

Abstract
Subclinical circulating bacterial endotoxin lipopolysaccharide (LPS) has been implicated as an important cofactor in the development and progression of nonalcoholic steatohepatitis (NASH), but the underlying mechanisms remain unclear. Here, we demonstrated that 4-week injection with super-low dose LPS significantly promoted neutrophils infiltration and accelerated NASH progression, including exacerbated macro-vesicular steatosis, inflammation and hepatocyte ballooning in high-fat diet fed apolipoprotein E knockout mice. This effect could sustain for a month after stoppage of LPS injection. LPS also significantly increased numbers of apoptotic nuclei in hepatocytes and expressions of pro-apoptotic regulators. Moreover, LPS sustained the low-grade activation of p38 mitogen-activated protein kinase and inhibited the expression of the upstream MAPK phosphatase 7. By applying selective inhibitors, we demonstrated that the activation of p38 MAPKs is required for neutrophil migration induced by super-low dose LPS in vitro. Together, these data suggest that super-low dose LPS may sustain the low-grade activation of p38 MAPKs and neutrophil infiltration, leading to the exacerbation of steatohepatitis.
Keywords: Low-grade inflammation, Mitogen-activated protein kinase, Neutrophil, Nonalcoholic fatty liver disease, Super-low dose endotoxin
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a condition ranging from simple lipid accumulation in the liver (steatosis) to steatosis combined with inflammation (nonalcoholic steatohepatitis, NASH) (1). Steatosis alone is considered a relatively benign and reversible condition. The transition toward NASH represents a key step in pathogenesis, because it will set the stage for further damage to the liver including fibrosis, cirrhosis, and liver cancer (2). The actual risk factors that drive hepatic inflammation during the progression to NASH remain largely unknown. Therefore, knowledge about events that induce steatohepatitis is of great importance for the diagnosis and treatment of NASH.
A growing body of experimental and clinical data suggests that subclinical low-levels of blood circulating endotoxin, likely derived from mucosal leakages, plays an important part in low-grade chronic inflammatory disease such as nonalcoholic liver injury (3). LPS may be capable of stimulating inflammation, cytokine production, and accumulation of inflammatory cells within the liver (4). In fact, slightly elevated circulating endotoxemia has been reported to be associated with a rise in tumor necrosis factor α (TNF-α) gene expression in the hepatic tissue, which supports a role of subclinical super-low dose endotoxemia in the development of steatohepatitis in obese individuals (5, 6). However, the pathophysiology of this phenomenon is not well understood. Neutrophils constitute the first line of defense against most microorganisms (7). Neutrophils actively recruited to liver sinusoids are a distinguishing feature of endotoxemia and sepsis (8). Their effector functions include phagocytosis, release of proteolytic enzymes, and regulation of the immune response (9). Although facilitating the elimination of invading organisms and their derived products, these functions can also cause severe tissue injury (10, 11). Despite these findings, the impact of super-low grade endotoxemia on liver neutrophil infiltration during NASH progression remains elusive. Therefore, we hypothesized that subclinical super-low level circulating LPS might sustain a low-grade systemic inflammation, facilitate the recruitment of neutrophils to the liver tissue, thereby promoting progression of NASH.
To address this question, we used a high-fat diet fed apolipoprotein E knockout (ApoE−/−) mice model, which results in graded steatosis, inflammation and fibrosis, closely resembling the pathophysiology of progressive NAFLD in humans (12). We studied the exacerbating effect of chronic super-low dose LPS injection on liver histopathology. We also determined whether steatohepatitis may be sustained when LPS injection is terminated. At the cellular level, the effect of super-low dose LPS on neutrophil migration was evaluated in vitro. We provide evidence that super-low dose LPS induces elevated neutrophil migration in vitro as well as liver neutrophil infiltration during the progression of NASH, through sustaining the low-grade activation of mitogen-activated protein kinases (MAPKs) p38. Our data support a model of sustained chronic low-grade inflammation and exacerbation of NASH pathogenesis in mice that experience super-low dose endotoxemia.
Materials and methods
Reagents
Antibodies against phospho-p38 MAPK (Thr180/Tyr182), p38 MAPK, phospho-JNK (Thr183/Tyr185) MAPK, JNK, phospho-ERK (Thr202/Tyr204) MAPK, ERK, MAPK phosphatase 7 (MKP7), poly (ADP-ribose) polymerase (PARP), and active caspase-3, and horseradish peroxidase conjugated secondary antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Mouse monoclonal antibody specific for GAPDH was purchased from Santa Cruz Biotech. (Santa Cruz, CA). Phycoerythrin (PE)-labeled anti-mouse Ly-6G antibody was provided by BioLegend, Inc. (San Diego, CA). Fat stain kit was provided by Newcomer Supply (Middleton, WI). All other chemicals, unless otherwise specified, were from Sigma–Aldrich (St. Louis, MO).
Animals
Specific pathogen-free male, C57BL/6J ApoE−/− mice were purchased from the Jackson Laboratory (Sacramento, CA) and used at 8–12 wk of age in the experiments described. Thirty-two mice were randomly divided into two groups: LPS group, injected intraperitoneally with 4 ng/kg body weight (~100 pg/mouse) purified LPS dissolved in endotoxin-free phosphate buffered saline (PBS); Control group, injected intraperitoneally with the same volume of PBS. Mice were injected i.p. every three days with either PBS or LPS. Upon acclimation with injection for 10 days, the mice were fed with a high-fat diet (TD90221; Harlan Tek-lad, Madison, WI) together with i.p. injection of either LPS or PBS every three-days for 4 weeks. A batch of mice were harvested for analyses. To determine whether the systemic effects of LPS injection may last post injection, another batch of mice were continually fed with HFD diet but without further LPS injection for an additional 4 weeks. Animals were housed under a 12:12-hour light/dark cycle and permitted ad libitum consumption of water and diet. Animal protocols were approved by the Animal Care and Use Committee of Virginia Polytechnic Institute and State University.
