

Abstract
Cardiovascular complications are major side effects of many anticancer drugs. Accumulated evidence indicates that oxidative stress in mitochondria plays an important role in cardiac injury, but how mitochondrial redox mechanisms are involved in cardiac dysfunction remains unclear. Here, we demonstrate that 4-hydroxy-2-nonenal (HNE) activates the translocation of the mitochondrial apoptosis inducing factor (AIFm2) and facilitates apoptosis in heart tissue of mice and humans. Doxorubicin treatments significantly enhance cardiac levels of HNE and AIFm2. HNE adduction of AIFm2 inactivates the NADH oxidoreductase activity of AIFm2 and facilitates its translocation from mitochondria. His 174 on AIFm2 is the critical target of HNE adduction that triggers this functional switch. HNE adduction and translocation of AIFm2 from mitochondria upon Doxorubicin treatment are attenuated by superoxide dismutase mimetics. These results identify a previously unrecognized role of HNE with important consequences for mitochondrial stress signaling, heart failure, and the side effects of cancer therapy.
Graphical Abstract
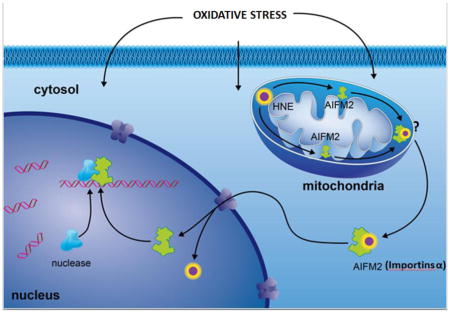
Introduction
Generation of reactive oxygen species (ROS) has been implicated in the toxicity of numerous cancer therapeutic drugs. It is well-documented that ROS including superoxide, hydrogen peroxide and nitric oxide are mediators of this toxicity, but the signaling role of ROS products remains obscure. ROS react with the polyunsaturated fatty acids of lipid membranes and induce lipid peroxidation. The end product of lipid peroxidation, α,β-unsaturated hydroxyalkenal, is considered to be a highly toxic product of ROS [1], leading to accretion of damaged/misfolded proteins [2], increased mutagenesis [3], inflammation [4, 5], and apoptosis.
Mitochondria not only power cells by producing ATP, they also are the major ROS producers and integrators of apoptosis mediators. Mitochondria engage in both caspase-dependent and caspase-independent apoptosis. One example of caspase-dependent apoptosis involves a well-known mitochondrial protein, cytochrome C (Cyt c). In healthy cells, Cyt c inhibits ROS formation, thus preventing apoptosis [6–9]. Under oxidative stress, Cyt c is released into the cytosol, initiating a cascade of caspase-dependent apoptosis. In the Cyt c/caspase-independent pathway, apoptosis inducing factor (AIF), a flavoprotein located within the mitochondrial membrane, participates in the apoptosis process [10]. In response to detrimental signals, AIF is released from the mitochondria into the nucleus and binds to nuclear DNA, thereby causing chromosomal condensation and large-scale DNA fragmentation [11, 12].
Several lines of evidence suggest that the AIF homologue, apoptosis inducing factor mitochondrion associated protein (AIFm2), may be a redox-responsive protein that resides in mitochondria and plays a central role in the caspase-independent cell death pathway [13–18]. AIFm2 is a p53 target gene. The expression of AIFm2 is relatively lower in tumor cells than in normal cells, suggesting a tumor suppressive effect of AIFm2 [19]. AIFm2 serves as an NADH-dependent oxidoreductase and is capable of non-sequence-specific DNA binding, resulting in DNA fragmentation, i.e., apoptosis, if the protein is translocated into the nucleus [15–18].
Our laboratory has recently shown that the absence of p53 significantly reduces cardiac injury in an animal model of anticancer therapy-induced cardiac toxicity. We showed that the potent anticancer drug doxorubicin (DOX) exerts less cardiac injury in p53 knockout mice compared to wild-type mice similarly treated, suggesting that p53 plays a critical role in mediating DOX-induced cardiac toxicity [20]. One of our prominent findings in that study was that the level of 4-hydroxy-2-nonenal (HNE) that was produced by lipid peroxidation was reduced in the cardiac mitochondria of p53-deficient mice, suggesting that mitochondrially localized, HNE-adducted proteins are likely to be involved in DOX-induced cardiac injury. Given that AIFm2 is a p53 target gene and a member of the AIF family, it is in a unique position to mediate the two-way communication between mitochondria and the nucleus under life and death conditions. The present study investigates the biochemical and molecular mechanisms underlying the role of AIFm2 in DOX-induced cardiac injury. The results identify a novel function of HNE in signaling of oxidative stress and a switch of AIFm2 functions in mitochondria-initiated apoptosis signaling.
Materials and Methods
Animals
Heterozygous mice (SOD2+/−) and wild-type (SOD2+/+) littermates were maintained in our laboratory. The SOD2+/− mice, designated Sod2 <tm1> Cje, were originally produced in the CD1 strain of mice; however, the mice described in this study were backcrossed to C57BL/6J mice for 14 generations. The genotype of the SOD2+/− mice was determined by PCR analysis as described [31]. Male mice between 8 and 12 weeks old were used in all studies. All procedures involving the mice were in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
Doxorubicin treatment and tissue collection
Mice were treated with a single dose of 20mg/kg of doxorubicin-adriamycin (DOXOrubicin HCl, from Bedford Laboratories, Inc., Bedford, OH) (DOX) or saline via intraperitoneal (IP) injection. Three days after treatment, mice were anesthetized using ketamine/xylazine (90–120 mg/kg and 10mg/kg, respectively,, IP). The heart was excised and immediately processed for ultrastructural studies and mitochondrial isolation or frozen in liquid nitrogen for molecular and biochemical studies.
SOD mimics
Three pentacationic Mn(III) N-substituted pyridylporphyrin-based of similarly high SOD-like potency were utilized in this study [21, 22] are highly potent SOD mimics [23] Such high ability to catalyze O2·− dismutation is based on their structure where cationic charges are located close to Mn site affording proper redox properties for the attraction of anionic superoxide and its dismutation. Importantly, that same high cationic charge drives the accumulation of these compounds in mitochondria. Somewhat lower effect in preventing AIFm2 nuclear translocation was observed in this study with MnTE-2-PyP5+ relative to MnTnHex-2-PyP5+ and MnTnBuOE-2-PyP5+. MnTE-2-PyP5+ is ~4 log units more hydrophilic than the other two analogs. Its only slightly lower efficacy points to the superior impact which cationic charges (relative to lipophilicity) have on its mitochondrial accumulation as demonstrated by mitochondria/cytosol ratios. While MnTE-2-PyP5+ accumulates in mouse heart mitochondria at 1.6 ratio relative to cytosol, that ratio is 3.6 and 3.0 for MnTnHex-2-PyP5+ and MnTnBuOE-2-PyP5+ [23].
Human heart atrial and ventricular tissue collection
Heart transplantation patients undergoing clinically indicated biopsy procedures as well as patients undergoing coronary artery bypass (CABG) surgery were enrolled in the study. No patients were excluded based on race or sex. All patients received standard care therapy without any interruptions during their enrollment in the study. All procedures were approved by the Institutional Review Board for human subjects at the University of Kentucky. Cardiothoracic surgeons routinely excised small pieces of tissue weighing a few milligrams during the right atrial appendage canulation and/or closure, and the tissue that otherwise would be discarded was used. Collected atrial and ventricular heart tissues were sliced into thin pieces and incubated in 1μM DOX for 3 days in a 37°C incubator and control tissues were similarly incubated; both were subjected to western blots and immunoprecipitation.
