediately following the cessation of the CS. All behavioral experiments were performed in an isolated behavioral suite under low light.
On days 4 and 5 after injury or sham operation, each animal was handled for 3 min by the experimenter. On day 6 animals underwent fear conditioning training in context A, a rectangular conditioning chamber (21.6 cm × 17.8 cm × 12.7 cm) with Plexiglas and metal walls, and a metal grid floor (Med Associates, St. Albans, VT, USA). Animals were allowed to freely explore the chamber for 1 min before experiencing 3 CS-US pairings (15s – 105s interstimulus interval, mean 60s). The mean of the first and second interstimulius intervals (ISI) was consistently 60s, with the actual duration of ISI 1 vs ISI 2 assigned pseudo-randomly within a group, but consistent across groups such that each ISI pair used in a sham animal was used in a subsequent LFPI animal. One min after the final CS-US pairing (5 min total), mice were removed from context A and placed back in their home cage (see Fig. 1 for schematic). Twenty four hours later (day 7) animals underwent behavioral testing to measure threat responses in context B, a custom made triangular conditioning chamber with black striped Plexiglas walls and a smooth, opaque black plastic floor, scented with organic vanilla extract. Mice were allowed to freely explore the chamber for 1 min before experiencing 3 presentations of the CS alone (60s, interstimulus interval). Again, animals were removed from context B after a total of 5min. During testing, freezing behavior was scan sampled every 5th second from the onset of the first CS presentation to the end of the trial (4 min total). Freezing was defined as a total lack of movement aside from respiration at the instant of every 5th second. The total instance of freezing was then divided by total observations to generate a freezing percentage per animal. Behavioral data was collected from 26 animals (17 sham; 9 LFPI).
Fig. 1. LFPI decreases freezing in a cued fear conditioning paradigm.

(A) Animals are trained in context A on day 6 after injury and tested in a novel context (context B) on day 7 (CS- 75dB white noise and yellow light pulses flickering at 4 Hz, 20 s; US- 1.05 mA footshock, 1.5 s). The inter stimulus interval (ISI) was between 15–105 s with a trial mean of 60s, meaning the first and second ISI always added up to 120 s. (B) Percent of time freezing after presentation of CS on day 7. LFPI animals freeze significantly less after injury (sham n=17, LFPI n=9, P <0.001***).
Electrophysiology
All electrophysiological experiments were performed on days 7–8 after injury or sham surgery. Slice preparation was performed as previously described (Johnson et al., 2014). Briefly, animals were anesthetized with isoflurane, the brain was dissected out and placed in ice-cold oxygenated (95%O2 / 5%CO2) sucrose-containing artificial cerebrospinal fluid (aCSF) containing (in mM): sucrose 202, KCl 3, NaH2PO4 2.5, NaHCO3 26, glucose 10, MgCl2 1, CaCl2 2. Coronal slices 300 μm thick were cut on a vibratome (VT1200S, Leica Microsystems, Buffalo Grove, IL, USA) and transferred to 33–37°C oxygenated (95%O2 / 5%CO2) control aCSF containing (in millimolar): NaCl 130, KCl 3, NaH2PO4 1.25, NaHCO3 26, glucose 10, MgCl2 1, CaCl2 2. Slices were allowed to incubate for at least 60 min before recording. VSD imaging and field potential recordings were performed in an interface chamber, intracellular recordings were performed in a submersion chamber, both with a flow rate of approximately 2.0 ml/min and maintained at 27–30°C. Brain slices for recording were consistently selected from the same rostral-caudal region of the amygdala that exhibited a pear shaped basolateral amygdala (BLA) contiguous with an ovoid central amygdala (CeA), as seen in Figs. 2&3. This rigid criterion resulted in 1-2 brain slices per animal. Brain slices were hemisected prior to recording and only slices from the hemisphere ipsilateral to the site of injury were used. Our laboratory has previously demonstrated that contralateral slices are altered by LFPI and thus do not serve as an appropriate control for the injured hemisphere (Tran et al., 2006). For single cell recordings, a maximum of 1 cell per brain slice was used for analysis.
Fig. 2. LFPI decreases lateral amygdala evoked basolateral amygdala fEPSP's.

(A) Representative fEPSP recordings from brain slices of sham (green) and LFPI (red) animals (200 μA stimulation intensity). (B) Average fEPSP slope input-output (I/O) curve (50-500 μA, 50 μA steps, stimulation). LFPI causes a significant decrease in the BLA I/O curve (sham n=16 slices / 10 animals, LFPI n=14 slices / 7animals, P <0.001***). Black line denotes repeated measures that are significantly different in post hoc comparisons. (C) Distribution of fEPSP slopes recorded simultaneously with VSD imaging experiments (200 μA stimulation). LFPI causes a significant decrease in the BLA fEPSP slope (sham n=17 slices / 10 animals, LFPI n=14 slices / 7animals, P <0.002**). (D) BLA fEPSP amplitude paired pulse ratio (200 μA stimulation, 70 ms inter stimulus interval). LFPI has no effect on BLA fEPSP paired puse ratio (sham n=14 slices / 8animals, LFPI n=12 slices / 7 animals, P= 0.999). (Experimental set-up) Representative brain slice showing amygdala subregions and electrode placements. “Stim” and “Record” arrows show placement of tip of stimulation and recording electrodes.
Fig. 3. Amygdala VSD imaging recording set-up and representative movie frames.

(A) Representative VSD movie frames from slices of sham and LFPI animals. Video frames show experimental set up and voltage response of the amygdala to LA stimulation. ΔF/F values at each spatiotemporal point are displayed as pseudocolor and stacked to create a video representation (see corresponding videos in Video S1 and Video S2) The pseudocolor scale bar below shows the correlation of colors displayed to ΔF/F values. The time points selected (6, 56, 306 ms) roughly correlate to the peak indices shown in C. (B) Representative slice showing example regions of interest and raster segmentation. (C) Representative group average, multi segment regional average line plot showing approximate location of the peaks used in analysis. The solid color line shows the mean values and the shaded area is a SEM ellipse.
Field excitatory post synaptic potential recording (fEPSP) electrodes were fabricated from borosilicate glass (World Precision Instruments, Sarasota, FL, USA, #1B150F-4) pulled to a tip resistance of 2-6 MΩ and filled with aCSF. Field potentials were recorded in the central region of the BLA (Fig. 2) with an Axoclamp 900A amplifier and pClamp10 data acquisition software (Molecular Devices, Sunnyvale, CA, USA), filtered at 2 kHz. Stimulating electrodes were non-concentric bipolar (World Precision Instruments, Sarasota, FL, USA, #ME12206) and placed in the dorsal most portion of the lateral amygdala (LA), just medial of the external capsule. Electrical stimuli were 100 μs in duration. Field potential input-output relationships (50-500 μA stim, 50 μA increments, 8 s inter stimulus interval) and paired-pulse ratios (200uA stim, 75 ms inter stimulus interval, 8 s inter-pair interval) were performed prior to VSD recordings. Field potential recordings were also recorded simultaneously with VSD recordings. The inter-stimulus interval for field potentials recorded during the VSD trials was 20 s. This duration was necessary to accommodate the every other trial subtraction method used for VSD analysis. Simultaneous VSD and field recordings were performed at 100 μA, 200 μA and 300 μA stimulation intensities in aCSF. Only the 200 μA stimulation intensity was used for experiments using various pharmacological manipulations. All figures and statistics reported in the manuscript are based on the 200 μA stimulation intensity data. However, see Table 1 for data and statistics generated from all stimulation intensities. The 200 μA stimulation intensity was chosen because it elicits an approximately half maximal field potential response, while providing a high enough signal to noise ratio for VSD analysis. A subset of brain slices underwent field potential recordings and/or VSD imaging in the presence of 50 μM D-(-)-2-Amino-5-phosphonopentanoic acid (APV) + 6 μM 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) to suppress ionotropic glutamatergic transmission, 0.4 μM TTX to block voltage-gated sodium channels, or 1 μM CGP 55845 to antagonize GABAB receptors. Field potential data was analyzed using pClamp10 and in house, custom written MATLAB scripts.
