

Abstract
The ubiquitin proteasome system (UPS) plays vital roles in maintaining protein equilibrium mainly through proteolytic degradation of targeted substrates. The archetypical SCF ubiquitin E3 ligase complex contains a substrate recognition subunit F-box protein that recruits substrates to the catalytic ligase core for its polyubiquitylation and subsequent proteasomal degradation. Several well characterized F-box proteins have been demonstrated that are tightly linked to neoplasia. There is mounting information characterizing F-box protein-substrate interactions with the rationale to develop unique therapeutics for cancer treatment. Here we review that how F-box proteins function in cancer and summarize potential small molecule inhibitors for cancer therapy.
Keywords: Ubiquitin, E3 ligase, F-box protein, small molecule inhibitor, cancer
The human genome sequence coupled with state-of-art proteomics and structural studies has uncovered potentially numerous proteins that could serve as prominent targets for drug therapy. Unfortunately, many of these targets are not considered druggable because of their lack of putative drug binding pockets or the indispensible nature of their roles in fundamental metabolic pathways precluding their chemical inhibition. Indeed, it is estimated that only 2% of drugs interact with proteins and 10–15% of proteins are druggable (1). Cellular protein equilibrium is maintained through dynamic gene expression and degradation. It is estimated that more than 80% of cellular proteins undergo ubiquitin mediated degradation, mainly through the 26S proteasome. Although first generation proteasome inhibitors are impacting the lifespan of some individuals with cancer, manipulating the upstream regulators of the proteasome for desired targets provides a promising opportunity for specific and efficacious drug development. Targeting upstream regulators may provide for more specific protein modulation, in contrast to broad and potentially off target effects of down-stream proteosome inhibition strategies used in cancer therapy. In addition, considering the high risk of failed drug development, pharmaceutical companies are increasingly attempting to identify gene-function pathways that validate the target pathway prior to expensive drug development.
The ubiquitin proteasome system (UPS) (Fig. 1) is a complex, hierarchical, and regulated cellular system that dominates selective protein degradation to modulate the abundance and activity of proteins in the cell (2,3). The majority of proteins are controlled by the UPS through the ATP-dependent enzymatic cascade, including the ubiquitin activating enzyme (E1), the ubiquitin conjugating enzyme (E2), and the ubiquitin ligase (E3). The 76-amino-acid protein ubiquitin is conjugated to a substrate through an isopeptide linkage of its last glycine residue to an internal lysine of the substrate. Ubiquitin chains linked through either lysine 48 or lysine 11 of each ubiquitin, tag substrates to the 26S proteasome for degradation to amino acids. Other non-Lys63 polyubiquitylation and multiple monoubiquitylation events within proteins have been implicated in ubiquitin-dependent degradation through alternative machinery or modulate nondegradative processes for individual proteins. Similar to other post-translational modifications, such as phosphorylation, ubiquitylation is reversible and linked with deubiquitylation mediated by deubiquitylating enzymes (DUBs). There are about 100 DUBs belonging to two classes of proteases: cysteine proteases and metalloproteases, and each DUB recognizes several substrates.
Figure 1.
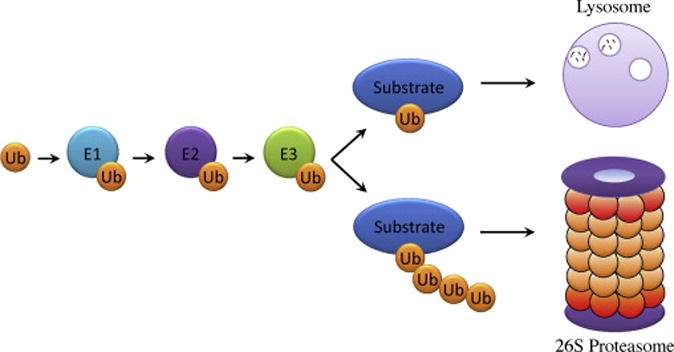
The recognition of a specific substrate in response to a specific stimulus is crucial for appropriate protein turnover that impacts cellular function. In humans, there are two E1 enzymes, approximately 30 E2 enzymes, and more than seven hundred E3 enzymes. Many E3 ligases are complexes formed by a core scaffold with interchangeable substrate-recognizing subunits. The Cullin–RING ligase (CRL) complexes represent the basic framework for the modular ubiquitin ligases, which contains eight members: CRL1, CRL2, CRL3, CRL4A, CRL4B, CRL5, CRL7 and CRL9 (4,5). In general, CRL E3s include a Cullin scaffold, an adaptor, a substrate receptor and a RING protein that recruits E2 enzyme. SCF is the best characterized CRL ligase, containing the scaffold Cul1, adaptor Skp1, substrate receptor F-box protein and RING protein Rbx1 (6–8). In the SCF complex, the F-box protein binds a specific substrate bringing it within intermolecular proximity to the ligase scaffold Cul1-Skp1 that bridges the RING domain protein Rbx1 catalyzing ubiquitin conjugation and extension (9–11).
F-box proteins are categorized within three families based on the recognizable domains beyond the F-box domain, which comprises the Leu-rich repeat (L) family (10 proteins), WD40 domain (W) family (21 proteins) and the F-box only (O) family (38 proteins) (8,12). In response to stimuli, F-box proteins typically recognize unique, short degradation peptidic signatures (termed a degron) in their substrates (13). Although newer members of the F-box protein family are being discovered and existing members better characterized, increasing evidence indicates that the tightly regulated interaction between the F-box protein and its substrate is often achieved by modifications to both proteins. To date, multiple recognition models have been proposed to be involved in F-box proteins targeting various substrates. Phosphorylation-dependent mode of substrate recognition is the canonical model for F-box protein binding to a phosphodegron within a substrate and recruiting it to the SCF core scaffold (14,15). The direct recognition of a phosphodegron is the most common mechanism for F-box proteins to recruit and degrade substrates (16–19). However, some phosphorylations of a substrate can block their interactions with an F-box protein to limit degradation (20,21). Post-translational modifications other than phosphorylation, such as glycosylation, and acetylation also have emerged as recognition signatures in mediating the interaction between F-box proteins and their substrates (22). In addition, an F-box protein is able to recognize an unmodified degron (23–27). Not surprisingly, F-box proteins themselves are often regulated at the transcriptional or posttranslational levels involving phosphorylation and proteolytic turnover (28,29). In summary, F box protein components are regulated subunits within SCF complexes that provide receptor-like selectivity in recognizing substrates for their elimination in cells. Their ability to identify and interact with specific motifs within targets offers promise for newer therapies in oncogenesis.
Malfunction of degradative mechanisms for oncoproteins or tumor suppressors can drive tumorogenesis and cancer progression. The FDA approved proteasome inhibitor Bortezomib provided validation for the UPS as an authentic target for the treatment of multiple myeloma and mantle cell lymphoma (30). One dose-limiting side effect, however, for clinical application of Bortezomib is peripheral neuropathy (31,32). Hence, recent studies have shifted to a focus on inhibiting UPS proteins upstream of the proteasome, particularly inhibitors specific to ubiquitin E3 ligases to reduce off-target effects (33–38). Emerging evidence demonstrate that F-box proteins play vital roles in cancer development and F-box proteins are overexpressed frequently within human cancers, suggesting that F-box proteins could serve as promising therapeutic anti-tumor targets.
In response to specific stimuli, one F-box protein can bind to multiple substrates with various biological functions in a temporally-controlled manner. Thus, activity-regulating small molecules need to interact with these highly specific and delicately controlled F-box protein-substrate interfaces (Fig. 2). In humans, small molecules possess overwhelming therapeutic advantages over biomolecules (e.g. peptides and nucleic acids), due to greater potential for oral delivery and bioavailability. Consequently, much effort has been dedicated to the discovery of small molecules that can disrupt the E3 ligase-substrate protein interface, among which an F-box has attracted tremendous attention due to its relatively specific and essential role in substrate recognition. Meanwhile, cell lines and animal models provide idealized platforms to evaluate these F-box proteins, their substrates, and drug effects in cancer models.
Figure 2.
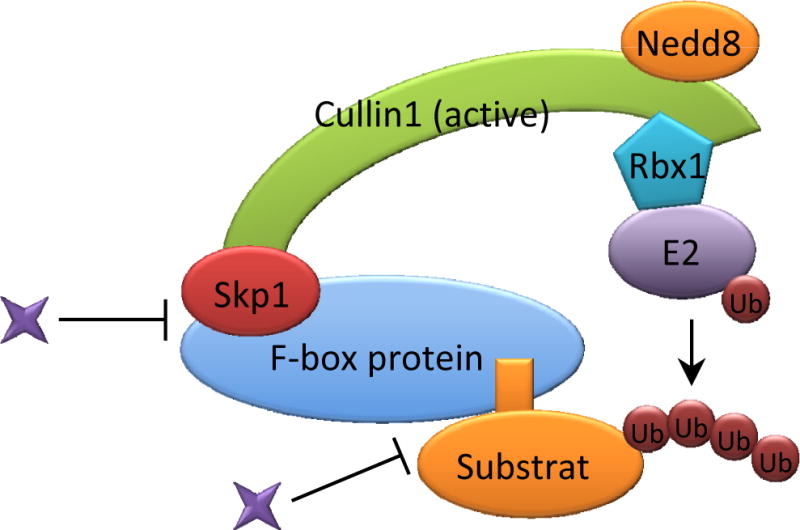
SCF E3 ligases have strong links to cancer owing to their control of the cell cycle, DNA replication, and DNA repair. Particularly, multiple F-box proteins act as attractive therapeutic targets for cancer treatment due to the correlation between their deregulation and tumorigenesis. SCF ligase small molecule inhibitors are needed for cancer that results from overexpressed or overactive F-box proteins. Meanwhile, different approaches are required to restore defective SCF function that leads to disease. Here we review the correlation of F-box proteins with their putative substrates that may lead to cancer development, and discuss the potential pharmacotherapies targeting F-box proteins in neoplasia.
1. Skp2
Cell cycle dysregulation has been considered as a cancer progression hallmark in most malignant tumors. Skp2, S-phase kinase associated protein 2, plays a crucial role in cell cycle progression, senescence, and metabolism. Skp2 is the classic oncogenic F-box protein because it promotes S phase entry and proliferation by inducing the ubiquitylation and degradation of several anti-proliferative cyclin-dependent kinase (CDK) inhibitors including p27, p21, and p57 (37,39–44). Skp2 exhibits high-level expression in human cancers and is implicated in several murine cancer models. Skp2 predominantly modulates p27, a cell cycle inhibitor that partakes in an essential role in mediating tumor suppression. Loss of p27 causes uncontrolled proliferation and tumor progression. In many aggressive tumors, the overexpression of Skp2 at both the mRNA and protein levels unequivocally correlates with p27 downregulation and a poor prognosis. Additionally, in mouse models, Skp2-knockout phenotypes can be rescued by p27 deletion, and Skp2 overexpression can be phenocopied by p27 deletion, which suggests that the oncogenic roles of Skp2 largely depend on p27 degradation (45,46). Thus, the Skp2-p27 axis presents an attractive target to generate p27 degradation inhibitors.
Advanced cancers also frequently develop resistance to chemotherapy and radiotherapy to another tumor suppressor, p53, due to its gene mutations with attendant loss of responsiveness to p53. Skp2 deficiency triggers p53-independent, p27-dependent cellular senescence and apoptosis (47). Therefore, developing cancer treatment strategies through boosting p53-independent senescence and apoptosis responses might provide a promising alternative for limiting cancer cell proliferation. In addition, cancer cells display high level of glycolysis regardless of the abundance of oxygen, suggesting that shifting metabolism to aerobic glycolysis might be an attractive strategy for reducing tumorogenesis. The kinase Akt plays a vital role in mediating aerobic glycolysis and SCFSkp2 mediates nonproteolytic K63-linked ubiquitylation of Akt (48). Overall, targeting Skp2 serves as a promising strategy for cancer treatment.