Biochemical analyses
The plasma and liver samples of each mouse were collected and stored at −80°C for further analysis. Plasma alanine aminotransferase (ALT), lactate dehydrogenase (LDH), free fatty acid (FFA), triglyceride (TG), and hepatic TG contents were determined using their respective detection commercial kits (Biovision, Milpitas, CA) in accordance with the manufacturer’s protocols. Myeloperoxidase (MPO) activities in liver extracts were quantified using Duoset ELISA development kit (R&D Systems, Minneapolis, MN). Briefly, 50-100 mg tissue was homogenized in 1 mL potassium phosphate buffer (50 mM, pH 6.0) on ice. Then, homogenates were centrifuged at 12,000 g for 10 min at 4°C. MPO activity and protein concentration (DC Protein Assay, Bio-Rad Laboratories Inc., Hercules, CA) in the supernatant was assayed according to the manufacturer’s instructions, respectively. The results are expressed as MPO ng/mg protein.
Histological assessment
Fresh liver tissue were embedded in Tissue Tek OCT and rapidly frozen in liquid nitrogen and then stored at −80°C for preparation of frozen sections. Five-micrometer cut sections were stained with hematoxylin and eosin (H&E) and Oil Red O for histological analysis and lipid under light microscopy (Olympus, Tokyo, Japan), respectively. The Pathology Committee of the NASH Clinical Research Network (13) provided guidance and recommendations for the NAFLD activity scoring (NAS) by semi-quantitatively evaluating the following histological features: steatosis (< 5% = 0; 5–33% = 1; 33–66% = 2; >66% = 3); lobular inflammation (none = 0; < 2 foci = 1; 2–4 foci = 2; >4 foci = 3); and hepatocellular ballooning (none = 0; few = 1; prominent = 2). All features were scored blindly based on at least 5 samples per group and 5 fields of vision in each sample.
Immunofluorescence and immunohistochemistry
Serial 5 μm sections were fixed with PBS containing 4% paraformaldehyde for 15 min. Slides were then incubated in serum blocking solution for another 30 min and incubated overnight in anti-Ly6G antibody or anti-MOMA2 antibody as specified in the figure legend. Following removal of the primary antibody, fluorescent secondary antibodies were used for immunofluorescence. DAPI (Roche, Germany) was utilized for nuclear staining and observed through Inverted Fluorescence Microscopy (Olympus, Tokyo, Japan). Ly6G expressing cells were then counted by ImageJ software (Research Services Branch of the NIH, Bethesda, MD).
Apoptotic cells in liver were detected by immunocytochemistry using Apo-BrdU-IHC in situ DNA fragmentation assay kit (Bio-Rad AbD Serotec Ltd., Raleigh, NC) according to the manufacturer’s protocols. Visualization of the reaction was performed using DAB as the chromogenic substrate and methyl green as a counter stain. Cells were considered positive for apoptosis as assessed by 2 criteria: a positive nuclear stain and, morphologic evidence of apoptotic cells. Five fields were counted per slide. The apoptotic index was calculated as follows: No. of apoptotic cells/(no. of apoptotic cell + no. of negative cells).
RNA extraction and real-time PCR
Total RNA was extracted from liver tissues using Trizol reagent and reverse transcription of mRNA was performed using the SuperScript Reverse Transcriptase reagent kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s recommendations. Quantitative PCR was performed using a real-time PCR system (Applied Biosystems CFX96), and reactions were performed using SYBR Green Master Mix (BioRad) with gene-specific primers. To normalize expression data, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control gene.
Neutrophil preparation and chemotaxis assay
Male C57BL/6J mice were purchased from the Jackson Laboratory and used at 6 weeks old. The mice were sacrificed by cervical dislocation. The femurs were removed, and the femoral bone marrow was harvested by flushing with 5 mL PBS buffer. The erythrocytes were removed using hypotonic shock and the leukocyte population was resuspended in PBS buffer. The purity of isolated neutrophils was routinely > 95% as assessed by light microscopic analysis of the cells stained with Diff-Quick (Wako Pure Chemical Industries, Osaka, Japan), and > 95% viable as assessed by a trypan blue exclusion test. Harvested neutrophils were pretreated with 20 μM SB203580 (p38 inhibitor) or 20 μM SP600125 (JNK inhibitor) for 2 h, then treated with or without 100 pg/mL LPS and incubate for 24 h.
Chemotaxis assays were performed using HTS Transwell-96 plates with 3-μm pores (Corning, Lowell, MA). After LPS stimulation, mouse neutrophils were added into the upper wells (2 × 105 cells/well) and PBS or 100 ng/mL of recombinant mouse chemokines CXCL2/MIP-2 (R&D Systems, Minneapolis, MN) was added to the bottom chamber. The plates were incubated at 37°C for 30 min. Because MIP-2 induces chemotaxis of neutrophils but not monocytes, only neutrophils will migrate into the matrigel (14). After removing the upper wells, migrated cells were stained with Wright-Giemsa and counted by light microscopy after coding the samples. For each experiment, the number of neutrophils in five fields (× 200) was counted and averaged at each incremental level.