Table 1.
| Region_Peak: | 100uA Stimulation | 200uA Stimulation | 300uA Stimulation | ANOVA Stats (by region) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ACSF | Sham (ΔF/F):Sham (ΔF/F): | LFPI (ΔF/F): | p-value: | Sham (ΔF/F):Sham (ΔF/F): | LFPI (ΔF/F): | p-value: | Sham (ΔF/F):Sham (ΔF/F): | LFPI (ΔF/F): | p-value: | F values | P-value |
| BLA(m)_FDP | 12.91 | 9.48 | 0.0012 | 17.77 | 12.42 | 0.0021 | 20.86 | 14.35 | 0.0033 | F(1,14)= 19.94 |
0.0005 |
| 95% CI | 11.94–14.14 | 7.74– 10.96 |
15.59–19.71 | 10.82– 14.79 |
18.07–11.84 | 11.84– 17.32 |
|||||
| BLA(m)_SDP | 6.87 | 5.89 | 0.0311 | 10.9 | 7.75 | 0.0115 | 10.53 | 7.69 | 0.0052 | F(1,14)= 10.83 |
0.0054 |
| 95% CI | 5.77–7.94 | 4.32– 6.57 |
8.72–11.62 | 6.96– 9.22 |
9.65–13.2 | 7.07– 9.4 |
|||||
| BLA(m)_SNP | −0.64 | −1.5 | 0.0962 | 0.87 | −0.59 | 0.0229 | 2.41 | 0.21 | 0.0079 | F(1,14)=7.5 | 0.016 |
| 95% CI | −1.22--.15 | −2.02-- .65 |
.09–2.65 | −1.5– 0.55 |
1.48–4.27 | −0.84– 1.59 |
|||||
| BLA(I)_FDP | 8.83 | 8.49 | 0.4524 | 12.44 | 9.81 | 0.0416 | 15.68 | 10.68 | 0.0164 | F(1,14)= 7.01 |
0.0191 |
| 95% CI | 8.02–10.1 | 5.34– 9.6 |
11.24–14.87 | 8.12– 12.4 |
13.89–17.71 | 8.79– 14.38 |
|||||
| BLA(I)_SDP | 6.97 | 6.1 | 0.5848 | 10.09 | 8.63 | 0.2463 | 11.23 | 8.97 | 0.0545 | F(1,14)= 3.26 |
0.0524 |
| 95% CI | 6.15–8.12 | 4.75– 8.28 |
8.77–11.62 | 7.55– 10.47 |
9.48–13.84 | 7.61– 10.32 |
|||||
| BLA(I)_SNP | −0.34 | −0.51 | 0.5543 | 0.33 | −0.16 | 0.0416 | 1.72 | 0.47 | 0.0229 | F(1,14)= 5.422 |
0.0354 |
| 95% CI | −.74–.14 | −1.05-- .1 |
.09–2.32 | −0.76– 0.43 |
1.07–3.97 | −.28–1.3 | |||||
| CeA_FDP | 8.27 | 5.4 | 0.0002 | 11.53 | 7.15 | 0.0012 | 12.95 | 7.56 | 0.0003 | F(1,14)= 33.08 |
<0.0001 |
| 95% CI | 7.41–9.44 | 4.38– 6.1 |
10.09–12.85 | 5.63– 8.64 |
11.69–14.37 | 6.16– 9.42 |
|||||
| CeA_SNP | 0.61 | −0.13 | 0.0545 | 1.75 | −0.21 | 0.0007 | 2.34 | −0.12 | 0.0002 | F(1,14)= 30.46 |
<0.0001 |
| 95% CI | −.11–.98 | −.7–.5 | 1.05–2.25 | −0.75– 0.34 |
1.94–2.98 | −.37–.76 | |||||
Whole-cell patch-clamp current-clamp recording electrodes were fabricated from borosilicate glass (World Precision Instruments, Sarasota, FL, USA, #1B150F-4) pulled to a tip resistance of 4–7 MΩ and filled with an internal solution containing (in milimolar): KGluconate 145, KCl 2.5, NaCl 2.5, HEPES 10, MgCl2 2, ATP.Mg 2, GTP.Tris 0.5, and BAPTA .1. Whole-cell recordings of BLA pyramidal neurons were performed using a Multiclamp 700B amplifier and pClamp10 data acquisition software (Molecular Devices, Sunnyvale, CA, USA). All current-clamp recording voltages were corrected for a liquid junction potential calculated at −14.5 mV (using the Junction Potential utility in Clampex 10.3), and a series voltage error of 2.2 mV. The resting membrane potential was measured immediately following break in, and then current was injected to bring cells to the target holding/resting membrane potential of −78mV. Target membrane potential value was chosen empirically based on the resting membrane potential of initial whole cell recordings. Measures of intrinsic excitability were gathered from a series of hyperpolarizing and depolarizing current injections through the whole-cell recording electrode (−50-225 pA, 25 pA increments, 8s inter step interval). BLA pyramidal neurons were identified on the basis of previously characterized morphological and physiological properties (Sah et al., 2003). Whole-cell current-clamp data was analyzed using pClamp10 software.
Voltage-Sensitive Dye Imaging Acquisition
Voltage-sensitive dye imaging (VSD) acquisition using Neuroplex software was performed as previously described (Johnson et al., 2014). Briefly, VSD dye di-3-ANEPPDHQ (Invitrogen) was prepared at a working concentration of 67 μg/ml in aCSF and applied to slices for 16 min. The slices were subsequently rinsed in standard aCSF and transferred to the interface recording chamber. The dye was excited by seven high-power green LEDs (Luxeon Rebel LXML-PM01-0100, Philips) coupled to a 535 ± 25 nm bandpass filter and 565 nm dichroic mirror. Fractional changes in VSD fluorescence (ΔF/F) were isolated with a 610 nm longpass filter and recorded at 1.0 kHz with a fast video camera with 80 × 80 pixel resolution (NeuroCCD, Redshirt Imaging, Decatur, GA, USA) through a reverse-lens macroscope with a 50 mm f/1.3 M46 lens (Dark Invader). Each camera pixel imaged a 25 μm × 25 μm region of tissue. The green light stimulus was triggered 230 ms prior to fluorescence acquisition, followed by electrical stimulation 170 ms after onset of fluorescence acquisition. All VSD recordings were 13 trials, 1.0 s in duration (1000 samples), with a 20 s interval between electrically stimulated trials. Each stimulated trial was followed 10 s later, by a non-electrically stimulated trial that was used to subtract photo-bleaching from the active signal (see below for details).
Voltage-Sensitive Dye Imaging Analysis
Each individual VSD trial used to create brain slice averages, was manually reviewed for ambient light contamination and gross abnormalities; if present, these trials were excluded from further analysis. Initial processing was performed as described in (Johnson et al., 2014). Fractional change in fluorescence values (ΔF/F) was calculated as follows: fluorescence values for each pixel in each trial were normalized according to the average fluorescence in the pixel during a 64 ms window immediately preceding the electrical stimulus. Then, the average from the corresponding pixel in the non-electrically stimulated trial was normalized and subtracted from the individual electrically stimulated trials to correct for photo-bleaching. VSD recordings were filtered in x and y spatial coordinates by convolution with a 5 pixels × 5 pixels Gaussian filter (Σ = 1.2 pixels), and in time by convolution with a five sample median filter. No additional filtering was applied to any of the images or analysis of VSD data. For VSD videos and representative movie frames, the ΔF/F for each pixel for the given sample (ms) is displayed as pseudocolor superimposed onto the image of the brain slice. Time points selected for the representative movie frames in (Fig. 3), correlate to florescence peaks analyzed in the regional average line plots (Fig. 3&4). Peaks maps (Fig. 5) were generated by pseudocoloring and plotting the maximum ΔF/F value of any sample at each pixel.