Through high-throughput screening (HTS), a variety of small molecules have been identified to stabilize p27, through multiple mechanisms including inhibiting 26S proteasome activity, preventing Skp2 from incorporating into the SCF core, or downregulating Skp2 mRNA (49–51). However, identification of an inhibitor that specifically and directly targets Skp2 is highly desirable to reduce off-target effects. Structure-based virtual library screening utilizes bioinformatics approaches to first identify hot spot residues and binding pockets that are used to select potential inhibitors that distort the protein–protein interactions; from these in silico interrogations, a workable number of molecules are predicted to bind most effectively and are selected for further experimental validation. Based on this approach, coupled with ligand based design strategies, in vitro functional screens and counterscreens, a series of compounds (C1, 2, 16, and 20) were identified that selectively inhibit ability of Skp2 to recruit p27 to the SCF E3 catalytic core (52). CDK regulatory subunit 1 (Cks1) is required for SCFSkp2-mediated p27 ubiquitylation through its binding to both Skp2 and p27. These compounds specifically disrupt the interaction between phospho-p27 and Skp2-Cks1 that leads to p27 stabilization. When tested in cells, these compounds inhibit cell cycle progression and proliferation in a variety of cancer cell lines in a Skp2-p27 dependent manner.
Utilizing the high throughput virtual screening approach, another Skp2 small molecule inhibitor, compound #25, has been recently developed to specifically block the Skp1-Skp2 interaction (47). The authors identified 19 ‘hot spot’ residues along the Skp1–Skp2 interaction surface. Compound #25 predicted from in silico high-performance computational-based molecule docking screens demonstrated that this chemical entity binds to Skp2, prevents Skp1-Skp2 interaction, stabilizes Skp2 substrates p27 and p21, and inhibits Akt activation, which subsequently suppresses the viability of cancer cells and cancer stem cells. Compound #25 also displays potent antitumor activity in animal studies (47). Remarkably, this compound does not affect activity of other F-box proteins, including Fbxw7 and β-TrCP.
Using a HTS tethered with SCFSkp2-mediated in vitro ubiquitylation studies, compound A (CpdA) was identified to prevent Skp2 from incorporating into the SCF core (49). CpdA interrupted SCFSkp2 E3 ligase function and induced specific accumulation of SCFSkp2 substrates including p21, which in turn triggered G1/S cell-cycle arrest and Skp2-p27-dependent cell killing. In multiple myeloma models, CpdA increased chemical sensitivity to Bortezomib and also functioned synergistically with this proteasome inhibitor. Prostate carcinoma often displays an inverse relationship between the levels of p27 and Skp2 (53). In the human prostate cancer cell line stably overexpressing engineered Skp2, a chemical genetic approach was employed to screen small compounds that restored p27 protein levels (51). SMIP001 and SMIP004 were identified to increase nuclear p27. Meanwhile, SMIPs upregulated p21, inhibited CDK2 activity, induced G1 delay and cell cycle arrest. This approach using nuclear p27 as an endpoint presents a means to identify bioactive small molecules with selective anticancer activity.
Hinokitiol (κ-thujaplicin), a tropolone-related compound, is another agent that decreases Skp2 protein levels and inhibits p27 phosphorylation at Thr187 mediated by cyclin E/CDK2 to prevent p27 degradation in FEM human melanoma cells (54). Hinokitiol also inhibited cell proliferation in human melanoma cells. In multiple cancers, hinokitiol exhibits anticancer activity by inhibiting cell proliferation. However, the in depth mechanisms for this compound remain elusive. Troglitazone was reported to downregulate Skp2 mRNA levels in human hepatocellular carcinoma cells, which consequently induces the accumulation of p27 and cell cycle arrest (55). Prodigiosin exhibits antiproliferative effects on human lung adenocarcinoma cells by suppressing transcriptional expression of Skp2, together with G1 cell cycle blockade and increased p27 and p21 levels (56). Mechanistically, prodigiosin engages PKB-mediated signaling to induce Skp2 transcriptional repression in an E2F1-independent manner. Sulforaphane, a cruciferous vegetable-derived isothiocyanate with anticancer activity, also induces proliferation arrest and apoptosis in multiple human colon adenocarcinoma cell lines. Through transcriptional repression, sulforaphane inhibited Skp2 promoter activity and Skp2 mRNA expression, leading to the stabilization of p27 (57).
BRAF is a mitogen-activated protein kinase (MAPK) frequently mutated in melanoma, lung carcinoma, glioma, colorectal and thyroid cancer. BRAF mutations lead to enhanced kinase activity and MAPK signaling activation that is closely linked to the malignant phenotype. The simultaneous inhibition of activated MAPK pathways and Skp2-dependent degradation of p27 reduced the melanoma malignant phenotype, suggesting that the combinatorial blockade of these pathways could be a useful therapeutic tool for cancer treatment (58).
One issue regarding Skp2 is that some downstream targets behave as tumor promoting proteins. For example, the ING (inhibitor of growth) tumor suppressor family member Ing3 is key component of the NuA4 histone acetyltransferase complex, regulating gene transcription, cell cycle progression and apoptosis. Dysregulated Ing3 expression has been observed in cancers including human head and neck squamous cell carcinomas (HNSCCs), numerous lymphoma originated cell lines, hepatocellular carcinoma, and melanoma (59). Reduced nuclear Ing3 significantly correlates with a poorer 5-year survival in patients with primary melanoma. Ing3 degradation is also under close regulation by the SCFSkp2 E3 ligase complex in melanoma cells and the inhibition of the activity of SCFSkp2 complex could restore Ing3 expression and promote UV-induced apoptosis (60).
2. β-TrCP
β-transducin repeat-containing protein (β-TrCP) is another prominent cancer-related F-box protein that has important roles in growth signaling throughout the cell cycle. β-TrCP includes two closely related homologs, β-TrCP1 (also known as Fbxw1) and β-TrCP2 (also known as Fbxw11) with indistinguishable biochemical properties in promoting in vitro ubiquitylation of substrates (61). β-TrCP mediates the degradation of pro-proliferative substrates upon mitogenic stimulation in G1, controls proteins within the feedback loops that regulate the mitotic progression. Through binding to a distinct phosphodegron motif (DSGXXS) in which two serines are phosphorylated, β-TrCP targets multi-functional substrates and functions as either an oncoprotein or tumor suppressor (15). Overexpressed β-TrCP1 or β-TrCP2 has been detected in multiple cancers, including gastrointestinal cancers, hepatoblastoma, colorectal cancer, pancreatic cancer, breast cancer and melanoma, suggesting a carcinogenic function for these two proteins. In these cases, β-TrCP targets a variety of cellular growth and survival inhibitors including IκB (inhibitor of κB) family members (NF-κB signaling pathway), BimEL (extra-long isoform of Bim) and Pdcd4 (programmed cell death 4). κ-catenin, which plays an important role in Wnt signaling and whose abnormal accumulation is observed in multiple cancers, is also targeted by SCFβ-TrCP for its degradation after its phosphorylation by glycogen synthase kinase 3 (GSK3κ).
Nuclear factor-kappa B (NF-κB) is a heterodimeric transcription factor known to regulate the expression of various genes involved in inflammatory responses, cell death, and survival (62). IκB associates with prototypical inactive NF-κB in the cytoplasm and prevents the translocation of NF-κB into the nucleus that in turn inhibits NF-κB transcriptional function (63). Therefore, NF-κB is canonically activated by the disposal of its inhibitory binding partner (IκB). As a major member of the IκB family, IκBα is phosphorylated by IκB kinase (IKK) and then polyubiquitylated by SCFβ-TrCP1 (64). GS143 interrupts the physical interaction between phosphorylated IκBα and SCFβ-TrCP1, an effect that suppresses IκBα ubiquitylation and subsequent degradation; however, GS143 dose not affect IκBα phosphorylation, MDM2-dictated p53 ubiquitylation or proteasome activity in vitro (65). GS143 markedly suppresses TNFα stimulated degradation of IκBα and a set of NF-κB downstream responses. However, GS143 does not cause accumulation of β-catenin, the other GSK3-regulated substrate of SCFβ-TrCP1 E3 ligase (66), which indicates that GS143 could serve as a specific and efficacious inhibitor of multiple pathways governed by NF-κB signaling.
Tumor suppressor Pdcd4 (programmed cell death 4) is a key factor that inhibits translation by interfering with the eukaryotic initiation factor (eIF) 4A activity. Therefore Pdcd4 attenuates neoplastic transformation, tumor cell intravasation, and invasion (67). Pdcd4 is lost in various tumors including lung, breast, ovarian and pancreatic cancer (68,69). β-TrCP mediated proteasomal degradation has been identified to also regulate Pdcd4 (70). In response to mitogens, the activated p70S6K1 consensus phosphorylation sequence within Pdcd4 directs β-TrCP to recognize its downstream binding motif for polyubiquitylation and degradation. Erioflorin, identified from a high throughput screen of natural product extract libraries, inhibits the interaction of Pdcd4 and β-TrCP1 thereby stabilizing Pdcd4 and suppressing proliferation of various cancer cell lines (71). Interestingly, erioflorin also stabilizes other SCFβ-TrCP substrates such as IκBα and β-catenin. Notably, the overexpressed oncogenic β-TrCP substrates β-catenin, Cdc25A, and Emi1 are also detected in cancers. However, it is not clear whether β-TrCP deficiency is causally linked to the overexpression of these tumorigenic proteins. Additionally, context-dependent functions of substrates also increase the complexity of context-dependent F-box protein function. For example, the β-TrCP substrate DEP-domain containing mammalian target of rapamycin (mTOR)-interacting protein (DEPTOR), appears to function as both a tumor suppressor and as an oncoprotein. As a tumor suppressor, DEPTOR generally blocks mTOR activity and inhibits protein synthesis and cell proliferation (72). Mitogenic stimulation induces β-TrCP-mediated DEPTOR degradation, which results in mTOR activation and subsequent cell proliferation (73–75). However in multiple myelomas, overexpressed DEPTOR is linked to improved survival in thalidomide treated patients (76). However, the upregulation of DEPTOR in multiple myelomas is largely due to increased transcription, and the precise role of β-TrCP-mediated DEPTOR degradation requires further study.
The first reported protein-targeting chimeric molecule 1 (PROTAC) was originally developed as a generic tool that could enable the inhibition of any desired protein target, in spite of its biochemical function (77). It contains a degron-mimic peptide at one end and a specific substrate capturing a small molecule on the other end, which effectively bridges the ligase to substrate for ubiquitylation and degradation. The first PROTAC, protac-1, linked the β-TrCP targeted NF-κB inhibitor-α (IκBα) phosphodegron to ovalicin that covalently was bound to MetAP-2 (non-endogenous substrate of SCFβ-TrCP). MetAP-2 is ubiquitylated and degraded in this synthesized PROTAC-dependent manner. This approach is then used for conditional destruction of disease-causing proteins, for example the estrogen receptor (ER) and androgen receptor (AR), which have been implicated in breast and prostate cancer progression, respectively (78). In addition to peptide–degron fusions, several PROTAC derived strategies, such as small molecules based on PROTAC, have proved effective in cell culture model systems. For example, p53 plays a critical role in the maintenance of genomic stability. The powerful anti-tumor molecule p53 is targeted by the E3 ligase MDM2 for its degradation. Nutlin was originally developed to bind MDM2 and inhibit p53-MDM2 interaction for cancer therapy. The heterobifunctional PROTAC, consisting of non-steroidal androgen receptor ligand (SARM) and nutlin (known as MDM2 ligand), can recruit the androgen receptor to MDM2 for degradation (79). This novel strategy has implications for developing pharmacological treatment of cancers with increased levels of the androgen receptor.