Mitochondria RT-PCR array
The heart tissues from SOD2+/+ and SOD2+/− mice were isolated. Total RNA was isolated from 10mg heart tissues using a MagNA Pure Compact RNA Isolation Kit (Roche). A reverse transcription reaction was performed using a RT2 First Strand Kit (SABiosciences Corp). The synthesized cDNA was further subjected to a Mouse Mitochondria RT-PCR Array (SABiosciences Corp.) using a LightCycler 480 Real-Time PCR System (Roche) according to the manufacturer’s protocol. The data were analyzed using a software packet provided by SABiosciences Corp.
Mitochondrial extract preparation
Heart mitochondria were isolated as described previously by Mela and Seitz [32]. Briefly, the hearts were collected, rinsed in ice-cold isolation buffer (0.225 M mannitol, 0.075 M sucrose, 1mM EGTA, pH 7.4), and cut into small pieces. The heart tissue was washed three times with the isolation buffer to remove any residual blood and was homogenized at 500 rpm with a chilled Teflon pestle in a glass cylinder with 10 strokes. The homogenate was centrifuged at 480×g at 4°C for 5 min in a Sorval SS 34 rotor. The resulting supernatant was filtered through a double-layered cheesecloth and was centrifuged at 7700×g at 4°C for 10 min. Supernatant was saved to check for leakage from mitochondria using MnSOD, a mitochondrial matrix enzyme, as an indicator by western blotting. The pellet was rinsed with 0.5 mL of the isolation buffer with gentle shaking to remove the “fluffy layer” (damaged mitochondria) on top of the pellet. The wall of the centrifuge tube was cleaned with cotton swabs to remove lipids. The pellet was washed by gentle resuspension in 3mL isolation buffer using the smooth surface of a glass rod and centrifuged at 7700×g at 4°C for 10 min. The supernatant was saved to check again for leakage from the mitochondria. The washing was repeated once. The resulting mitochondria were collected for further analysis. The purity of mitochondria was examined using LaminB (a nuclear protein) and IκB-α (a cytoskeletal protein) as indicators by western blotting. Protein content in the lysate was determined by BCA protein assay (Pierce, Rockford, IL).
Double Immunogold labeling of AIFm2 and 4HNE
Heart tissues from the left ventricle were fixed, embedded, and processed for immunogold electron microscopy as described previously in detail [33]. Two embedded blocks from each heart for each mouse were sectioned and transferred to nickel grids. Only longitudinal sections of cardiac muscle were used for the study. Grids were rinsed with TBS, blocked with bovine serum albumin-C, and then washed with TBS. Rabbit anti-AIFm2 antibody (dilution at 1:50, obtained from Santa-Cruz) was pre-incubated with 15nm gold conjugated anti-rabbit antibody, whereas rabbit anti-4HNE modified proteins antibody (dilution at 1:40, obtained from Dr. Luke Szweda, Case Western University, Cleveland, OH) was pre-incubated with 10 nm gold conjugated anti-rabbit at room temperature for 3 hr. The grids were then incubated with these pre-incubated primary antibodies at 4°C overnight in a humidified chamber. Grids were rinsed in TBS, counterstained with uranyl acetate, observed, and photographed with an electron microscope (Hitachi H-600) operated at 75 kV. For relative quantification of the immunoreactive protein of interest, 4HNE modified proteins and AIFm2, grids from experimental group versus a control group were stained simultaneously under the same conditions. Random sampling was achieved by scanning the grid at low magnification so that immunogold beads could not be seen, yet gross sample artifacts (folds in tissues, dust particles, etc.) could be avoided. Grids were scanned systematically from top to bottom and from left to right, and then photographs of entire cardiomyocyte cells were taken at ×12,300 magnifications for every 10–15 grid fields. Photographs of 30 cardiomyocyte cells were taken from each mouse group. The areas of each compartment (mitochondria, mitochondrial membranes, cytoplasm, and nucleus) were outlined and measured by Image J analysis software. Gold beads within specific subcellular compartments were then counted manually, 15 nm and 10 nm gold beads (at least 4 total gold beads) within a 25nm radius of each other were counted as a colocalization of AIFm2 and 4HNE modified proteins.
Immunoprecipitation and immunoblotting
Heart tissues were homogenized with lysis buffer (10 mM Tris–HCl pH 7.2, 1% Nonidet P-40, 158 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate) and lysates centrifuged at 16,000×g for 10 min. Immunoblotting (40 μg protein/well) was performed according to the instructions accompanying the Odyssey infrared imaging system (LI-COR, Lincoln, NE) with antibodies against AIFm2, γH2Ax, cardiolipin and importins, with actin and Lamin B as the loading control (Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies were conjugated with Alexa680 (Molecular Probes, Carlsbad, CA) or IRdye800 (Rockland Immunochemicals, Gilbertsville, PA), and were detected and quantified using the Odyssey infrared imaging system (LI-COR, Lincoln, NE).
For immunoprecipitation, 250 μg of protein were taken, and antibodies (2.5 μg/mL) were incubated for 2 h at 4°C with protein G-conjugated agarose beads (30 μL) and then washed five times with lysis buffer. To immunoprecipitate endogenous HNE, immunoprecipitates were subjected to SDS-PAGE (Santa Cruz Biotechnology, Santa Cruz, CA) and detected by immunoblotting. Inputs control were run for the immunoprecipitaion reactions.
Expression constructs
cDNA containing the complete human AIFm2 sequence was obtained from a publicly accessible cDNA collection. The Gateway cloning system (Invitrogen, Inc.) was used to generate plasmid and viral expression vectors for transient expression in cardiomyocytes according to the manufacturer’s instructions.
Lentiviral expression constructs
pLenti-AIFm2-GFPc was then constructed based on AIFm2 wild-type and mutant cDNA plasmids. The inserted sequence was PCR amplified by Pfu Turbo DNA polymerase using forward primer 5′-ACG CGT CGA CAA GAT GGG GTC CCA GGT CTC GGT -3′ and reverse primer 5′-GCT CTA GAC AGC AGC AGA GCC GGG GAC AAA GC -3′, incorporating SalI and XbaI restriction sites (underlined), respectively. The product was then cloned into SalI- and XbaI-digested pLenti-III-GFP-C to yield a C-terminal green fluorescent protein-tagged expression clone of AIFm2. Clones were found to be free of random mutations following automated DNA sequencing.
Site directed mutagenesis
A murine AIFm2 clone containing full-length AIFm2 cDNA was purchased from Open Biosystems (clone ID 1395873) for use as the template for protein synthesis. Amino acid substitution was performed by constructing H174R and C187T mutant clones using Stratagene’s QuikChange™ site-directed mutagenesis kit. The AIFm2 cDNA template was PCR-amplified using Pfu Turbo DNA polymerase and site specific mutagenesis primer. The PCR contained 50 ng of cDNA clone, 125 ng of each primer, 5 μL of 10X reaction buffer, 0.25 mM dNTPs, and 3 units of Pfu Turbo DNA polymerase. Cycling parameters were 16–18 cycles at 95°C for 50 s, 58°C for 1 min, and 68°C for 1 min per kb of plasmid length. The resultant fragment was then digested by DpnI restriction enzyme (10 U/μL) at 37°C for 1 h.