Western Blot
Liver tissues or neutrophils lysates containing 30 μg of total protein were loaded onto a 10% SDS polyacrylamide gels. After 90 min of electrophoresis, the proteins were transferred onto a polyvinylidene difluoride membrane in ice for 2 h. The membrane was blocked with 5% skim milk which dissolved in trihydroxymethyl aminomethane buffer salt + Tween-20 (TBST). The membrane was next incubated at 4°C overnight with primary antibodies. Secondary antibodies conjugated with horseradish peroxidase were incubated for 2 h at room temperature. Signals were detected by enhanced chemiluminescence (Thermo Fisher Scientific, Waltham, MA). GAPDH was used for normalization. The density of the specific bands was quantified using ImageJ software.
Statistics
The data are expressed as mean ± standard error of the mean (SEM). Statistical differences were analyzed by Student's t-test using SPSS version 18.0 (SPSS Inc., Chicago, IL). Differences were considered as statistically significant if P < 0.05.
Results
Subclinical super-low dose LPS exacerbates lipid accumulation in liver and plasma
Although recent studies have suggested potential connection between low-grade endotoexmia and chronic inflammation, dosages of LPS injection in animal models tend to be in the mg/kg range (15-17), which are significantly higher than the clinical concentrations (~ng/kg) routinely observed in mice and humans with chronic conditions (18, 19). In order to mimic the pathophysiological effects of low-grade endotoxemia, the dosage of LPS used in this study were much lower than previous studies. We set up the i.p. injection of ApoE deficient mice with either PBS or LPS (4 ng/kg body weight), as the i.p. route was known to cause low-level circulating endotoxemia (20, 21). Mice were fed with a high-fat diet and continued with the injection of PBS or LPS every three-days for 4 weeks. 50% of the animals in each group were sacrificed and the rest continued on the same diet but without injection for another 4 weeks, to further examine the potential lasting “memory” effects of low-grade endotoxemia. Liver pathology, neutrophil infiltration, potential molecular mechanisms in vivo and in vitro were systematically evaluated.
As shown in Figure 1A, Oil Red O staining clearly indicated that the high-fat diet feeding induced lipid accumulation in the liver, especially in hepatocytes. In line with a previous report with chronic injection of LPS (22), we documented that 4-week injection with super-low dose LPS exacerbated high-fat diet induced lipid accumulation in the liver (Figure 1B). The enhanced liver triglyceride (TG) accumulation in the LPS group was transient and not sustained after the stoppage of LPS injection at the 8 wk time point. There was no significant difference in plasma TG content between the two groups (Figure 1C). The plasma FFA levels were significantly elevated in the LPS injected group at the 4 wk time point as compared to the PBS group. Strikingly, the significantly elevated plasma FFA levels in the LPS injected group were sustained even one month after the stoppage of LPS injection at the 8 wk time points (Figure 1D).
Figure 1. Liver and plasma lipid content in high-fat diet fed ApoE−/− mice.

(A) Frozen sections (5 μm thick) were stained with Oil Red O, which marked neutral lipid (200 × magnification). (B, C) TG levels were measured from all frozen liver specimens (B) and plasma (C). (D) Plasma FFA concentrations. Results are shown as mean ± SEM (n = 8) or as representative images from at least three independent experiments. *P < 0.05 versus the control group at each time point.
Chronic injection of super-low dose LPS did not affect the body weight gain in high-fat diet fed ApoE−/− mice, accompanied by no significant difference in liver weights (% of body weight) during the experimental period (Table 1). Compared with the PBS control group, the plasma levels of LDH were transiently increased in the LPS group at wk 4 as compared to the PBS group. The plasma ALT activities in the LPS group were also higher than that of control group, but without significant difference.
Table 1.
Effect of LPS on animal characteristics in high-fat diet fed ApoE−/− mice.
| LPS | Control | P value | |
|---|---|---|---|
| Initial body weight (g) | 22.04 ± 0.29 | 22.26 ± 0.41 | 0.669 |
| 4 wk | |||
| Body weight (g) | 30.22 ± 1.59 | 32.34 ± 1.05 | 0.282 |
| Liver/body weight (%) | 6.00 ± 0.10 | 5.66 ± 0.26 | 0.290 |
| Plasma LDH (U/L) | 23.25 ± 2.07 | 10.93 ± 0.90 | < 0.001 |
| Plasma ALT (U/L) | 38.01 ± 3.27 | 31.52 ± 1.56 | 0.102 |
| 8 wk | |||
| Body weight (g) | 35.33 ± 1.25 | 35.34 ± 1.84 | 0.995 |
| Liver/body weight (%) | 5.97 ± 0.13 | 5.79 ± 0.21 | 0.456 |
| Plasma LDH (U/L) | 16.38 ± 5.45 | 13.94 ± 3.73 | 0.315 |
| Plasma ALT (U/L) | 43.22 ± 2.72 | 41.55 ± 2.13 | 0.304 |
ALT, alanine aminotransferase; LDH, lactate dehydrogenase.
Values represent mean ± SEM, n = 16 for initial body weight and n = 8 for the other parameters.
Super-low dose LPS initiates sustained chronic inflammation and exacerbates liver steatohepatitis
To determine the effect of super-low dose LPS on hepatic morphology, we performed H&E staining of the liver sections. In general, the histopathological features required for a diagnosis of NASH in humans include macrovascular steatosis (hepatocyte fat accumulation), lobular inflammation around hepatocytes and hepatic sinusoids and hepatocyte ballooning (23). At wk 4, mice injected with PBS did not show significant inflammation (Figure 2A and 2B). In contrast, ApoE−/− mice injected with super-low dose LPS developed more severe hepatic inflammation compared to controls as reflected by increased inflammatory cell infiltration and significantly elevated expression of inflammatory markers such as TNF-α, monocyte chemotactic protein-1 (MCP-1), interleukin-6 (IL-6) (Figure 2B). Strikingly, the significant elevation of liver inflammatory mediators was remarkably sustained, one month after the stoppage of LPS injection at the 8 wk measurement point, suggesting an inflammatory “memory” effect following the chronic injection of super-low dose LPS (Figure 2B).