Tumor suppressor Bcl-2 interacting mediator of cell death (Bim) is a potent proapoptotic member of the Bcl-2 protein family (80). Alternative mRNA splicing generates three major isoforms, including short (BimS), long (BimL), and a predominant extra long (BimEL). Bim directly binds and inhibits antiapoptotic Mcl1 and Bcl-XL, which restrains the ability of Bak and Bax to promote apoptosis (81). In response to survival signals, BimEL is rapidly phosphorylated by Rsk1/2 and degraded via β-TrCP (82). In non-small cell lung cancer (NSCLC) cells often resistant to gefitinib (a tyrosine kinase inhibitor that induces apoptosis via BimEL), silencing of β-TrCP can restore BimEL level and BimEL-dependent gefitinib sensitivity. In addition, Drosophila Slimb (a homolog of mice β-TrCP1) targets Polo-like kinase 4 (Plk4) for ubiquitylation and subsequent proteasomal degradation (83,84). Plk4 plays a crucial role in initiating centriole duplication that controls centrosome duplication essential for genomic integrity and genome maintenance. Abnormalities in centrosome number causes spindle formation errors that lead to ensuing chromosome missegregation. Extra centrosomes are frequently observed in variety of cancers. Thus, understanding the mechanisms underlying β-TrCP ability to regulate the centrosome via unique mitotic targets opens up new avenues to develop rationally designed therapy for cancers that are characterized by defective Plk4 activity.
β-TrCP is constitutively expressed and active. The diversity of β-TrCP substrates is partially supported by its recognition of a phosphodegron, which allows stimulus-induced recognition and subsequent degradation. For example, when GSK3 is activated, β-catenin gets degraded but IκBα remains stable. When the IKK complex is active, IκBα is degraded and β-catenin remains stable. Thus, some kinase inhibitors might synergize with other modalities as an effective strategy to manipulate degradation of substrates for cancer treatment.
3. Fbxw7
Fbxw7 predominantly functions as a tumor suppressor by mediating the degradation of multiple important oncoproteins including c-Myc, c-Jun, cyclin E, Hypoxia inducible factor-1α (HIF-1α), mTOR (mammalian target of rapamycin), Myeloid cell leukemia-1 (Mcl-1), NF-κB2, Notch-1 and other substrates (85). Given its crucial role in tumorogenesis, Fbxw7 is tightly governed at the gene expression and activity levels. Recent studies indicate that in human malignancies, Fbxw7 expression is under the control of several regulators including microRNAs (miR-27a and miR-223) (86,87), p53 (88,89), and CCAAT/enhancer-binding protein-d (C/EBP-δ) (90). Overexpression of miR-223 decreases Fbxw7 mRNA level and upregulates Fbxw7 substrate cyclin E activity, which leads to increased genomic instability. Additionally, miR-27a inhibits Fbxw7-mediated ubiquitylation and degradation of cyclin E. These data suggest that a post-transcriptional Fbxw7 regulator could affect Fbxw7 abundance and substrate degradation.
Myc family transcription factors regulate cell growth, proliferation, differentiation, and apoptosis and are frequently overexpressed in a variety of human cancers. The tumor suppressor Fbxw7 recognizes a phosphodegron generated by GSK-3 mediated phosphorylation of c-Myc on threonine-58 that promotes UPS dependent c-Myc turnover (91). Thr58 is the most frequent c-Myc mutation site in lymphoma cells and c-Myc activation is one of the key tumorigenic consequences of Fbxw7 loss in cancers. Intriguingly, Skp2 has also been shown to partake in c-Myc turnover (92,93). Instead of targeting Thr58, Skp2 binds to c-Myc via its MB2 and HLH-Zip domains for degradation. Although the timing and subcellular location of c-Myc ubiquitylation mediated by Fbxw7 or Skp2 seem to be similar, the Fbxw7-mediated ubiquitylation is governed by phosphorylation of c-Myc, whereas Skp2-mediated ubiquitylation is mainly dependent on the Skp2 expression pattern (94). This dual regulation mechanism increases the complexity to develop cancer therapy targeting c-Myc. Oridonin, the diterpenoid compound extracted from plants displays potent activity to induce apoptosis in a variety of cancer cells (95). The anticancer mechanisms of oridonin include increasing Fbxw7 expression and GSK-3 activity, which is accompanied by downregulation of c-Myc, mTOR and cyclin E, among which the decrease in c-Myc is the most significant (96). Fbxw7-mediated degradation of c-Myc triggered by oridonin induces cell growth inhibition and apoptosis. The other natural compound genistein, a major isoflavonoid isolated from soybean, has been shown to inhibit oncogenic progression in various human cancers including pancreatic cancer (PC). Genistein inhibits miR-223 expression and subsequently upregulates Fbxw7, one of the targets of miR-223, consequently inhibiting cell growth and inducing apoptosis in PC cells (87). In addition, treatment of PC cells with genistein leads to the elevation of miR-34a, resulting in Notch-1 downregulation, which correlates with inhibition of cell growth and induction of apoptosis (97). These observations shed light on the application of these natural compounds through the Fbxw7-c-Myc axis in cancer treatment.
Intriguingly, the inhibitors targeting tumor suppressor F-box proteins have been proposed to sensitize cancer stem cells to chemotherapies. For example, imatinib (Gleevec or Glivec; Novartis) is highly effective in tumor killing in chronic myelogenous leukemia, however, the drug fails to kill the cancer-initiating cells frequently quiescent in the bone marrow (98). The cancer-initiating cell population is a reservoir that accelerates generation of inhibitor resistance mutations within specific cell populations, which results in relapse of cancer once therapy is discontinued. Consequently, one potential approach could be use of strategies facilitating cancer stem cell re-entry into the cell cycle to facilitate susceptibility to chemotherapy. Indeed, tyrosine kinase inhibitors act in a manner to eradicate leukemia-initiating cells in the Fbxw7 deletion mouse model of chronic myelogenous leukemia. The effect is probably dependent on an increase in c-Myc, suggesting that Fbxw7 inhibition or inhibition of Fbxw7-mediated c-Myc degradation might synergize actions of chemotherapies. However, the long-term effect of Fbxw7 inhibition remains unclear. Specifically, the interaction between an F-box protein and substrate can also be affected by substrate mutations. For example, a GSK3 targeted phosphodegron site (Thr58) of c-Myc is frequently mutated in patients with Burkitt’s lymphoma, which distorts Fbxw7 recognition and stabilizes c-Myc (99).
In a variety of human cancers, Fbxw7 is often deleted or identified as a loss-of-function mutant. The mechanism underlying Fbxw7 depletion in tumors is not well addressed. Most tumorigenic missense Fbxw7 point mutations occur in residues crucial to substrate binding regions. These mutations interrupt oncogenic substrate recruitment and subsequent turnover. Of the cancer related Fbxw7 mutations, 43% involve two Arg mutations that directly affect the phosphodegron for substrate recognition. Other Fbxw7 inaction mechanisms result from mutations that terminate Fbxw7 translation prematurely, interfere with Fbxw7 localization, or mutate the F-box domain. In the case of a simple Fbxw7 deletion, the strategy could be replacing Fbxw7 function with another E3 ligase and reconstitute the degradation of Fbxw7 substrates, for example using PROTAC as discussed above.
Last, although in many cancers Fbxw7 primarily functions as a tumor suppressor, it can also mediate p100 (inhibitor of NF-κB signaling) degradation and act as a pro-survival factor in multiple myelomas where cancer cell growth and proliferation require p100 degradation and NF-κB activity (100). Fbxw7 targets p100 using a GSK3-mediated phosphodegron. Preventing substrate modification to indirectly inhibit F-box protein-dependent degradation could also be a potentially promising strategy to enhance tumor killing by impairing F-box recognition of its substrate.
4. Fbxl2
Fbxl2 is an emerging F-box protein that regulates mitosis and cellular proliferation and therefore might play an integral role in tumorigenesis. The first authenticated Fbxl2 substrate is phosphocholine cytidylyltransferase (CCTα), an indispensable enzyme needed for membrane phosphatidylcholine production (24). SCFFbxl2 mediates CCTα monoubiquitylation in a calcium-dependent manner. Calmodulin (CaM) is a highly conserved calcium-sensing protein crucial to control cytokinesis. Both CaM and Fbxl2 competitively interact to a canonical IQ motif (I/LQXXXRGXXXR) within CCTα. CaM binding protects CCTα from degradation. This exclusive competition model reveals that CaM functions as an antagonist of substrate ubiquitylation mediated by SCFFbxl2.
Fbxl2 also targets several cell cycle proteins. Cyclin D2, a dominant D-type cyclin in leukemic cells, translocates the cyclin-dependent kinase inhibitor p27 out of the nucleus, which is essential for G0/G1 phase progression. In B-lymphocytes and leukemic cells, SCFFbxl2 mediated degradation of cyclin D2 results in G0 arrest and apoptosis (26). Cyclin D3 directly interacts with CDK11p58 thereby controlling both centrosome maturation and bipolar spindle formation. Fbxl2 also ubiquitylates cyclin D3 for proteasomal degradation that in turn impairs G2/M transition (25). Intriguingly, cyclin D2 and cyclin D3 both harbor IQ motifs characteristic of calcium-independent CaM binding proteins. The glutamine residue within IQ motif appears essential for Fbxl2 targeting.
Tumor necrosis factor receptor-associated factors (TRAFs) play crucial role in transducing cell surface signals to the NF-κB pathway, to modulate innate and adaptive immune responses and apoptotic programs (101). In this manner, the TRAF proteins (TRAF1–6) regulate cytokine synthesis through a wide range of cell surface immunoreceptors. Notably, TRAFs mediate signals emanating from the TNFR superfamily and TLR/IL-1R family. In addition, TRAF family proteins tether with IL-1R, CD40, RANK, I-TAC, and the p75 NGF receptor to transduce divergent signals. Particularly, TRAF2, TRAF5, and TRAF6 act as adaptor proteins that mediate cell surface receptors to activate NF-κB, which in turn, potently and rapidly triggers cytokine gene expression. Exuberant cytokine release mediated by TRAF leads to severe effects of edema and multiorgan failure. Inhibition of TRAFs appears to have emerged as one potential pharmacological therapy to lessen pro-inflammatory host responses. Notably, TRAF proteins also contain a conserved CaM-binding motif by which Fbxl2 targets TRAFs for polyubiquitylation and degradation. Intriguingly, another F-box protein, Fbxo3, mediates the proteasomal degradation of Fbxl2 (28,102). Based on the prokaryotic C-terminal ApaG molecular signature within the poorly characterized Fbxo3, the highly selective small molecular inhibitor BC-1215 has been developed as an Fbxo3 antagonist. BC-1215 distorts the interaction interface of Fbxo3 and Fbxl2 to protect Fbxl2 from ubiquitylation mediated by SCFFbxo3 and subsequent degradation. Indeed, BC-1215 sufficiently destabilizes TRAF proteins and profoundly alleviates NF-κB activation and inflammation through TNF signaling. The unique broad-spectrum activity of BC-1215 has also been confirmed by reduced inflammation in multiple murine models of cytokine-driven tissue injury. It is unclear whether BC-1215 possesses anti-neoplastic activities by its ability to modulate Fbxl2 substrates such as cyclin D2, cyclin D3, or Aurora B. Nevertheless, inhibitors targeting upstream regulators of Fbxl2 represent a unique layer of highly specific class of therapeutics development.
SCFFbxl2 additionally mediates the ubiquitin-dependent degradation of Aurora B that involves in mitotic progression (103). It appears that CaM, a calcium-binding messenger protein, competes with Fbxl2 to access the IQ motif within Aurora B thereby preventing its turnover. In this molecular competition model, CaM serves as a sensor of chromosome bridges and protects Aurora B from degradation at the midbody, which stabilizes the ingressed furrow for delayed abscission. The CaM inhibitor calmidazolium dissociates CaM from Aurora B and promotes Aurora B degradation to inhibit cell proliferation and growth, displaying potent tumor killing activity. BC-1258, an inhibitor of Fbxo3, increases Fbxl2 protein levels, accompanied by decreased Fbxl2 substrates including Aurora B in a variety of human cancer cell lines including leukemic monocyte U937 cells, erythroleukemic K562 and acute monocytic leukemia THP1 cells. The antitumor activity of BC-1258 and off-target effects need to be further assessed in larger tumorigenic models to fully ascertain its therapeutic potential.
In conclusion, these studies have identified that multiple CaM-binding proteins are SCFFbxl2 substrates that are essential for cell proliferation, growth, or innate immunity. Indeed, Fbxl2 protein levels are significantly reduced in peripheral blood cells in leukemia patients, where its substrates cyclin D2, cyclin D3, and Aurora B are highly expressed. Fbxl2 recognizing a CaM-binding signature represents a unique molecular mechanism and provides a potential therapeutic strategy for cancer drug development.
