

Abstract
Major depressive disorder (MDD) is a leading cause of disability worldwide. Current first line therapies target modulation of the monoamine system. A large variety of agents are currently available that effectively alter monoamine levels; however, approximately one third of MDD patients remain treatment refractory after adequate trials of multiple monoamine based therapies. Therefore, patients with treatment-resistant depression (TRD) may require modulation of pathways outside of the classic monoamine system. The purpose of this review was thus to discuss novel targets for TRD, to describe their potential mechanisms of action, the available clinical evidence for these targets, the limitations of available evidence as well as future research directions. Several alternate pathways involved in the patho-etiology of TRD have been uncovered including the following: inflammatory pathways, the oxidative stress pathway, the hypothalamic-pituitary-adrenal (HPA) axis, the metabolic and bioenergetics system, neurotrophic pathways, the glutamate system, the opioid system and the cholinergic system. For each of these systems, several targets have been assessed in preclinical and clinical models. Preclinical models strongly implicate these pathways in the patho-etiology of MDD. Clinical trials for TRD have been conducted for several novel targets; however, most of the trials discussed are small and several are uncontrolled. Therefore, further clinical trials are required to assess the true efficacy of these targets for TRD. As well, several promising novel agents have been clinically tested in MDD populations, but have yet to be assessed specifically for TRD. Thus, their applicability to TRD remains unknown.
Keywords: Brain derived neurotrophic factor (BDNF), cytokines, glutamate, infliximab, insulin, ketamine, riluzole, pioglitazone, treatment-resistant depression.
INTRODUCTION
Major depressive disorder (MDD) is a leading cause of disability worldwide with significant impact on patient’s function and quality of life [1, 2]. Current first line treatments are only effective for approximately one third of MDD patients, as measured by remission, indicated by a score ≤ 7 on the 17-item Hamilton depression rating scale (HDRS) [3]. Furthermore, after multiple antidepressant trials, approximately one-third of patients still remain treatment refractory with significant functional impairments [3].
Treatment-resistant depression (TRD) is thus a topic of high relevance as it greatly affects a sizeable proportion of MDD patients. While several definitions of TRD have been proposed, for the purpose of this review, TRD will be used to broadly refer to an inadequate response to a single adequate trial of a first-line antidepressant. Of note, a lack of a unified definition for TRD limits the progress of the field. Various definitions and grading systems are reviewed elsewhere [4].
Several evidence-based approaches have been proposed for the treatment of TRD using conventional psychotropic medications (selective serotonin reuptake inhibitors (SSRIs), selective serotonin norepinephrine reuptake inhibitors (SNRIs), second generation antipsychotics (SGAs), monoamine oxidase inhibitors (MAOIs), tricyclic anti-depressants (TCA), etc.) and are reviewed elsewhere [4, 5]. The primary pathway targeted by all currently available agents is the monoamine pathway. The monoamine pathway has been the center of psychopharmacology for the past five decades and has been targeted from numerous angles with numerous agents, being fully exhausted of its pharmacologic potential [6]. After the advent of dozens of agents targeting the same pathway used in a variety of combinations and dosages, TRD remains a public health problem [3, 7, 8]. Therefore, the impetus for discovering genuinely novel agents acting outside of the monoamine pathways is compelling.
A variety of pathways that may be involved in the patho-etiology of MDD and, more specifically, TRD are a subject of current interest. Investigation of these pathways is yielding numerous, new promising neurotherapeutic targets for TRD. Novel pathways of particular interest are the following: the inflammatory pathways, the oxidative and nitrosative stress (O&NS) pathways, the hypothalamic-pituitary-adrenal (HPA) axis, neurotrophic signaling pathways, the metabolic and bioenergetics system, the glutamate system, the opioid system and the cholinergic systems [5, 9-14].
Numerous current conventional antidepressants have been noted to have some effects on these systems [15-17]; however, of more interest has been new agents currently being investigated for the treatment of TRD that primarily target one or more of these novel pathways instead of primarily the monoamine pathway. The purpose of this paper is therefore to review novel targets for TRD (i.e. drugs that act outside the classic monoamine system), to describe their proposed mechanisms of action, the available clinical evidence for these targets, the limitations of available evidence and the future direction of investigations. Therefore, the evaluation of novel psychotherapies, electroconvulsive therapy (ECT), deep brain stimulation (DBS) and repetitive transcranial magnetic stimulations (rTMS) for the management of TRD are out of the scope of this review. Also of note, the novel manipulation of the melatoninergic system in TRD is not discussed, as melatonin is a classic monoamine and thus also falls out of the scope of this review. The interested reader is directed to some recent reviews on this specific topic [18, 19].
METHODS
For this narrative review, the MEDLINE/PubMed, EMBASE, Google Scholar and ClinicalTrials.gov databases were searched from inception through August 2014 for published randomized-controlled trials, open label trials, meta-analyses and systematic reviews for novel targets of TRD. Searches included various combinations of the following terms: treatment resistant depression (TRD), novel targets, infliximab, cytokines, interleukin (IL), IL-1β, IL-6, tumor necrosis factor alpha (TNF-alpha), anti-TNF-alpha, pioglitazone, creatine, non-steroidal anti-inflammatory drugs (NSAIDs), celecoxib, acetylsalicylic acid (ASA), omega-3 polyunsaturated fatty acid (O3PUFA), curcumin, glutamate, opioid, opiate, MDD, ketamine, riluzole, oxidative stress, reactive oxygen species (ROS), cholinergic, HPA axis, cortisol, metabolic syndrome, diabetes, CP-101, AZD6765, D-cycloserine, EVT 101, GLYX-13, scopolamine, mecamylamine, LY2456302, buprenorphine, oxytocin, tibolone, cysteamine, one-carbon cycle, L-methylfolate, S-andenosylmethionine (SAMe) and novel treatments. Reference lists from included papers were also manually searched for additional pertinent references. Ongoing clinical trials for TRD were also searched for on ClinicalTrials.gov and Google Scholar databases.
RESULTS
Novel Pathways and Targets
Several pathways are described in the literature as having potential, novel targets for the management of TRD. Pre-clinical and clinical data in support of these pathways have been reported and further investigation is currently underway for several of these targets. The evidence for each of these pathways and corresponding targets will be discussed in turn.
Inflammatory System
Julius Wagner-Jauregg, one of the only psychiatrist who won a Nobel Prize (1927), was the first to describe a potential link between inflammation and mood disorders in 1887 with his observation of the psychiatric manifestations of fever [20]. His theory was abandoned, however, with the advent of tricyclic anti-depressants, monoamine oxidase inhibitors and selective serotonin reuptake inhibitors [6]. In recent years his theory has been revisited as the link between inflammation and MDD has become more apparent [9].
The increased co-prevalence of inflammatory comorbidities, including auto-immune diseases, cardiovascular diseases, diabetes, obesity and metabolic syndrome, asthma and allergies with MDD is one epidemiologic observation that prompted further investigation [21, 22]. Indeed, epidemiologic studies have repeatedly shown this phenomenon, alerting investigators to a potential link [9]. In further support of a connection, levels of inflammatory cytokines (TNF-alpha, IL-1B, IL-6) have repeatedly been correlated with mood symptoms [23, 24]. Moreover, the induction of an inflammatory state in pre-clinical and clinical models has repeatedly demonstrated mood symptoms, namely, poor mood, poor cognition, poor sleep and anhedonia [9, 21, 24-28]. Interestingly, elevated levels of inflammatory cytokines has also been shown to be predictive of TRD indicating that inflammation may induce a separate mood pathway unaffected by monoamine modulation [29].
Several mechanisms have been proposed linking inflammation with MDD (summarized in Fig. 1). The most supported theories include the effect of inflammatory cytokines on the following elements: (1) central tryptophan and serotonin levels, (2) microglial function, (3) the HPA axis, (4) neuroplasticity and (5) glutamate receptor activation.
Fig. (1).
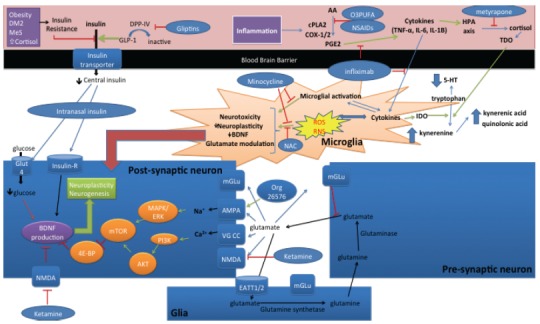
Potential pathways and novel targets of TRD. (1) Inflammation increases levels of prostaglandins leading to increased cytokine levels and microglial activation leading to ROS and RNS formation which leads to neurotoxicity, decreased neuroplascticity, decreased BDNF levels and glutamate modulation. Cytokine production also leads to IDO activation, which in turn converts tryptophan to its catabolites and decreases serotonin (5-HT) levels. HPA axis activation also activates TDO, which has the same enzymatic effect of IDO. NSAIDS and O3PUFA can prevent the production of PGE2 thereby inhibiting this inflammatory-mood pathway. Similarly, infliximab can stop the pathway at the level of TNF-alpha. Minocycline can also prevent inflammation and oxidative stress. NAC can prevent oxidative stress and thus prevent the complications of ROS and RNS production. Metyrapone can prevent the production of cortisol thereby preventing TDO activation and decreasing the risk of insulin resistance. (2) Metabolic dysfunction can lead to insulin resistance and thus decreased transport of insulin across the blood brain barrier leading to decreased central levels of insulin and decreased neuronal trophic effects of insulin. GLP-1 or DPP-IV inhibitors or other insulin sensitizers such a TZDs (ex. Pioglitazone) can prevent this effect, thereby increasing the trophic effects of insulin and increasing the levels of intracellular glucose via insulin dependent uptake of glucose at Glut4 transporters. Intranasal insulin can also have this direct effect. (3) Glutamate is produced in the pre-synaptic neuron and reuptake occurs in the surrounding glia cell which converts glutamate to glutamine by glutamine synthetase and transports glutamine back to the pre-synaptic neuron where it is converted back to glutamate via the action of glutaminase. Increased synaptic glutamate levels and inhibition of NMDA by ketamine can lead to increased influx of sodium via AMPA and calcium via voltage gated calcium channels (VG CC) leading to PI3K and MAPK/ERK activation which activates mTOR, thus inhibiting 4E-BP, preventing the inhibition of BDNF production and ultimately leading to increase neuroplasticity and neurogenesis. AMPA may be directly activated by Org 26576 having a similar effect.
Starting with the familiar monoamine system, increased levels of inflammatory cytokines increase the activation of indolamine 2,3-dioxygenase (IDO) which increases the conversion of tryptophan to kynurenine and consequently decrease the production of serotonin leading to decreased central serotonin levels and increased production of tryptophan catabolites (TRYCATs) known to have depressogenic and anxiogenic properties [30, 31]. Therefore, inflammation has a direct effect on central serotonin levels, providing one direct link to mood symptoms from cytokine production.
Microglial over-activation has also been identified as a potential culprit mediating mood symptoms during elevated levels of inflammatory cytokines. More specifically, TNF-α and IL-1β are potent activators of microglia as part of the innate immune system [32]. The over-activation of microglia may lead to over-pruning of neural circuits, decreased neuroplasticity and ultimately decreased function of the neuronal circuits leading to impaired cognition and emotional regulation on a functional level [9, 32-37]. Further, microglial activation impairs glutamate metabolism leading to alteration of glutamate levels and glutamate receptor activation, a common feature of TRD [12, 13, 38].