1D gel electrophoresis
To identify the HNE-modified amino acid, purified AIFm2 protein was exposed to HNE and subjected to tandem mass spectrometric analysis (MS/MS). The native as well as the HNE-adducted protein mixtures were subjected to SDS-PAGE electrophoresis and stained with Coomassie blue.
In-gel trypsin digestion
Briefly, protein bands identified as significantly altered in the native and the modified proteins were excised from 1D-gels with a clean, sterilized blade and transferred to Eppendorf microcentrifuge tubes. Gel plugs were then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at room temperature (RT) for 15 min, followed by incubation with 100% acetonitrile at RT for 15 min. After solvent removal, gel plugs were dried in their respective tubes under a flow hood at RT. Plugs were incubated for 45 min in 20 μL of 20 mM DTT in 0.1 M NH4HCO3 at 56°C. The DTT/NH4HCO3 solution was then removed and replaced with 20 μL of 55 mM IA in 0.1 M NH4HCO3 and incubated with gentle agitation at RT in the dark for 30 min. Excess IA solution was removed and plugs were incubated for 15 min with 200 μL of 50 mM NH4HCO3 at RT. A volume of 200 μL of 100% acetonitrile was added to this solution and incubated for 15 min at RT. Solvent was removed and gel plugs were allowed to dry for 30 min at RT under a flow hood. Plugs were rehydrated with 20 ng/μL of modified trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 in a shaking incubator overnight at 37°C. Enough trypsin solution was added in order to completely submerge the gel plugs.
Mass spectrometry
Salts and contaminants were removed from tryptic peptide solutions using C18 ZipTips (Sigma-Aldrich, St. Louis, MO), reconstituted to a volume of ~15 μL in a 50:50 water:acetonitrile solution containing 0.1% formic acid. Tryptic peptides were analyzed with automated Nanomate electrospray ionization (ESI) (Advion Biosciences, Ithaca, NY) using an Orbitrap XL MS (ThermoScientific, Waltham, MA) platform. The Orbitrap MS was operated in a data-dependent mode whereby the eight most intense parent ions appearing in the Fourier transform (FT) at 60,000 resolution were selected for ion trap fragmentation with the following conditions: injection time 50 ms, 35% collision energy; FT displayed at 7500 resolution; and dynamic exclusion set for 120 s. Each sample was acquired for a total of ~2.5 min. MS/MS spectra were searched against the International Protein Index (IPI) database using SEQUEST and the following parameters: 2 trypsin miscleavages, fixed carbamidomethyl modification, variable methionine oxidation, parent tolerance 10 ppm, and fragment tolerance of 25 mmu or 0.01 Da. Results were filtered with the following criteria: Xcorr >1.5, >2.0,>2.5, and >3.0 for +1, +2, +3, and +4 charge states, respectively; delta CN >0.1; and P-value (protein and peptide) <0.01. IPI accession numbers were cross-correlated with SwissProt accession numbers for final protein identification. It should be noted that proteins identified with a single peptide were kept for further analyses if multiple spectral counts (i.e., more than one MS/MS spectrum) were observed in a single analysis or if the peptide was identified in a separate analysis and workup of the same protein spot.
Modeling analysis
A three-dimensional model of AIFm2 was generated using the Phyre2 server [34]. The NADH-dependent persulfide reductase (Npsr) from S. loihica (PDB=3NTA) [35] was utilized as the template structure. It has 17% sequence identity and 96% backbone coverage, resulting in a model with 100% fold probability confidence. Bound coenzymes were positioned using B. anthracis Coenzyme A-Disulfide Reductase (CoADR) (PDB=3CGD [36], as the template structure. Structural superimposition was performed using the DALI server [37]. Molecular graphics were prepared using MolMol [38].
NADH oxidoreductase activity
Oxidoreductase activity was assayed spectrophotometrically at 25°C as a decrease of absorption at 340 nm with 100 μM NADH and 30 mM FAD as the substrates in the presence of HNE-modified and unmodified forms of recombinant AIFm2 wild-type and mutant proteins. The assay buffer was 100 mM potassium phosphate pH8.0 containing 100 mM NaCl. Specific activity of the protein was calculated as the units of enzyme that can oxidize 1.0 μmole of β-NADH per minute at pH 8.0 and 25°C.
Statistical analysis
The percent changes from morphometric quantization immunogold, immunoreactive protein detection of HNE adducts, and all the enzymatic assays were analyzed using Tukey’s HSD test or the Holm’s procedure to control for multiple comparisons (GraphPad Prism-4). A P-value of less than 0.05 was considered a significant difference.
Results
AIFm2 is a mediator of oxidative stress signaling
Manganese superoxide dismutase (SOD2), a major antioxidant enzyme in mitochondria, is essential for the survival of all aerobic organisms as well as for defending against superoxide radicals in high energy demanding tissues. To characterize the mitochondria of mice heterozygous for superoxide dismutase (SOD2+/−), we used the mouse mitochondria PCR Array to generate (i) a cluster heat map showing the expression of 84 genes involved in the biogenesis and function of mitochondria, and (ii) quantification showing AIFm2 as a gene that increased in the heart tissue of SOD2+/− mice (Supplementary Fig. 1). This result suggests that AIFm2 is highly sensitive to oxidative stress-inducing conditions in mitochondria.
AIFm2 is in a unique position to mediate the two-way communication between mitochondria and the nucleus under oxidative stress conditions, but how it potentially mediates signals in response to mitochondrial stress is unknown. As the first step in testing the potential for AIFm2 to be a direct mediator of ROS-mediated signaling between mitochondria and the nucleus during cancer therapy, we determined the mRNA and protein levels of AIFm2 in cardiac tissue after treatment with DOX, which is known to generate ROS (with superoxide radical as the initiating reactive species) in mitochondria and cause cardiac injury [24]. Consistent with the results from PCR array analyses, we found that the level of AIFm2 is greater in cardiac tissues of saline treated SOD2+/− mice compared to SOD2+/+ mice. We also found that mRNA and protein levels of AIFm2 are increased in cardiac tissues after DOX treatment in both genotypes (Fig. 1AI, & ii).
Figure 1.

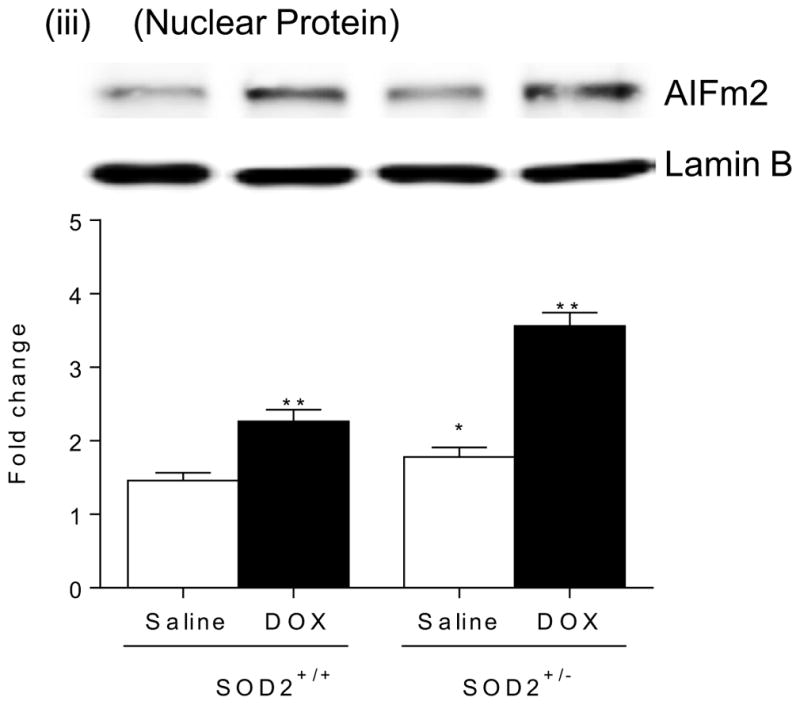
Figure 1A. Identification of AIFm2 as a mitochondrial resident target of oxidative stress mediated signaling.