5. Fbxl7
Mitotic spindle formation and proper chromosome segregation are critical to cellular mitosis. During these processes, spindle checkpoint proteins ensure that sister chromatids distribute correctly before completing cytokinesis by abscission (104,105). By phosphorylating structural and motor proteins, Aurora A plays major roles in mitotic spindle formation, chromosome alignment and separation (106). Aurora A amplification has been observed in variety of cancers including colorectal, gastric, prostate, and breast cancers. AURKA Phe31Ile polymorphism is linked to an increased risk for esophageal, ovarian and non–small-cell lung cancers. Fbxl7-mediated centrosomal Aurora A degradation disrupts normal mitotic spindle formation which induces mitotic arrest and apoptosis (107). Fbxl7 also targets survivin for ubiquitylation and degradation (108). The apoptosis inhibitor survivin is highly expressed in most cancers and linked to chemotherapy resistance, tumor recurrence, and a shorter survival. Aiming to downregulate survivin by elevating upstream Fbxl7 might be an attractive cancer treatment strategy. Intriguingly, Fbxl18 recruits Fbxl7 for ubiquitin-mediated turnover and the FQ domain within Fbxl7 is important for Fbxl18 targeting (29). A small molecule inhibitor distorting the interface surrounding the FQ domain might preserve Fbxl7 protein from degradation mediated by SCFFbxl18. Additionally, elevated NF-κB signaling induces the expression of Fbxl7 (unpublished data). Thus, identifying transcriptional regulators of the FBXL7 gene might provide an alternative approach for cancer treatment through the Fbxl7-Aurora A/survivin axis.
6. Fbxo31
Fbxo31 was originally identified as a tumor suppressor gene in breast, ovarian, prostate and hepatocellular cancers. As part of the pre-replication complex, Cdt1 DNA replication licensing factor is essential to maintain genomic integrity. In the cell cycle G2 phase, Fbxo31 specifically targets the NH2-terminus of Cdt1 to mediate its ubiquitin-dependent proteolysis (109). Fbxo31 depletion that results in Cdt1 stabilization leads to re-replication.
Fbxo31 plays crucial roles in the DNA damage response and oncogenesis. Ectopic expression of Fbxo31 has been reported to mediate the proteolysis of cyclin D1, an important regulator of G1 to S phase progression, resulting in cell cycle arrest in G1. The phosphorylation on Thr286 within the cyclin D1 phosphodegron is required for Fbxo31 recognition (110). In human primary gastric cancer (GC) tissue, Fbxo31 mRNA and protein dramatically decrease. Overexpression of Fbxo31 significantly induces G1 phase arrest and downregulates cyclin D1 levels in GC cells. MiR-20a and miR-17 reduces the expression of Fbxo31, and there exists a significant negative association between miR-20a/miR-17 and Fbxo31 in GC samples. Therefore, effective therapy targeting the miR-20a /miR-17-Fbxo31-cyclin D1 axis may help control GC progression. Notably, Fbxo4 also targets cyclin D1 for ubiquitin-dependent degradation via diverged pathway (111). In NIH3T3 cells, ATM activation further potentiates the GSK3β-mediated cyclin D1 phosphorylation at Thr286. The cyclin D1 phosphodegron is then recognized by the F-box Fbxo4/αB crystalline complex within the SCF E3 ubiquitin ligase core, leading to intra-S-phase checkpoint control. Therefore, there may exist potential therapeutic value by targeting Fbxo31-cyclin D1 through a tissue or cell type specific approach.
In unstressed cells, the tumor suppressor protein p53 is maintained at low levels through MDM2-mediated ubiquitylation and proteolysis (112). Under cellular stress, increased p53 levels induce a variety of protective responses to prevent cancer. In response to genotoxic stress, Fbxo31 is phosphorylated by the DNA damage serine/threonine kinase ATM. Fbxo31 then interacts with ATM-phosphorylated MDM2 and directs its degradation (113). Fbxo31-mediated loss of MDM2 increases levels of p53 and leads to growth arrest. These data indicate a tumor suppressing function of Fbxo31 by its ability to regulate p53 levels following DNA damage.
However, overexpression of Fbxo31 is also observed in lung cancer (114). Higher expression of Fbxo31 significantly correlates to tumor size and infiltration, clinical stages and metastasis. In human lung carcinoma A549 cells, ectopic expression of Fbxo31 promotes cell growth, metastasis and invasion, whereas specific siRNA-mediated Fbxo31 silencing inhibits these processes. Furthermore, tumorigenicity assays demonstrate that Fbxo31 promotes tumor growth in nude mice. In esophageal squamous cell carcinoma, the group with high expression of Fbxo31 was shown to have a significantly poorer prognosis, suggesting that Fbxo31 might function as an oncogenic protein. MAPK (p38) plays pivotal roles in inflammatory responses and stress responses. In response to growth factors, pro-inflammatory cytokines and environmental stresses, p38 MAPK is activated by dual phosphorylation on the “TGY” motif. Mkk6 is among p38 activators responding to a variety of external stimuli. Long-term activation of the Mkk6-p38 can induce cell apoptosis. By recognizing phosphorylated Mkk6 and triggering its ubiquitylation and proteolysis, Fbxo31 exerts antiapoptotic effects on cancer cells in response to stress stimuli (115). The stimuli and temporal specificity displayed by Fbxo31 on tumorigenesis increase the complexity of targeting Fbxo31 for cancer therapeutics. However, this highly specific approach may also increase the accuracy of precision therapeutics.
7. Cyclin F
Cyclin F (also known as Fbxo1) is the first described F-box protein from which the F-box protein family obtained its name (9). Despite its essential nature in mouse development and the founding member status in the F-box protein family, until recently cyclin F remained as an orphan protein with unknown function. Cyclin F localizes to both centrosomes and within the nucleus. During cell cycle progression, cyclin F protein level oscillates, peaking in G2 (116). CP110 is an integral centrosomal component and essential for centrosome duplication. During the G2 phase, cyclin F associates to CP110 on the centrioles and mediates its ubiquitylation and subsequent turnover (23). Centrosomal cyclin F targets CP110 to mediate its degradation to ensure that duplication of the centrosome only occurs once per cell cycle. Through high throughput screening based on genetic and pharmacologic studies, NUSAP1, a key cell-cycle-regulated microtubule-binding protein important in chromosome congression and segregation was identified as an SCFcyclin F target for ubiquitylation and proteasomal degradation (23). Like CP110, NUSAP1 is involved in regulating the microtubule cytoskeleton. Thus, cyclin F mediated degradation of both factors is essential to maintain chromosome stability.
The function of nuclear cyclin F is highly related to the degradation of the ribonucleotide reductase family member 2 (RRM2). Ribonucleotide reductase (RNR) converts ribonucleotides to deoxyribonucleotides (dNTPs), which is required for replicative and repair DNA synthesis (117). During G2, CDK-phosphorylated RRM2 (on Thr33) is degraded via SCFcyclin F E3 ligase to preserve delicate dNTP pools and genomic stability (27). Following DNA damage, cyclin F is downregulated to allow RRM2 accumulation. In response to genotoxic stress, this biochemical pathway controls dNTPs abundance and ensures efficient DNA repair.
Although cyclin F is implicated in controlling centrosome duplication and maintaining genome stability, its expression and significance in cancer remain elusive. A recent study indicates that cyclin F is noticeably decreased in hepatocellular carcinoma tissue at both mRNA and protein levels (118). Low cyclin F expression is an independent poor prognostic marker for survival, significantly correlated with tumor size, clinical stage, serum alpha-fetoprotein level and tumor multiplicity. However, the true roles of cyclin F in cancer require further characterization.
8. Fbxo11
The proto-oncogene product Bcl6 is implicated in human B-cell lymphomas pathogenesis (119). Through binding to specific DNA sequences, Bcl6 regulates the transcription of multiple genes important in B-cell development, differentiation and activation (120). Bcl6 is frequently overexpressed in patients with aggressive diffuse large B-cell lymphoma (DLBCL). Transgenic mice with constitutively expressed Bcl6 in B cells develop DLBCLs. SCFFbxo11 targets Bcl6 for ubiquitylation and proteasomal degradation (121). Meanwhile, the gene encoding Fbxo11 is often deleted or mutated in a variety of DLBCL cell lines and primary DLBCLs, and Fbxo11 dysfunction correlates with increased Bcl6 protein stability and abundance. Additionally, tumor-derived Fbxo11 mutants display deficiency in degrading Bcl6. In Fbxo11-deleted DLBCL cells, restoring Fbxo11 expression inhibits cell proliferation and induces cell death by promoting Bcl6 ubiquitylation and degradation. Fbxo11 reconstitution also suppresses tumorigenicity in immunodeficient mice with tumors generated by Fbxo11-deleted DLBCL cells.
The tumor suppressing function of Fbxo11 is further demonstrated through Fbxo11 targeting WD40 repeat-containing DDB1 and Cul4-associated factors (DCAF) proteins (20,21). DCAFs are the substrate binding subunits of Cul4-RING ubiquitin ligase (CRL4). Cdt2 belongs to DCAF family and plays fundamental roles in regulating the cell cycle S phase by mediating the degradation of Cdt1, Set8, and p21 under both normal and stress conditions (122–128). Cdt2 is elevated in various cancers including liver, breast, gastric, and colon cancers, and its overexpression is associated with advanced cancer, metastasis, and poor patient survival (129,130). Fbxo11 recruits Cdt2 to the SCF core to promote its proteasomal degradation. Different from most SCF substrates, which exhibit phosphorylation-dependent binding to F-box proteins, CDK-mediated Thr464 phosphorylation within the Cdt2 degron inhibits its recognition by Fbxo11 (21). The Set8 stabilization resulting from Cdt2 degradation is important to suppress the phospho-Smad2 response to TGF-β and to promote cell migration (20). These observations indicate that reconstitution of the tumor suppressing function of Fbxo11 may represent a unique strategy for lymphoma treatment. Furthermore, the ubiquitin-dependent degradation of Cdt2 through SCFFbxo11 provides an interesting example of cross-regulation between two ubiquitin E3 ligases. In the future, it will be important to identify the specific cellular context under which Cdt2 is degraded to control cell differentiation.
MicroRNAs (miRNAs) have been demonstrated to have crucial roles in regulating cancer cell proliferation, survival and sensitivity to chemotherapy. The recent study on microRNA miR-21 seems to further demonstrate the tumor suppressing activity of Fbxo11. MiR-1 is overexpressed in most human cancers and it functions as an oncogene. By microarray analysis and quantitative PCR, Fbxo11 has been identified and validated as a miR-21 target gene (131). Fbxo11 promotes apoptosis and the degradation of the oncogenic protein Bcl6, and silencing Fbxo11 in miR-21 knockout cancer cells restores their high tumorigenicity. The expression of miR-21 and Fbxo11 are inversely correlated in tumor tissue. High Fbxo11 expression correlates with lower tumor grade and better patient survival, which is consistent with its tumor suppressor activity. However, Fbxo11 is also reported as a direct target of the other microRNA miR-621 (132). Overexpression of miR-621 promotes apoptosis and increases chemosensitivity in cultured breast cancer cells and in xenograft tumor model. High Fbxo11 expression significantly correlates with poor survival in breast cancer patients. Mechanistically, in breast cancer cells, Fbxo11 binds to p53 and facilitates its neddylation that in turn inhibits p53 transactivity. MiR-621 enhances chemosensitivity of breast cancer cells through its suppression of Fbxo11-mediated p53 inhibition. The function of Fbxo11 in tumorigenesis and cancer progression is under further investigation. Nevertheless, Fbxo11 highlights the context-specific utilization of F-box protein inhibitors.
Small molecule inhibitors developed in cancer treatment are summarized in Table 1.
Table 1.