Inflammatory cytokines also stimulate activation of the HPA axis and prevent inhibitory negative feedback loops, thus leading to hypercortisolemia. An elevated level of cortisol (endogenous or exogenous) has been repeatedly shown to induce mood symptoms and is thus another clear potential link between inflammation and MDD [39]. Further, HPA axis activation leads to increased activity of tryptophan 2,3-dioxygenase (TDO) which also degrades tryptophan leading to elevated levels of TRYCATs and decreased production of serotonin [40]. As well, hypercortisolemia may lead to metabolic dysfunction, insulin resistance and glutamate modulation, which are all potent causes of neuronal dysfunction and impaired neuroplasticity [40].
Targets of interest in the inflammatory pathway include, but are not limited to TNF-alpha antagonists, non-steroidal anti-inflammatory drugs (NSAIDs), natural anti-inflammatory agents (omega-3 polyunsaturated fatty acids (O3PUFA) and curcumin) and tetracycline antibiotics (minocycline and doxycycline). Of these agents, TNF-α antagonists (infliximab) have been evaluated for treatment of TRD. Activated macrophages are chiefly responsible for the production TNF-α leading to a systemic inflammatory response and activation of other immune cells including microglia centrally [33]. Currently, TNF-α antagonists are being used clinically in autoimmune disorders to prevent a systemic inflammatory response. The reduction in microglia activation with TNF-α antagonists use may be particular relevant for TRD [33]. A proof-of-concept randomized controlled trial (RCT) tested the efficacy of intravenous (IV) infliximab (5 mg/Kg) or placebo administered at baseline and at weeks 2, 4 and 6 of a 12-week trial in a sample of 60 patients with TRD. The trial showed no difference between groups in the primary outcome (17-item HDRS); however, when patients were stratified based on the levels of their inflammatory state (based on [hsCRP]>5mg/L), a significant anti-depressant effect was shown as 62% of TRD patients obtained >50% reduction in the HDRS scores versus only 33% in the placebo group [41]. While these results are promising that infliximab may be effective for a subset of TRD patients, namely, patients with increased inflammatory biomarkers, clearly additional trials are required to confirm or refute this effect. As well, the side effect profile may be a limiting factor as infliximab increases the risk of infection due to its potent anti-inflammatory effect [42].
Also of interest have been NSAIDs, specifically acetyl-salicylic acid (ASA) and celecoxib. Acetyl-Salicylic Acid irreversibly inhibits cyclooxygenase-1 and -2 (COX-1 and COX-2) thereby decreasing prostaglandin and thromboxane levels and thus decreasing TNF-α and IL-6 [43]. Berk et al. (2013) recently extensively reviewed the potential role of ASA in the treatment of mental illness discussing studies from 1996–2012 showing the great potential in the treatment of MDD as an adjunct to conventional therapy [44]. Similarly, celecoxib a selective COX-2 inhibitor has been studied as an adjunctive therapy to conventional antidepressant therapy. Indeed, several clinical and pre-clinical studies and two meta-analyses have shown the addition of celecoxib to anti-depressants to increase therapeutic efficacy and lower serum IL-6 and IL-1β levels [45-49]. While both agents show promise for MDD, neither has been assessed for TRD. Also of note, significant potential side effects including renal impairment and peptic ulcer disease may be a limiting factors for a broader application of NSAIDs in MDD [50].
A significant interest in natural anti-inflammatory agents for TRD has grown as well. Omega-3 polyunsaturated fatty acids are dietary fatty acids that cannot be endogenously produced by humans [51]. It exhibits an anti-inflammatory effect by competing with arachadonic acid (AA) for COX enzymes thereby decreasing prostaglandin E2 (PGE2) levels and thus decreasing pro-inflammatory cytokine production [52]. Omega-3 polyunsaturated fatty acids with >60% EPA content has shown the greatest promise with several trials and meta-analyses showing a significant antidepressant effect of O3PUFA as an adjunct to SSRI therapy in the treatment of MDD [9, 53-55]. Curcumin, an Asian spice known to have potent anti-inflammatory and antioxidant effects has also shown promise in preclinical studies; however randomized controlled trials (RCTs) of curcumin in MDD have provided conflicting results thus far [56-59]. Currently no trials have been conducted to evaluate curcumin or O3-PUFA in TRD samples; therefore, their application to this population is still unknown. These agents are particularly attractive due to their benign tolerability and safety profiles.
Tetracycline antibiotics, doxycycline and minocycline, also have potent anti-inflammatory effects and are currently being evaluated for the treatment of MDD. Preclinical data for minocycline indicated that it could exert antidepressant effects through its anti-inflammatory, antioxidant, anti-glutamatergic and neuroprotective effects [10, 60, 61]. Clinical trials are currently underway to investigate minocycline use as an adjunctive therapy for MDD (NCT01574742) and bipolar depression (NCT01429272, NCT01514422, NCT01403662) [10, 60, 61]. Doxycycline has also now shown promise as recent pre-clinical data shows amelioration of depressive-like behavioural manifestationsin inflammatory mouse models [62]. Currently no clinical trials for tetracycline antibiotics for TRD have been reported; however, on their preclinical effects, these pleiotropic agents may represent a target for TRD.
Taken together, several of the previously mentioned anti-inflammatory agents (NSAIDS, tetracycline antibiotics, O3PUFA and curcumin) show promise in the treatment of MDD however have yet to be evaluated for TRD, with the exception of infliximab. Infliximab showed efficacy for the subpopulation of TRD patients with elevated inflammatory markers. Similar to infliximab, the other anti-inflammatory agents may have a role in TRD in the subpopulation with a heightened inflammatory state, however, further clinical trials are still required to determine what population is suitable and what effect could be expected.
Oxidative and Nitrosative Stress
Inexorably linked to inflammatory dysregulation is oxidative and nitrosative stress (O&NS). Both inflammation and O&NS are necessary and physiologic when appropriately regulated; however, an excess of inflammation and/or O&NS can be extremely problematic for cellular function [17, 63-65]. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are normally produced during cellular respiration and are counterbalanced by anti-oxidant defense pathways composed of endogenously produced anti-oxidant molecules, such as glutathione, glutathione peroxidase, catalase, superoxide dismutase (SOD), melatonin and coenzyme Q10 [66-68].
In normal cellular respiration, glucose is used to produce chemical energy in the form of adenosine triphosphate (ATP). Initially, glucose undergoes glycolysis to produce a minimal amount of ATP through anaerobic respiration. The end product of glycolysis, pyruvate, subsequently is used in aerobic respiration to produce 90-95% of total ATPvia the tricarboxylic acid (TCA) cycle and subsequent oxidative phosphorylation via the electron transport chain (ETC) in the mitochondria [69]. In the mitochondria, electrons are transported via a series of redox reactions with the final product of reduced oxygen. Reduced oxygen is chemically unstable and usually reacts with hydrogen ions to form H2O (further review of aerobic respiration by Rich [70]). However, at times, the ROS may react with other molecules such as lipids, proteins or genetic material (for example, mitochondrial or nuclear DNA) causing damage to these macromolecules and potentially leading to mitochondrial and cellular dysfunction [17]. Increased cellular energy demands leads to increased utilization of the ETC and consequently increases the amount of ROS produced [17]. The brain is particularly vulnerable to high levels of oxidative stress as it consumes 20% of the body’s available oxygen and 25% of its glucose, while only accounting for 2% of the body’s weight [71]. The high level of consumption relates to the high-energy demands of maintaining neuronal ion gradients, producing action potentials and neurotransmitter transport, release and uptake [65, 72, 73]. Of note, inflammatory states and increased excitatory activity increases energy demands and subsequently increases production of ROS [65]. Further, in several disease processes, such as autoimmune as well as metabolic diseases, either there is an increase in the generation of ROS, which cannot be balanced by the anti-oxidant reserve, or there is a decrease in anti-oxidant reserve leading to oxidative stress [65, 72, 73].
Nitric oxide (NO) is produced from L-arginine by a variety of nitric oxide synthase (NOS) isozymes [74]. Of note, neuronal NOS (nNOS) is expressed in response to increase intracellular calcium as seen in a neuronal excitatory state [72], while inducible NOS (iNOS) is induced by pro-inflammatory cytokines [65]. After NO is formed, it may react with superoxide to form peroxynitrite, which is a highly reactive RNS. At physiologic levels, however, SOD competes with NO for superoxide, thus limiting the production of peroxynitrite [75]. When SOD levels are decreased or NO levels are greatly increased (as seen with increase neuronal excitation or inflammation), peroxynitrite levels rise [75]. Therefore, increased neuronal excitation and inflammation lead to increased production NO and peroxynitrite, which reacts with and damages lipids, proteins and DNA/RNA leading to mitochondrial and cellular dysfunction [65, 72, 75].
In sum, the O&NS can be damaging at a gene, protein, lipid, cell and ultimately systems level [66, 72, 76]. Centrally, increased O&NS has been shown to cause neurotoxicity and impair neuroplasticity and neurogenesis [63, 73]. Not surprisingly, increased levels of O&NS has been linked to several psychiatric disorders, including MDD, as shown in a recent meta-analysis [66, 76]. Therefore, the use of anti-oxidants may present another novel therapeutic target separate from the monoamine pathway.
One particularly promising antioxidant target is N-acetylcysteine (NAC). N-acetylcysteine is a glutathione precursor that has multiple potential antidepressant effect through its pleiotropic actions, including decreasing inflammatory cytokines, modulating glutamate, promoting neurogenesis, and decreasing cellular apoptosis [77]. N-acetylcysteine has been shown to have anti-oxidant effects in the anterior cingulate cortex in a multicenter RCT of MDD participants treated with adjunctive NAC. Using spectroscopy, higher glutamate-glutamine (Glx) and N-acetyl-aspartate (NAA) levels were shown in patients treated with NAC, indicative of an anti-oxidant effect [78].
Further, in a recent large RCT (n=252), MDD patients were randomized to conventional therapy with placebo versus conventional therapy with NAC for 12 weeks. At the 12-week endpoint, there was no statistically significant difference in symptoms, however, by week 16, NAC was shown to be superior. The endpoint of the study was 12 weeks, thus making it a negative trial, however, the delayed effect may suggest a latency of effect or potential preventative mechanism of further damage [79]. Further studies are required to assess NAC in the TRD population on a more long-term basis.
As previously discussed, minocycline is currently being investigated as novel target of TRD through its anti-inflammatory effects [10]. Minocycline has also been shown to have potent anti-oxidant effects through decreasing free radical generation, decreasing ROS and RNS levels, decreasing lipid peroxidation, increasing catalase activity and through direct scavenging of free radicals [10]. Therefore, minocycline could have anti-depressant effects through its anti-oxidant properties as well.
Hypothalamic Pituitary Adrenal Axis
The HPA axis serves as an essential control system for numerous physiologic processes. Numerous feedback loops are in place to regulate the levels of adrenal hormones as an excess or a shortfall may have profound effects such as seen in Cushing’s syndrome and Addison’s disease, respectively [80]. Dysregulation of the HPA axis and failure of negative feedback loops has been noted in TRD [40, 81]. Of particular interest have been the mood effects of hypercortisolemia, one of the end products of the HPA axis [81]. Elevated levels of cortisol may be caused iatrogenicaly (through exogenous administration of steroids) or endogenously through over-activation of the HPA axis without appropriate inhibitory feedback. As previously discussed, inflammation is one cause of a blunted negative feedback loop of the HPA axis. Regardless of having an endogenous or exogenous source, cortisol has been shown to have potent negative effects on mood [82]. Several mechanisms may be at play including the increased activity of TDO leading to decreased serotonin and increased levels of TRYCATs as shown in Fig. 1. Recently, potent effects on the glutamate system have also been noted which might facilitate some of the deleterious mood effects observed [83]. Therefore, preventing hypercortisolemia may serve as another novel target. Of particular interest has been metyrapone, a cortisol synthesis inhibitor (inhibitor of 11-β hydroxylase, the enzyme that catalyses the conversion of 11-deoxycortisol to cortisol) [39]. Several open label trials showed a positive effect of metyrapone on TRD as reviewed by Sigalas et al. [39]. Further, in a RCT with sixty-three inpatients with severe MDD, augmentation of nefazodone or fluvoxamine for three weeks with placebo versus 1 g of metyrapone once daily was assessed. The metyrapone group showed a significantly greater improvement compared with the placebo group (effect size of 0.6) using response (a decrease in HDRS score by 50%, 5 weeks post initiation of treatment) as the outcome measure [84]. One additional RCT have been completed, however results have yet to be published (NCT01375920).