(i) RNA levels of AIFm2 were assessed by quantitative reverse transcription–polymerase chain reaction (RT-PCR) analysis in heart tissues of age-matched SOD2+/+ and SOD2+/− mice with and without DOX treatment. The cardiac AIFm2 mRNA expression levels were normalized by an 18S ribosomal RNA. All results were graphed from 3 experiments and are presented as mean ± SEM. The expression level of WT mice was set to 1.*P<0.01;**P<0.001 compared to SOD2+/+ saline treated. (ii) Protein levels of AIFm2 were assessed by immunoblot analysis after normalization to β-tubulin expression (n=5 per group). Results are presented as mean ± SEM. *P<0.05;**P<0.001 compared to saline treated SOD2+/+ mice.
Figure 1B. Immunoblot analysis of AIFm2 levels in mitochondria (i), cytosol(ii), and nucleus(iii) of cardiac tissues (n=5 per group). Results are presented as mean ± SEM. *P<0.05;**P<0.001 compared to saline treated SOD2+/+ mice.
To determine the localization of AIFm2 in mitochondria, we performed subcellular organelle isolation. The results indicate that the mitochondrial protein level of AIFm2 in DOX-treated mice is significantly decreased with a concomitant increase of AIFm2 protein levels in the cytosolic and nuclear fractions (Fig. 1B, i, ii, iii). These results are consistent with the possibility that AIFm2 migrates from mitochondria to the nuclear compartment of a cell.
Sequence homology identifies AIFm2 as a member of the AIF family of proteins. It possesses an oxidoreductase domain, which is present in the AIF protein. However, conflicting data [17] suggest that during apoptosis, AIFm2, unlike AIF, does not translocate to the nucleus. Despite its name, AIFm2 lacks a mitochondrial or nuclear localization sequence, and it has been suggested it is associated with the mitochondrial membrane [18]. Thus, the localization of AIFm2 and its role in apoptosis remain ambiguous. To address questions concerning the localization of AIFm2, we used two complementary techniques to localize and quantify it. First, we used the antibody against AIFm2 coupled with electron microscopy procedures developed in the Oberley laboratory [25]. Representative immunogold electron micrographs (Fig. 2i) from left ventricular tissues of wild-type mice demonstrate labeling of AIFm2 in mitochondria (M) but not in myofilaments (Myo) in cardiomyocytes. Electron-dense beads indicate positive staining for AIFm2 protein (arrow). Second, to further confirm that AIFm2 is resident in mitochondria, we used immunofluorescence to determine the cellular locations of AIFm2 by transfecting H9C2 cells with mammalian expression vectors encoding AIFm2-GFP fusion proteins. As shown in Fig. 2ii, AIFm2 was co-localized with MitoTracker, a marker for mitochondria. These results clearly establish the localization of AIFm2 to mitochondria but do not explain why AIFm2 was found in the cytosol and nucleus after exposure to DOX.
Figure 2. Localization of the AIFm2 protein.

(i) Representative immunogold electron micrographs showing the localization of AIFm2 in cardiomyocytes. Electron dense beads indicate positive staining for AIFm2 protein (arrows). Left ventricular tissues from saline treated SOD2+/+ mice demonstrate strong labeling of AIFm2 in mitochondria (M) and low labelingbut not in myofilaments (Myo). (ii) H9C2 cells were transfected with GFP-tagged AIFm2-expressing lentivirus, and ~72 h later were co-stained with MitoTracker to identify the mitochondria and DAPI to show the nucleus. Localization of AIFm2 was visualized by indirect immunofluoroscence microscopy.
HNE activates AIFm2 translocation after DOX treatment
Lipid peroxidation yields a variety of electrophilic, nonradical products [26, 27] including HNE. Our previous studies document that mitochondrial oxidative damage precedes nitrative damage in cardiac tissues following DOX treatment. We also found that p53 selectively enhances HNE levels in mitochondria of DOX-treated p53 mice and that lack of p53 is protective against DOX-induced cardiac injury [20]. Since AIFm2 is a p53 target gene, we probed whether AIFm2 is a target for HNE adduction. Tandem mass spectrometric analysis (MS/MS) of the native and HNE-modified AIFm2 (supplementary Fig. 2a and 2b) showed His174 and Cys187, both located in the oxidoreductase domain of AIFm2, to be the HNE modification sites.
Associations between AIFm2 and HNE were further demonstrated by immunoprecipitation assays. Cellular proteins pulled down by monoclonal HNE antibody were analyzed with western blot assays using anti-AIFm2 polyclonal antibody. Densitometric quantification of AIFm2 protein levels in the HNE-protein complex showed that, in both wild type and SOD2+/− mice, the significant level of HNE adduction increases upon DOX treatment (Fig. 3A). A low level of HNE-adducted AIFm2 is detectable in the saline-treated SOD2+/− tissue, indicating a low level of oxidative stress in SOD2-deficient mice. These results show that AIFm2 is oxidatively modified by HNE in vivo.
Figure 3. HNE adduction linked to translocation of AIFm2.


A. Total heart lysates from SOD2+/+ and SOD2+/− mice with and without DOX treatment were immunoprecipitated with HNE and immunoblotted for AlFm2. Results are presented as mean ± SEM. *P<0.05;**P<0.001 compared to saline treated SOD2+/+ mice.
B. Immunoprecipitation with HNE and immunblotting with AlFm2 in SOD2+/+ and SOD2+/− mice with and without DOX treatment in heart mitochondria(i), cytosol(ii), and nucleus(iii) (n=5 per group). Results are presented as mean ± SEM. *P<0.05;**P<0.001 compared to saline treated SOD2+/+ mice.
C. Immunoprecipitation with HNE and immunblotting with importin alpha, importin beta and AlFm2 in SOD2+/+ and SOD2+/− mice with and without DOX treatment in total heart cytosolic fraction (n=5 per group).
D. Expression of AIFm2 in human heart cell lysates from tissues treated with and without DOX was detected by western blotting. Immunoprecipitation and immunblotting showed that AlFm2 in human heart tissues treated with DOX were adducted with HNE.
To evaluate whether HNE modification of AIFm2 is linked to the translocation of AIFm2 from mitochondria, we performed subcellular organelle isolation and reciprocal immunoprecipitation–western blot analysis of antibodies to HNE-adducted proteins and AIFm2, respectively. The results show that HNE-adducted AIFm2 was nearly absent in mitochondria, but robust levels of HNE-adducted AIFm2 were observed both in cytosolic and nuclear extracts (Fig. 3BI, ii, iii). These results suggest that HNE adduction to AlFm2 is a prerequisite for AlFm2 to leave mitochondria.
To determine whether the HNE-adducted AIFm2 protein interacts with cytosolic transport proteins, we assayed for the presence of importins after immunoprecipitation with the antibody to HNE. The results indicate that AIFm2 and HNE co-immunoprecipiate with importins α2 and β3, which are present at only a low level in untreated heart tissue but increase after DOX treatment (Fig. 3C). These results suggest that the HNE-adducted AIFm2 is recognized by these transport proteins.