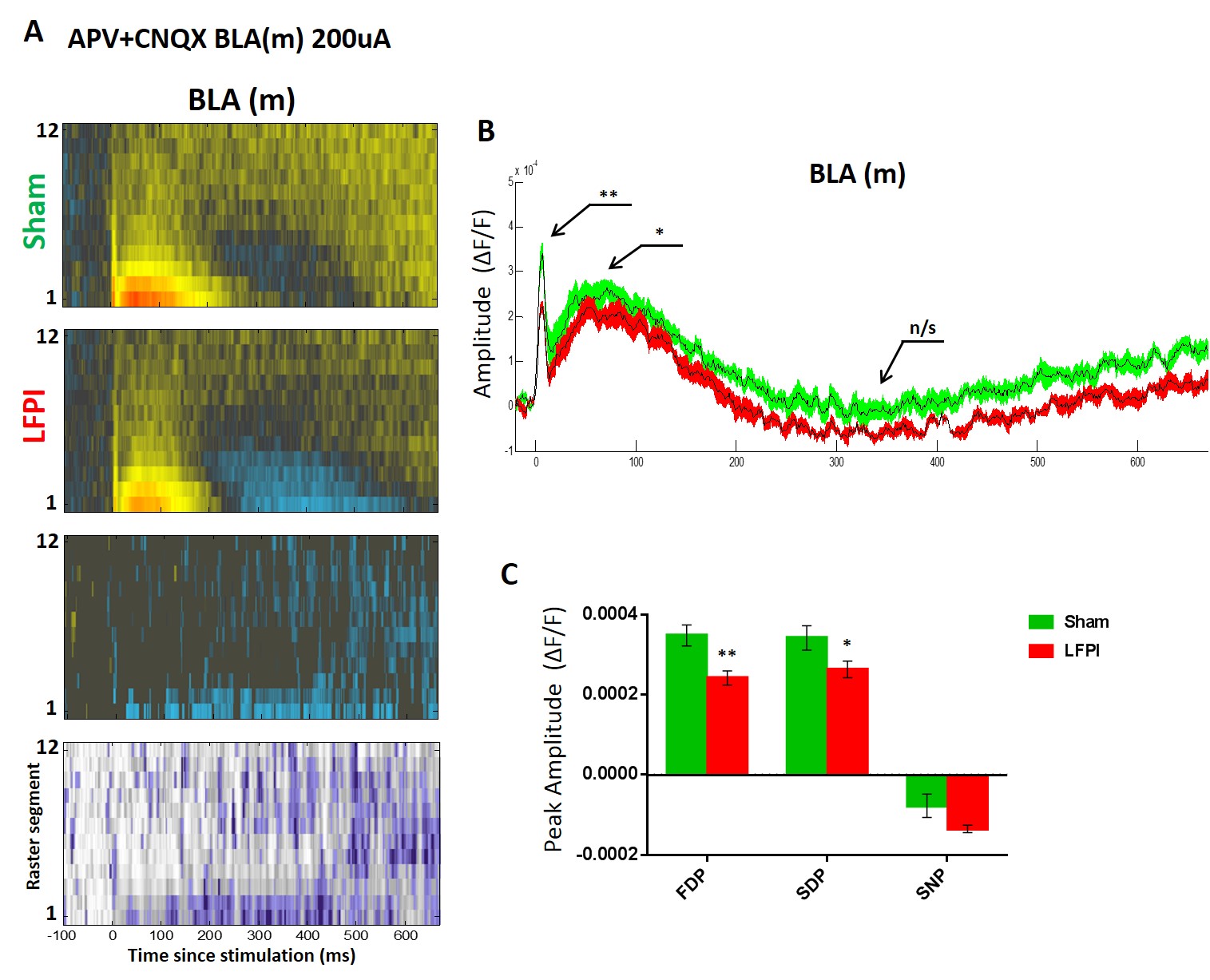
Fig. 4. VSD average raster plots and multi segment regional averages showing LFPI-induced alterations in amygdala activation.

(A) Rows 1 and 2: group average raster representation of sham (n=16) and LFPI (n=14) VSD responses. Row 3: difference raster showing magnitude of differences between sham and LFPI rasters. Sham raster was subtracted from the LFPI raster, and only the ΔF/F differences at sites with p = <0.05 are plotted. Row 4: P value heat map showing the results of a permutation test used to compare sham and LFPI rasters, displaying a P value for each spatiotemporal site. (B–D) Group average multi segment regional average line plots and graphs of peak ΔF/F values from sham (n=16 slices / 9 animals) and LFPI (n=14 slices / 7 animals) animals. (B) Central amygdala (CeA) (segments 1-3) from LFPI animals exhibit a decrease in depolarization (FDP, P=0.001**) and an increase in late hyperpolarization (SNP, P <0.001***). (C) Medial basolateral amygdalae (BLA(m)) (segments 2-6) from LFPI animals exhibit a decrease in depolarization (FDP, P=0.002**; SDP, P= 0.011*) and an increase in late hyperpolarization (SNP, P= 0.023*). (D) Lateral basolateral amygdalae (BLA(l)) (segments 2-6) from LFPI animals exhibit a decrease in depolarization (FDP, P= 0.042**) and an increase in late hyperpolarization (SNP, P= 0.042).
Fig. 5. Amygdala VSD peak maps and regional activation ratios showing decreased propagation in amygdala circuit following LFPI.

(A) Representative peak maps showing the maximum ΔF/F recorded at each spatial site at any time point, for brain slices from sham and LFPI animals. From left to right: slice image showing amygdala anatomy followed by peak response to 100, 200, and 300 μA stimulation intensities. (B) LA-to-BA activation ratio. BA FDP amplitude/LA FDP amplitude reveals a significant decrease in propagation of activation in LFPI animals (sham n= 16 slices / 9 animals, LFPI n=14 slices / 7 animals, P= 0.005**). (C) LA-to-CeA activation ratio. CeA FDP amplitude/LA FDP amplitude reveals a significant decrease in propagation of activation from lateral to central amygdala in LFPI animals (sham n= 16 slices / 9 animals, LFPI n=14 slices / 7 animals, P= 0.003**).
Raster plot construction, analysis, and statistics were performed using the MATLAB VSD analysis toolbox provided by and described in (Bourgeois et al., 2014). To begin raster construction, regions of interest (anatomical regions) were defined for segmentation. In these experiments, the anatomical boundaries of central amygdala (CeA) and basolateral amygdala (BLA) were clearly visible and used to draw region boundaries. There was no clear, reliable anatomical boundary separating lateral and basal amygdala in our images, thus they were combined into the “BLA” region for analysis, unless specifically stated. However, the BLA was further divided into medial (BLA(m)) and lateral (BLA(l)) sections. Medial and lateral BLA were analyzed separately as they are physiologically distinct, in that they receive differential inputs from other brain regions and sit in a different position along the general dorsoventral, lateromedial directionality of amygdala inter-nuclear signaling (Duvarci and Pare, 2014; Sah et al., 2003). Regions were then split into 100 μm segments along the dorsoventral axis, numbering segments in ascending order from dorsal to ventral location (see Fig. 3). Average ΔF/F vales were calculated for each spatiotemporal site and plotted as pseudocolor in the distance from stimulating electrode versus time raster plots. To account for slight variability in region of interest size when creating average raster plots, all individual slice raster plots were stretched or compressed to have the same number of segments. That number was set as the mean number of segments from the individual slices (CeA = 6, BLA(m) = 12, BLA(l) = 12).