Figure 2. Super-low dose LPS exacerbates and sustains steatohepatitis in high-fat diet fed ApoE−/− mice.

(A) Hepatic histological analysis of H&E staining (200 × magnification). Inflammatory infiltration (black arrow) could be observed in liver sections from the LPS group. (B) Real-time PCR was applied to measure mRNA expression of inflammatory markers. (C) Histological scores of liver tissues. Results are shown as mean ± SEM (n = 8) or as representative images from at least three independent experiments. *P < 0.05, **P < 0.01 versus the control group.
Together, collective disease scores that combine the evaluation of steatosis, inflammatory neutrophil infiltration, and hepatocyte ballooning reveal a significant elevation of disease scores in the liver of LPS injected mice at the 4 wk time point. The exacerbated liver pathology can still be detected one month after the stoppage of LPS injection at the 8 wk time point (Figure 2C).
Sustained neutrophil infiltration in liver of mice injected with super-low dose LPS
Neutrophils and monocytes represent one of the most prominent components of the innate immune system (9). To further explore the hypothesis that neutrophils are activated in response to LPS stimulation, we assessed the activation marker of neutrophils, Ly6G, in liver tissues using immunofluorescence (Figure 3A). The Ly6G-positive cells were significantly elevated in LPS treated mice as compared to control mice (Figure 3B). In line with this, biochemical analysis revealed that the injection of super-low dose LPS caused a 2.7-fold increase of liver MPO levels in high-fat diet fed ApoE−/− mice as compared to PBS injected ones (Figure 3C). Significantly, the elevation of liver neutrophils and MPO levels was sustained after one month after the stoppage of LPS injection at the 8 wk time points. To further address whether the increased neutrophil infiltration may due to elevated levels of neutrophil chemoattractants, we performed ELISA analyses of MIP2 and KC, Indeed, we found significantly higher levels of plasma KC and MIP2 as well as higher levels of liver MIP2 at the 8 week time point, long after the withdrawal of LPS injection, further supporting the lasting memory effects of subclinical low-dose endotoxemia (Figure 3D). Furthermore, we also observed elevated levels of macrophages within the liver tissues injected with subclinical dose endotoxin both at the 4 wk and the 8 wk time points (Figure 3E). Together, our data suggest that super-low dose LPS challenge may establish sustained “memory” inflammatory state in mice, leading to hepatic inflammation.
Figure 3. Super-low dose LPS promotes and sustains liver neutrophil and macrophage infiltration in high-fat diet fed ApoE−/− mice.

(A, B) Ly6G immunofluorescence reveals infiltration of neutrophils into the liver after 4-weeks of LPS treatment. Representative photographs (A; 200 × magnification) and the proportion of Ly6G-positive cells (B) are shown. (C) Total liver MPO content assessed by ELISA was almost three-fold increased by 4-weeks of LPS-injection. (D) The plasma levels of MIP2 and KC as well as liver levels of MIP2 were measured by ELISA. (E) The liver levels of macrophages were stained with MOMA2 antibody, and the relative MOMA2 positive staining areas were represented. Results are shown as mean ± SEM or as representative images from at least three independent experiments. *P < 0.05, **P < 0.01 versus the control group.
Elevated cellular apoptosis in liver tissues from mice injected with super-low dose LPS
Neutrophil-derived MPO has powerful pro-apoptotic effects, partly attributable to the generation of reactive oxygen species (24). Given sustained neutrophil infiltration and elevated liver MPO levels in mice pre-conditioned with super-low dose LPS, we next tested the hypothesis that super-low dose LPS challenge may cause a sustained apoptotic response in liver. Indeed, we observed a significantly increased number of apoptotic nuclei within liver hepatocytes with classical cubical shapes in LPS injected mice as compared to control mice (Figure 4A and 4B). Such effect was not only apparent in mice harvested at the 4 wk time point, but also in mice harvested one month after the stoppage of LPS injection at the 8 wk time point.
Figure 4. Super-low dose LPS promotes and sustains apoptotic response in the liver of high-fat diet fed ApoE−/− mice.

(A) TUNEL staining of the apoptotic cells in liver sections. Positive nuclei staining is indicated by brown, and the background nuclei staining is methyl green. Representative photographs were taken at 200 × magnifications. Representative cubical shaped hepatocytes with positive TUNEL staining were shown in an enlarged panel on the side. (B) Numerical data on apoptotic hepatocyte nuclei are presented. (C) Real-time PCR was applied to measure mRNA expression of apoptotic markers. (D) Western blotting was applied to determine the protein expression of caspase-3, and cleavage of PARP. Results are shown as mean ± SEM (n = 8) or as representative images from at least three independent experiments. *P < 0.05, **P < 0.01 versus the control group.
We then determined the gene expression levels of several apoptotic markers such as membrane apoptotic receptor Fas and its ligand FasL, intracellular pro-apoptotic regulator Bax and anti-apoptotic regulator Bcl-2. We observed a lasting and sustained elevation of Fas, FasL and Bax mRNAs in liver of mice injected with super-low dose LPS at both the 4 wk and 8 wk time point (Figure 4C). Our immunoblotting data demonstrated that high-fat diet fed ApoE−/− mice injected with super-low dose LPS had elevated levels of active caspase-3, and increased cleavage of poly (ADP-ribose) polymerase (PARP), as compared to vehicle-treated mice, both at the 4 wk and 8 wk time points (Figure 4D). These results reveal that super-low dose LPS conditioning may lead to a lasting “memory” effect of liver cell apoptosis.