Oxytocin has also been shown to be a potent suppressor of the HPA axis [85]. Intranasal oxytocin is therefore currently being investigated in a RCT for TRD (NCT01239888). Of note, a previous trial showed that oxytocin and cortisol levels were unaffected by SSRI therapy (NCT00168493) [86].
Also of interest have been anti-inflammatory agents (previously discussed) as inflammation is a potent activator of the HPA axis, therefore, decreasing inflammation serves to also decrease HPA activation and thus would lower cortisol levels [9]. Of note, this interaction may seem paradoxical as cortisol has potent anti-inflammatory effects. Therefore, one may hypothesize that the deleterious mood effects of cortisol may outweigh the potential positive mood effects that may occur through cortisol’s anti-inflammatory effects [35, 87].
Repurposing of Anti-diabetic Agents
Epidemiologic data has unequivocally shown a correlation between MDD and metabolic dysfunction [88]. In particular, diabetes, obesity and metabolic syndrome have been shown to have a bidirectional relationship with mood disorders [89-91]. Mechanistically, metabolic dysfunction has several pathways by which brain function may be impaired in the realms of mood and cognition. Pre-clinical models have shown that insulin resistance may decrease the transport of insulin across the blood brain barrier leading to decreased central insulin levels [90-93]. Insulin serves as a potent growth factor in the brain, increasing levels of brain derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF), notably having trophic effects on the amygdala, hippocampus, and prefrontal cortex, areas essential to the regulation of emotion and cognition [91]. Therefore, insulin resistance may lead to decreased central insulin levels leading to impaired neuroplasiticy, atrophy, and impaired function of these key brain regions. As well, glucose uptake is partly insulin dependent in the hippocampus, amygdala and cerebral cortex via the Glut4 insulin dependent glucose transporter [91]. Therefore, decreased central insulin levels may lead to decreased intracellular glucose levels in these brain areas as shown in Fig. 1. The decrease in energy supply may also lead to decreased long-term potentiation, decreased neural plasticity and ultimately decreased neural function leading to deficits in emotional as well as cognitive functions [91].
Taken together, poor neuronal function secondary to insulin resistance may serve as a potential cause of TRD as this pathway is minimally affected by monoamine based therapies. This hypothesis has led to the repurposing of anti-diabetic medications for treatment of MDD. Of particular interest has been the use of incretins, insulin sensitizers and insulin [94]. Currently no clinical trials have been conducted using anti-diabetic medications for the treatment of TRD, however, clinical trials showing the effective use of these medications as an adjunct to conventional anti-depressant therapies for MDD provide the impetus for evaluation of their use in TRD, especially in the large subpopulation of TRD patients with comorbid metabolic dysfunction. Particularly promising agents include intranasal insulin, glucagon like peptide-1 (GLP-1) analogs and pioglitazone.
Intravenous insulin has been shown to increase hippocampal neural activity and improve mood and cognition in preclinical and clinical models [95, 96]. Recently interest has shifted to intranasal insulin as it allows for the direct increase of central insulin levels without altering systemic glucose or insulin levels [97, 98]. This increase in central insulin levels may potentially improve neuronal function and thus cognition and mood through the previously described central mechanism of insulin as a growth factor and glucose uptake facilitator. Moreover, intranasal insulin has been shown to improve mood and cognition reproducibly in healthy, obese and Alzheimer’s disease patients [99]. Currently a double-blind, placebo controlled RCT assessing the affect of intranasal insulin on MDD symptoms is currently underway (NCT00570050).
Glucagon like peptide-1 is an incretin that serves to delay gastric emptying and improves insulin sensitivity. This increase in insulin sensitivity may also serve to improve cognition and mood through the previously described mechanisms. Preclinical models have shown improvement in cognition and decreased depressive-like behaviours with administration of stable GLP-1 analogues [11]. Glucagon like peptide-1 as a potential target for MDD and TRD was recently reviewed by McIntyre et al. [11]. Another potential target in the same system are gliptins (for example, sitagliptin and saxigliptin), which inhibit dipeptidyl peptidase-4 (DPP-4), an enzyme that degrades GLP-1.
Pioglitazone is a thiazolidinedione (TZD) drug used primarily in the diabetic population as an insulin sensitizer. The primary mechanism of action is through stimulation of peroxisome proliferator-activated receptor gamma (PPARγ). Stimulation of PPARγ has anti-inflammatory, anti-oxidant and neuroprotective effects [100]. Pre-clinical studies also suggest modulation of N-Methyl-D-asparate receptors (NMDARs) by pioglitazone [101].
Gold et al. discuss the multiple neuroprotective effects of PPARγ stimulation as mediated through modulation of the parainflammatory and endoplasmic reticulum (ER) stress responses [102]. Parainflammation is an inflammatory response to ‘new’ stimuli of the modern era that have not been seen earlier in evolution and therefore appropriate responses have yet to be selected for [103, 104]. Examples of such stimuli include exposure to novel foodstuffs and chemicals, alteration in light-dark exposure patterns, obesity, type 2 diabetes, sedentary lifestyle, atherosclerosis, aging and emotional stress (i.e. stressors that were not present for our evolutionary predecessors) [103, 104]. These stimuli prompt the attempt to restore homeostasis, which leads to chronic activation of acute stress pathways including the activation of the innate immune system and subsequent release of inflammatory cytokines discussed previously (i.e. TNF-α, IL-1β, IL-6) [103]. The PPARγ system when activated prevents the perpetuation of the parainflammatory response and is thus neuroprotective against the deleterious effects of a prolonged inflammatory response [102].
Endoplasmic reticulum stress presents when there is increased demand on the ER to produce more proteins [103, 104]. This increased demand may be secondary to an inflammatory response or an increase in neuronal stimulation through the glutamate system [101, 104]. The increased demand of the ER leads to a stress response that either leads to appropriate up-regulation of chaperon proteins to allow for increased capacity to adequately fold increased levels of protein or may lead to release of calcium from the ER (the major storage organelle for calcium intracellular) leading to an apoptotic pathway and ultimately neurodegeneration [102]. Stimulation of the PPARγ system may facilitate an appropriate ER stress response and prevent calcium release and apoptosis [102].
Clinical data in support of pioglitazone’s use for MDD has also been accumulating. In a 12-week, open-label, flexible-dose study, 23 patients with MDD received pioglitazone monotherapy or adjunctive therapy initiated at 15 mg daily. Pioglitazone decreased depression symptom severity from a total Inventory of Depressive Symptomatology (IDS) score of 40.3±1.8 to 19.2±1.8 at Week 12 (p<.001) and a significant reduction in inflammation as measured by log highly- sensitive C-reactive protein (-0.87±0.72; p<.001). Also of note, the majority of participants (74%, n=17) had already failed at least one antidepressant trial [105]. These results were replicated in a double-blind, placebo controlled RCT with 40 patients with MDD randomized to citalopram plus pioglitazone (15 mg every 12 h) or citalopram plus placebo for 6 weeks. Pioglitazone showed superiority over placebo during the course of the trial [106]. These results were also replicated in a six-week double-blinded RCT with 50 patients with comorbid polycystic ovarian syndrome (PCOS) and MDD [107]. It is important to note though that the mechanisms of action may be multifactorial. As shown by Kashani et al. [107], the anti-depressant effect of pioglitazone was greater than metformin even though metformin is a more potent insulin sensitizer. Pioglitazone may also be having anti-depressant effects through its anti-inflammatory, anti-oxidant and neuroprotective properties. While there has yet to be a trial specific for TRD, there has been one case report for TRD treated effectively with pioglitazone [108].
Taken together, the metabolic-brain axis may serve as another novel pathway for the management of TRD. Currently, no clinical trials have been conducted to evaluate these targets specifically for TRD, however, results from clinical trials evaluating these targets for MDD show promise and merit investigation of their efficacy for TRD, specifically in patients with insulin resistance. These pathways would be of particular interest in TRD as the metabolic-brain axis is largely unaffected by monoamine agents. Therefore, insulin sensitivity and metabolic dysfunction could represent a subgroup of TRD patients that monoamine therapy would be particularly not useful for. Indeed, epidemiologic data suggests that metabolic dysfunction is a major risk factor of TRD in further support of this hypothesis [88]. Limitations of use of these targets largely relates to selecting the subpopulation of TRD patients that may benefit (i.e. TRD patient with perfect metabolic function and insulin sensitivity might not benefit from insulin based therapies). Of note, these targets are usually extremely well tolerated with mild side effect profiles well studied in the diabetic population of patients.
Mitochondrial Modulators
Also of interest have been alterations in intracellular bioenergetics as a pathogenic mechanism of MDD [65, 72, 73]. Central to intracellular bioenergetics is mitochondrial function. Mitochondrial dysfunction has been repeatedly linked to MDD and other psychiatric disorders [65, 72, 73, 109]. As previously discussed, mitochondria are responsible for the majority (90-95%) of ATP production, thus controlling intracellular energy availability [69]. Mitochondria are also the major potential contributors of ROS, which may have extremely damaging effects, if not appropriately counterbalanced by intracellular anti-oxidants [72]. Mitochondria also modulate intracellular calcium stores and apoptotic pathways [63, 69, 72]. Taken together, neuronal mitochondrial dysfunction may impair ATP production, promote ROS formation, inappropriately increase intracellular calcium and induce neuronal apoptosis [17, 64, 65, 92]. Therefore, mitochondrial dysfunction may potently affect neuronal function and thus mitochondrial modulators may present more novel targets for MDD and TRD [65, 72, 73].
As previously discussed, the use of anti-oxidants presents one important target of mitochondrial dysfunction that is modifiable in the pathogenesis of TRD. Also of interest has been increasing intracellular energy stores to buffer potential mitochondrial dysfunction leading to decreased ATP levels [110-112]. Of particular interest have been observed decreased levels of creatine monohydrate in MDD patients [110-112]. Further, supplementation of creatine leads to a shift in creatine kinase activity and thus increased phosphocreatine (PCr), a high-energy molecule, production and use as intracellular energy [110]. In theory, this increased availability of energy in brain cells may buffer potential mitochondrial dysfunctionthus allowing for improved neuroplasticity and neuronal function [110, 111]. Indeed, preclinical studies have shownmolecular and functional improvements with creatine supplementation leading to antidepressant-like and heightened motivation in animal models (reviewed by Allen et al. [110]).
Furthermore, one adult and one adolescent open-label trial have shown improved mood in TRD patients with the addition of 3-5g of creatine to treatment regimens [113, 114]. Roitman et al. [113] showed in their small, preliminary, open label study of creatine monohydrate that significant improvement in seven out of the ten TRD patients enrolled significantly improved HDRS, HARS, and Clinical Global Impression scores, at weeks 1, 2, 3 and 4. Kodo et al. [114] studied five female adolescents TRD to fluoxetine using 4g of creatine in addition to fluoxetine and observed improved depression score and a significant increase in brain Phosphocreatine (PCr) concentration (p=0.02) on follow-up (31) P MRS brain scans. Clearly, for both of these studies the small sample size and lack of a placebo control group limit the interpretation of the data. Several randomized controlled trials using creatine supplementation for TRD are currently underway (NCT00729755, NCT01175616, NCT00313417, NCT00851006, NCT01601210 NCT02134808). Furthermore, the benign side effect profile of creatine supplementation is another appealing factor to allow wider investigation in TRD populations [110].