Statistical comparison of group raster plots was performed at each individual spatiotemporal site using a permutation test (t=1000). Permutation is a nonparametric test that randomly resamples data to generate a null distribution describing variability in the data. Significant differences (α < .05) are registered at sites where the experimental versus control groupings explain the variability in the data. The p-values generated are displayed in pseudocolor on a p-value heat-map (Fig. 4A row 4). To clarify areas of significant difference between average raster plots, the sham raster plot was subtracted from the LFPI raster plot, and only the ΔF/F differences at sites with p = < 0.05 are plotted (Fig. 4A row 3).
Multi-segment regional averages were calculated and used to create line plots (Fig. 3&4). Line plots show the average ΔF/F (dark line) surrounded by SEM ellipses (shaded surrounding). The segments selected for line plots were the same between conditions and consistent by region (CeA = seg 1-3, BLA(m) = seg 2-6, BLA(l) = seg 2-6). To look at signal propagation through the amygdala circuit we assessed the ratio of activation in the lateral amygdala (LA) compared to the basal amygdala (BA) or CeA. The quantification of activation for these ratios, seen in (Fig. 5B&C) is based on fast depolarizing peak (FDP) values from multi-segment averages within each region. The LA region was defined as a combination of segments 1-3 of both BLA(m) and BLA(l). The BA region was defined as a combination of segments 7-10 of both BLA(m) and BLA(l). The CeA region was viewed in its entirety (segments 1-6) for this analysis, as we were interested in both the magnitude and distance of signal propagation. VSD analysis was performed using a combination of Neuroplex, custom MATLAB scripts, and GraphPad Prism. VSD/electrophysiological data was collected in 74 slices prepared from 41 animals.
Statistical Procedures
All statistical analyses and calculations were performed using MATLAB and/or GraphPad Prism. A priori power calculations were performed using G*Power (F. Faul et al., 2007) based on variability from similar previous experiments. All statistical tests for significance were conducted using Mann-Whitney U-tests, two-way repeated-measures ANOVA with Sidak's multiple comparison test in order to test for injury effect and stimulation intensity effect or the permutation test described in (Bourgeois et al., 2014), where applicable. Statistical significance was P <0.05 (P <0.05*, P <0.01**, P <0.001***). N values are reported per brain slice and per animal, where appropriate, with a maximum of 2 slices per animal. For physiological experiments (fEPSP and VSD) when multiple brain slices from a single animal were used, data from individual brain slices generated from the same animal were averaged yielding a single animal value for each given measure. The value generated was used in group analysis and statistics, based on the animal N. For pharmacological manipulation pre vs. post drug wash comparisons, slices were considered independent samples, used for analysis and statistics, based on the slice N. Data in the figures are presented as group means ± SEM.
Results
LFPI causes a decreased threat response to cued fear conditioning
In order to assess the potential effects of LFPI on amygdala dependent behaviors, mice were tested using a combined auditory and visually cued fear conditioning response paradigm. It is well established that amygdala activity plays an essential role in the acquisition and expression of light and/or tone cued threat response (Duvarci and Pare, 2014; Ledoux, 2000; Newton et al., 2004; Phillips and Ledoux, 1992). While it is traditional to refer to the behavioral response in this paradigm as fear, the authors note that freezing behavior is better defined as a threat response based on survival instinct, rather than a subjective or conscious feeling of fear (LeDoux, 2012). Mice were randomly assigned to, and underwent, either sham or LFPI procedures prior to fear conditioning and retrieval testing. On day 6 after injury animals underwent a conditioning period in context A where they were exposed to 3 presentations of the CS-US (light/tone-foot shock) pairing. On day 7 animals were placed in a novel context (context B) and exposed to 3 presentations of the CS (light/tone) alone. Freezing behavior was assessed from the onset of the first CS presentation to the end of the trial and expressed as percent of time freezing (Fig. 1A) (see methods for full details). During the testing period LFPI animals froze significantly less than sham animals, indicating a deficit in acquisition and/or expression of threat response (Fig. 1B, sham = 50%, 95% CI 46.47–57.94% n=17, LFPI = 33.33%, 95% CI 27.9–37.38% n=9; P <0.001).
Decreased network excitability in the amygdala following LFPI
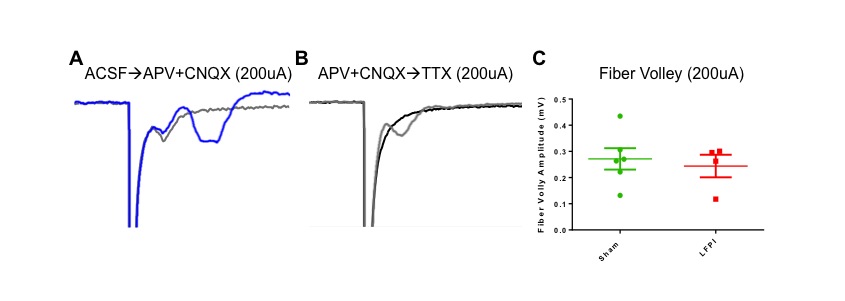
After identifying LFPI-induced deficits in amygdala-dependent behaviors we investigated amygdala function for putative physiological substrates contributing to the observed behavioral deficits. To begin assessing the effects of LFPI on amygdala physiology we recorded ex vivo extracellular field excitatory post synaptic potentials (fEPSP) from the central region of the basolateral amygdala (BLA) in coronal brain slices. Field EPSP recordings contain both a fiber volley (FV), representing the presynaptic action potentials and an extracellular field potential response, which is predominantly the postsynaptic dendritic component of the signal. BLA field potentials were evoked via electrical stimulation of the lateral amygdala (LA) and external capsule, the primary input of sensory information into the amygdala (Ledoux, 2000; Sah et al., 2003) (Fig. 2, Experimental set-up). Field EPSP responses were recorded over a range of stimulation intensities to construct input-output curves (I/O), and simultaneous to VSD recordings. Representative field potential recordings in Fig. 2A show the drastic decrease observed in fEPSPs in slices from LFPI animals. Brain slices from LFPI animals demonstrate a significant downward shift in the I/O curve (Fig. 2B, sham n=16 slices / 10 animals, LFPI n=14 slices / 7 animals, two-way repeated-measures ANOVA, (injury effect) F (1,15) = 18.66; P <0.001). Additionally, brain slices from LFPI animals showed a significant decrease in fEPSPs recorded simultaneously with VSD recordings (Fig. 2C, sham 0.458 95% CI 0.368–0.678 mV/ms n=17 slices / 10 animals, LFPI 0.148 95% CI 0.049–0.305 mV/ms n=14 slices / 7 animals; P =0.002). Interestingly, LFPI does not appear to alter transmitter release probabilities as there was no significant difference in the paired pulse ratio when a second stimuli was delivered shortly after the first (Fig. 2D, sham 0.470 95% CI 0.289–0.645 n=14 slices / 8 animals, LFPI 0.424 95% CI 0.302–0.661 n=12 slices / 7 animals, P= 0.999). Bath application of APV (50 μM) and CNQX (6 μM) decreased fEPSPs to a level indistinguishable from noise (fEPSP slope post APV/CNQX = 0.005 95% CI −0.002–0.008 mV/ms n=10 slices) and isolated the presynaptic FV responses. There was no significant difference in FV amplitude recorded in the presence of APV/CNQX between slices from sham and LFPI animals (Fig. S1, sham 0.267 95% CI 0.167–0.376 mV n=8 slices / 6 animals, LFPI 0.279 95% CI 0.107–0.381 mV n=6 slices / 4 animals; P= 0.695). This suggests that LFPI has little or no effect on the number of neurons or axons directly activated by the stimulating electrode. Both FV and fEPSP responses were blocked by bath application of TTX (0.4 μM) (Fig. S1).