Summary of small molecule targeting F-box protein in cancer
| F-box Protein | Disease Link | Compound | Structure | Mechanism | Reference |
|---|---|---|---|---|---|
| Skp2 | Multiple myeloma | Compound A (CpdA) |
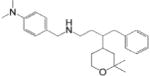
|
Interferes Skp2-Skp1 interaction and stabilizes p27 | (49) |
| Prostate cancer | SMIP004 |
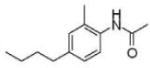
|
Downregulates Skp2 | (51) | |
| Metastatic melanoma, prostate, breast, ovarian, lung cancer | C1, C2, C16, C20 |
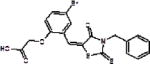
|
Binds to a pocket formed by Skp2 and Cks1 to block substrate binding | (52) | |
| Cancer, general | Compound #25 |
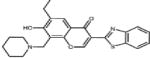
|
Binds to Skp2 and prevents Skp2-Skp1 interaction | (47) | |
| Breast, prostate cancer | Curcumin |
|
Natural agents, inhibit Skp2 expression | (146–148) | |
| Quercetin |
|
||||
| Lycopene |
|
||||
| silibinin |
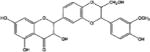
|
||||
| EGCG |
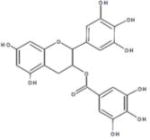
|
||||
| Vitamin D |
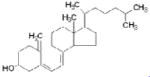
|
||||
| κ-TrCP | Inflammation | GS143 |
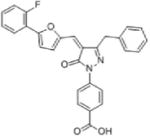
|
Disrupts interaction between phospho-IκBκκ and κ-TrCP and suppress IκBκ ubiquitylation | (65) |
| Cancer, general | Erioflorin |
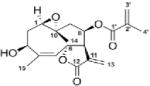
|
Inhibits the interaction between κ-TrCP and tumor suppressor Pdcd4 | (71) | |
| Fbxw7 | Pancreatic cancer | SINE KPT-185 |
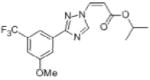
|
Inhibits nuclear export of Fbxw7, enhances nuclear retention of Fbxw7 and degrades Notch1 | (149) |
| Leukemia, lymphoma | Oridonin |
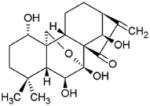
|
Increases Fbxw7 expression, activates GSK3 and facilitates c-Myc turnover | (150) | |
| Genistein |
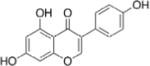
|
Inhibits miR-223 expression and elevates its target Fbxw7 expression | (87) | ||
| Fbxl2 | Inflammation | BC-1215 |
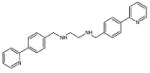
|
Inhibits the Fbxo3 and Fbxl2 binding | (28) |
| BC-1258 |
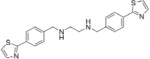
|
Inhibits binding between Fbxo3 and Fbxl2, stabilizes Fbxl2 and promotes Aurora B degradation | (103) | ||
| Fbxl3 | circadian-related disorders (sleep disorder, cancer, cardiovascular and metabolic diseases | KL001 |
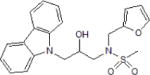
|
Competes for binding in the FAD pocket of CRYs and prevents Fbxl3 binding | (151,152) |
Others. Fbxw8 (also called Fbx29) assembles the E3 ligase with Cul7, Skp1 and Roc1, in which Cul7 directly interacts with Fbxw8 rather than Skp1 (133). As a linker, Fbxw8 tethers Cul7 and Cul1 to form a heterodimeric complex (134). Cyclin D1 regulates G1 progression and it undergoes increased degradation during S phase in a variety of cancer cells. Fbxw8 can mediate the degradation of phosphorylated cyclin D1 (on Thr286) via SCF E3 or CRL7 E3 ligase (135). Depletion of Fbxw8 causes significant cyclin D1 accumulation and potently inhibits cell proliferation. Thus, Fbxw8 plays an essential role in cancer cell proliferation through the proteolysis of cyclin D1, indicating that Fbxw8 might function as an oncogenic protein. TBC1D3 is a hominoid-specific oncogene and it is overexpressed in several human cancers including prostate, breast, pancreatic cancer, and gall bladder tumors. Expression of TBC1D3 enhances growth factor receptor signaling that in turn promotes cellular proliferation and survival. Fbxw8 recruits phosphorylated TBC1D3 for Cul7-mediated ubiquitylation and degradation, supporting a tumor suppressing function of Cul7/Fbxw8 (136). Hematopoietic progenitor kinase 1 (Hpk1) is a member of Ste20-like serine/threonine kinases and functions as a MAP4K to activate MAP3Ks. Hpk1 is expressed in normal pancreatic ducts but is missing in >95% of pancreatic cancers. The loss of Hpk1 is strongly associated with the progression to invasive pancreatic cancer. The Cul7/Fbxw8 ubiquitin ligase mediates Hpk1 degradation that is dependent upon Hpk1 autophosphorylation and is reversely regulated by the protein phosphatase 4 (PP4) (137). The complex role of Fbxw8 linked to both SCF and CRL7 E3 ligases in tumorigenesis will need further characterization. Nevertheless, Fbxw8 may present a new target for cancer therapy.
Fbxo4 is the specificity factor for an SCF E3 ligase that directs timely proteolysis of cyclin D1 (111). In mice Fbxo4 deficiency induces melanoma and it depends on cyclin D1 accumulation, which underscores the importance of Fbxo4 as a tumor suppresser. Furthermore, a substrate-binding mutation, Fbxo4 I377M, was identified to selectively disrupt cyclin D1 degradation. In addition, the proteolysis of TRF1, another known Fbxo4 substrate, is preserved. Fbxo4 deficiency and I377M mutations lead to cyclin D1 nuclear accumulation, a key event in tumorigenesis. Collectively, cyclin D1 overexpression induced by Fbxo4 dysfunction may be an important contributor to some human malignancies.
As integral components of mTORC1 and mTORC2, the Tel2 and Tti1 proteins control the cellular abundance of phosphatidylinositol 3-kinase-related kinases (PIKKs) (138–141). SCFFbxo9 targets Tel2 and Tti1 for degradation to modulate mTOR signaling in response to growth factor availability (142). Upon growth factor deprivation, cytoplasmic protein kinase CK2 mediates mTORC1-specific phosphorylation of Tel2/Tti1. In turn, mTORC1 is inactivated to restrain protein translation and cell growth. Primary human multiple myelomas express high levels of Fbxo9, which is essential for PI(3)K/TORC2/Akt signalling and survival of multiple myeloma cells. This mTORC1-specific Fbxo9 mediated degradation of Tel2/Tti1 provides targets for the unique treatment of multiple myeloma with high levels of Fbxo9.
Dlc1 (deleted in liver cancer 1) is a RhoA GTPase-activating protein and tumor suppressor. Loss of Dlc1 expression by genomic deletion or epigenetic silencing and loss of gene transcription is implicated in liver, breast, lung, ovarian, kidney, colon, stomach, prostate, and other cancers. In non-small cell lung cancer (NSCLC) cell lines and tumor tissue, Dlc1 is ubiquitylated and degraded by the Cullin 4A-RING E3 ligase (CRL4A) complex, interestingly, where Fbxw5 serves as the substrate receptor (143). Inhibition of Fbxw5 leads to a Dlc1-dependent decrease in cell proliferation. This CRL4A-Fbxw5-Dlc1-linkage associated oncogenesis pathway provides a novel set of targets for potential drug design.
Another F-box protein, Fbxo5, functions as a pseudo-substrate inhibitor of the anaphase promoting complex/cyclosome (APC/C). Overexpression of Fbxo5 has been found in in a variety of malignant tumors including ovarian clear cell carcinoma. Through APC/C misregulation, Fbxo5 overexpression causes mitotic catastrophe and genomic instability and potentially contributes to tumorigenesis (144,145). Fbxo18 has been often found to be deleted or mutated in melanoma and lung cancers. These largely uncharacterized F-box proteins might also serve as unique therapeutic targets by modulating downstream abundance of substrates involved in neoplasia.
Potentially druggable F-box proteins involved in cancer treatment are summarized in Table 2.
Table 2.
Summary of emerging F-box proteins in cancer
| F-box Protein | Disease Link | Abnomalities | Mechanism | Reference |
|---|---|---|---|---|
| Fbxl10 | Pancreatic ductal adenocarcinoma | overexpression | Stimulates histone H2A ubiquitylation in fruit fly Drosophila | (153,154) |
| Fbxl20 | Colorectal adenocarcinoma | overexpression | Promotes carcinogenesis through regulating Wnt signaling and caspase activation, mediates Vps34 ubiquitylation and autophagy, facilitates E-cadherin ubiquitylation and induces invasion | (155–160) |
| Fbxw8 | 3-M syndrome | Assembles ubiquitin E3 ligase with Cul7, Skp1 and Roc1, tethers the SCF and CRL7 E3 hetermeric complex | (161–165) | |
| Fbxo4 | Cancer, general | mutate | Targets cyclin D1 and TRF1 for degradation | (166–169) |
| Cyclin F | Hepatocellular carcinoma | low expression | Targets CP110 and RRM2 to control centrosome duplication and maintains genome stability | (23,27,116,118) |
| Fbxo5 | Ovarian clear cell carcinoma | overexpression | Inhibitor of APCCDH1 and APCCDC20 | (145,170,171) |
| Fbxo9 | Multiple myelomas | overexpression | Targets degradation of Tel2 and Tti1 and regulates mTOR signaling | (172) |
| Fbxo10 | Associate with breast cancer susceptibility | (173–175) | ||
| Fbxo11 | B cell lymphoma/breast cancer | mutate or delete | Degrades Bcl6, Cdt2, Snail | (131,132,176) |
| Fbxo18 | melanoma, lung cancer (deleted) | delete or mutate | DNA-dependent ATPase and DNA-unwinding activities, possesses both pro- and anti-recombinogenic activities | (177–179) |
| Fbxo31 | Breast cancer, hepatocellular carcinoma, esophageal squamous cell carcinoma | controversial | Inhibits proliferation and tumorigenesis in breast cancer, hepatocellular carcinoma and ovarian cancer, but overexpressed in lung cancer | (114,180,181) |
The growing details in understanding of SCF based protein targeting of a diverse array of fundamental substrates provide a great opportunity for cancer therapeutic development targeting F-box proteins. Meanwhile, the complexity of one F-box protein recruiting multiple targets and each substrate being recognizing by several E3 ligases provides therapeutic challenges. There remains an unmet need to devise novel, highly potent and selective small molecules that exhibit limited off-targeting side effects that will necessitate even deeper characterization of the F-box protein/substrate pair interacting mechanisms. Particularly, the majority of F-box proteins largely remain functionally mysterious. Except for the biological activity in physiologic and disease models, the F-box protein/substrate complex 3D structure is especially critical for high throughput in silico small molecule screening. This will facilitate efficient selection of candidates from millions of compound-rich libraries based on the unique interaction surface pocket formed by an F-box protein and its putative substrate. The successful application of these small compounds to cancer therapy also inspires more rigorous study of F-box proteins in other diseases and facilitates specific therapeutic development.
Acknowledgments
The authors apologize for the omission of many colleagues’ work owing to space constraints. Work in the Mallampalli laboratory is partly supported by the US National Institutes of Health R01 Grant HL096376 and P01 HL114453 (to R.K.M.), and is partly supported by the United States Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development and by a Merit Review award from the United States Department of Veterans Affairs (to R.K.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None.