Super-low dose LPS initiates sustained activation of the p38 MAPKs signaling axis in liver, through reducing the negative feedback phosphatase MKP7
Mitogen-activated protein kinases (MAPKs) play essential roles during the processes of neutrophil migration as well as cellular apoptosis (25). Next, we tested whether super-low dose LPS injection may cause sustained activation of MAPKs in liver tissues. Indeed, Western-blotting analyses revealed that there were significantly elevated levels of phosphorylated p38 and JNK in liver tissues from mice injected with super-low dose LPS (Figure 5). Intriguingly, the “memory” effect of super-low dose LPS injection on the phosphorylation of p38 and JNK was still apparent one month after the stoppage of LPS injection.
Figure 5. Super-low dose LPS sustains the low-grade activation of the p38/JNK MAPKs in the liver of high-fat diet fed ApoE−/− mice.

The expression and phosphorylation of MAPKs (p38, JNK, ERK) and expression of the upstream MAPK phosphatase 7 (MKP7) were detected by Western blotting. Images are representative of, and protein expression profiles are pooled from, 3 independent experiments. *P < 0.05, **P < 0.01 versus the control group.
Based on systems analyses of analogous systems, one of the potential hypotheses for maintaining sustained memory and activation is the removal of negative feedbacks. In the context of MAPK activation, the negative feedback that dampens its sustained activation is the MAPK phosphatases (26). Indeed, we observed that super-low dose LPS potently reduced the proteinlevels of MAPK phosphatase-7 (MKP7), a selective p38/JNK MAPK phosphatase (27). The suppression of MKP7 was apparent one month after the stoppage of supper-low dose LPS injection. Our data suggest that super-low dose LPS may sustain a “memory” state of MAPK activation in liver tissues through the removal of negative modulator MKP7.
Sustained activation of p38 MAPKs are involved in enhanced neutrophil migration induced by super-low dose LPS in vitro
Consistent with in vivo results, we found that p38 MAPKs were activated in isolated neutrophils after challenges with super-low dose LPS for 24 h (Figure 6C). To identify whether the enhanced neutrophil migration may be due to, or at least partly, the sustained activation of p38 MAPKs by super-low dose LPS, we investigated the effects of the p38 MAPKs inhibitors on neutrophil migration in vitro. As expected, super-low dose LPS exposure induced a significant migration of neutrophils toward the chemoattractant MIP-2 (Figure 6A and 6B). Application of either SB203580 (p38 inhibitor) or SP600125 (JNK inhibitor) potently inhibited the chemotactic activity of neutrophils challenged with super-low dose LPS (Figure 6A and 6B).
Figure 6. Super-low dose LPS promotes neutrophil migration via the p38 MAPKs pathway.

(A) Chemotaxis of neutrophils pretreated with 20 μM SB203580 (p38 inhibitor) or 20 μM SP600125 (JNK inhibitor) (2 h prior to the assay) against chemokine MIP-2. Representative images were shown. (B) Quantification of neutrophils that migrated into the gels toward MIP-2. (C) Phosphorylation of p38 and JNK MAPKs and expression of MKP7 in neutrophils treated with or without LPS for 24 h. Images are representative of, and protein expression profiles are pooled from, 3 independent experiments. *P < 0.05, **P < 0.01 versus the control group. (D) In vivo activated memory state of neutrophils. HFD-fed ApoE−/− mice injected were injected with either PBS or super-low dose LPS twice-weekly for one month as described in the Materials and Methods. Following the stoppage of injection, mice were continually fed with HFD for an additional month. Peripheral blood leukocytes and bone marrow cells were stained with fluorescent antibodies, and the expression levels of CD14 on CD11b+Ly6G+ neutrophils gated within the Ly6C− population were examined by flow cytometry. Error bars show means ± s.e.m.;** P <0.01; student t-test.
To further evaluate whether neutrophil may retain an “activated” memory state in vivo in mice pre-conditioned with super-low dose LPS, we examined the CD14 levels of circulating neutrophils in HFD-fed ApoE−/− mice pre-conditioned with either PBS or super-low dose LPS. ApoE−/− mice fed with HFD were injected with either PBS or LPS for one month. Following the stoppage of injection, mice were continually fed on HFD for an additional month. Blood and bone marrow were harvested and neutrophil activation status were evaluated by flow cytometry through the surface expression levels of CD14. Such ex vivo identification of activated neutrophil may serve as important evidence of in vivo programming of neutrophils by super-low dose LPS. As shown in Figure 6D, HFD-fed mice pre-conditioned with super-low dose LPS retain the activated neutrophil memory state as reflected in significantly higher levels of CD14 expression.
Discussion
In this report, we provided evidence that reveal a “memory” state of low grade inflammation in liver tissues of high-fat diet fed ApoE−/− mice chronically injected with super-low dose LPS. Super-low dose LPS may sustain and exacerbate the low-grade inflammatory state via sustained p38 MAPKs signaling circuit due to the removal of negative modulator MKP7, subsequently enhanced leukocyte infiltration, MPO release, and hepatocytes apoptosis. These mutually propagating events may lead to the exacerbation of NASH progression.