To determine whether this novel finding can occur in human cardiac tissues, we incubated human ventricular tissues with or without DOX and examined AIFm2 levels. A robust increase occurred in the expression of AIFm2 upon treatment with DOX, and the resulting proteins were adducted by HNE (Fig. 3D).
Because subcellular fractionation may cause structural injury to mitochondrial membranes, resulting in leakage of HNE-adducted protein into the cytosol, we further verified the translocation of HNE-adducted AIFm2 from mitochondria using immunogold labeling and quantitative immunohistochemistry at the electron microscopy level. Figure 4A depicts the distribution of AIFm2 in cardiac tissues three days after DOX treatment in SOD+/+ and SOD2+/− mice. Quantitative analysis of AIFm2 in various compartments demonstrated more HNE-adducted AIFm2 in the cytoplasm than in the mitochondria in DOX-treated SOD2+/+ and SOD2+/− mice, with the highest level in the cytoplasm of SOD2+/− mice (Fig. 4A).
Figure 4. Localization of AIFm2 in the HNE-adducted protein complex.





A. (i) Photographs present colocalization of AIFm2 and 4HNE-adduct immunogold labeling (30000× magnification) in SOD2+/− mice treated with saline (left) and DOX (right). M=Mitochondria, Myo=Myofibrils, arrows indicate 15 nm gold beads staining the AIFm2 protein while arrow heads indicate 10 nm gold beads staining the 4HNE-adduct protein.
(ii)The bar graphs shows the quantification of immunogold labeling in left ventricular tissue of SOD2+/+ and SOD2+/− mice with and without DOX treatment, expressed as a percentage of colocalization. *p<0.01; #p<0.001 compared to respective mitochondria within the same genotype.
B. Immunofluorescence images of H9C2 cardiomyocytes transfected with lentiviral wild-type or mutant (H174 and C187) AIFm2 protein linked to GFP (green column). MitoTracker was used to stain mitochondria (red column), and DAPI to stain the nuclei (blue column) to track the translocation of the AlFm2 proteins.
C. Immunoblots of mitochondrial (i), cytosolic (ii), and nuclear fractions (iii) of H9C2 cardiomyocytes transfected with lentiviral wild-type and mutant AlFm2 with and without DOX treatment.
D. Immunoprecipitation with HNE and immunoblotting of AlFm2 from mitochondrial, cytosolic, and nuclear fractions of H9C2 cardiomyocytes transfected with lentiviral wild-type and mutant AlFm2. Cells were treated with SOD mimetics before DOX treatment.
E. Immunofluorescence images of H9C2 cardiomyocytes transfected with lentiviral wild-type AIFm2 or H174 and C187 mutants, treated with DOX and SOD mimetics. AIFm2 proteins were linked to GFP (green). MitoTracker was used to stain mitochondria (red), and DAPI to stain nuclei (blue).
His174 is essential for AIFm2 translocation
To further confirm that HNE adduction of AIFm2 is important for AlFm2 translocation, we used several additional complementary approaches. First, we expressed wild-type or mutant AIFm2 protein linked to GFP in H9C2 cardiomyocytes in confocal microscope studies. The results (Fig. 4B) clearly demonstrate that under normal conditions the wild-type as well as the mutant AIFm2 proteins are localized in mitochondria. Interestingly, under stress conditions including treatment with DOX, translocation from the mitochondria to the nucleus occurred in the wild-type AIFm2 as well as the C187T mutant, while the similarly treated H174R mutant failed to be expelled from mitochondria.
To further elucidate the effect of DOX on HNE adduction and AIFm2 translocation, H9C2 cells were transfected with wild-type and mutant protein, incubated with DOX, and subjected to subcellular fractionation followed by western analysis. Consistent with immunogold analysis and confocal studies, the wild-type and the C187T mutant, but not the H174R mutant, were expelled from mitochondria (Fig. 4c) and were found in the cytosol and the nucleus. These results suggest that H174 of AIFm2 is essential for oxidative stress-mediated AIFm2 expulsion from mitochondria.
Adduction and expulsion of AIFm2 are blocked by SOD mimetics
To verify if HNE adduction of H174 is required for AIFm2 expulsion, we used SOD mimetics that are known to be effective in mitochondria [28] to block HNE adduction to AIFm2. As shown in Figure 4Dii & iii, HNE adduction is absent when H174 is changed to arginine. To further validate the role of ROS in mediating expulsion of AIFm2 from the mitochondria and to probe the possibility of potential therapeutic intervention, we used a series of SOD mimetics, including MnTE-2-PyP, MnTnHex-2-PyP5+, and MnTnBu-2-PyP5+ to block AIFm2 expulsion after DOX treatment. Immunoblots, cell fractionation studies and confocal studies all show that SOD mimetics blocked the translocation of oxidatively modified protein from the mitochondria to the cytosol and nucleus after DOX treatment (Fig. 4D). Colocalization with confocal microscopy clearly showed that SOD mimetics inhibited the expulsion of wild-type and C187T AIFm2 from mitochondria after DOX treatment (Fig. 4E). Together, these results demonstrate that oxidative stress precipitated by HNE adduction of AIFm2 plays a novel role in directing protein traffic out of mitochondria and that H174 is the critical target for HNE adduction and AIFm2 expulsion.
HNE adduction inactivates the oxidoreductase activity of AIFm2
AIFm2 contains the typical amino acid sequence of the AIF family of proteins in its NADH oxidoreductase domain. To further understand the location of H174 and C187 and the potential effects if these residues were modified, a model of AIFm2 with bound coenzymes was constructed (Fig 5). H174 is located in the central coenzyme binding region of AIFm2, with its side chain in direct contact with adenosine ribose (Fig. 5A, red), which suggests that H174 may be part of a conserved adenosine binding pocket in AIFm2. Indeed, significant conservation of AIFm2 and CoADR residues located in the pocket was observed. For example, R176 of AIFm2 is located in a position similar to R181 of CoADR, which has the highest single residue NADH contact (62 Å2 buried surface area), stacking with the adenine moiety. This further suggests a mechanism by which HNE adduction of H174 could regulate the function of AIFm2. Because the side chain of H174 and the NADH are within direct van der Waals contact, modification of the side chain by HNE would be predicted to inhibit or disrupt NADH binding. Modification of H174 could also directly impact DNA binding by AIFm2, since it has been shown that the binding of nicotinamide coenzymes and DNA is mutually exclusive, suggesting at least partially overlapping binding sites [29]. In contrast, C187 is located on an exposed surface region of the protein, remote from all known functional regions (Fig. 5A, purple). Thus, HNE adduction of C187 is predicted to have minimal effects on the function of AIFm2.
Figure 5. HNE adduction inactivates the NADH oxidoreductase activity of AIFm2.

A. Molecular model of AIFm2 with bound NADH (green) and FAD (gold). H174 (red) is located in the central adenosine binding region, whereas C187 (purple) is solvent exposed on the periphery of the structure.
B. Oxidoreductase activity of the AlFm2 wild-type and mutant proteins, using an NADH catalytic activity assay. Oxidation of NADH can be detected via the decrease in absorption at 340 nm, and generation of FADH as an increase in absorption at 450 nm. The spectrophotometric analysis at left shows the loss in catalytic activity after the wild-type protein was modified by HNE. Quantitative results of the oxidoreductase assay of AlFm2 wild-type and mutant proteins are shown in the bar graph. All results are from 3 experiments and are represented as mean ± SEM.*P<0.01;***P<0.001 compared to the respective untreated group.