In order to more fully understand and visualize the effects of LFPI on amygdala physiology and circuit function, we performed a series of voltage sensitive dye (VSD) imaging experiments. VSD imaging allows for the recording of changes in voltage in hundreds or thousands of neurons in all sub-regions of the amygdala simultaneously. This not only allows for the evaluation of the spatiotemporal dynamics of amygdala circuit activation, but to also directly view inhibition as areas of regional hyperpolarization. VSD shows relative changes in voltage by recording fractional changes in fluorescence (ΔF/F) emitted by voltage sensitive dyes. When used in the hippocampal brain region, this voltage sensitive dye (di-3-ANEPPDHQ) fluorescence has a linear correlation to neuronal membrane voltage (Vm), where 1 × 10−4 ΔF/F equals a roughly 1 mV change in Vm (Ang et al., 2006). For this reason all ΔF/F values are reported in (10−4) notation. Figure 3A again shows the experimental setup and representative VSD movie frames from slices from sham and LFPI animals (see also Video S1 and Video S2 in supplemental materials). A post injury decrease in the intensity and duration of depolarization throughout the amygdala can be seen in the representative movie frames as fewer red and orange pixels at 6 ms and 56 ms. An increased delayed hyperpolarization is also apparent as an increase in blue pixels in BLA at the 306 ms time point. In order to interpret group data, VSD videos were used to create regional raster plots and multi segment regional average line plots for analysis (see methods for full details). Figure 3B shows an example of how anatomical amygdala regions were defined and segmented for rasterization. The amygdala was separated into three regions for analysis, central amygdala (CeA), medial basolateral amygdala (BLA(m)) and lateral basolateral amygdala (BLA(l)). The same rostral-caudal region of amygdala (1-2, 300 μm brain slices) that exhibited a pear shaped BLA contiguous with an ovoid CeA was used in every animal. Figure 3C shows a typical VSD regional average line plot response profile, in a slice from a sham animal, containing the three distinct peaks ultimately used in analysis. There is an initial fast depolarizing peak (FDP = max ΔF/F 0–35 ms), followed by a secondary slow depolarizing peak (SDP = max ΔF/F 36–136 ms) and a delayed hyperpolarization or slow negative-going peak (SNP = minimum ΔF/F 137–600 ms). In all regional line plots the dark line is the average ΔF/F surrounded by a shaded standard error of the mean (SEM) ellipse.
The group data presented in Figure 4 (VSD response to 200 μA stimulation of LA) reveals the profound alterations in amygdala physiology following LFPI. Rows one and two of Figure 4A show the average group raster plots for slices from sham and LFPI animals in all three amygdala subregions. In these raster plots warm colors (yellow to red) represent depolarization and cool colors (blue to purple) represent hyperpolarization. Row three is a difference raster showing the magnitude and direction of difference between sham and LFPI raster plots at each spatiotemporal site. Row four shows the results of a permutation test used to compare sham and LFPI raster plot data, displaying a P value at each spatiotemporal site to denote where differences between slices from sham and LFPI animals are significant (see methods for full details). All three amygdala regions show an LFPI-induced decrease in the intensity and duration of the depolarizing response to electrical stimulation and an increase in delayed hyperpolarization 200–400 ms after stimulation. This is particularly evident in the BLA(m) raster plots. We then measured and compared the aforementioned peak ΔF/F values from each amygdala region in response to 200 μA stimulation. In CeA, LFPI caused a significant decrease of the FDP and SNP (Fig. 4B, Table 1). Interestingly there is no clear SDP in CeA, thus it was not analyzed. In BLA(m) LFPI caused a significant decrease of the FDP, SDP and SNP (Fig. 4C, Table 1). In BLA(l) LFPI caused a significant decrease in the FDP and SNP, but had no effect on the SDP (Fig. 4D, Table 1). These experiments were repeated at 100 μA and 300 μA stimulation intensities, both of which maintained a similar trend of decreased activation in LFPI animals. All peak values and statistical comparisons are reported in Table 1.
In order to isolate and remove the ionotropic glutamatergic component of the VSD signal a separate set of VSD experiments were performed in the presence of glutamatergic antagonists (APV 50 μM, CNQX 6 μM). When glutamatergic transmission was suppressed the magnitude of the ΔF/F difference between sham and LFPI animals, at both depolarizing peaks, was drastically decreased throughout and eliminated in certain regions (Table S1, Fig S2). While the blockade of excitatory neurotransmission substantially reduces the magnitude of the depolarization difference between brain slices from sham and LFPI animals, a significant depression of the fast depolarization in BLA(m) in brain slices from LFPI animals remains (Fig. S2B&C, sham 3.54 95% CI 2.84–4.12 × 10−4 ΔF/F n=10 slices / 7 animals, LFPI 2.41 95% CI 1.96–2.87 × 10−4 ΔF/F n=12 slices / 6 animals; P= 0.008). This suggests an additional component of the remaining VSD signal, possibly GABAergic inhibition, is also altered by LFPI.
Amygdala circuit dysfunction following LFPI
Diminished amygdala network excitability as expressed by a decrease in fEPSP and VSD depolarization could be due to a decrease in activation of the primary input into the amygdala (LA/external capsule), a decrease in propagation of that signal through the amygdala circuit, or a combination of both. While the LFPI-induced decrease in depolarization adjacent to the stimulating electrode (visible in representative movie frames & and group raster plots) demonstrates a decrease in activation of the primary input, it is still unclear if propagation through the amygdala circuit is affected by LFPI. Therefore, to better understand the spatial component or spread of the stimulus evoked response, peak maps were created to show the max ΔF/F at any time point in each pixel to isolate the pattern of maximal depolarization through the amygdala. Further, by using multiple stimulation intensities we can determine how increasing activation of the input affects the spatial component of activation in brain slices from sham and LFPI animals. Reduced depolarization and spread of activation from lateral amygdala to basal or central amygdala can be seen in the representative peak maps of slices from LFPI animals (Figure 5A).
Based on the apparent decrease in spread of depolarization after LFPI, we next sought to examine the propagation of activity through the amygdala circuit. In order to normalize differences in activation of the primary input (lateral amygdala) and isolate propagation of activity, we used the FDP from the different amygdala regions to create ratios of activation, a method originally described in (Avrabos et al., 2013). The amygdala circuit propagates signals in a dorsoventral, lateromedial fashion, beginning in the lateral amygdala (LA) and moving towards the medial portion of CeA. Thus, an important measure of amygdala circuit function or strength is what percentage of depolarization in LA is propagated to basal amygdala (BA) and/or CeA. For this analysis, LA was defined as a combination of raster segments 1-3 of both BLA(m) and BLA(l), the BA region was defined as a combination of raster segments 7-10 of both BLA(m) and BLA(l), and the CeA region was viewed in its entirety (segments 1-6). BA and CeA FDP response amplitudes to various stimulation intensities were divided by the corresponding LA FDP amplitude to create activation ratios that express the relative strength of LA-to-CeA or LA-to-BA signaling in slices from sham and LFPI animals. Amygdalae of LFPI animals exhibit a significant decrease in the ratio of activation between LA-to-BA (Fig. 5B, sham n=16 slices / 9 animals, LFPI n=14 slices / 7 animals, two-way repeated-measures ANOVA, (injury effect) F (1,14) = 10.89; P = 0.005) and LA-to-CeA (Fig. 5C, sham n=16 slices / 9 animals, LFPI n=14 slices / 9 animals, two-way repeated-measures ANOVA, (injury effect) F (1,14) = 12.77; P= 0.003). Taken together with previous measures of network excitability, this data suggests there is both a decrease in the excitability of the primary input to the amygdala together with a decrease in signal propagation through the amygdala circuit following LFPI.