Our study shed light on a novel pro-inflammatory circuitry in animals challenged with subclinical super-low dose endotoxin, a phenomenon increasingly recognized in the biomedical field (28). Although empirical knowledge reckons that subclinical super-low dose LPS may be capable of inducing liver inflammation, almost all existing studies have utilized significantly higher dosages of LPS that may bear little patho-physiological relevance in the context of low-grade chronic inflammatory disease (15-17). Most of these studies conclude a transient inflammatory effect triggered by LPS, followed by a “tolerant” anti-inflammatory state (29). Therefore, these studies could not reconcile the sustained low-grade inflammation seen in clinical settings. Our previous in vitro study reveal an important paradigm of “priming” and “tolerance” in innate leukocytes challenged with varying dosages of LPS (30). Instead of causing a transient inflammatory response in monocytes treated with a higher dose LPS, as commonly reported in the literature (31), a sustained non-resolving low-grade inflammatory state can be established in cells treated with a subclinical super-low dose LPS (32, 33). Capitalizing on this new concept, we extend our mechanistic in vitro studies in vivo, and demonstrated herein that chronic injection of super-low dose LPS can sustain a “memory” low-grade pro-inflammatory state in liver tissues of high-fat diet fed ApoE−/− mice. We used the well-established steatosis-prone model of ApoE deficient mice, as previous studies suggest that hyperlipidemia is the critical “first hit” and an essential driver for the development of steatosis and atherosclerosis (34, 35), and that the low-grade circulatory inflammatory components are subsequent facilitators not capable of initiating the development of steatosis or atherosclerosis in the absence of hyperlipidemia. We employed the subclinical endotoxemia model with chronic injection of super-low dose endotoxin, compatible with circulating levels of endotoxemia reported in humans and experimental animals with chronic inflammatory diseases such as atherosclerosis and diabetes (36-39). Our data clearly support the role of circulating chronic low-grade inflammatory agent endotoxemia during the exacerbation of liver steatosis though elevating systemic low-grade inflammation, sustaining low-grade non-resolving p38 activation, and facilitating liver tissue leukocyte infiltration.
It is also interesting to note that subclinical endotoxemia not only sustained chronic low-grade inflammation, but also elevated tissue levels of free fatty acid (FFA). Unlike conjugated triglyceride (TG) that preferentially affect cellular metabolism, FFA have been shown to have additional important roles in inflammation. Due to their small and soluble nature, FFA serve as de facto inflammatory mediators through the activation of key inflammatory pathways that include NFκB, PKC and others in both inflammatory cells and hepatocytes (40-42). In turn, these inflammatory processes may further facilitate the generation of FFA and perpetuating the non-resolving low-grade inflammation, even after the withdrawal of subclinical endotoxemia.
Our data suggest a fundamental biochemical mechanism responsible for the sustained “memory” in liver tissues, in that super-low dose LPS may establish sustained activation of p38 MAPKs through the removal of the negative modulator MKP7. It was previously reported that MKP7 shuttles between the nucleus and the cytoplasm and specifically suppresses the activation of p38 instead of ERK (43, 44). In this study, we observed in vivo that decreased MKP7 expression is accompanied by sustained p38 MAPKs activation in liver of mice chronically injected with super-low dose LPS. Our complementary in vitro studies with isolated neutrophils further confirmed this notion.
Functionally, this study reveals an integrated circuit that couples the sustained activation of p38 MAPKs signaling circuit with sustained recruitment and sequestration of neutrophils. There is increasing appreciation for the role of elevated neutrophil recruitment in chronic liver disease (9). Neutrophil recruitment is an elaborate process in which circulating neutrophils are attracted to the site of inflammation by following chemokine trails laid out by microbes or host cells (11). MAPKs such as p38 have been implicated in the enhanced neutrophil chemotaxis (25, 45). Our current study provides evidence that supports a critical role of sustained MAPK signaling in enhanced neutrophil migration following challenges with super-low dose LPS both in vivo and in vitro.
The activation of cellular p38 MAPKs circuit is also responsible for the release of neutrophil MPO, an additional potent neutrophil recruiting mediator (46). Furthermore, MPO-derived oxidants may diffuse into hepatocytes and trigger intracellular oxidative stress. Moreover, neutrophils can express Fas ligand and kill hepatocytes through an apoptosis induced mechanism (47). Here we demonstrated that liver tissues from mice chronically injected with super-low dose LPS exhibit sustained elevation of several inflammatory and apoptotic genes, even long after the stoppage of LPS injection. Our study provides functional evidence that support the innate “memory” inflammatory state in vivo (Figure 7).
Figure 7. A schematic summary of the potential dynamics involved in the sustained systemic inflammation due to subclinical endotoxemia.
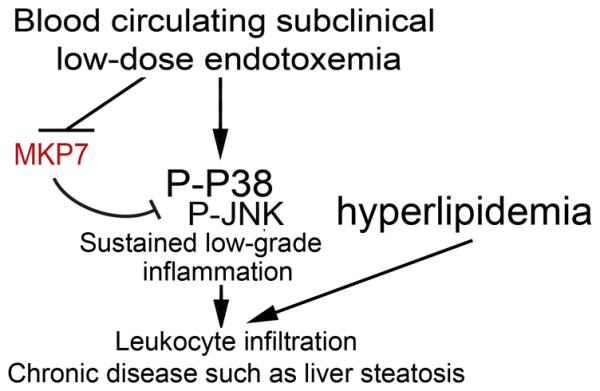
Systemic subclinical super-low dose endotoxin may establish a sustained “memory” state of low-grade liver inflammation through the removal of MKP7, sustained activation of the p38/JNK MAPKs signaling circuit, which eventually facilitate the propagation of neutrophil infiltration, MPO release, hepatocyte death, and NASH progression.
Due to the heterogeneous nature of NAFLD, it is unlikely that NASH pathogenesis is dependent upon any one single factor. Currently, the “multiple parallel hits” theory reflects more precisely the pathophysiological factors behind the progression from steatosis to NASH (48). The present study suggests that subclinical circulating super-low dose endotoxin may be involved in sustaining the inflammatory responses, sustaining leukocyte infiltration, contributing to subsequent hepatocyte apoptosis and steatohepatitis. We realize that other un-identified factors may also be affected by subclinical circulating endotoxemia and further sustain the systemic inflammation and subsequent liver damage. In additional to activated inflammatory mediators such as cytokines, chemokine, and FFA, sustained inflammatory environment may also alter the overall landscape of gut microbiota, which then may further favor the mucosal leakages and endotoxemia. Future studies are warranted to systemically examine the microbiota dynamics during the initiation and maintenance of non-resolving low-grade inflammation and related complications.