To validate the above prediction, we tested whether HNE adduction would alter the enzymatic function of wild-type and mutant AIFm2 proteins. As shown in Figure 5B, the NADH/FAD spectrum and the specific NADH oxidoreductase activity were significantly altered in wild-type AIFm2 and the C187T mutant after DOX treatment. Mutation of H174 alone drastically reduced the NADH oxidoreductase activity of the protein and there was no further reduction after DOX treatment.
DOX-mediated HNE adduction of AIFm2 triggers cell death
To test the significance of the observed effect of HNE adduction on AIFm2, we treated wild-type and SOD-deficient mice with DOX and examined the presence of DNA damage, manifested by an increase in γH2AX levels, which are indicative of DNA fragmentation. The results, shown in Figure 6A, demonstrate that treatment with DOX led to increased γH2AX levels in both wild-type and SOD-deficient mice and that a slightly higher level occurred in SOD2+/− mice. These results are consistent with the higher basal level of HNE-adducted AIFm2 protein in SOD2+/− mice. To determine the role of H174 in DNA damage, we expressed wild-type and AIFm2 mutants in H9C2 cardiomyocytes and determined their role in γH2AX accumulation. The results suggest that the wild-type and C187T mutant AIFm2 were capable of causing DNA damage after DOX treatment, but the H174R mutant was not (Fig. 6B). The role of HNE adduction was further investigated by Hoechst 33342 nucleic acid counterstaining that emits blue fluorescence when bound to dsDNA and is used to distinguish condensed pyknotic nuclei in cells undergoing cell death. The results clearly show that when H9C2 cells were transfected with wild-type and C187T mutant protein and treated with DOX, there were significantly more truncated or pyknotic nuclei than appeared in the cells infected with saline-treated control and DOX-treated H174R mutant (Fig. 6C). Quantitative analysis of live and dead cells confirmed that cells expressing the histidine mutant are less susceptible to DOX-induced cytotoxicity (Fig. 6D).
Figure 6. HNE adduction of AIFm2 triggers cell death.

A. Western blot analysis of γH2AX as a marker for cell death in heart tissue nuclear extracts from SOD2+/+ and SOD2+/− mice treated with saline or DOX. Quantitative analysis represents the mean ± SEM, with n=5 in each group. *P<0.01; **P<0.001 compared to saline treated SOD2+/+ mice.
B. Western blot analysis of γH2AX as a marker for cell death in nuclear extracts from H9C2 cardiomyocytes transfected with wild-type AlFm2 and mutants treated with or without DOX.
C. H9C2 cells were transfected with lentiviral vectors for expression of wild-type and mutant AlFm2, and examined by fluorescence microscopy.
D. Truncated or pyknotic nuclei-containing cells were identified by visualization of DAPI fluorescence (blue) and counted. The data shown are mean ± SEM numbers of cells/field for 3–5 fields of cells from the same slide.
Discussion
The present study reveals a novel role of oxidative protein modification in cell signaling. We demonstrate that HNE adduction of AIFm2 shifts the function of AIFm2 from an NADH oxidoreductase to a proapototic protein. Our results identify HNE adduction of AIFm2 as a critical step of mitochondrial stress signaling. HNE is an important product of oxidative stress in cardiac mitochondria. HNE adduction resulted in the translocation of AIFm2 from mitochondria to the cytosol. In the cytosol, HNE-adducted AIFm2 is recognized by importins that can deliver it to the nucleus, which leads to cell death.
Our previous studies demonstrated that the level of HNE-adducted protein is significantly reduced in the mitochondria of DOX- treated p53-deficient mice and that DOX causes significantly less cardiac injury in p53-deficient mice, suggesting that HNE adduction of p53 target gene products may contribute to the observed cardiotoxic effect of DOX. Here, we identify AIFm2 as the p53 target gene product modified by HNE adduction in mitochondria. This was supported by several complementary experimental approaches including: 1) the localization of endogenous AIFm2 protein and HNE by immunogold with electron microscopy, 2) the presence of AIFm2 in immunocomplex with HNE antibody, 3) the identification of HNE-adducted sites on the AIFm2 protein by MS analysis, and 4) the localization of ectopically expressed AIFm2 protein in cardiomyocytes.
We did in fact see a HNE bound to the appropriate peptide in AIFm2, so clearly the bond was stable to mass spectrometric analysis. However, this has to be viewed in context of reactivity, stability, and mass spectrometric identification. In terms of reactivity, the kinetics for Michael adducts formation by HNE is at least one order of magnitude faster than that of Schiff base formation. Moreover in stability, while both products are covalent bonds, the single bond of the Michael adduct has bond energy of about 100 kcal/mol and is therefore a strong bond. In contrast, the imine (Schiff base) double bond between C and N of Lys is not stable to pH and unless it is converted to a secondary amine by reduction could be reversible and is inherently less stable than the Michael adduct. In the mass spectrometer, it is conceivable that this less stable imine could fall apart and the Schiff base not detected. In contrast, the Michael adduct, being a strong bond, would be detectable. All these considerations strongly support the high probability that the HNE is covalently bound as a Michael adduct.
Recent studies have suggested that ROS may be a mechanism by which AIF, a homologue of AIFm2, causes apoptosis. It has been shown that upon apoptotic stimulation, AIF translocates into the nucleus and induces apoptosis by an unknown mechanism [11, 30]. Post-translational modification of AIF is critical for its cleavage by calpain [31]. The present study provides direct experimental evidence for a novel function of HNE-adducted protein. Although AIFm2 shares significant homology with AIF, it lacks the mitochondrial localization signal (MLS) and nuclear localization signal (NLS) found in the AIF protein. Using immunogold coupled to electron microscopy analysis, we have demonstrated that, under physiological conditions, unmodified AIFm2 is located in the mitochondria. However, upon exposure to oxidative stress conditions, the level of the protein increased and the protein translocated from mitochondria. This was initially observed at a low level in SOD-deficient mice. The extent of AIFm2 translocation from mitochondria clearly increased after DOX treatment, as demonstrated by immunogold coupled to EM and cell fractionations followed by immunoprecipitation studies. These findings were confirmed by the data from confocal microscope studies, in which exogenous AIFm2 expressed in H9C2 cells was found to colocalize with mitochondrial markers in untreated cells and to colocalize with nuclear markers in DOX- treated cells. Thus, AIFm2 is a mitochondrial protein that is expelled from mitochondria upon exposure to oxidative stress-inducing conditions.
In addition to HNE, numerous secondary products of ROS, including peroxynitrite, are produced under oxidative stress conditions. Our previous studies indicate that the levels of other secondary products of ROS, including nitrotyrosine-adducted proteins, also increased in cardiac tissues of DOX-treated mice. However, the relative increase of nitrotyrosine adducts is lower and occurs later than the HNE adducts observed in the same animals [32]. The formation of oxidized lipid has been widely observed in several human and animal models of cardiac injury [33] including ischemic and failing heart, supporting the hypothesis that the generation of lipid peroxidation products is an important instigator of cardiac injury. Our results suggest that HNE is likely to be a key player, but we cannot rule out the possibility that reactive electrophilic fatty acids may modulate additional proteins in the same manner. It is possible that oxidative modification of AIFm2 by HNE and the consequent effect on AIFm2 translocation represent a prototype of post-translational modification resulting in activation of a novel function.