The One-Carbon Cycle
The one-carbon cycle is a metabolic pathway that facilitates the methylation of intracellular molecules. The cycle is required in the synthesis of monoamine neurotransmitters including serotonin, dopamine and norepinephrine (reviewed by Papakostas et al. [115]). Therefore, dysfunction of the one-carbon cycle has been investigated as another potential cause of MDD. Of particular interest in MDD has been S-adenosyl-methionine (SAMe) and L-methyl-folate, key naturally occurring compounds involved in the one carbon cycle [115, 116]. Indeed, decreased levels of folate and SAMe has been linked to MDD [115-117]. Further, supplementation of folate and SAMe has been shown repeatedly in RCTs to improve MDD symptoms [115-120]. Mechanistically, supplementation of SAMe and folate provide donor methyl groups thus increasing the synthesis of monoamines (further reviewed by Papakostas [116]).
S-adenosyl-methionine is natural supplement that has been available in Europe for decades and recently has become available in North America. As discussed, the main mechanism of action is believed to be through increased monoamine production [115]; however, SAMe is also upstream of creatine production and thus may be partly exerting its anti-depressant effects on TRD patients through this bioenergetics pathway as reviewed by Papakostas et al. [115]. Several RCTs and meta-analyses have shown a positive effect of SAMe for MDD [115]. Recently, Papakostas et al. [119] studied 73 TRD patients in a six-week, double-blind, RCT of adjunctive oral SAMe (target dose: 800 mg/twice daily). The HDRs response rate was significantly higher for participants treated with adjunctive SAMe (36.1% vs 17.6%) with a number needed to treat (NNT) of seven. The results are highly suggestive of SAMe having a positive effect for TRD, but more studies are certainly warranted. Similarly, L-methyl-folate has been shown to increase monoamine levels and improve MDD symptoms in several studies and has been extensively reviewed elsewhere [116, 121-124]. Of note, several other mechanisms of action have been proposed for SAMe and L-methyl-folate including modulation of oxidative stress as well anti-inflammatory effects [110, 122].
Neurotrophin Signaling Pathways
Neurotrophins are a family of four signaling molecules (nerve growth factor (NGF), BDNF and neurotrophin-3 (NT-3) and neurotrophin-4 (NT-4)) shown to be vital for neurogenesis, neuroplasticity, dendrite formation and synaptogenesis throughout development and in adulthood [125]. A relative decrease in neurotrophins centrally and peripherally has been implicated in the pathoetiology of MDD [126]. Moreover, the decrease in neurotrophin levels in MDD has been implicated in the observed decrease in volume and synaptic connectivity of the prefrontal cortex (PFC), amygdala and hippocampus, areas notably involved in mood and cognition [127]. Mechanistically, this decrease in connectivity and volume may lead to less functional neuronal circuits and ultimately poorer emotional and cognitive function, as observed in MDD. Of note, the neurotrophic hypothesis of MDD has been extensively reviewed elsewhere [126-128].
Of particular interest in relation to MDD have been BDNF levels. In fact, decreased levels of BDNF appear to be associated with MDD [126-128]. Further, many of the previously discussed pathways (inflammation, oxidative stress, metabolic-brain axis, glutamate system) appear to converge on the BDNF pathway, exerting their antidepressant effects via improved neurogenesis and neuroplasticity [126-128]. As well, several conventional anti-depressants have been shown to increase BDNF levels [126-128]. More recently, ketamine has been shown to potently increase BDNF levels and have a strong immediate anti-depressant effect in TRD patients (vida infra), further pointing to a key role of BDNF in the pathoetioogy of TRD [12].
Also of interest has been erythropoietin (EPO), an endogenously produced glycoprotein hormone well known for its physiologic effect of increasing erythropoiesis. Erythropoietin is also produced in the brain [129] where is has potent neuroprotective effects mediated through signaling pathways increasing BDNF levels [130]. Centrally, EPO has also been shown to activate anti-apoptotic pathways, promote dendritic sprouting and neurogenesis and have antioxidant and anti-inflammatory effects in neurons, glial, and cerebrovascular endothelial cells [129, 130]. Of note, systemically administered EPO has been shown to cross the blood–brain barrier and exert the aforementioned downstream effects [129-131]. Therefore, systemic administration of EPO has been evaluated for its effect on TRD.
In a double-blind, placebo-controlled, parallel-group design, forty TRD patients were randomized to eight weekly EPO (Eprex®; 40 000 IU) or saline infusions [131]. Patients were assessed at baseline and at weeks 5, 9, and 14 for improvement in depressive symptoms and cognition. Hamilton depression rating scale scores and remission rates did notreveal beneficial effects of EPO over saline at week 9; however, EPO improved BDI and WHOQOL-BREF scores, and this effect was maintained at follow-up week 14. As well, EPO enhanced verbal recall and recognition, which was sustained at follow-up [131]. Although the trial had a negative primary outcome (no HRDS improvement compared to control), improvement in other depression scores and cognitive function provide an impetus for further investigating EPO’s therapeutic effects in TRD in larger trials. Of note, the clinical relevance is low for improved depression scores without remission. The more notable effect of EPO is its potential role in improved cognition for TRD patients [131].
The Glutamate System
Glutamate was recognized as a neurotransmitter in the early 1980s, much later than the monoamine neurotransmitters [12, 13]. Glutamate has now been recognized as the major excitatory neurotransmitter of the brain [38]. The regulation of the glutamate “tripartide” synapse involves presynaptic and post synaptic neurons and glia [14]. Multiple different receptors regulate glutamate levelsas well as the transmission of downstream effects. The following are key targets involved in determining glutamate levels and effects: excitatory amino acid transporters (EAATs), postsynaptic density proteins, (alpha-amino-3-hydroxy-5-methylisoxazole methylisoxazole-4-propionic acid) AMPA receptors, N-methyl-D-aspartate (NMDA) receptors, kainate (KA) receptors and cognate metabotropic glutamate (mGlu) receptors (as shown in Fig. 1) [12-15, 38, 83]. Further, modulations of all of these receptors havebeen implicated in preclinical models of mood disorders [12, 13]. Therefore, pharmaceutical modulation of these targets is currently being investigated for treatment of TRD.
Of particular interest has been modulation of inotropic glutamate receptors, specifically NMDA and AMPA. Current preclinical and clinical evidence suggests that decreased activation of NMDA and increased activation of AMPA receptors leads to a favorable mood outcomes [13]. In brief, increased AMPA stimulation relative to NMDA stimulation leads to an influx of calcium and sodium and a intracellular cascade ultimately leading to increased BDNF expression and release, thus facilitating improved neuroplasticity and neuronal function as shown in Fig. 1 [12-14, 38]. The mammalian target of rapamycin (mTOR) pathway has been implicated as a point of convergence of these intracellular pathways as mTOR has been shown to inhibit 4E-binding protein (4E-BP), which is known to inhibit BDNF production [132, 133]. Notably, mTOR has been shown to be down-regulated in MDD patients and when up-regulated is associated with antidepressant effects [132]. AMPA activation is associated with activation of the mTOR pathway [132]. Taken together, modulation of the glutamate inotropic receptors to increase AMPA stimulation and decrease NMDA stimulation may lead to downstream effects of increased BDNF, improved neuroplasticity and ultimately improved mood and cognition. Assessing targets with these mechanisms of action have shown great promise for the treatment of TRD [12-15, 38, 83].
Ketamine is a voltage dependent NMDA antagonist currently used clinically as an anesthetic agent. Interest in ketamine for other purposes, including MDD, has greatly grown in the past decade [134]. In a recent meta-analysis [135], seven RCTs using an IV infusion and one RCT using intranasal ketamine were assessed for ketamine’s anti-depressant effect for MDD and BD patients. Ketamine was associated with higher rates of clinical remission relative to comparator (saline or midazolam) at 24 hours (OR 7.06, NNT = 5), 3 days (OR 3.86, NNT = 6), and 7 days (OR 4.00, NNT = 6), as well as higher rates of clinical response at 24 hours (OR 9.10, NNT = 3), 3 days (OR 6.77, NNT = 3), and 7 days (OR 4.87, NNT = 4). Ketamine was associated with transient psychotomimetic effects; however, no persistent psychosis or affective switches were noted [135]. Therefore, ketamine presents as a potent, fast acting anti-depressant for BD and MDD, completely different from conventional antidepressants, which take days to weeks before observing an initial anti-depressant effect [133].
Further, several clinical trials have also shown an effect of ketamine in the setting of TRD. Ina randomized, placebo-controlled, double-blinded crossover study, 18 TRD patients were given an IV infusion of either ketamine hydrochloride (0.5 mg/kg) or placebo on two test days, a week apart. Patients receiving ketamine showed significant improvement in depression ratings compared with subjects receiving placebo. The effect was noted within 110 minutes after injection and remained significant throughout the following week [136]. Similar results were observed in several other clinical trials [137-140] and are summarized in Table 1. Of note, two additional RCTs have shown an anti-suicidal effect of ketamine in the TRD population [141, 142].
Table 1.
Summary of key published clinical trials of novel targets for TRD.Abbreviations: intravenous (IV), oral (PO), double-blinded randomized controlled trial (DB-RCT), open label (OL).