In summary, our present study reveal that a novel circuit triggered by circulating subclinical super-low dose LPS that involves sustained low-grade activation of p38, systemic low-grade inflammation, and leukocyte infiltration into liver tissues (Figure 7). Coupled with high fat diet, this secondary systemic low-grade inflammation may be critically involved in exacerbating the pathogenesis of liver steatosis.
Acknowledgement
HG and LL designed the study. HG, ND, RY and KC carried out experiments and data analysis; HG and LL were involved in writing the paper. All authors read and approved the content of the final version of the manuscript.
This work was supported by grants from the National Institute of Health R01 HL135835 to LL, and the National Natural Science Foundation (81372994, 81172655), and China Scholarship Council (201308440005) to HG.
Abbreviations
- ALT
alanine aminotransferase
- FFA
free fatty acid
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MKP7
MAPK phosphatase 7
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- MPO
myeloperoxidase
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- TG
triglyceride
References
- 1.Musso G, Gambino R, Cassader M. Non-alcoholic fatty liver disease from pathogenesis to management: an update. Obes. Rev. 2010;11:430–445. doi: 10.1111/j.1467-789X.2009.00657.x. [DOI] [PubMed] [Google Scholar]
- 2.Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin. Liver Dis. 2004;24:99–106. doi: 10.1055/s-2004-823104. [DOI] [PubMed] [Google Scholar]
- 3.Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. The Journal of physiology. 2012;590:447–458. doi: 10.1113/jphysiol.2011.219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int. 2010;4:659–672. doi: 10.1007/s12072-010-9219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vyberg M, Ravn V, Andersen B. Pattern of progression in liver injury following jejunoileal bypass for morbid obesity. Liver. 1987;7:271–276. doi: 10.1111/j.1600-0676.1987.tb00355.x. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz AG, Casafont F, Crespo J, Cayon A, Mayorga M, Estebanez A, Fernadez-Escalante JC, Pons-Romero F. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes. Surg. 2007;17:1374–1380. doi: 10.1007/s11695-007-9243-7. [DOI] [PubMed] [Google Scholar]
- 7.Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, Sivridis E, Koffa M, Giatromanolaki A, Boumpas DT, Ritis K, Kambas K. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014;233:294–307. doi: 10.1002/path.4359. [DOI] [PubMed] [Google Scholar]
- 8.Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, Lopes GA, Russo RC, Avila TV, Melgaco JG, Oliveira AG, Pinto MA, Lima CX, De Paula AM, Cara DC, Leite MF, Teixeira MM, Menezes GB. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. 2012;56:1971–1982. doi: 10.1002/hep.25801. [DOI] [PubMed] [Google Scholar]
- 9.Xu R, Huang H, Zhang Z, Wang FS. The role of neutrophils in the development of liver diseases. Cell. Mol. Immunol. 2014;11:224–231. doi: 10.1038/cmi.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, Malle E, Greve JW, Buurman WA. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol. 2009;175:1473–1482. doi: 10.2353/ajpath.2009.080999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) 2009;17:2014–2018. doi: 10.1038/oby.2009.113. [DOI] [PubMed] [Google Scholar]
- 12.Tous M, Ferre N, Camps J, Riu F, Joven J. Feeding apolipoprotein E-knockout mice with cholesterol and fat enriched diets may be a model of non-alcoholic steatohepatitis. Mol. Cell. Biochem. 2005;268:53–58. doi: 10.1007/s11010-005-2997-0. [DOI] [PubMed] [Google Scholar]
- 13.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 14.Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J. Immunol. 2001;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- 15.Thomsen KL, Hebbard L, Glavind E, Clouston A, Vilstrup H, George J, Gronbaek H. Non-alcoholic steatohepatitis weakens the acute phase response to endotoxin in rats. Liver Int. 2014;34:1584–1592. doi: 10.1111/liv.12547. [DOI] [PubMed] [Google Scholar]
- 16.Jia X, Iwanowycz S, Wang J, Saaoud F, Yu F, Wang Y, Hu J, Chatterjee S, Wang Q, Fan D. Emodin attenuates systemic and liver inflammation in hyperlipidemic mice administrated with lipopolysaccharides. Exp Biol Med (Maywood) 2014;239:1025–1035. doi: 10.1177/1535370214530247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, Kato S, Mawatari H, Shibata W, Kitani H, Ikejima K, Kirikoshi H, Nakajima N, Saito S, Maeyama S, Watanabe S, Wada K, Nakajima A. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16:44–54. doi: 10.1016/j.cmet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 18.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 19.Clemente-Postigo M, Queipo-Ortuno MI, Murri M, Boto-Ordonez M, Perez-Martinez P, Andres-Lacueva C, Cardona F, Tinahones FJ. Endotoxin increase after fat overload is related to postprandial hypertriglyceridemia in morbidly obese patients. J. Lipid Res. 2012;53:973–978. doi: 10.1194/jlr.P020909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu M, Munford RS. The transport and inactivation kinetics of bacterial lipopolysaccharide influence its immunological potency in vivo. J Immunol. 2011;187:3314–3320. doi: 10.4049/jimmunol.1004087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao B, Lu M, Katz SC, Varley AW, Hardwick J, Rogers TE, Ojogun N, Rockey DC, Dematteo RP, Munford RS. A host lipase detoxifies bacterial lipopolysaccharides in the liver and spleen. J Biol Chem. 2007;282:13726–13735. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 22.Fukunishi S, Sujishi T, Takeshita A, Ohama H, Tsuchimoto Y, Asai A, Tsuda Y, Higuchi K. Lipopolysaccharides accelerate hepatic steatosis in the development of nonalcoholic fatty liver disease in Zucker rats. Journal of clinical biochemistry and nutrition. 2014;54:39–44. doi: 10.3164/jcbn.13-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, Enders F, Saksena S, Burt AD, Bida JP, Lindor K, Sanderson SO, Lenzi M, Adams LA, Kench J, Therneau TM, Day CP. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846–854. doi: 10.1002/hep.21496. [DOI] [PubMed] [Google Scholar]