It is generally thought that oxidative modifications lead to inactivation of protein function. HNE is known to be preferentially reactive toward histidine, cysteine, and lysine. Our data, which demonstrate that the NADH oxidoreductase activity of HNE-modified AIFm2 is reduced and that His174 and Cys187 are the sites of HNE adductions, are consistent with these properties. Intriguingly, HNE modification of AIFm2 is required for its translocation from mitochondria to trigger cell death. The data from the DNA damage analyses demonstrate that His174 is the site from which HNE adduction leads to activation of the apoptotic function of AIFm2. The finding that HNE adduction at His174 but not Cys187 triggers the switch between NADH oxidoreductase activity and apoptosis activity of AIFm2 provides support for the importance of the site of HNE adduction. The C187T mutant of AIFm2 does show decreased nuclear translocation and oxidoreductase activity. While modeling can predict the effect on NADH activity when histidine and cysteine are modified because the side chain of H174 and the NADH are within direct van der Waals contact, modification of the side chain by HNE would be predicted to inhibit or disrupt NADH binding. In contrast, C187 is located on an exposed surface region of the protein, remote from all known functional regions (Fig. 6A), the novel gain of function is not anticipated by structural prediction analyses. Thus, it would be interesting if a future study could directly compare the crystal structure of the native and HNE-adducted AIFm2 proteins.
Mitochondria are vital for many metabolic activities, contributing important constituent enzymes for diverse functions such as β-oxidation of fatty acids, the urea cycle, the citric acid cycle, and ATP synthesis. Mitochondria are also major sites of heme synthesis, iron metabolism, and integration of apoptosis and inflammatory mediators. A shift in mitochondrial redox status toward an oxidizing condition can activate mitochondrial stress signaling, a pathway of communication from mitochondria to the nucleus, leading to activation of adaptive response or cell death pathways [20]. Our data, which demonstrate that DNA fragmentation and apoptosis occurred in animal and human tissues treated with DOX, suggest that HNE adduction of AIFm2 in mitochondria signals apoptosis. Our study sheds new light on a mechanism by which ROS generated in mitochondria activate retrograde signaling, an area of emerging interest and importance for our understanding of metabolic stress and cell death. The results of this study are the first to link oxidative modification to protein translocation and may have important implications for common human diseases, including cancer, cardiovascular diseases and metabolic syndrome.
Supplementary Material
Supplemenatry Figure 1. Identification of a potential candidate for retrograde signaling in vivo. The cluster heat map shows the expression of 84 genes involved in the biogenesis and function of mouse mitochondria. The rows of the heat map represent genes and the columns represent samples. Each cell is colored based on the level of expression of that gene in that sample. The expression levels of each gene have been normalized to mean 0 and standard deviation 1. Red indicates a high level of expression relative to the mean value; green indicates a low level of expression; and white indicates no data available. Genes are ordered based on the results of a hierarchical cluster analysis. The bar graph shows the quantification of the AlFm2 RNA from microarray data in SOD2+/+ and SOD2 +/− heart tissues. Results are presented as mean ± SEM (n=4 per group).
Supplementary Figure 2a and 2b. Mass sprctrometric analysis of native and HNE adducted AIFm2.
(a) Tandem mass spectrometric analysis (MS/MS) of the native and oxidatively modified AIFm2 showing His174 to be the HNE modification site. (ii) & (iii) AlFm2 sequence (row 1, AlFm2) and the tryptic digested peptide sequence targeted by HNE showing the specific target amino acid residue (row 2, AIFm2 His).
(b) Tandem mass spectrometric analysis (MS/MS) of the native and oxidatively modified AIFm2 showing Cys187 to be the HNE modification site. (ii) & (iii) AlFm2 sequence (row 1, AlFm2) and the tryptic digested peptide sequence targeted by HNE showing the specific target amino acid residue (row 2, AIFm2 Cys).
Supplementary Figure 3a,b and c. Western blot of inputs control for the immunoprecipitation reactions shown in Fig. 3 and Fig 4.
Acknowledgments
This work is supported by NIH grants CA 139843, CA 049797, CA143428, and the Edward P. Evans Foundation. The authors thank Dr. Carol Beach of the Free Radical Biology Core (FRBC) at the University of Kentucky for her help with the mass spectrometry analysis. This work is dedicated to the late Dr. Terry Oberley, for his critical contribution to the ultrastructural immunogold analysis, and for his leadership in the filed of redox biology.
Footnotes
Author contributions
S.M., C.Y., D.K.S. and V.P. designed the study and drafted the manuscript; L.C. performed ultrastructral immunogold analysis; Y.X. prepared lentiviral constructs; T.N. performed animal injection and tissue harvested; A.T. and I.B.H. synthesized SOD mimics; A.A.L. and H.R. obtained and provided human discarded tissue; M.P. performed inmaging experiments; W.C. analysed array results; C.V.A. performed structural analysis. D.A.B., M.V. and J.M. designed experiments and discussed the results.
Competing financial interests
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stadtman ER. Protein oxidation in aging and age-related diseases. Annals of the New York Academy of Sciences. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- 2.Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. The EMBO journal. 2005;24:1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutation research. 2011;711:193–201. doi: 10.1016/j.mrfmmm.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 4.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell research. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 6.Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nature reviews. Molecular cell biology. 2008;9:532–542. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 7.Gnaiger E. Oxygen conformance of cellular respiration. A perspective of mitochondrial physiology. Advances in experimental medicine and biology. 2003;543:39–55. doi: 10.1007/978-1-4419-8997-0_4. [DOI] [PubMed] [Google Scholar]
- 8.Lenaz G, Genova ML. Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxidants & redox signaling. 2010;12:961–1008. doi: 10.1089/ars.2009.2704. [DOI] [PubMed] [Google Scholar]
- 9.Santucci R, Sinibaldi F, Patriarca A, Santucci D, Fiorucci L. Misfolded proteins and neurodegeneration: role of non-native cytochrome c in cell death. Expert review of proteomics. 2010;7:507–517. doi: 10.1586/epr.10.50. [DOI] [PubMed] [Google Scholar]
- 10.Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? Journal of cell science. 2002;115:4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 11.Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prevost MC, Leber B, Andrews D, Penninger J, Kroemer G. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2000;14:729–739. [PubMed] [Google Scholar]
- 12.Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G, Wu H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nature structural biology. 2002;9:680–684. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- 13.Bilyy R, Kit Y, Hellman U, Stoika R. AMID: new insights on its intracellular localization and expression at apoptosis. Apoptosis: an international journal on programmed cell death. 2008;13:729–732. doi: 10.1007/s10495-008-0198-5. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Sun L, Liang Q, Wang J, Mo W, Zhou B. Yeast AMID homologue Ndi1p displays respiration-restricted apoptotic activity and is involved in chronological aging. Molecular biology of the cell. 2006;17:1802–1811. doi: 10.1091/mbc.E05-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall KR, Gong M, Wodke L, Lamb JH, Jones DJ, Farmer PB, Scrutton NS, Munro AW. The human apoptosis-inducing protein AMID is an oxidoreductase with a modified flavin cofactor and DNA binding activity. The Journal of biological chemistry. 2005;280:30735–30740. doi: 10.1074/jbc.M414018200. [DOI] [PubMed] [Google Scholar]