| Target (Novel Pathway) |
Route | Study Type (n) | Summarized Trial Results |
|---|---|---|---|
| Infliximab (inflammation) | IV | DB-RCT (n=60) | Trial showed no difference between placebo and infliximab in the primary outcome (17-item HDRS); however, when patients were stratified based on the levels of their inflammatory state (based on [hsCRP]>5mg/L), a significant anti-depressant effect was shown as 62% of TRD patients obtained >50% reduction in HAM-D scores versus only 33% in the placebo group [41]. |
| Creatine (bioenergetics) |
PO | OL (n=10) | Trial showed significant improvement in depression scores at weeks 1, 2, 3 and 4 in seven out of the ten TRD patients [113]. |
| PO | OL (n=5) | Kodo et al. studied five female adolescents with TRD using 4g of creatine in addition to fluoxetine (which they were already taking) and observed improved depression score and a significant increase in brain Phosphocreatine (PCr) concentration (p=0.02) on follow-up (31) P MRS brain scan [114]. | |
| SAMe (bioenergetics) | PO | DB-RCT (n=73) | Trial showed significant improvement in depression scores after a 6-week trial of adjunctive oral SAMe (target dose: 800 mg/twice daily) with a NNT of seven [119]. |
| Ketamine (glutamate/ NMDA) |
IV | DB-RCT (n=18) | Zarate et al. (2006) showed that patients receiving IV infusion of ketamine hydrochloride (0.5 mg/kg) on 2 test days, a week apart showed significant improvement in depression ratings compared with subjects receiving placebo within 110 minutes after injection, which remained significant throughout the following week [136]. |
| IV | DB-RCT (n=73) | TRD patients were randomly assigned to receive a single IV infusion of ketamine or midazolam in a 2:1 ratio. The primary outcome was change in depression severity 24 hours after drug administration, as assessed by the Montgomery-Asberg Depression Rating Scale (MADRS). After adjustment for baseline scores and site, the MADRS score was lower in the ketamine group than in the midazolam group by 7.95 points. The likelihood of response at 24 hours was greater with ketamine than with midazolam with response rates of 64% and 28%, respectively [137]. | |
| IV | OL (n=24) | TRD patients underwent a washout of antidepressant medication followed by a series of up to six IV infusions of ketamine (.5 mg/kg) administered open-label three times weekly over a 12-day period. Participants meeting response criteria were monitored for relapse for up to 83 days from the last infusion. The overall response rate at study end was 70.8%. There was a large mean decrease in MARDS score at 2 hours after the first ketamine infusion (18.9 +/- 6.6, p < .001), and this decrease was largely sustained for the duration of the infusion period. Response at study end was strongly predicted by response at 4 hours (94% sensitive, 71% specific) [139]. | |
| Riluzole (glutamate) | PO | OL (n=19) | Riluzole (100-200mg/day oral) was used as a monotherapy for six weeks after a one-week drug free period. Significant improvement in depression scores occurred during weeks 3 through 6 for all patients [145]. |
| PO | OL (n=10) | Riluzole was used as an augmenting agent for 6-12 weeks showing significant improvement in depression scores at the end of the first week of treatment, which persisted for the 12-week duration of the study [146]. | |
| EPO | IV | DB-RCT (n=40) | TRD patients were randomized to eight weekly EPO (Eprex; 40 000 IU) or saline infusion. Patients were assessed at baseline and at weeks 5, 9, and 14 for improvement in depressive symptoms and cognition. Hamilton depression rating scores and remission rates showed no effects of EPO over saline at week 9; however, EPO improved the BDI and WHOQOL-BREF scores, and this was maintained at follow-up week 14. As well, EPO enhanced verbal recall and recognition, which was sustained at follow-up [131]. |
| CP-101,606 (glutamate) | IV | DB-RCT (n=30) | Intravenous CP-101,606 was usedas an adjunct to paroxetine versus paroxetine alone for four weeks. Significant improvement was found in depression scales with seventy-eight percent of CP-101,606-treated responders maintaining response status for at least 1 week after the infusion [147]. |
| AZD6765 (glutamate) | IV | DB-RCT (n=22) | A single infusion of AZD6765 (150 mg) on 2 test days 1 week apart was associated with rapid but short-lived improvement in depression scores versus infusion of placebo [173]. |
| Buprenorphine (opioid) | PO | OL (n=10) | Buprenorphinewas tolerated by 7/10 participants for whom significant improvement in depression scales was shown within the first week and persisted for the duration of the trial (four to six weeks) [161]. |
| Target (Novel Pathway) |
Route | Study Type (n) | Summarized Trial Results |
| Metyrapone (HPA axis) | PO | DB-RCT (n=63) |
Inpatients with TRD received augmentation of nefazodone or fluvoxamine for 3 weeks with placebo versus 1 g of metyrapone once daily. The metyrapone group showed a significant improvement in depression scores compared with the placebo group [84]. |
| Biperiden (muscarinic) | PO | OL (n=10) | Trial showed significant improvement in depression scores for TRD inpatients given an average dose of 12mg biperiden per day for thirty days [164]. |
| Scopolamine (muscarinic) | IV | DB-RCT (n=18) | Sample included both bipolar and unipolar treatment refractory depression. Trial showed a rapid and robust improvement in depression scores with intravenous administration of scopolamine versus placebo [167]. |
| IV | DB-RCT (n=23) | This trial was a replication of the above trial except was exclusive for unipolar TRD. Trial showed a similar rapid and robust improvement in depression scores with intravenous administration of scopolamine versus placebo [168] | |
| Mecamylamine (nicotinic) | PO | DB-RCT (n=23) | Trial used mecamylamine as an augmenter of SSRIs for treatment of TRD, significantly improving depression scores compared to SSRI therapy alone during an 8-week trail [171] |
| Varenicline (nicotinic) | PO | OL (n=18) | Adult smokers with TRD were givenvarenicline, another nicotinic antagonist, as an adjunct to conventional therapy, lowering depression scores and facilitate smoking cessation [170, 172]. |
Several other trials of ketamine for TRD are currently underway as interest grows in NMDA modulation (NCT01920555, NCT01304147, NCT01179009, NCT00768430, NCT01945047, NCT01627782, NCT01582945, NCT01613820). Of note, intranasal ketamine has also been developed and shown to be effective in the treatment of MDD in a double-blind RCT [143]. This easier route of administration may further increase the feasibility of treatment, especially in an outpatient setting. While numerous trials have been conducted using ketamine, the clinical applicability is currently extremely limited due to the short duration of effect.
Long-term side effects of ketamine are yet to be assessed. Concern over side effects observed in patients abusing ketamine has prompted concern and stigmatization to the treatment [134]. However, it is important to note that observational data in these substance abusers should not be directly applied as the dosage varies greatly and it is unclear if these patients are co-administering other substances of abuse, altering the perceived side-effect profile interpreted from this patient populations.
Several other NMDA modulators have also been assessed for TRD. Riluzole, a glutamate modulator approved by the FDA for the treatment of amyotrophic lateral sclerosis has been of great interest [144]. Riluzole blocks voltage-gated sodium channels, thereby blocking glutamate release and enhancing astrocytic uptake of glutamate [144]. In a small open-label trail with nineteen TRD patients, riluzole (100-200mg/day oral) was used as a monotherapy for six weeks after a one-week drug free period. Significant improvement occurred during weeks three through six for all patients [145]. Another small open-label trial used riluzole as an augmenting agent for six to twelve weeks for ten TRD patients and showed a significant decline in HDRS and HARS ratings. The effect of riluzole was significant at the end of the first week of treatment and persisted for the twelve-week duration of the study [146]. While both of these studies yielded promising results, larger sample sizes and placebo controls are still required. Currently, Sanacora et al. are conducting a double-blinded eight-week RCT of adjunctive riluzolefor TRD (NCT01204918).
CP-101,606, a NR2-subunit specific NMDA receptor antagonist is also of interest. In a double-blinded RCT (n=30) using IV CP-101,606 as an adjunct to paroxetine for four weeks for TRD, significant improvement was found as the HDRS response rate was 60% for CP-101,606 versus 20% for placebo. Seventy-eight percent of CP-101,606-treated responders maintained this response status for at least one week post-infusion [147].
AZD6765, a low-trapping NMDA channel blocker was shown in a double-blinded RCT with 22 TRD patients that a single infusion of either AZD6765 (150 mg) on two test days one week apart was associated with rapid but short-lived antidepressant effects as shown through lowering of depression scores [96]. This effect has yet to be replicated in other studies.
Clinical trials have also been completed for D-cyclosporine (NMDA partial agonist), EVT 101 (NR2B subtype-selective NMDA receptor antagonist) and GLYX-13 (NMDA receptor glycine-site functional partial agonist), however, no results are available from these trials at the present time (ongoing trials summarized in Table 2). Taken together, several agents modulating NMDA show promise in the treatment of TRD; however, further trials are required to evaluate the efficacy and safety of long-term use.
Table 2.
Unpublished clinical trails(currently underway or completed without results available).
| Target (pathway) | Description with clinical trial identifier from ClinicalTrials.gov |
|---|---|
| Creatine (bioenergetics) | Several randomized controlled trials using creatine supplementation for TRD are currently underway. (NCT00729755, NCT01175616, NCT00313417, NCT00851006) NCT01601210 NCT02134808) |
| Ketamine (glutamate/NMDA) | Several other trials of ketamine for TRD are currently underway as interest grows in NMDA modulation (NCT01920555, NCT01304147, NCT01179009, NCT00768430, NCT01945047, NCT01627782, NCT01582945, NCT01613820). |
| Riluzole (glutamate) | Sanacora et al. are conducting a randomized, double-blind, placebo-controlled, 8 week trial of adjunctive riluzole for TRD (NCT01204918). |
| D-cycloserine (glutamate) | D-cycloserine, is a broad spectrum antibiotic that acts as a partial agonist at the NMDAR-associated GLY site. Randomized placebo controlled trial of conventional therapy with adjuvant with D-cycloserine, (up to 1 g/day) versus conventional therapy with placebo (NCT00408031). |
| EVT 101 (glutamate) | EVT101 is an orally active NR2B subtype-selective NMDA receptor antagonist. This clinical trial has been completed, however, no results have been published yet (NCT01128452). |
| GLYX-13 (IV) (glutamate) | GLYX-13 is an intravenous NMDA receptor glycine-site functional partial agonist. Clinical trial for TRD has been completed however no results have been published yet (NCT01234558). |
| RO4917523 (glutamate) | Trial assessed treatment of TRD with RO4917523,a mGluR5 allosteric antagonist however no results have been published yet (NCT01437657). |
| Buprenorphine (opioid) | Karp et al. has completed a double-blinded RCT of buprenorphine for TRD however results are currently unavailable (NCT01407575). |
| LY2456302 (opioid) | LY2456302 is a specific KOR antagonist currently being trialed for TRD in a double-blinded, placebo-controlled, proof-of-concept (POC) trial (NCT01913535). |
| Metyrapone (HPA axis) | Metyrpaone was assessed for TRD in a double-blinded, placebo-controlled twenty four week trial, however results have not yet been published (NCT01375920). |
| Oxytocin (HPA axis) | Intranasal oxytocin and tibolone are being trialed in a three-arm trial of oxytocin versus placebo versus oxytocin plus tibolone (NCT01239888). |
| Cysteamine (BDNF) | Cysteamine is a FDA approved agent for nephropathic cystinosis. It increases BDNF in the brain and promotes neuronal growth. Clinical trial for TRD has been terminated with no results available (NCT00715559). |
| Scopolamine + Ketamine (glutamate and muscarinic) | A combination intravenous injection of scopolamine with ketamine for TRD is currently being investigated in an open label trial (NCT01613820). |
AMPA modulation has also shown some promise. Preclinical models have shown that the anti-depressant affect of NMDA antagonists may be through antagonism of NMDA receptors on GABAergic interneurons which reduce the inhibition of pyramidal cells leading to increased release of glutamate on non-NMDA receptors, notably AMPA receptors, in the prefrontal cortex [148]. Therefore, the role of direct and specific AMPA stimulation has been questioned as another potential target for TRD. One potential target is Org 26576 that acts by increasing glutamate stimulation of AMPA receptors. Currently, safety and tolerability is still being assessed and no trials have yet been conducted in the TRD population [149].
Also of interest has been modulation of mGlu receptors. Metabotropic glutamate receptors are G-protein receptors located pre and post-synaptically. Of particular interest for TRD has been Group 1 mGlu receptors (mGluR1,5) as they are functionally coupled to NMDA receptors and preclinical data shows their antagonism inhibits depressive-like manifestations [15]. Several targets have emerged in preclinical models that bind to the various mGlu receptors, however, these results are yet to have been adequately translated into clinical trials [150]. Only one clinical trial targeting the mGlu system has been completed, to our knowledge, for the treatment of TRD with RO4917523, an mGluR5 allosteric antagonist (NCT01437657) and results are currently unavailable. The mechanism of action of these agents is currently unclear, however, preclinical models suggest that the final molecular effect may be similar to that of NMDA antagonists leading to mTOR activation and increased BDNF levels leading to neuro-protection and improved neuroplasticity [132, 150]. Therefore, targets of the mGlu receptors may also present novel opportunities in the treatment of TRD; however, due to a lack of clinical trials, their potential efficacy, safety and applicability remains unclear.