References
- 1.Stockwell B. Outsmarting Cancer. A biologist talks about what makes disease-causing proteins so difficult to target with drugs. Scientific American. 2011;305:20. [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A. The ubiquitin system. Annual review of biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 3.Komander D, Rape M. The ubiquitin code. Annual review of biochemistry. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 4.Deshaies RJ, Joazeiro CAP. RING Domain E3 Ubiquitin Ligases. Annual review of biochemistry. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 5.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nature reviews. Molecular cell biology. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 6.Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 7.Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 8.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nature reviews. Molecular cell biology. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 9.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 10.Cenciarelli C, Chiaur DS, Guardavaccaro D, Parks W, Vidal M, Pagano M. Identification of a family of human F-box proteins. Current biology: CB. 1999;9:1177–1179. doi: 10.1016/S0960-9822(00)80020-2. [DOI] [PubMed] [Google Scholar]
- 11.Winston JT, Koepp DM, Zhu C, Elledge SJ, Harper JW. A family of mammalian F-box proteins. Current biology: CB. 1999;9:1180–1182. doi: 10.1016/S0960-9822(00)80021-4. [DOI] [PubMed] [Google Scholar]
- 12.Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes & development. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nature reviews. Molecular cell biology. 2008;9:679–690. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lau AW, Fukushima H, Wei W. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Frontiers in bioscience. 2012;17:2197–2212. doi: 10.2741/4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta- TrCP: tipping the scales of cancer. Nature reviews. Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 17.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 18.Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Molecular cell. 2003;12:381–392. doi: 10.1016/s1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 19.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Abbas T, Mueller AC, Shibata E, Keaton M, Rossi M, Dutta A. CRL1-FBXO11 promotes Cdt2 ubiquitylation and degradation and regulates Pr-Set7/Set8-mediated cellular migration. Molecular cell. 2013;49:1147–1158. doi: 10.1016/j.molcel.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossi M, Duan S, Jeong YT, Horn M, Saraf A, Florens L, Washburn MP, Antebi A, Pagano M. Regulation of the CRL4(Cdt2) ubiquitin ligase and cell-cycle exit by the SCF(Fbxo11) ubiquitin ligase. Molecular cell. 2013;49:1159–1166. doi: 10.1016/j.molcel.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nature reviews. Molecular cell biology. 2013;14:369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, Dynlacht B, Pagano M. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466:138–142. doi: 10.1038/nature09140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen BB, Coon TA, Glasser JR, Mallampalli RK. Calmodulin antagonizes a calcium-activated SCF ubiquitin E3 ligase subunit, FBXL2, to regulate surfactant homeostasis. Molecular and cellular biology. 2011;31:1905–1920. doi: 10.1128/MCB.00723-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen BB, Glasser JR, Coon TA, Mallampalli RK. F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest. Oncogene. 2012;31:2566–2579. doi: 10.1038/onc.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen BB, Glasser JR, Coon TA, Zou C, Miller HL, Fenton M, McDyer JF, Boyiadzis M, Mallampalli RK. F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood. 2012;119:3132–3141. doi: 10.1182/blood-2011-06-358911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–1034. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y, Zou C, Ellis B, Sciurba FC, Zhang Y, Mallampalli RK. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nature immunology. 2013;14:470–479. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Lear T, Zhao Y, Zhao J, Zou C, Chen BB, Mallampalli RK. F-box protein Fbxl18 mediates polyubiquitylation and proteasomal degradation of the pro-apoptotic SCF subunit Fbxl7. Cell death & disease. 2015;6:e1630. doi: 10.1038/cddis.2014.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. The New England journal of medicine. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 31.Richardson PG, Briemberg H, Jagannath S, Wen PY, Barlogie B, Berenson J, Singhal S, Siegel DS, Irwin D, Schuster M, Srkalovic G, Alexanian R, Rajkumar SV, Limentani S, Alsina M, Orlowski RZ, Najarian K, Esseltine D, Anderson KC, Amato AA. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:3113–3120. doi: 10.1200/JCO.2005.04.7779. [DOI] [PubMed] [Google Scholar]
- 32.Kouroukis TC, Baldassarre FG, Haynes AE, Imrie K, Reece DE, Cheung MC. Bortezomib in multiple myeloma: Systematic review and clinical considerations. Current Oncology. 2014;21:e573–e603. doi: 10.3747/co.21.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie CM, Wei W, Sun Y. Role of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases in skin cancer. Journal of genetics and genomics = Yi chuan xue bao. 2013;40:97–106. doi: 10.1016/j.jgg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Sun Y. Cullin-RING Ligases as attractive anti-cancer targets. Current pharmaceutical design. 2013;19:3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weathington NM, Mallampalli RK. Emerging therapies targeting the ubiquitin proteasome system in cancer. The Journal of clinical investigation. 2014;124:6–12. doi: 10.1172/JCI71602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nature reviews. Drug discovery. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nature reviews. Cancer. 2014;14:233–247. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Shaik S, Dai X, Wu Q, Zhou X, Wang Z, Wei W. Targeting the ubiquitin pathway for cancer treatment. Biochimica et biophysica acta. 2015;1855:50–60. doi: 10.1016/j.bbcan.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Current biology: CB. 1999;9:661–664. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 41.Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nature cell biology. 1999;1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 42.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin- mediated degradation of the CDK inhibitor p27. Nature cell biology. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 43.Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nature cell biology. 2001;3:321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 44.Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, Reed SI. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Molecular cell. 2001;7:639–650. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 45.Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S-i, Natsume T, Nakayama KI. Skp2-Mediated Degradation of p27 Regulates Progression into Mitosis. Developmental cell. 2004;6:661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- 46.Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, Malek NP. Skp2- dependent degradation of p27kip1 is essential for cell cycle progression. Genes & development. 2004;18:2602–2607. doi: 10.1101/gad.321004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, Logothetis CJ, Hung MC, Zhang S, Lin HK. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, Hazle JD, Yu D, Wei W, Sarbassov D, Hung MC, Nakayama KI, Lin HK. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149:1098–1111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, Corral LG, Krenitsky VP, Xu W, Moutouh-de Parseval L, Webb DR, Mercurio F, Nakayama KI, Nakayama K, Orlowski RZ. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111:4690–4699. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nickeleit I, Zender S, Sasse F, Geffers R, Brandes G, Sorensen I, Steinmetz H, Kubicka S, Carlomagno T, Menche D, Gutgemann I, Buer J, Gossler A, Manns MP, Kalesse M, Frank R, Malek NP. Argyrin a reveals a critical role for the tumor suppressor protein p27(kip1) in mediating antitumor activities in response to proteasome inhibition. Cancer cell. 2008;14:23–35. doi: 10.1016/j.ccr.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 51.Rico-Bautista E, Yang CC, Lu L, Roth GP, Wolf DA. Chemical genetics approach to restoring p27Kip1 reveals novel compounds with antiproliferative activity in prostate cancer cells. BMC biology. 2010;8:153. doi: 10.1186/1741-7007-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chemistry & biology. 2012;19:1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang G, Ayala G, De Marzo A, Tian W, Frolov A, Wheeler TM, Thompson TC, Harper JW. Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:3419–3426. [PubMed] [Google Scholar]
- 54.Liu S, Yamauchi H. p27-Associated G1 arrest induced by hinokitiol in human malignant melanoma cells is mediated via down-regulation of pRb, Skp2 ubiquitin ligase, and impairment of Cdk2 function. Cancer letters. 2009;286:240–249. doi: 10.1016/j.canlet.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 55.Koga H, Harada M, Ohtsubo M, Shishido S, Kumemura H, Hanada S, Taniguchi E, Yamashita K, Kumashiro R, Ueno T, Sata M. Troglitazone induces p27Kip1-associated cell- cycle arrest through down-regulating Skp2 in human hepatoma cells. Hepatology. 2003;37:1086–1096. doi: 10.1053/jhep.2003.50186. [DOI] [PubMed] [Google Scholar]
- 56.Hsieh HY, Shieh JJ, Chen CJ, Pan MY, Yang SY, Lin SC, Chang JS, Lee AY, Chang CC. Prodigiosin down-regulates SKP2 to induce p27(KIP1) stabilization and antiproliferation in human lung adenocarcinoma cells. British journal of pharmacology. 2012;166:2095–2108. doi: 10.1111/j.1476-5381.2012.01921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung YK, Chi-Hung Or R, Lu CH, Ouyang WT, Yang SY, Chang CC. Sulforaphane down-regulates SKP2 to stabilize p27(KIP1) for inducing antiproliferation in human colon adenocarcinoma cells. Journal of bioscience and bioengineering. 2015;119:35–42. doi: 10.1016/j.jbiosc.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 58.Sumimoto H, Hirata K, Yamagata S, Miyoshi H, Miyagishi M, Taira K, Kawakami Y. Effective inhibition of cell growth and invasion of melanoma by combined suppression of BRAF (V599E) and Skp2 with lentiviral RNAi. International journal of cancer. Journal international du cancer. 2006;118:472–476. doi: 10.1002/ijc.21286. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Dai DL, Martinka M, Li G. Prognostic significance of nuclear ING3 expression in human cutaneous melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:4111–4116. doi: 10.1158/1078-0432.CCR-07-0408. [DOI] [PubMed] [Google Scholar]
- 60.Chen G, Wang Y, Garate M, Zhou J, Li G. The tumor suppressor ING3 is degraded by SCF(Skp2)-mediated ubiquitin-proteasome system. Oncogene. 2010;29:1498–1508. doi: 10.1038/onc.2009.424. [DOI] [PubMed] [Google Scholar]
- 61.Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 62.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nature reviews. Molecular cell biology. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 63.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nature cell biology. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396:590–594. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 65.Nakajima H, Fujiwara H, Furuichi Y, Tanaka K, Shimbara N. A novel small- molecule inhibitor of NF-kappaB signaling. Biochemical and biophysical research communications. 2008;368:1007–1013. doi: 10.1016/j.bbrc.2008.01.166. [DOI] [PubMed] [Google Scholar]
- 66.Nakayama K, Hatakeyama S, Maruyama S-i, Kikuchi A, Onoé K, Good RA, Nakayama KI. Impaired degradation of inhibitory subunit of NF-κB (IκB) and β-catenin as a result of targeted disruption of the β-TrCP1 gene. Proceedings of the National Academy of Sciences. 2003;100:8752–8757. doi: 10.1073/pnas.1133216100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, Lockett SJ, Sonenberg N, Colburn NH. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Molecular and cellular biology. 2003;23:26–37. doi: 10.1128/MCB.23.1.26-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mudduluru G, Medved F, Grobholz R, Jost C, Gruber A, Leupold JH, Post S, Jansen A, Colburn NH, Allgayer H. Loss of programmed cell death 4 expression marks adenoma- carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer. 2007;110:1697–1707. doi: 10.1002/cncr.22983. [DOI] [PubMed] [Google Scholar]