- 16.Ohiro Y, Garkavtsev I, Kobayashi S, Sreekumar KR, Nantz R, Higashikubo BT, Duffy SL, Higashikubo R, Usheva A, Gius D, Kley N, Horikoshi N. A novel p53-inducible apoptogenic gene, PRG3, encodes a homologue of the apoptosis-inducing factor (AIF) FEBS letters. 2002;524:163–171. doi: 10.1016/s0014-5793(02)03049-1. [DOI] [PubMed] [Google Scholar]
- 17.Varecha M, Amrichova J, Zimmermann M, Ulman V, Lukasova E, Kozubek M. Bioinformatic and image analyses of the cellular localization of the apoptotic proteins endonuclease G, AIF, and AMID during apoptosis in human cells. Apoptosis: an international journal on programmed cell death. 2007;12:1155–1171. doi: 10.1007/s10495-007-0061-0. [DOI] [PubMed] [Google Scholar]
- 18.Wu M, Xu LG, Li X, Zhai Z, Shu HB. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. The Journal of biological chemistry. 2002;277:25617–25623. doi: 10.1074/jbc.M202285200. [DOI] [PubMed] [Google Scholar]
- 19.Wu M, Xu LG, Su T, Tian Y, Zhai Z, Shu HB. AMID is a p53-inducible gene downregulated in tumors. Oncogene. 2004;23:6815–6819. doi: 10.1038/sj.onc.1207909. [DOI] [PubMed] [Google Scholar]
- 20.Velez JM, Miriyala S, Nithipongvanitch R, Noel T, Plabplueng CD, Oberley T, Jungsuwadee P, Van Remmen H, Vore M, St Clair DK. p53 Regulates oxidative stress-mediated retrograde signaling: a novel mechanism for chemotherapy-induced cardiac injury. PloS one. 2011;6:e18005. doi: 10.1371/journal.pone.0018005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batinic-Haberle I. Manganese porphyrins and related compounds as mimics of superoxide dismutase. Methods in enzymology. 2002;349:223–233. doi: 10.1016/s0076-6879(02)49337-8. [DOI] [PubMed] [Google Scholar]
- 22.Rajic Z, Tovmasyan A, Spasojevic I, Sheng H, Lu M, Li AM, Gralla EB, Warner DS, Benov L, Batinic-Haberle I. A new SOD mimic, Mn(III) ortho N-butoxyethylpyridylporphyrin, combines superb potency and lipophilicity with low toxicity. Free Radic Biol Med. 2012;52:1828–1834. doi: 10.1016/j.freeradbiomed.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Batinic-Haberle I, Tovmasyan A, Roberts ER, Vujaskovic Z, Leong KW, Spasojevic I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal. 2014;20:2372–2415. doi: 10.1089/ars.2012.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. The Journal of clinical investigation. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oberley TD. Ultrastructural localization and relative quantification of 4-hydroxynonenal-modified proteins in tissues and cell compartments. Methods in enzymology. 2002;352:373–377. doi: 10.1016/s0076-6879(02)52034-6. [DOI] [PubMed] [Google Scholar]
- 26.Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- 27.Roberts LJ, 2nd, Salomon RG, Morrow JD, Brame CJ. New developments in the isoprostane pathway: identification of novel highly reactive gamma-ketoaldehydes (isolevuglandins) and characterization of their protein adducts. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1999;13:1157–1168. doi: 10.1096/fasebj.13.10.1157. [DOI] [PubMed] [Google Scholar]
- 28.Tovmasyan A, Sheng H, Weitner T, Arulpragasam A, Lu M, Warner DS, Vujaskovic Z, Spasojevic I, Batinic-Haberle I. Design, mechanism of action, bioavailability and therapeutic effects of mn porphyrin-based redox modulators. Medical principles and practice: international journal of the Kuwait University, Health Science Centre. 2013;22:103–130. doi: 10.1159/000341715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong M, Hay S, Marshall KR, Munro AW, Scrutton NS. DNA binding suppresses human AIF-M2 activity and provides a connection between redox chemistry, reactive oxygen species, and apoptosis. J Biol Chem. 2007;282:30331–30340. doi: 10.1074/jbc.M703713200. [DOI] [PubMed] [Google Scholar]
- 30.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 31.Norberg E, Gogvadze V, Vakifahmetoglu H, Orrenius S, Zhivotovsky B. Oxidative modification sensitizes mitochondrial apoptosis-inducing factor to calpain-mediated processing. Free radical biology & medicine. 2010;48:791–797. doi: 10.1016/j.freeradbiomed.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 32.Chaiswing L, Cole MP, Ittarat W, Szweda LI, St Clair DK, Oberley TD. Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute adriamycin-induced mitochondrial toxicity. Molecular cancer therapeutics. 2005;4:1056–1064. doi: 10.1158/1535-7163.MCT-04-0322. [DOI] [PubMed] [Google Scholar]
- 33.Pantke U, Volk T, Schmutzler M, Kox WJ, Sitte N, Grune T. Oxidized proteins as a marker of oxidative stress during coronary heart surgery. Free radical biology & medicine. 1999;27:1080–1086. doi: 10.1016/s0891-5849(99)00144-6. [DOI] [PubMed] [Google Scholar]
- 34.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warner MD, Lukose V, Lee KH, Lopez K, MHS, Crane EJ., 3rd Characterization of an NADH-dependent persulfide reductase from Shewanella loihica PV-4: implications for the mechanism of sulfur respiration via FAD-dependent enzymes. Biochemistry. 2011;50:194–206. doi: 10.1021/bi101232y. [DOI] [PubMed] [Google Scholar]
- 36.Wallen JR, Paige C, Mallett TC, Karplus PA, Claiborne A. Pyridine nucleotide complexes with Bacillus anthracis coenzyme A-disulfide reductase: a structural analysis of dual NAD(P)H specificity. Biochemistry. 2008;47:5182–5193. doi: 10.1021/bi8002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holm L, Kaariainen S, Wilton C, Plewczynski D. Using Dali for structural comparison of proteins. Curr Protoc Bioinformatics. 2006;Chapter 5(Unit 5):5. doi: 10.1002/0471250953.bi0505s14. [DOI] [PubMed] [Google Scholar]
- 38.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemenatry Figure 1. Identification of a potential candidate for retrograde signaling in vivo. The cluster heat map shows the expression of 84 genes involved in the biogenesis and function of mouse mitochondria. The rows of the heat map represent genes and the columns represent samples. Each cell is colored based on the level of expression of that gene in that sample. The expression levels of each gene have been normalized to mean 0 and standard deviation 1. Red indicates a high level of expression relative to the mean value; green indicates a low level of expression; and white indicates no data available. Genes are ordered based on the results of a hierarchical cluster analysis. The bar graph shows the quantification of the AlFm2 RNA from microarray data in SOD2+/+ and SOD2 +/− heart tissues. Results are presented as mean ± SEM (n=4 per group).
Supplementary Figure 2a and 2b. Mass sprctrometric analysis of native and HNE adducted AIFm2.
(a) Tandem mass spectrometric analysis (MS/MS) of the native and oxidatively modified AIFm2 showing His174 to be the HNE modification site. (ii) & (iii) AlFm2 sequence (row 1, AlFm2) and the tryptic digested peptide sequence targeted by HNE showing the specific target amino acid residue (row 2, AIFm2 His).
(b) Tandem mass spectrometric analysis (MS/MS) of the native and oxidatively modified AIFm2 showing Cys187 to be the HNE modification site. (ii) & (iii) AlFm2 sequence (row 1, AlFm2) and the tryptic digested peptide sequence targeted by HNE showing the specific target amino acid residue (row 2, AIFm2 Cys).
Supplementary Figure 3a,b and c. Western blot of inputs control for the immunoprecipitation reactions shown in Fig. 3 and Fig 4.