Interestingly, lithium, an old and well-established psychotropic medication for the treatment of bipolar disorder and an effective augmenting agent for TRD has also been shown to have modulatory effects on the glutamate system [15, 151-154]. More specifically, lithium has been shown to alter glutamate uptake and glutamate receptor expression and function. The mechanism of action of lithium has never been fully understood, however, initial hypotheses related to increased serotonin neurotransmission. Currently, several other pathways have been implicated including the glutamate system, intracellular signaling via glycogen synthase kinase-3 (GSK-3) leading to increased BDNF and potential anti-inflammatory effects [15, 151]. The multiple targets outside of the monoamine system may explain why lithium is such an effective augmenting agent in TRD and reinforce the importance of these novel pathways.
Opioid System
The primary therapeutic effect of analgesia from opiates has long been known and used to relieve pain since pre-historic times. The potent euphoric effects and high density of opioid receptors in the limbic brain areas point to a significant role of the opioid system in reward and mood regulation [155]. Opiates were also used to treat MDD until the early 1950s [156]. More recently, with the advent of long half-life mixed agonist/antagonist opiates that decrease the abuse and tolerance potential, modulation of opioid receptors for the treatment of MDD has been revisited [157]. Preclinical studies have pointed to a role of opioid mu, delta and kappa receptors (MORs, DORs and KORs) in the pathoetiology of MDD [157]. DOR agonists and KOR antagonists are currently being investigated for their role in TRD.
Animal models have shown that modulation of DOR, KOR and MOR may affect mood through several pathways including but not limited to alterations in monoamine and BDNF levels, neurogenesis and neuroplasticity, HPA axis stimulation and inflammatory pathways [157]. DOR agonists and KOR antagonists appear to favorably alter these systems (i.e. increase monoamine and BDNF levels and promote neuroplasticity) [158, 159] while the role of MOR is more unclear as MOR agonists appear to initially improve mood, however, chronic use may impair mood [157]. Interestingly, in patients with comorbid opiate addiction with MDD, some clinical observations suggest that chronic controlled opiate administration for maintenance therapy (stimulating primarily MOR) might increase the risk of TRD [157, 160]. Therefore, some investigators have proposed that while stimulating MOR (for maintenance therapy), co-administration of KOR antagonist and DOR agonists may serve to treat the comorbid MDD while not interfering with the treatment of the substance use disorder [157, 160]. With this high level of evidence from preclinical models in support of a role of the opioid system in the pathogenesis of MDD (further reviewed by Lutz [157]), several agents are currently being investigated as novel treatments of TRD.
Buprenorphine, a partial MOR agonist and KOR antagonist, was shown to have some effect for a subgroup of TRD patients in a small open label trial [161]. Karp et al. has completed a double-blinded RCT of buprenorphine for TRD however results are currently unavailable (NCT01407575). LY2456302 is a specific KOR antagonist currently also being evaluated for TRD (NCT01913535). Taken together, targets of the opioid system are currently being evaluated. Preclinical data shows promise for KOR antagonists and DOR agonists. While the role of MOR in TRD remains unclear. Further preclinical and clinical studies are required to determine the mechanism of action, safety of use and efficacy in TRD patients.
Cholinergic System
Janowsky (1972) conceived the “cholinergic-adrenergic hypothesis” of mood disorders several decades ago [162]. Briefly, the hypothesis supposed that an imbalanced increase in the parasympathetic (cholinergic) or sympathetic (adrenergic) systems could lead to depressive or manic symptoms, respectively. In recent years this theory has been revisited, most notably with the use of anti-cholinergic agents for the treatment of MDD [163]. Promising results in the treatment of MDD has led to further trials using anti-cholinergic agents for TRD [164]. The anticholinergic biperiden was shown in 1981 to induce significant improvement in a small open label study of TRD inpatients (n=10) [164]. More recently, anti-cholinergics including scopolamine, mecamylamine and varenicline have been and continue to be investigated.
Scopolamine is an anti-muscarinic agent that has also been shown to potentially modulate the glutamate system [165, 166]. Notably, animal models have shown the anti-muscarinic effects of scopolamine may decrease NMDAR gene expression, thereby having a similar downstream effect as ketamine and preventing glutamate mediated neurotoxicity [165]. Further, scopolamine was shown to induce the mTOR pathway with a similar magnitude and timing of ketamine leading to an increase in BDNF [165]. Anti-inflammatory and anti-oxidant effects of scopolamine have also been suspected [165]. Therefore, scopolamine may exert anti-depressant effects through several novel pathways.
Scopolamine was initially evaluated for its effect on cognition during which profound mood improvement was noted [165]. It was therefore evaluated in a double-blinded RCT showing a rapid and robust anti-depressant effect of intravenous scopolamine, as measured by improved depression ratings [167]. These results were replicated by a second RCT (n=23) [168]. Also of interest was the recent use of oral scopolamine as an augmenting agent for moderate to severe MDD. In this RCT, Khajavi et al. (2012) showed the safe and efficacious use of oral scopolamine, thus greatly increasing the feasibility of use in an outpatient setting [169]. Of note, a combination intravenous injection of scopolamine with ketamine for TRD is also currently being investigated in an open label trial (NCT01613820).
Antagonism of nicotinic acetylcholine receptors has also been shown in preclinical and clinical models to exert antidepressant effects [170]. Mecamylamine, a nicotinic antagonist was shown to be an efficacious augmenter of SSRIs for treatment of TRD in a small (n=23) eight-week RCT, showing significant improvement in HRDS scores compared to SSRI therapy alone [171]. As well, in a small open label trial with eighteen adult smokers with TRD, varenicline, another nicotinic antagonist, was shown to lower depression scores and facilitate smoking cessation [170, 172].
CONCLUSION
The need for novel targets acting outside of the monoamine system is of great importance as rates of TRD are high while using monoamine modulators alone. Several pathways have been presented which may yield novel targets for TRD. Many of these pathways are connected as shown in Fig. 1. For example, increased inflammation may lead to increased oxidative stress and over activity of the HPA axis, which has modulatory effects on the glutamate system, autonomic nervous system and monoamine system. Agents targeting each of these pathways were discussed. Of note, several of the agents discussed have been shown to affect multiple pathways directly and indirectly.
For many of the targets discussed, preclinical data is highly encouraging and informative of potential mechanisms underlying TRD. Clinical data for these agents is still greatly limited as many of the trials discussed were open label and/or had small numbers of participants. Therefore, the generalized clinical applicability in TRD remains uncertain. Further, the authors cannot recommend the clinical use of any of the agents discussed based on the evidence presented. Several clinical trials for these agents are currently underway and as such there is still hope for more definitive evidence in the near future in support for or against the targets discussed. Our review is narrative and this is an inherent limitation to our work. However, every effort was made to summarize available evidences from both preclinical studies as well as from both published and ongoing trials for TRD.
Another theme that emerged while reviewing the evidence for targets of the various pathways, was the importance of personalized medicine in TRD. Currently, algorithms for treatment of TRD are not personalized and are broadly applied to TRD patients, largely based on what agents have already been trialed. In using novel targets acting outside of the monoamine pathways, identifying the reason for TRD on an individual basis may allow correction of the specific cause of the TRD in the specific patient instead of simply using trial and error until an effective agent or combination of agents is found. The findings of Raison et al. [41] showing that infliximab had anti-depressant properties only for TRD patients with elevated inflammatory markers illustrated this concept. Therefore, the use of biomarkers to identify the dysfunctional pathway causing non-response to conventional therapies may inform which target is most likely to have success in a specific TRD patient. This form of treatment selection may also inform future clinical trials for TRD to more adequately choose a patient population of interest rather than grouping all TRD patients together purely based on poor treatment response to conventional therapies.
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- 4E-BP
= 4E binding protein
- AMPA
= alpha-amino-3-hydroxy-5-methylisoxazole methylisoxazole-4-propionic acid
- ASA
= acetylsalic acid
- ATP
= adenosine triphosphate
- BDI
= Beck Depression Inventory
- BDNF
= brain derived neurotropic factor
- COX-1 and COX-2
= cyclooxygenase-1 and -2
- DBS
= deep brain stimulation
- DPP-4
= dipeptidyl peptidase-4
- EAATs
= excitatory amino acid transporters
- ECT
= electroconvulsive therapy
- EPO
= erythropoietin
- ER
= endoplasmic reticulum
- ETC
= electron transport chain;
- GLP-1
= glucagon like peptide-1
- Glx
= glutamate-glutamine
- GSK-3
= glycogen synthase kinase-3
- HAM-D
= Hamilton Depression Rating Scale
- HARS
= Hamilton anxiety rating scale
- HDRS
= Hamilton depression rating scale
- HPA
= hypothalamic-pituitary-adrenal
- IDO
= indolamine 2,3-dioxygenase
- IDS
= Inventory of Depressive Symptomatology
- IL
= interleukin
- iNOS
= inducible NOS
- IV
= intravenous
- KA
= kainate
- MAOIs
= monoamine oxidase inhibitors
- MDD
= Major depressive disorder
- mGlu
= metabotropic glutamate
- MORs, DORs and KORs
= opioid mu, delta and kappa receptors
- mTOR
= mammalian target of rapamycin
- NAA
= N-acetyl-aspartate
- NAC
= N-acetylcysteine
- NADPH
= nicotinamide adenine dinucleotide phosphate
- NGF
= nerve growth factor
- NMDARs
= N-Methyl-D-asparate receptors
- nNOS
= neuronal NOS
- NNT
= number needed to treat
- NO
= nitric oxide
- NOS
= nitric oxide synthase
- NSAIDS
= non-steroidal anti-inflammatory drugs
- NT-3
= neurotrophin-3
- NT-4
= neurotrophin-4
- O&NS
= oxidative and nitrosative stress
- O3PUFA
= omega-3 polyunsaturated fatty acid
- OL
= open label
- PCOS
= polycystic ovarian syndrome
- PGE2
= prostaglandin E2
- PO
= oral
- PPARγ
= peroxisome proliferator-activated receptor gamma
- RCT
= randomized controlled trial
- RNS
= reactive nitrogen species
- ROS
= reactive oxygen species
- rTMS
= repetitive transcranial magnetic stimulations
- SAMe
= S-adenosyl-methionine
- SGAs
= second generation anti-psychotics
- SNRIs
= selective serotonin norepinephrine reuptake inhibitors
- SOD
= superoxide dismutase
- SSRIs
= selective serotonin reuptake inhibitors
- TCA
= tricarboxylic acid
- TCA
= tricyclic anti-depressants
- TDO
= tryptophan 2,3-dioxygenase
- TNF-alpha
= tumor necrosis factor alpha
- TRD
= treatment refractory depression
- TRYCATs
= tryptophan catabolites
- TZD
= thiazolidinedione
- VEGF
= vascular endothelial growth factor
- WHOQOL-BREF
= World Health Organization Quality of life-BREF
CONFLICTS OF INTEREST
JR, AFC and GSA have no conflicts of interest to declare. RSM declares that he has been on advisory boards and/orreceived honoraria for educational activities and/or research grants from AstraZeneca, Bristol-Myers Squibb, Janssen-Ortho, Eli Lily, Forest, Lundbeck, Pfizer, Shire, Merck, Sepracor and Otsuka. KNFhas received support concerning travel and accommodation expenses from various pharmaceutical companies to participate in medical congresses. He has also received honoraria for lectures and participated in advisory boards from Astra-Zeneca, Janssen-Cilag and Eli-Lilly. KNF also received two research grants from Pfizer Foundation.
REFERENCES
- 1.Kessler R.C., Akiskal H.S., Ames M., Birnbaum H., Greenberg P., Hirschfeld R.M., Jin R., Merikangas K.R., Simon G.E., Wang P.S. Prevalence and effects of mood disorders on work performance in a nationally representative sample of U.S. workers. 2006. [DOI] [PMC free article] [PubMed]
- 2.WHO. WHO, Depression Fact Sheet - http://www.who.int/mediacentre/ factsheets/fs369/en/. Updated 2012, accessed August 2014. 2012.