- 69.Wei NA, Liu SS, Leung TH, Tam KF, Liao XY, Cheung AN, Chan KK, Ngan HY. Loss of Programmed cell death 4 (Pdcd4) associates with the progression of ovarian cancer. Molecular cancer. 2009;8:70. doi: 10.1186/1476-4598-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 71.Blees JS, Bokesch HR, Rubsamen D, Schulz K, Milke L, Bajer MM, Gustafson KR, Henrich CJ, McMahon JB, Colburn NH, Schmid T, Brune B. Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase beta-TrCP1. PloS one. 2012;7:e46567. doi: 10.1371/journal.pone.0046567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, Pagano M. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Molecular cell. 2011;44:317–324. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, Lyssiotis CA, Gygi SP, Toker A, Cantley LC, Asara JM, Harper JW, Wei W. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Molecular cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Molecular cell. 2011;44:304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boyd KD, Walker BA, Wardell CP, Ross FM, Gregory WM, Davies FE, Morgan GJ. High expression levels of the mammalian target of rapamycin inhibitor DEPTOR are predictive of response to thalidomide in myeloma. Leukemia & lymphoma. 2010;51:2126–2129. doi: 10.3109/10428194.2010.509893. [DOI] [PubMed] [Google Scholar]
- 77.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Molecular & cellular proteomics: MCP. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 79.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic & medicinal chemistry letters. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. The EMBO journal. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fletcher JI, Huang DC. Controlling the cell death mediators Bax and Bak: puzzles and conundrums. Cell cycle. 2008;7:39–44. doi: 10.4161/cc.7.1.5178. [DOI] [PubMed] [Google Scholar]
- 82.Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, Pagano M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Molecular cell. 2009;33:109–116. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rogers GC, Rusan NM, Roberts DM, Peifer M, Rogers SL. The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. The Journal of cell biology. 2009;184:225–239. doi: 10.1083/jcb.200808049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cunha-Ferreira I, Rodrigues-Martins A, Bento I, Riparbelli M, Zhang W, Laue E, Callaini G, Glover DM, Bettencourt-Dias M. The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Current biology: CB. 2009;19:43–49. doi: 10.1016/j.cub.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 85.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O, Spruck C. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer research. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 86.Spruck C. miR-27a regulation of SCF(Fbw7) in cell division control and cancer. Cell cycle. 2011;10:3232–3233. doi: 10.4161/cc.10.19.17125. [DOI] [PubMed] [Google Scholar]
- 87.Ma J, Cheng L, Liu H, Zhang J, Shi Y, Zeng F, Miele L, Sarkar FH, Xia J, Wang Z. Genistein down-regulates miR-223 expression in pancreatic cancer cells. Current drug targets. 2013;14:1150–1156. doi: 10.2174/13894501113149990187. [DOI] [PubMed] [Google Scholar]
- 88.Kimura T, Gotoh M, Nakamura Y, Arakawa H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer science. 2003;94:431–436. doi: 10.1111/j.1349-7006.2003.tb01460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 90.Balamurugan K, Wang JM, Tsai HH, Sharan S, Anver M, Leighty R, Sterneck E. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. The EMBO journal. 2010;29:4106–4117. doi: 10.1038/emboj.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c- Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc- regulated transcription. Molecular cell. 2003;11:1189–1200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]
- 93.Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Molecular cell. 2003;11:1177–1188. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- 94.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. The EMBO journal. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou GB, Chen SJ, Wang ZY, Chen Z. Back to the future of oridonin: again, compound from medicinal herb shows potent antileukemia efficacies in vitro and in vivo. Cell research. 2007;17:274–276. doi: 10.1038/cr.2007.21. [DOI] [PubMed] [Google Scholar]
- 96.Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, Wen JZ, Zhou H, Qu LH. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Molecular cancer therapeutics. 2012;11:1155–1165. doi: 10.1158/1535-7163.MCT-12-0066. [DOI] [PubMed] [Google Scholar]
- 97.Xia J, Duan Q, Ahmad A, Bao B, Banerjee S, Shi Y, Ma J, Geng J, Chen Z, Rahman KM, Miele L, Sarkar FH, Wang Z. Genistein inhibits cell growth and induces apoptosis through up-regulation of miR-34a in pancreatic cancer cells. Current drug targets. 2012;13:1750–1756. doi: 10.2174/138945012804545597. [DOI] [PubMed] [Google Scholar]
- 98.Reavie L, Buckley SM, Loizou E, Takeishi S, Aranda-Orgilles B, Ndiaye-Lobry D, Abdel- Wahab O, Ibrahim S, Nakayama KI, Aifantis I. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer cell. 2013;23:362–375. doi: 10.1016/j.ccr.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Molecular and cellular biology. 2000;20:2423–2435. doi: 10.1128/mcb.20.7.2423-2435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O’Connor O, Hoffmann A, Elenitoba-Johnson KS, Pagano M. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nature cell biology. 2012;14:375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, Yamamoto T. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Experimental cell research. 2000;254:14–24. doi: 10.1006/excr.1999.4733. [DOI] [PubMed] [Google Scholar]
- 102.Mallampalli RK, Coon TA, Glasser JR, Wang C, Dunn SR, Weathington NM, Zhao J, Zou C, Zhao Y, Chen BB. Targeting F box protein Fbxo3 to control cytokine-driven inflammation. Journal of immunology. 2013;191:5247–5255. doi: 10.4049/jimmunol.1300456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen BB, Glasser JR, Coon TA, Mallampalli RK. Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates Aurora B to inhibit tumorigenesis. Cell death & disease. 2013;4:e759. doi: 10.1038/cddis.2013.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Margolis RL. Tetraploidy and tumor development. Cancer cell. 2005;8:353–354. doi: 10.1016/j.ccr.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 105.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 106.Cimini D, Wan X, Hirel CB, Salmon ED. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Current biology : CB. 2006;16:1711–1718. doi: 10.1016/j.cub.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 107.Coon TA, Glasser JR, Mallampalli RK, Chen BB. Novel E3 ligase component FBXL7 ubiquitinates and degrades Aurora A, causing mitotic arrest. Cell cycle. 2012;11:721–729. doi: 10.4161/cc.11.4.19171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu Y, Lear T, Iannone O, Shiva S, Corey C, Rajbhandari S, Jerome J, Chen BB, Mallampalli RK. The Proapoptotic F-box Protein Fbxl7 Regulates Mitochondrial Function by Mediating the Ubiquitylation and Proteasomal Degradation of Survivin. The Journal of biological chemistry. 2015;290:11843–11852. doi: 10.1074/jbc.M114.629931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Johansson P, Jeffery J, Al-Ejeh F, Schulz RB, Callen DF, Kumar R, Khanna KK. SCF-FBXO31 E3 ligase targets DNA replication factor Cdt1 for proteolysis in the G2 phase of cell cycle to prevent re-replication. The Journal of biological chemistry. 2014;289:18514–18525. doi: 10.1074/jbc.M114.559930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722–725. doi: 10.1038/nature08011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Molecular cell. 2006;24:355–366. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nature reviews. Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Malonia SK, Dutta P, Santra MK, Green MR. F-box protein FBXO31 directs degradation of MDM2 to facilitate p53-mediated growth arrest following genotoxic stress. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8632–8637. doi: 10.1073/pnas.1510929112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huang HL, Jiang Y, Wang YH, Chen T, He HJ, Liu T, Yang T, Yang LW, Chen J, Song ZQ, Yao W, Wu B, Liu G. FBXO31 promotes cell proliferation, metastasis and invasion in lung cancer. American journal of cancer research. 2015;5:1814–1822. [PMC free article] [PubMed] [Google Scholar]
- 115.Liu J, Han L, Li B, Yang J, Huen MS, Pan X, Tsao SW, Cheung AL. F-box only protein 31 (FBXO31) negatively regulates p38 mitogen-activated protein kinase (MAPK) signaling by mediating lysine 48-linked ubiquitination and degradation of mitogen-activated protein kinase kinase 6 (MKK6) The Journal of biological chemistry. 2014;289:21508–21518. doi: 10.1074/jbc.M114.560342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fung TK, Siu WY, Yam CH, Lau A, Poon RY. Cyclin F is degraded during G2- M by mechanisms fundamentally different from other cyclins. The Journal of biological chemistry. 2002;277:35140–35149. doi: 10.1074/jbc.M205503200. [DOI] [PubMed] [Google Scholar]
- 117.Nordlund P, Reichard P. Ribonucleotide reductases. Annual review of biochemistry. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 118.Fu J, Qiu H, Cai M, Pan Y, Cao Y, Liu L, Yun J, Zhang CZ. Low cyclin F expression in hepatocellular carcinoma associates with poor differentiation and unfavorable prognosis. Cancer science. 2013;104:508–515. doi: 10.1111/cas.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ci W, Polo JM, Melnick A. B-cell lymphoma 6 and the molecular pathogenesis of diffuse large B-cell lymphoma. Current opinion in hematology. 2008;15:381–390. doi: 10.1097/MOH.0b013e328302c7df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Advances in immunology. 2005;87:163–208. doi: 10.1016/S0065-2776(05)87005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, Chapuy B, Shipp M, Chiarle R, Pagano M. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481:90–93. doi: 10.1038/nature10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Higa LA, Banks D, Wu M, Kobayashi R, Sun H, Zhang H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell cycle. 2006;5:1675–1680. doi: 10.4161/cc.5.15.3149. [DOI] [PubMed] [Google Scholar]
- 123.Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Molecular cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Molecular cell. 2010;40:22–33. doi: 10.1016/j.molcel.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Molecular cell. 2010;40:364–376. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes & development. 2011;25:1568–1582. doi: 10.1101/gad.2068611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jorgensen S, Eskildsen M, Fugger K, Hansen L, Larsen MS, Kousholt AN, Syljuasen RG, Trelle MB, Jensen ON, Helin K, Sorensen CS. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. The Journal of cell biology. 2011;192:43–54. doi: 10.1083/jcb.201009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hall JR, Bereman MS, Nepomuceno AI, Thompson EA, Muddiman DC, Smart RC. C/EBPalpha regulates CRL4(Cdt2)-mediated degradation of p21 in response to UVB-induced DNA damage to control the G1/S checkpoint. Cell cycle. 2014;13:3602–3610. doi: 10.4161/15384101.2014.962957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, Hsu HC. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell cycle. 2006;5:2676–2687. doi: 10.4161/cc.5.22.3500. [DOI] [PubMed] [Google Scholar]
- 130.Ueki T, Nishidate T, Park JH, Lin ML, Shimo A, Hirata K, Nakamura Y, Katagiri T. Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA- regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene. 2008;27:5672–5683. doi: 10.1038/onc.2008.186. [DOI] [PubMed] [Google Scholar]
- 131.Yang CH, Pfeffer SR, Sims M, Yue J, Wang Y, Linga VG, Paulus E, Davidoff AM, Pfeffer LM. The Oncogenic MicroRNA-21 Inhibits the Tumor Suppressive Activity of FBXO11 to Promote Tumorigenesis. Journal of Biological Chemistry. 2015;290:6037–6046. doi: 10.1074/jbc.M114.632125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xue J, Chi Y, Chen Y, Huang S, Ye X, Niu J, Wang W, Pfeffer LM, Shao Zm, Wu ZH, Wu J. MiRNA-621 sensitizes breast cancer to chemotherapy by suppressing FBXO11 and enhancing p53 activity. Oncogene. 2015 doi: 10.1038/onc.2015.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: A DOC domain-containing cullin selectively binds Skp1κFbx29 to form an SCF-like complex. Proceedings of the National Academy of Sciences. 2002;99:16601–16606. doi: 10.1073/pnas.252646399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsunematsu R, Nishiyama M, Kotoshiba S, Saiga T, Kamura T, Nakayama KI. Fbxw8 Is Essential for Cul1-Cul7 Complex Formation and for Placental Development. Molecular and cellular biology. 2006;26:6157–6169. doi: 10.1128/MCB.00595-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A Critical Role for FBXW8 and MAPK in Cyclin D1 Degradation and Cancer Cell Proliferation. PloS one. 2006;1:e128. doi: 10.1371/journal.pone.0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kong C, Samovski D, Srikanth P, Wainszelbaum MJ, Charron AJ, Liu J, Lange JJ, Chen PI, Pan ZQ, Su X, Stahl PD. Ubiquitination and Degradation of the Hominoid-Specific Oncoprotein TBC1D3 Is Mediated by CUL7 E3 Ligase. PloS one. 2012;7:e46485. doi: 10.1371/journal.pone.0046485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang H, Chen Y, Lin P, Li L, Zhou G, Liu G, Logsdon C, Jin J, Abbruzzese JL, Tan TH, Wang H. The CUL7/F-box and WD Repeat Domain Containing 8 (CUL7/Fbxw8) Ubiquitin Ligase Promotes Degradation of Hematopoietic Progenitor Kinase 1. Journal of Biological Chemistry. 2014;289:4009–4017. doi: 10.1074/jbc.M113.520106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Takai H, Wang RC, Takai KK, Yang H, de Lange T. Tel2 regulates the stability of PI3K-related protein kinases. Cell. 2007;131:1248–1259. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 139.Horejsi Z, Takai H, Adelman CA, Collis SJ, Flynn H, Maslen S, Skehel JM, de Lange T, Boulton SJ. CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Molecular cell. 2010;39:839–850. doi: 10.1016/j.molcel.2010.08.037. [DOI] [PubMed] [Google Scholar]