Late hyperpolarization in VSD signal mediated via GABAB
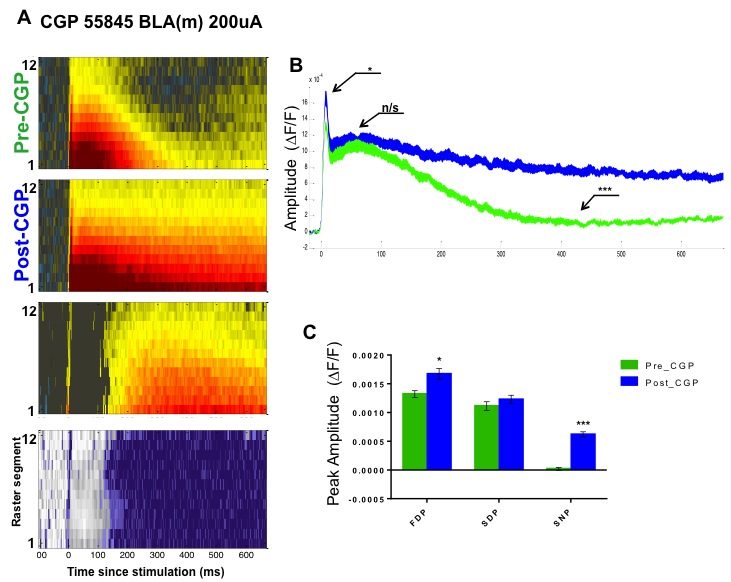
To further investigate the putative contribution of GABAA and B to the late hyperpolarization component of the VSD signal, VSD recordings were performed in brain slices from naïve animals treated with the GABAB antagonist GCP55845 (1 μM). As seen in Table S1 and Supplementary figure S3, application of CGP55845 completely abolishes the late hyperpolarization, revealing a persistent depolarization similar to the transmembrane voltage response of stimulated glia (Leung and Zhao, 1995). Analysis of the average ΔF/F signal shows that CGP55845 has a small but significant effect on the FDP (Fig. S3B&C, pre-CGP 13.3 95% CI 11.6–14.8 × 10−4 ΔF/F, post-CGP 17.1 95% CI 14.1–19.3 × 10−4 ΔF/F n=5 slices; P= 0.03), perhaps due to the presynaptic activity of GABAB (Yamada et al., 1999), and a drastic effect on the SNP (Fig. S3B&C, pre-CGP .571 95% CI −.471–.86 × 10−4 ΔF/F, post-CGP 6.0 95% CI 4.88–7.47 × 10−4 ΔF/F n=5; P= 0.008).
BLA pyramidal neuron intrinsic excitability is unaltered by LFPI
To determine if the LFPI induced differences seen in VSD imaging and fEPSP recordings were due to a change in the intrinsic properties of amygdala principal neurons, we performed a series of whole-cell current-clamp recordings of BLA pyramidal neurons in brain slices from sham and LFPI animals. To begin, we assessed the passive membrane properties of amygdala neurons that effect the fractional changes in fluorescence measured in VSD recordings. Resting membrane voltage (Vm) measured immediately following break in was unaffected by LFPI (Fig. 6C, sham −78.05 95% CI −80.97--75.28 mV n=8, LFPI −79.45 95% CI −81.8--77.1 mV n=8; P= 0.49). Furthermore, there was no significant difference in the input resistance (Rin) of BLA pyramidal neurons in slices from sham and LFPI animals (Fig. 6D, sham 118.6 95% CI 99.56–151.7 MΩ n=8, LFPI 133 95% CI 112.2–144.9 MΩ n=8; P= 0.78). Next, a series of depolarizing current injections were used to assess intrinsic excitability. Action potential threshold (Fig. 6E, sham −53.79 95% CI −56.4--52.18 mV n=8, LFPI −52.78 95% CI −54.5--51.18 mV n=8; P= 0.32) and frequency in response to depolarizing current were not significantly different in BLA neurons from sham and LFPI slices (Fig. 6B, sham n=8 LFPI n=8, two-way repeated-measures ANOVA, (injury effect) F (1,14) = .09628; P = 0.76).
Fig. 6. BLA Pyramidal neuron intrinsic excitability.

(A) Representative voltage response to current injection in cells from sham and LFPI animals (−50 & 200 pA current steps). (B) AP frequency in response to depolarizing current (−50-225pA) (P= 0.76). (C) Resting membrane potential measured immediately following break in (P= 0.49). (D) Input resistance calculated from hyperpolarizing current injections (P= 0.78). (E) Action potential threshold: Vm at which 1st evoked AP initiated independent of current step (P= 0.32). All comparisons were made between Sham (n=8 cells / 4 animals) and LFPI (n=8 cells / 5 animals).
Discussion
Neuropsychiatric symptoms are highly prevalent in mTBI patients and represent some of the most prolonged and debilitating symptoms associated with mTBI (Jorge et al., 2004; Malkesman et al., 2013; McAllister, 1992; Tateno et al., 2003). In this study we have taken the first steps towards understanding how mTBI disrupts circuit level function in the amygdala, a brain region crucial for processing emotional stimuli. Initially, we explored amygdala-dependent behaviors 7 days post-LFPI, demonstrating a significant deficit in threat response in a cued fear conditioning paradigm. We next characterized LFPI-induced alterations in amygdala physiology using a combination of electrophysiological and voltage sensitive dye imaging (VSD) techniques. We discovered significant decreases in network excitability and activation throughout several amygdala sub-regions. Furthermore, leveraging the spatial information provided by VSD imaging we establish that in addition to the primary input to the amygdala being less excitable, there is also a congruent decrease in signal propagation of activation through the amygdala circuit. Finally, we preformed a series of whole-cell current-clamp recordings demonstrating that the intrinsic excitability and membrane properties of BLA pyramidal neurons are unaltered by LFPI. Together these data suggest that LFPI causes robust circuit level dysfunction in the amygdala associated with an altered behavioral response to threatening stimuli.
Many previous reports have shown opposing effects in amygdala associated behaviors following mTBI (Malkesman et al., 2013; Reger et al., 2012). A recent publication by Reger et al. reported enhanced fear conditioning 4 days post LFPI in rats. Potential reasons for the heterogeneity in results using animal models is likely due to subtle differences in technique, experimental time point, injury models, and animal strains used. Similarly, the array of neuropsychiatric symptoms present in mTBI patients is likely the consequence of the heterogeneity of physical insults suffered by the brain that result in mTBI and a variety of other environmental factors. It is certainly feasible that different animal brain injury protocols are simply modeling different mTBI patient populations or post-injury time points, rather than producing contradictory results. For our study we chose a well-established behavioral task (cued fear conditioning), in which the threat response (freezing behavior) is directly associated with activation of the medial portion of central amygdala (Duvarci and Pare, 2014; Ledoux, 2000; Phillips and Ledoux, 1992). Our injury model produced a consistent and robust decrease in freezing behavior in a cued fear conditioning paradigm at 7 days post-LFPI. While increases in anxiety and fear response are often associated with mTBI it is important to note that depression/apathy is at least as prevalent as anxiety disorders in human mTBI patients (Jorge et al., 2004; Malkesman et al., 2013). Furthermore, it is conceivable that the observed behavioral deficit is only present acutely (7 days after injury) and could progress to heightened fear response or anxiety at protracted time points.