- 3.Gaynes B.N., Warden D., Trivedi M.H., Wisniewski S.R., Fava M., Rush A.J. Neuropsychological testing of cognitive impairment in euthymic bipolar disorder: an individual patient data meta-analysis. Acta Psychiatr. Scand. 2009;128:149–162. doi: 10.1111/acps.12133. [DOI] [PubMed] [Google Scholar]
- 4.McIntyre R.S., Filteau M.J., Martin L., Patry S., Carvalho A., Cha D.S., Barakat M., Miguelez M. Treatment-resistant depression: definitions, review of the evidence, and algorithmic approach. 2014. [DOI] [PubMed]
- 5.Carvalho A.F., Berk M., Hyphantis T.N., McIntyre R.S. The integrative management of treatment-resistant depression: a comprehensive review and perspectives. Psychother. Psychosom. 2014;83(2):70–88. doi: 10.1159/000357500. [DOI] [PubMed] [Google Scholar]
- 6.López-Muñoz F., Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr. Pharm. Des. 2009;15(14):1563–1586. doi: 10.2174/138161209788168001. [DOI] [PubMed] [Google Scholar]
- 7.Rizvi S.J., Grima E., Tan M., Rotzinger S., Lin P., Mcintyre R.S., Kennedy S.H. Treatment-resistant depression in primary care across Canada. Can. J. Psychiatry. 2014;59(7):349–357. doi: 10.1177/070674371405900702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas L., Kessler D., Campbell J., Morrison J., Peters T.J., Williams C., Lewis G., Wiles N. Prevalence of treatment-resistant depression in primary care: cross-sectional data. Br. J. Gen. Pract. 2013;63(617):e852–e858. doi: 10.3399/bjgp13X675430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenblat J.D., Cha D.S., Mansur R.B., McIntyre R.S. Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2014;53:23–34. doi: 10.1016/j.pnpbp.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Soczynska J.K., Mansur R.B., Brietzke E., Swardfager W., Kennedy S.H., Woldeyohannes H.O., Powell A.M., Manierka M.S., McIntyre R.S. Novel therapeutic targets in depression: minocycline as a candidate treatment. Behav. Brain Res. 2012;235(2):302–317. doi: 10.1016/j.bbr.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 11.McIntyre R.S., Powell A.M., Kaidanovich-Beilin O., Soczynska J.K., Alsuwaidan M., Woldeyohannes H.O., Kim A.S., Gallaugher L.A. The neuroprotective effects of GLP-1: possible treatments for cognitive deficits in individuals with mood disorders. 2013. [DOI] [PubMed]
- 12.Hashimoto K. The role of glutamate on the action of antidepressants. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2011;35(7):1558–1568. doi: 10.1016/j.pnpbp.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res. Brain Res. Rev. 2009;61(2):105–123. doi: 10.1016/j.brainresrev.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Machado-Vieira R., Manji H.K., Zarate C.A. The role of the tripartite glutamatergic synapse in the pathophysiology and therapeutics of mood disorders. Neuroscientist. 2009;15(5):525–539. doi: 10.1177/1073858409336093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Machado-Vieira R., Zarate C.A., Jr Proof of concept trials in bipolar disorder and major depressive disorder: a translational perspective in the search for improved treatments. Depress. Anxiety. 2011;28(4):267–281. doi: 10.1002/da.20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leonard B.E. Impact of inflammation on neurotransmitter changes in major depression: an insight into the action of antidepressants. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2014;48:261–267. doi: 10.1016/j.pnpbp.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 17.Lee S.Y., Lee S.J., Han C., Patkar A.A., Masand P.S., Pae C.U. Oxidative/nitrosative stress and antidepressants: targets for novel antidepressants. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2013;46:224–235. doi: 10.1016/j.pnpbp.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Boyce P., Hopwood M. Unmet needs in the treatment of schizophrenia: new targets to help different symptom domains. J. Clin. Psychiatry. 2013;75(Suppl 1):21–26. doi: 10.4088/JCP.13049su1c.04. [DOI] [PubMed] [Google Scholar]
- 19.Lanfumey L., Mongeau R., Hamon M. Biological rhythms and melatonin in mood disorders and their treatments. 2013. [DOI] [PubMed]
- 20.Raju T.N. The Nobel chronicles. 1927: Julius Wagner-Jauregg (1857-1940). Lancet. 1998;352((9141)):1714. doi: 10.1016/s0140-6736(05)61500-0. [DOI] [PubMed] [Google Scholar]
- 21.Brydon L., Walker C., Wawrzyniak A., Whitehead D., Okamura H., Yajima J., Tsuda A., Steptoe A. Synergistic effects of psychological and immune stressors on inflammatory cytokine and sickness responses in humans. Brain Behav. Immun. 2009;23(2):217–224. doi: 10.1016/j.bbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker J.R., Graff L.A., Dutz J.P., Bernstein C.N. Psychiatric disorders in patients with immune-mediated inflammatory diseases: prevalence, association with disease activity, and overall patient well-being. J. Rheumatol. Suppl. 2011;88:31–35. doi: 10.3899/jrheum.110900. [DOI] [PubMed] [Google Scholar]
- 23.Steptoe A., Hamer M., Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav. Immun. 2007;21(7):901–912. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 24.McNamara R.K., Lotrich F.E. Elevated immune-inflammatory signaling in mood disorders: a new therapeutic target? 2012. [DOI] [PMC free article] [PubMed]
- 25.Reichenberg A., Yirmiya R., Schuld A., Kraus T., Haack M., Morag A., Pollmächer T. Cytokine-associated emotional and cognitive disturbances in humans. Arch. Gen. Psychiatry. 2001;58(5):445–452. doi: 10.1001/archpsyc.58.5.445. [DOI] [PubMed] [Google Scholar]
- 26.Wright C.E., Strike P.C., Brydon L., Steptoe A. A. Acute inflammation and negative mood: mediation by cytokine activation. Brain Behave. Immun. 2005;19(4):345–350. doi: 10.1016/j.bbi.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Raison C.L., Broadwell S.D., Borisov A.S., Manatunga A.K., Capuron L., Woolwine B.J., Jacobson I.M., Nemeroff C.B., Miller A.H. Depressive symptoms and viral clearance in patients receiving interferon-alpha and ribavirin for hepatitis C. Brain Behav. Immun. 2005;19(1):23–27. doi: 10.1016/j.bbi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Dunn A.J., Swiergiel A.H., de Beaurepaire R. Cytokines as mediators of depression: what can we learn from animal studies? Neurosci. Biobehav. Rev. 2005;29(4-5):891–909. doi: 10.1016/j.neubiorev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 29.Carvalho L.A., Torre J.P., Papadopoulos A.S., Poon L., Juruena M.F., Markopoulou K., Cleare A.J., Pariante C.M. Lack of clinical therapeutic benefit of antidepressants is associated overall activation of the inflammatory system. J. Affect. Disord. 2013;148(1):136–140. doi: 10.1016/j.jad.2012.10.036. [DOI] [PubMed] [Google Scholar]
- 30.Capuron L., Neurauter G., Musselman D.L., Lawson D.H., Nemeroff C.B., Fuchs D., Miller A.H. Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol. Psychiatry. 2003;54(9):906–914. doi: 10.1016/S0006-3223(03)00173-2. [DOI] [PubMed] [Google Scholar]
- 31.Capuron L., Ravaud A., Neveu P.J., Miller A.H., Maes M., Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol. Psychiatry. 2002;7(5):468–473. doi: 10.1038/sj.mp.4000995. [DOI] [PubMed] [Google Scholar]
- 32.Harry G.J., Kraft A.D. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology. 2012;33(2):191–206. doi: 10.1016/j.neuro.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ekdahl C.T. 2012.
- 34.Lifschytz T., Segman R., Shalom G., Lerer B., Gur E., Golzer T., Newman M.E. Basic mechanisms of augmentation of antidepressant effects with thyroid hormone. Curr. Drug Targets. 2006;7(2):203–210. doi: 10.2174/138945006775515482. [DOI] [PubMed] [Google Scholar]
- 35.Miller A.H., Haroon E., Raison C.L., Felger J.C. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress. Anxiety. 2013;30(4):297–306. doi: 10.1002/da.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raison C.L., Miller A.H. Malaise, melancholia and madness: the evolutionary legacy of an inflammatory bias. Brain Behav. Immun. 2013;31:1–8. doi: 10.1016/j.bbi.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stertz L., Magalhaes P.V., Kapczinski F. Is bipolar disorder an inflammatory condition? The relevance of microglial activation. 2013. [DOI] [PubMed]
- 38.Hashimoto K., Malchow B., Falkai P., Schmitt A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2013;263(5):367–377. doi: 10.1007/s00406-013-0399-y. [DOI] [PubMed] [Google Scholar]
- 39.Sigalas P.D., Garg H., Watson S., McAllister-Williams R.H., Ferrier I.N. Metyrapone in treatment-resistant depression. 2012. [DOI] [PMC free article] [PubMed]
- 40.Cowen P.J. Not fade away: the HPA axis and depression. Psychol. Med. 2010;40(1):1–4. doi: 10.1017/S0033291709005558. [DOI] [PubMed] [Google Scholar]
- 41.Raison C.L., Rutherford R.E., Woolwine B.J., Shuo C., Schettler P., Drake D.F., Haroon E., Miller A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramiro S., Gaujoux-Viala C., Nam J.L., Smolen J.S., Buch M., Gossec L., van der Heijde D., Winthrop K., Landewé R. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2013 update of the EULAR recommendations for management of rheumatoid arthritis. Ann. Rheum. Dis. 2014;73(3):529–535. doi: 10.1136/annrheumdis-2013-204575. [DOI] [PubMed] [Google Scholar]
- 43.Vane J.R., Botting R.M. The mechanism of action of aspirin. Thromb. Res. 2003;110(5-6):255–258. doi: 10.1016/S0049-3848(03)00379-7. [DOI] [PubMed] [Google Scholar]
- 44.Berk M., Dean O., Drexhage H., McNeil J.J., Moylan S., O’Neil A., Davey C.G., Sanna L., Maes M. Aspirin: a review of its neurobiological properties and therapeutic potential for mental illness. BMC Med. 2013;11:74. doi: 10.1186/1741-7015-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldstein B.I., Kemp D.E., Soczynska J.K., McIntyre R.S. Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J. Clin. Psychiatry. 2009;70(8):1078–1090. doi: 10.4088/JCP.08r04505. [DOI] [PubMed] [Google Scholar]
- 46.Müller N. COX-2 inhibitors as antidepressants and antipsychotics: clinical evidence. Curr. Opin. Investig. Drugs. 2010;11(1):31–42. [PubMed] [Google Scholar]
- 47.Abbasi S.H., Hosseini F., Modabbernia A., Ashrafi M., Akhondzadeh S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J. Affect. Disord. 2012;141(2-3):308–314. doi: 10.1016/j.jad.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 48.Faridhosseini F., Sadeghi R., Farid L., Pourgholami M. Celecoxib: a new augmentation strategy for depressive mood episodes. A systematic review and meta-analysis of randomized placebo-controlled trials. Hum. Psychopharmacol. 2014;29(3):216–223. doi: 10.1002/hup.2401. [DOI] [PubMed] [Google Scholar]
- 49.Na K.S., Lee K.J., Lee J.S., Cho Y.S., Jung H.Y. Efficacy of adjunctive celecoxib treatment for patients with major depressive disorder: a meta-analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2014;48:79–85. doi: 10.1016/j.pnpbp.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 50.Green G.A. Understanding NSAIDs: from aspirin to COX-2. Clin. Cornerstone. 2001;3(5):50–60. doi: 10.1016/S1098-3597(01)90069-9. [DOI] [PubMed] [Google Scholar]
- 51.Simopoulos A.P. Omega-3 fatty acids in health and disease and in growth and development. Am. J. Clin. Nutr. 1991;54(3):438–463. doi: 10.1093/ajcn/54.3.438. [DOI] [PubMed] [Google Scholar]
- 52.Calder P.C. The relationship between the fatty acid composition of immune cells and their function. 2008. [DOI] [PubMed]
- 53.Gertsik L., Poland R.E., Bresee C., Rapaport M.H. Omega-3 fatty acid augmentation of citalopram treatment for patients with major depressive disorder. J. Clin. Psychopharmacol. 2012;32(1):61–64. doi: 10.1097/JCP.0b013e31823f3b5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jazayeri S., Tehrani-Doost M., Keshavarz S.A., Hosseini M., Djazayery A., Amini H., Jalali M., Peet M. Comparison of therapeutic effects of omega-3 fatty acid eicosapentaenoic acid and fluoxetine, separately and in combination, in major depressive disorder. Aust. N. Z. J. Psychiatry. 2008;42(3):192–198. doi: 10.1080/00048670701827275. [DOI] [PubMed] [Google Scholar]
- 55.Peet M., Horrobin D.F. A dose-ranging study of the effects of ethyl-eicosapentaenoate in patients with ongoing depression despite apparently adequate treatment with standard drugs. Arch. Gen. Psychiatry. 2002;59(10):913–919. doi: 10.1001/archpsyc.59.10.913. [DOI] [PubMed] [Google Scholar]
- 56.Lopresti A.L., Hood S.D., Drummond P.D. 2012.