- 140.Takai H, Xie Y, de Lange T, Pavletich NP. Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes & development. 2010;24:2019–2030. doi: 10.1101/gad.1956410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K, Iemura S, Natsume T, Mizushima N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. The Journal of biological chemistry. 2010;285:20109–20116. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fernandez-Saiz V, Targosz BS, Lemeer S, Eichner R, Langer C, Bullinger L, Reiter C, Slotta-Huspenina J, Schroeder S, Knorn AM, Kurutz J, Peschel C, Pagano M, Kuster B, Bassermann F. SCFFbxo9 and CK2 direct the cellular response to growth factor withdrawal via Tel2/Tti1 degradation and promote survival in multiple myeloma. Nature cell biology. 2013;15:72–81. doi: 10.1038/ncb2651. [DOI] [PubMed] [Google Scholar]
- 143.Kim TY, Jackson S, Xiong Y, Whitsett TG, Lobello JR, Weiss GJ, Tran NL, Bang YJ, Der CJ. CRL4A-FBXW5-mediated degradation of DLC1 Rho GTPase-activating protein tumor suppressor promotes non-small cell lung cancer cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16868–16873. doi: 10.1073/pnas.1306358110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hsu JY, Reimann JDR, Sorensen CS, Lukas J, Jackson PK. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APCCdh1. Nature cell biology. 2002;4:358–366. doi: 10.1038/ncb785. [DOI] [PubMed] [Google Scholar]
- 145.Gutgemann I, Lehman NL, Jackson PK, Longacre TA. Emi1 protein accumulation implicates misregulation of the anaphase promoting complex/cyclosome pathway in ovarian clear cell carcinoma. Mod Pathol. 2008;21:445–454. doi: 10.1038/modpathol.3801022. [DOI] [PubMed] [Google Scholar]
- 146.Huang HC, Lin CL, Lin JK. 1,2,3,4,6-penta-O-galloyl-beta-D-glucose, quercetin, curcumin and lycopene induce cell-cycle arrest in MDA-MB-231 and BT474 cells through downregulation of Skp2 protein. Journal of agricultural and food chemistry. 2011;59:6765–6775. doi: 10.1021/jf201096v. [DOI] [PubMed] [Google Scholar]
- 147.Huang HC, Way TD, Lin CL, Lin JK. EGCG stabilizes p27kip1 in E2-stimulated MCF-7 cells through down-regulation of the Skp2 protein. Endocrinology. 2008;149:5972–5983. doi: 10.1210/en.2008-0408. [DOI] [PubMed] [Google Scholar]
- 148.Yang ES, Burnstein KL. Vitamin D Inhibits G1 to S Progression in LNCaP Prostate Cancer Cells through p27Kip1 Stabilization and Cdk2 Mislocalization to the Cytoplasm. Journal of Biological Chemistry. 2003;278:46862–46868. doi: 10.1074/jbc.M306340200. [DOI] [PubMed] [Google Scholar]
- 149.Gao J, Azmi AS, Aboukameel A, Kauffman M, Shacham S, Abou-Samra AB, Mohammad RM. Nuclear retention of Fbw7 by specific inhibitors of nuclear export leads to Notch1 degradation in pancreatic cancer. Oncotarget. 2014;5:3444–3454. doi: 10.18632/oncotarget.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, Wen JZ, Zhou H, Qu LH. Triggering Fbw7-Mediated Proteasomal Degradation of c-Myc by Oridonin Induces Cell Growth Inhibition and Apoptosis. Molecular cancer therapeutics. 2012;11:1155–1165. doi: 10.1158/1535-7163.MCT-12-0066. [DOI] [PubMed] [Google Scholar]
- 151.Nangle S, Xing W, Zheng N. Crystal structure of mammalian cryptochrome in complex with a small molecule competitor of its ubiquitin ligase. Cell research. 2013;23:1417–1419. doi: 10.1038/cr.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, Pongsawakul PY, Sonntag T, Welsh DK, Brenner DA, Doyle FJ, Schultz PG, Kay SA. Identification of Small Molecule Activators of Cryptochrome. Science. 2012;337:1094–1097. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lagarou A, Mohd-Sarip A, Moshkin YM, Chalkley GE, Bezstarosti K, Demmers JA, Verrijzer CP. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes & development. 2008;22:2799–2810. doi: 10.1101/gad.484208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tzatsos A, Paskaleva P, Ferrari F, Deshpande V, Stoykova S, Contino G, Wong KK, Lan F, Trojer P, Park PJ, Bardeesy N. KDM2B promotes pancreatic cancer via Polycomb- dependent and -independent transcriptional programs. The Journal of clinical investigation. 2013;123:727–739. doi: 10.1172/JCI64535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xiao J, Zhang T, Xu D, Wang H, Cai Y, Jin T, Liu M, Jin M, Wu K, Yuan J. FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes & development. 2015;29:184–196. doi: 10.1101/gad.252528.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Takagi H, Setou M, Ito S, Yao I. SCRAPPER Regulates the Thresholds of Long-Term Potentiation/Depression, the Bidirectional Synaptic Plasticity in Hippocampal CA3-CA1 Synapses. Neural Plasticity. 2012;2012:7. doi: 10.1155/2012/352829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dobie F, Craig AM. A Fight for Neurotransmission: SCRAPPER Trashes RIM. Cell. 2007;130:775–777. doi: 10.1016/j.cell.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, MacGregor GR, Tanaka K, Setou M. SCRAPPER- Dependent Ubiquitination of Active Zone Protein RIM1 Regulates Synaptic Vesicle Release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhu Li, Dong, Chen Role of FBXL20 in human colorectal adenocarcinoma. Oncology Reports. 2012;28:2290–2298. doi: 10.3892/or.2012.2065. [DOI] [PubMed] [Google Scholar]
- 160.Zhu J, Deng S, Duan JIE, Xie X, Xu S, Ran M, Dai X, Pu YU, Zhang X. FBXL20 acts as an invasion inducer and mediates E-cadherin in colorectal adenocarcinoma. Oncology Letters. 2014;7:2185–2191. doi: 10.3892/ol.2014.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Huber C, Dias-Santagata D, Glaser A, O’Sullivan J, Brauner R, Wu K, Xu X, Pearce K, Wang R, Uzielli MLG, Dagoneau N, Chemaitilly W, Superti-Furga A, Santos HD, Megarbane A, Morin G, Gillessen-Kaesbach G, Hennekam R, Burgt IVd, Black GCM, Clayton PE, Read A, Merrer ML, Scambler PJ, Munnich A, Pan ZQ, Winter R, Cormier-Daire V. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. 2005;37:1119–1124. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- 162.Ponyeam W, Hagen T. Characterization of the Cullin7 E3 ubiquitin ligase — Heterodimerization of cullin substrate receptors as a novel mechanism to regulate cullin E3 ligase activity. Cellular Signalling. 2012;24:290–295. doi: 10.1016/j.cellsig.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 163.Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PloS one. 2006;1:e128. doi: 10.1371/journal.pone.0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, Zhu W, Nakajima H, Nakajima HO, Field LJ, Wang R, Pan ZQ. The CUL7 E3 Ubiquitin Ligase Targets Insulin Receptor Substrate 1 for Ubiquitin-Dependent Degradation. Molecular cell. 2008;30:403–414. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Litterman N, Ikeuchi Y, Gallardo G, O’Connell BC, Sowa ME, Gygi SP, Harper JW, Bonni A. An OBSL1-Cul7<sup>Fbxw8</sup> Ubiquitin Ligase Signaling Mechanism Regulates Golgi Morphology and Dendrite Patterning. PLoS Biol. 2011;9:e1001060. doi: 10.1371/journal.pbio.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Barbash O, Zamfirova P, Lin DI, Chen X, Yang K, Nakagawa H, Lu F, Rustgi AK, Diehl JA. Mutations in Fbx4 Inhibit Dimerization of the SCFFbx4 Ligase and Contribute to Cyclin D1 Overexpression in Human Cancer. Cancer cell. 2008;14:68–78. doi: 10.1016/j.ccr.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lin DI, Barbash O, Kumar KGS, Weber JD, Harper JW, Klein-Szanto AP, Rustgi A, Fuchs S, Diehl JA. Phosphorylation-Dependent Ubiquitination of Cyclin D1 by the SCFFBX4-αB Crystallin Complex. Molecular cell. 2006;24:355–366. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyctin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 169.Lee TH, Perrem K, Harper JW, Lu KP, Zhou XZ. The F-box Protein FBX4 Targets PIN2/TRF1 for Ubiquitin-mediated Degradation and Regulates Telomere Maintenance. Journal of Biological Chemistry. 2006;281:759–768. doi: 10.1074/jbc.M509855200. [DOI] [PubMed] [Google Scholar]
- 170.Chen JY, Wang MC, Hung WC. Bcr-Abl-induced tyrosine phosphorylation of Emi1 to stabilize Skp2 protein via inhibition of ubiquitination in chronic myeloid leukemia cells. Journal of Cellular Physiology. 2011;226:407–413. doi: 10.1002/jcp.22346. [DOI] [PubMed] [Google Scholar]
- 171.Lehman NL, Tibshirani R, Hsu JY, Natkunam Y, Harris BT, West RB, Masek MA, Montgomery K, van de Rijn M, Jackson PK. Oncogenic Regulators and Substrates of the Anaphase Promoting Complex/Cyclosome Are Frequently Overexpressed in Malignant Tumors. The American journal of pathology. 2007;170:1793–1805. doi: 10.2353/ajpath.2007.060767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fernández-Sáiz V, Targosz BS, Lemeer S, Eichner R, Langer C, Bullinger L, Reiter C, Slotta-Huspenina J, Schroeder S, Knorn AM, Kurutz J, Peschel C, Pagano M, Kuster B, Bassermann F. SCFFbxo9 and CK2 direct the cellular response to growth factor withdrawal via Tel2/Tti1 degradation and promote survival in multiple myeloma. Nature cell biology. 2013;15:72–81. doi: 10.1038/ncb2651. [DOI] [PubMed] [Google Scholar]
- 173.Smits BMG, Traun BD, Devries TL, Tran A, Samuelson D, Haag JD, Gould M. An insulator loop resides between the synthetically interacting elements of the human/rat conserved breast cancer susceptibility locus MCS5A/Mcs5a. Nucleic Acids Research. 2012;40:132–147. doi: 10.1093/nar/gkr610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xu X, Powell DW, Lambring CJ, Puckett AH, Deschenes L, Prough RA, Poeschla EM, Samuelson DJ. Human MCS5A1 candidate breast cancer susceptibility gene FBXO10 is induced by cellular stress and correlated with lens epithelium-derived growth factor (LEDGF) Molecular Carcinogenesis. 2014;53:300–313. doi: 10.1002/mc.21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Samuelson DJ, Hesselson SE, Aperavich BA, Zan Y, Haag JD, Trentham-Dietz A, Hampton JM, Mau B, Chen KS, Baynes C, Khaw KT, Luben R, Perkins B, Shah M, Pharoah PD, Dunning AM, Easton DF, Ponder BA, Gould MN. Rat Mcs5a is a compound quantitative trait locus with orthologous human loci that associate with breast cancer risk. Proceedings of the National Academy of Sciences. 2007;104:6299–6304. doi: 10.1073/pnas.0701687104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco Mario A, Ren G, Zhou T, Storz P, Wang HY, Kang Y. PKD1 Phosphorylation-Dependent Degradation of SNAIL by SCF-FBXO11 Regulates Epithelial-Mesenchymal Transition and Metastasis. Cancer cell. 2014;26:358–373. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jeong YT, Cermak L, Guijarro M, Hernando E, Pagano M. FBH1 protects melanocytes from transformation and is deregulated in melanomas. Cell cycle. 2013;12:1128–1132. doi: 10.4161/cc.24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jeong YT, Rossi M, Cermak L, Saraf A, Florens L, Washburn MP, Sung P, Schildkraut CL, Pagano M. FBH1 promotes DNA double-strand breakage and apoptosis in response to DNA replication stress. The Journal of cell biology. 2013;200:141–149. doi: 10.1083/jcb.201209002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fugger K, Mistrik M, Danielsen JR, Dinant C, Falck J, Bartek J, Lukas J, Mailand N. Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. The Journal of cell biology. 2009;186:655–663. doi: 10.1083/jcb.200812138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Huang HL, Zheng WL, Zhao R, Zhang B, Ma WL. FBXO31 is down-regulated and may function as a tumor suppressor in hepatocellular carcinoma. Oncology Reports. 2010;24:715–720. doi: 10.3892/or_00000912. [DOI] [PubMed] [Google Scholar]
- 181.Kogo R, Mimori K, Tanaka F, Komune S, Mori M. FBXO31 determines poor prognosis in esophageal squamous cell carcinoma. International Journal of Oncology. 2011;39:155–159. doi: 10.3892/ijo.2011.1018. [DOI] [PubMed] [Google Scholar]