As a decrease in freezing behavior in a cued fear response paradigm is typically associated with decreased network excitability in the amygdala, we hypothesized that amygdala excitability would be diminished as a result of LFPI. In order to test this hypothesis we performed a series of electrophysiological and VSD experiments. Basolateral amygdala (BLA) fEPSP responses evoked by lateral amygdala (LA) stimulation, revealed a significant decrease in network excitability, as measured by fEPSP input/output curves following LFPI. Interestingly, paired-pulse stimulation was not significantly altered by LFPI, suggesting that a change in probability of vesicular release did not occur. Furthermore, the pre-synaptic fiber volley was also unchanged by LFPI, implying that the number of neurons activated by the stimulating electrode was not different in slices derived from sham and LFPI animals. The lack of in injury effect on the intrinsic properties of BLA pyramidal neurons further supports the conclusion that a similar number of principal cells were activated in brain slices from sham and LFPI animals. Overall these results suggest that LFPI disrupts the balance between excitation and inhibition in the amygdala via mechanisms other than a change in probability of release or large scale excitatory cell loss, likely through a perturbation of the critical balance between glutamatergic and GABAergic transmission seen in other brain regions following LFPI (Cole et al., 2010b; Witgen et al., 2005).
To further probe amygdala circuit function and examine the spatiotemporal dynamics of activation throughout all amygdala sub-regions, we performed a series of VSD experiments. These experiments corroborated fEPSP data revealing a strong LFPI-induced decrease in the fast depolarizing component in all amygdala sub-regions. Once we determined that LFPI causes a significant decrease in amygdala network excitability, we were further interested in determining if this decrease was due to a decreased ability to excite the primary input of the amygdala (LA), a decrease in the propagation of the signal through the amygdala circuit, or both. The LFPI-induced decrease in depolarization adjacent to the stimulating electrode (visible in representative movie frames & and group raster plots) shows a decrease in activation of the primary input. By normalizing the amount of activation adjacent to the stimulation electrode we were able to determine the percentage of activation of the primary input of the amygdala (LA) that was propagated to distal regions of BA or the primary output of the amygdala i.e., the CeA. This revealed that not only is the LA less excitable after injury, but the relative circuit strength of LA-to-CeA and LA-to-BA projections are also weakened by LFPI.
The fast depolarizing response of the VSD signal is thought to be a mixed signal including action potential firing, glutamatergic and GABAergic transmission (Carlson and Coulter, 2008; Tominaga et al., 2000). Notably, this decrease in FDP was present in the central amygdala, which projects directly to autonomic brain stem structures responsible for the freezing behavior measured in the cued fear conditioning paradigm (Duvarci and Pare, 2014; Sah et al., 2003). The typical observed VSD response had two or three distinct peaks, depending on region, the aforementioned fast depolarizing peak (FDP), a protracted slow depolarizing peak (SDP), and a late slow negative peak (SNP) (Fig. 3C). The FDP is most commonly assessed as a measure of activation/excitation and is the best characterized portion of the VSD signal. The SDP and SNP are likely heavily influenced by gliotic activation, which is proportional to the initial depolarization (Leung and Zhao, 1995). Thus, differences in these peaks could simply be a consequence of the differences observed in the preceding FDP. However, GABAB is playing a large role in the generation of the SNP (see Fig. S3) and could be contributing to the differences in late hyperpolarization between slices from sham and LFPI animals. Further, the observation that an LFPI-induced decrease in the FDP and SDP persists in the presence of glutamatergic antagonists suggests that other intrinsic and/or extrinsic components of the amygdala circuit are affected by LFPI. While the current study does not directly assess GABAergic inhibition in the amygdala, previous findings from our laboratory demonstrated an increase in inhibition from CCK positive interneurons in area CA1 of the hippocampus, 7 days post LFPI(Johnson et al., 2014). It is possible that CCK positive inhibitory interneurons present in the BLA have a similar response to LFPI and contribute to the decrease in amygdala network excitability demonstrated in this study.
We next examined the intrinsic properties of BLA pyramidal neurons to determine if the LFPI-induced decrease in excitability seen in fEPSP and VSD recordings was a consequence of changes in the intrinsic excitability of the principal excitatory cell type within the BLA. Whole-cell current-clamp recordings of BLA pyramidal neurons revealed no changes in passive membrane properties or measures of intrinsic excitability following LFPI. As the amygdala nuclei are somewhat distal from the site of injury this finding, while not completely unexpected, does further elucidate a possible mechanism of the decrease in amygdala activation following LFPI. Taken together with FV and VSD in the presence of glutamatergic antagonists results, these experiments suggest that LFPI decreases excitability in the amygdala via a perturbation of glutamatergic transmission and likely other neurotransmitter/neuromodulator systems.
In summary, our data demonstrates that LFPI causes substantial perturbation of the balance between excitation and inhibition within the amygdala circuit, resulting in a decrease in network excitability of the basolateral amygdala and weakening of dorsoventral-lateromedial internuclear amygdala projections. Further, this decrease in amygdala network excitability correlates to a depression in threat response behavior. To our knowledge this is the first study demonstrating the comprehensive effects of LFPI on amygdala circuit function and strength. This work highlights promising new sites of study towards understanding the mechanism LFPI-induced amygdala dysfunction and for the development of therapeutics targeted at mitigating or ameliorating mTBI-induced amygdala circuit dysfunction.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
Brain injury (LFPI) caused a decrease in amygdala-dependent cued threat response
LFPI induced a decrease in network excitability of the basolateral amygdala
Strength of internuclear amygdala projections was significantly weakened by LFPI
LFPI had no effect on the intrinsic excitability of amygdala principal neurons
Acknowledgments
This work was supported in part by NIH grants R37 HD059288 and RO1 NS069629. The authors would like to thank Drs. Gordon Barr and Colin Smith for critiquing and earlier version of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ang CW, Carlson GC, Coulter DA. Massive and Specific Dysregulation of Direct Cortical Input to the Hippocampus in Temporal Lobe Epilepsy. Journal of Neuroscience. 2006;26:11850–11856. doi: 10.1523/JNEUROSCI.2354-06.2006. doi:10.1523/JNEUROSCI.2354-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrabos C, Sotnikov SV, Dine J, Markt PO, Holsboer F, Landgraf R, Eder M. Real-Time Imaging of Amygdalar Network Dynamics In Vitro Reveals a Neurophysiological Link to Behavior in a Mouse Model of Extremes in Trait Anxiety. Journal of Neuroscience. 2013;33:16262–16267. doi: 10.1523/JNEUROSCI.2397-13.2013. doi:10.1523/JNEUROSCI.2397-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazarian JJ, Cernak I, Noble-Haeusslein L, Potolicchio S, Temkin N. Long-term neurologic outcomes after traumatic brain injury. J Head Trauma Rehabil. 2009;24:439–451. doi: 10.1097/HTR.0b013e3181c15600. doi:10.1097/HTR.0b013e3181c15600. [DOI] [PubMed] [Google Scholar]