- 24.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat. Med. 2011;17:1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, Ye RD, Xu J. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nature immunology. 2012;13:457–464. doi: 10.1038/ni.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda K, Shima H, Watanabe M, Kikuchi K. MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J. Biol. Chem. 2001;276:39002–39011. doi: 10.1074/jbc.M104600200. [DOI] [PubMed] [Google Scholar]
- 28.Morris M, Li L. Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch. Immunol. Ther. Exp. (Warsz.) 2012;60:13–18. doi: 10.1007/s00005-011-0155-9. [DOI] [PubMed] [Google Scholar]
- 29.Frasinariu OE, Ceccarelli S, Alisi A, Moraru E, Nobili V. Gut-liver axis and fibrosis in nonalcoholic fatty liver disease: an input for novel therapies. Dig. Liver Dis. 2013;45:543–551. doi: 10.1016/j.dld.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 30.Morris MC, Gilliam EA, Button J, Li L. Dynamic modulation of innate immune response by varying dosages of lipopolysaccharide (LPS) in human monocytic cells. J. Biol. Chem. 2014;289:21584–21590. doi: 10.1074/jbc.M114.583518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 32.Baker B, Maitra U, Geng S, Li L. Molecular and cellular mechanisms responsible for cellular stress and low-grade inflammation induced by a super-low dose of endotoxin. J. Biol. Chem. 2014;289:16262–16269. doi: 10.1074/jbc.M114.569210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maitra U, Li L. Molecular mechanisms responsible for the reduced expression of cholesterol transporters from macrophages by low-dose endotoxin. Arterioscler. Thromb. Vasc. Biol. 2013;33:24–33. doi: 10.1161/ATVBAHA.112.300049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma KL, Z. Ruan X, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology. 2008;48:770–781. doi: 10.1002/hep.22423. [DOI] [PubMed] [Google Scholar]
- 35.Kerenyi L, Mihalka L, Csiba L, Bacso H, Bereczki D. Role of hyperlipidemia in atherosclerotic plaque formation in the internal carotid artery. J Clin Ultrasound. 2006;34:283–288. doi: 10.1002/jcu.20233. [DOI] [PubMed] [Google Scholar]
- 36.Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86:1286–1292. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- 37.Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, Willeit J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. Journal of the American College of Cardiology. 1999;34:1975–1981. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 38.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 39.Goto T, Eden S, Nordenstam G, Sundh V, Svanborg-Eden C, Mattsby-Baltzer I. Endotoxin levels in sera of elderly individuals. Clin Diagn Lab Immunol. 1994;1:684–688. doi: 10.1128/cdli.1.6.684-688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, Xiang X, Luo Z, Ruderman N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes. 2005;54:3458–3465. doi: 10.2337/diabetes.54.12.3458. [DOI] [PubMed] [Google Scholar]
- 41.Hommelberg PP, Plat J, Langen RC, Schols AM, Mensink RP. Fatty acid-induced NF-kappaB activation and insulin resistance in skeletal muscle are chain length dependent. Am J Physiol Endocrinol Metab. 2009;296:E114–120. doi: 10.1152/ajpendo.00436.2007. [DOI] [PubMed] [Google Scholar]
- 42.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 43.Zhu J, Shen W, Gao L, Gu H, Shen S, Wang Y, Wu H, Guo J. PI3K/Akt-independent negative regulation of JNK signaling by MKP-7 after cerebral ischemia in rat hippocampus. BMC neuroscience. 2013;14:1. doi: 10.1186/1471-2202-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masuda K, Shima H, Watanabe M, Kikuchi K. MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J. Biol. Chem. 2001;276:39002–39011. doi: 10.1074/jbc.M104600200. [DOI] [PubMed] [Google Scholar]
- 45.Cara DC, Kaur J, Forster M, McCafferty DM, Kubes P. Role of p38 mitogen-activated protein kinase in chemokine-induced emigration and chemotaxis in vivo. J. Immunol. 2001;167:6552–6558. doi: 10.4049/jimmunol.167.11.6552. [DOI] [PubMed] [Google Scholar]
- 46.Klinke A, Nussbaum C, Kubala L, Friedrichs K, Rudolph TK, Rudolph V, Paust HJ, Schroder C, Benten D, Lau D, Szocs K, Furtmuller PG, Heeringa P, Sydow K, Duchstein HJ, Ehmke H, Schumacher U, Meinertz T, Sperandio M, Baldus S. Myeloperoxidase attracts neutrophils by physical forces. Blood. 2011;117:1350–1358. doi: 10.1182/blood-2010-05-284513. [DOI] [PubMed] [Google Scholar]
- 47.Jaeschke H, Ho YS, Fisher MA, Lawson JA, Farhood A. Glutathione peroxidase-deficient mice are more susceptible to neutrophil-mediated hepatic parenchymal cell injury during endotoxemia: importance of an intracellular oxidant stress. Hepatology. 1999;29:443–450. doi: 10.1002/hep.510290222. [DOI] [PubMed] [Google Scholar]
- 48.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]