- Bourgeois EB, Johnson BN, McCoy AJ, Trippa L, Cohen AS, Marsh ED. A Toolbox for Spatiotemporal Analysis of Voltage-Sensitive Dye Imaging Data in Brain Slices. PLoS ONE. 2014;9:e108686. doi: 10.1371/journal.pone.0108686. doi:10.1371/journal.pone.0108686.s011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell WS, Maris DO, McCall T, Grady MS. Adaptation of the fluid percussion injury model to the mouse. Journal of Neurotrauma. 1998;15:217–229. doi: 10.1089/neu.1998.15.217. [DOI] [PubMed] [Google Scholar]
- Cardinal RN, Parkinson JA, Hall J, Everitt BJ. Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev. 2002;26:321–352. doi: 10.1016/s0149-7634(02)00007-6. [DOI] [PubMed] [Google Scholar]
- Carlson GC, Coulter DA. In vitro functional imaging in brain slices using fast voltage-sensitive dye imaging combined with whole-cell patch recording. Nat Protoc. 2008;3:249–255. doi: 10.1038/nprot.2007.539. doi:10.1038/nprot.2007.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JT, Mitala CM, Kundu S, Verma A, Elkind JA, Nissim I, Cohen AS. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proceedings of the National Academy of Sciences. 2010a;107:366–371. doi: 10.1073/pnas.0910280107. doi:10.1073/pnas.0910280107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JT, Mitala CM, Kundu S, Verma A, Elkind JA, Nissim I, Cohen AS. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proceedings of the National Academy of Sciences. 2010b;107:366–371. doi: 10.1073/pnas.0910280107. doi:10.1073/pnas.0910280107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depue BE, Olson-Madden JH, Smolker HR, Rajamani M, Brenner LA, Banich MT. Reduced Amygdala Volume Is Associated with Deficits in Inhibitory Control: A Voxel- and Surface-Based Morphometric Analysis of Comorbid PTSD/Mild TBI. BioMed Research International. 2014;2014:1–11. doi: 10.1155/2014/691505. doi:10.1523/JNEUROSCI.5606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dębiec J, Díaz-Mataix L, Bush DEA, Doyère V, LeDoux JE. The amygdala encodesspecific sensory featuresof an aversive reinforcer. Nature Publishing Group. 2010;13:536–537. doi: 10.1038/nn.2520. doi:10.1038/nn.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, Pare D. Amygdala Microcircuits Controlling Learned Fear. Neuron. 2014;82:966–980. doi: 10.1016/j.neuron.2014.04.042. doi:10.1016/j.neuron.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang A-G, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- Faul M, Coronado V. Epidemiology of traumatic brain injury. Handb Clin Neurol. 2015;127:3–13. doi: 10.1016/B978-0-444-52892-6.00001-5. doi:10.1016/B978-0-444-52892-6.00001-5. [DOI] [PubMed] [Google Scholar]
- Johnson BN, Palmer CP, Bourgeois EB, Elkind JA, Putnam BJ, Cohen AS. Augmented Inhibition from Cannabinoid-Sensitive Interneurons Diminishes CA1 Output after Traumatic Brain Injury. Front. Cell. Neurosci. 2014;8:435. doi: 10.3389/fncel.2014.00435. doi:10.3389/fncel.2014.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge RE, Robinson RG, Moser D, Tateno A, Crespo-Facorro B, Arndt S. Major depression following traumatic brain injury. Arch. Gen. Psychiatry. 2004;61:42–50. doi: 10.1001/archpsyc.61.1.42. doi:10.1001/archpsyc.61.1.42. [DOI] [PubMed] [Google Scholar]
- Juranek J, Johnson CP, Prasad MR, Kramer LA, Saunders A, Filipek PA, Swank PR, Cox CS, Ewing-Cobbs L. Mean diffusivity in the amygdala correlates with anxiety in pediatric TBI. Brain Imaging and Behavior. 2011;6:36–48. doi: 10.1007/s11682-011-9140-5. doi:10.1007/s11682-011-9140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- LeDoux J. Perspective. Neuron. 2012;73:653–676. doi: 10.1016/j.neuron.2012.02.004. doi:10.1016/j.neuron.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux JE. Emotion circuits in the brain. Annu. Rev. Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. doi:10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Leung LS, Zhao D. Glial potentials evoked by single afferent pulses in hippocampal CA1 area in vitro. Brain Research. 1995;697:262–265. doi: 10.1016/0006-8993(95)00976-w. [DOI] [PubMed] [Google Scholar]
- Malkesman O, Tucker LB, Ozl J, McCabe JT. Traumatic brain injury - modeling neuropsychiatric symptoms in rodents. Front. Neur. 2013;4:157. doi: 10.3389/fneur.2013.00157. doi:10.3389/fneur.2013.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister TW. Neuropsychiatric sequelae of head injuries. Psychiatric Clinics of North America. 1992 [PubMed] [Google Scholar]
- Newton JR, Ellsworth C, Miyakawa T, Tonegawa S, Sur M. Acceleration of visually cued conditioned fear through the auditory pathway. Nat Neurosci. 2004;7:968–973. doi: 10.1038/nn1306. doi:10.1038/nn1306. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the Amygdala to Emotion Processing: From Animal Models to Human Behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. doi:10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Phillips RG, Ledoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behavioral Neuroscience. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Rao V, Rosenberg P, Bertrand M, Salehinia S, Spiro J, Vaishnavi S, Rastogi P, Noll K, Schretlen DJ, Brandt J, Cornwell E, Makley M, Miles QS. Aggression after traumatic brain injury: prevalence and correlates. J Neuropsychiatry Clin Neurosci. 2009;21:420–429. doi: 10.1176/appi.neuropsych.21.4.420. doi:10.1176/appi.neuropsych.21.4.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger ML, Poulos AM, Buen F, Giza CC, Hovda DA, Fanselow MS. Concussive Brain Injury Enhances Fear Learning and Excitatory Processes in the Amygdala. Biological Psychiatry. 2012;71:335–343. doi: 10.1016/j.biopsych.2011.11.007. doi:10.1016/j.biopsych.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggio S. Traumatic Brain Injury and Its Neurobehavioral Sequelae. Psychiatric Clinics of North America. 2010;33:807–819. doi: 10.1016/j.psc.2010.08.004. doi:10.1016/j.psc.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Rutland-Brown W, Langlois JA, Thomas KE, Xi YL. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehabil. 2006;21:544. doi: 10.1097/00001199-200611000-00009. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ESL, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol. Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. doi:10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Tateno A, Jorge RE, Robinson RG. Clinical correlates of aggressive behavior after traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2003;15:155–160. doi: 10.1176/jnp.15.2.155. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. Journal of Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. doi:10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Tominaga Y, Yamada H, Matsumoto G, Ichikawa M. Quantification of optical signals with electrophysiological signals in neural activities of Di-4-ANEPPS stained rat hippocampal slices. Journal of Neuroscience Methods. 2000;102:11–23. doi: 10.1016/s0165-0270(00)00270-3. [DOI] [PubMed] [Google Scholar]
- Tran LD, Lifshitz J, Witgen BM, Schwarzbach E, Cohen AS, Grady MS. Response of the contralateral hippocampus to lateral fluid percussion brain injury. Journal of Neurotrauma. 2006;23:1330–1342. doi: 10.1089/neu.2006.23.1330. doi:10.1089/neu.2006.23.1330. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Corcoran KA, Jovasevic V, Radulovic J. Fear conditioning and extinction: emotional states encoded by distinct signaling pathways. Trends in Neurosciences. 2012;35:145–155. doi:10.1016/j.tins.2011.10.003. [Google Scholar]
- Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang SL, Grady MS, Cohen AS. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: A systems, network and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. doi:10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]
- Wolff SBE, Gründemann J, Tovote P, Krabbe S, Jacobson GA, Müller C, Herry C, Ehrlich I, Friedrich RW, Letzkus JJ, Lüthi A. Amygdala interneuron subtypes control fear learning through disinhibition. Nature. 2014;509:453–458. doi: 10.1038/nature13258. doi:10.1038/nature13258. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. doi:10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Saitow F, Satake S, Kiyohara T, Konishi S. GABA(B) receptor-mediated presynaptic inhibition of glutamatergic and GABAergic transmission in the basolateral amygdala. Neuropharmacology. 1999;38:1743–1753. doi: 10.1016/s0028-3908(99)00126-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.