- 57.Bergman J., Miodownik C., Bersudsky Y., Sokolik S., Lerner P.P., Kreinin A., Polakiewicz J., Lerner V. Curcumin as an add-on to antidepressive treatment: a randomized, double-blind, placebo-controlled, pilot clinical study. Clin. Neuropharmacol. 2013;36(3):73–77. doi: 10.1097/WNF.0b013e31828ef969. [DOI] [PubMed] [Google Scholar]
- 58.Sanmukhani J., Satodia V., Trivedi J., Patel T., Tiwari D., Panchal B., Goel A., Tripathi C.B. Efficacy and safety of curcumin in major depressive disorder: a randomized controlled trial. 2014. [DOI] [PubMed]
- 59.Lopresti A.L., Maes M., Maker G.L., Hood S.D., Drummond P.D. Curcumin for the treatment of major depression: a randomised, double-blind, placebo controlled study. 2014. [DOI] [PubMed]
- 60.Levine J., Cholestoy A., Zimmerman J. Possible antidepressant effect of minocycline. Am. J. Psychiatry. 1996;153(4):582. doi: 10.1176/ajp.153.4.582b. [DOI] [PubMed] [Google Scholar]
- 61.Savitz J., Preskorn S., Teague T.K., Drevets D., Yates W., Drevets W. Minocycline and aspirin in the treatment of bipolar depression: a protocol for a proof-of-concept, randomised, double-blind, placebo-controlled, 2x2 clinical trial. BMJ Open. 2012;2(1):e000643. doi: 10.1136/bmjopen-2011-000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mello B.S., Monte A.S., McIntyre R.S., Soczynska J.K., Custódio C.S., Cordeiro R.C., Chaves J.H., Vasconcelos S.M., Nobre H.V., Jr, Florenço de Sousa F.C., Hyphantis T.N., Carvalho A.F., Macêdo D.S. Effects of doxycycline on depressive-like behavior in mice after lipopolysaccharide (LPS) administration. J. Psychiatr. Res. 2013;47(10):1521–1529. doi: 10.1016/j.jpsychires.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 63.Rawdin B.J., Mellon S.H., Dhabhar F.S., Epel E.S., Puterman E., Su Y., Burke H.M., Reus V.I., Rosser R., Hamilton S.P., Nelson J.C., Wolkowitz O.M. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav. Immun. 2013;31:143–152. doi: 10.1016/j.bbi.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gill R., Tsung A., Billiar T. 2010.
- 65.Anderson G., Berk M., Dean O., Moylan S., Maes M. Role of immune-inflammatory and oxidative and nitrosative stress pathways in the etiology of depression: therapeutic implications. CNS Drugs. 2014;28(1):1–10. doi: 10.1007/s40263-013-0119-1. [DOI] [PubMed] [Google Scholar]
- 66.Pompella A., Visvikis A., Paolicchi A., De Tata V., Casini A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003;66(8):1499–1503. doi: 10.1016/S0006-2952(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 67.Aboul-Fotouh S. Coenzyme Q10 displays antidepressant-like activity with reduction of hippocampal oxidative/nitrosative DNA damage in chronically stressed rats. Pharmacol. Biochem. Behav. 2013;104:105–112. doi: 10.1016/j.pbb.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 68.Morris G., Anderson G., Berk M., Maes M. Coenzyme Q10 depletion in medical and neuropsychiatric disorders: potential repercussions and therapeutic implications. Mol. Neurobiol. 2013;48(3):883–903. doi: 10.1007/s12035-013-8477-8. [DOI] [PubMed] [Google Scholar]
- 69.Acuña-Castroviejo D., Martín M., Macías M., Escames G., León J., Khaldy H., Reiter R.J. Melatonin, mitochondria, and cellular bioenergetics. J. Pineal Res. 2001;30(2):65–74. doi: 10.1034/j.1600-079X.2001.300201.x. [DOI] [PubMed] [Google Scholar]
- 70.Rich P.R. The molecular machinery of Keilin’s respiratory chain. Biochem. Soc. Trans. 2003;31(Pt 6):1095–1105. doi: 10.1042/bst0311095. [DOI] [PubMed] [Google Scholar]
- 71.Belanger M., Allaman I., Magistretti P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. 2011. [DOI] [PubMed]
- 72.Moylan S., Berk M., Dean O.M., Samuni Y., Williams L.J., O’Neil A., Hayley A.C., Pasco J.A., Anderson G., Jacka F.N., Maes M. Oxidative & nitrosative stress in depression: why so much stress? Neurosci. Biobehav. Rev. 2014;45:46–62. doi: 10.1016/j.neubiorev.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 73.Moylan S., Maes M., Wray N.R., Berk M. The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol. Psychiatry. 2013;18(5):595–606. doi: 10.1038/mp.2012.33. [DOI] [PubMed] [Google Scholar]
- 74.Zhou L., Zhu D.Y. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20(4):223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 75.Eu J.P., Liu L., Zeng M., Stamler J.S. An apoptotic model for nitrosative stress. Biochemistry. 2000;39(5):1040–1047. doi: 10.1021/bi992046e. [DOI] [PubMed] [Google Scholar]
- 76.Palta P., Samuel L.J., Miller E.R., III, Szanton S.L. Depression and oxidative stress: results from a meta-analysis of observational studies. 2014. [DOI] [PMC free article] [PubMed]
- 77.Berk M., Malhi G.S., Gray L.J., Dean O.M. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci. 2013;34(3):167–177. doi: 10.1016/j.tips.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 78.Das P., Tanious M., Fritz K., Dodd S., Dean O.M., Berk M., Malhi G.S. Metabolite profiles in the anterior cingulate cortex of depressed patients differentiate those taking N-acetyl-cysteine versus placebo. Aust. N. Z. J. Psychiatry. 2013;47(4):347–354. doi: 10.1177/0004867412474074. [DOI] [PubMed] [Google Scholar]
- 79.Berk M., Dean O.M., Cotton S.M., Jeavons S., Tanious M., Kohlmann K., Hewitt K., Moss K., Allwang C., Schapkaitz I., Robbins J., Cobb H., Ng F., Dodd S., Bush A.I., Malhi G.S. The efficacy of adjunctive N-acetylcysteine in major depressive disorder: a double-blind, randomized, placebo-controlled trial. J. Clin. Psychiatry. 2014;75(6):628–636. doi: 10.4088/JCP.13m08454. [DOI] [PubMed] [Google Scholar]
- 80.Miller G.E., Chen E., Zhou E.S. If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychol. Bull. 2007;133(1):25–45. doi: 10.1037/0033-2909.133.1.25. [DOI] [PubMed] [Google Scholar]
- 81.McAllister-Williams R.H., Ferrier I.N., Young A.H. Mood and neuropsychological function in depression: the role of corticosteroids and serotonin. Psychol. Med. 1998;28(3):573–584. doi: 10.1017/S0033291798006680. [DOI] [PubMed] [Google Scholar]
- 82.Murphy B.E. Steroids and depression. J. Steroid Biochem. Mol. Biol. 1991;38(5):537–559. doi: 10.1016/0960-0760(91)90312-S. [DOI] [PubMed] [Google Scholar]
- 83.Sanacora G., Treccani G., Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62(1):63–77. doi: 10.1016/j.neuropharm.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jahn H., Schick M., Kiefer F., Kellner M., Yassouridis A., Wiedemann K. Metyrapone as additive treatment in major depression: a double-blind and placebo-controlled trial. Arch. Gen. Psychiatry. 2004;61(12):1235–1244. doi: 10.1001/archpsyc.61.12.1235. [DOI] [PubMed] [Google Scholar]
- 85.Cardoso C., Kingdon D., Ellenbogen M.A. A meta-analytic review of the impact of intranasal oxytocin administration on cortisol concentrations during laboratory tasks: moderation by method and mental health. Psychoneuroendocrinology. 2014;49:161–170. doi: 10.1016/j.psyneuen.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 86.Keating C., Dawood T., Barton D.A., Lambert G.W., Tilbrook A.J. Effects of selective serotonin reuptake inhibitor treatment on plasma oxytocin and cortisol in major depressive disorder. BMC Psychiatry. 2013;13:124. doi: 10.1186/1471-244X-13-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller A.H., Maletic V., Raison C.L. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry. 2009;65(9):732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vancampfort D., Correll C.U., Wampers M., Sienaert P., Mitchell A.J., De Herdt A., Probst M., Scheewe T.W., De Hert M. Metabolic syndrome and metabolic abnormalities in patients with major depressive disorder: a meta-analysis of prevalences and moderating variables. Psychol. Med. 2013;•••:1–12. doi: 10.1017/S0033291713002778. [DOI] [PubMed] [Google Scholar]
- 89.Kozumplik O., Uzun S. Metabolic syndrome in patients with depressive disorder--features of comorbidity. Psychiatr. Danub. 2011;23(1):84–88. [PubMed] [Google Scholar]
- 90.Kan C., Silva N., Golden S.H., Rajala U., Timonen M., Stahl D., Ismail K. A systematic review and meta-analysis of the association between depression and insulin resistance. Diabetes Care. 2013;36(2):480–489. doi: 10.2337/dc12-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McNay E.C., Recknagel A.K. Brain insulin signaling: a key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. 2011. [DOI] [PMC free article] [PubMed]
- 92.Karunakaran U., Park K.G. A systematic review of oxidative stress and safety of antioxidants in diabetes: focus on islets and their defense. Diabetes Metab. J. 2013;37(2):106–112. doi: 10.4093/dmj.2013.37.2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Katon W., von Korff M., Ciechanowski P., Russo J., Lin E., Simon G., Ludman E., Walker E., Bush T., Young B. Behavioral and clinical factors associated with depression among individuals with diabetes. Diabetes Care. 2004;27(4):914–920. doi: 10.2337/diacare.27.4.914. [DOI] [PubMed] [Google Scholar]
- 94.Mansur R.B., Cha D.S., Woldeyohannes H.O., Soczynska J.K., Zugman A., Brietzke E., McIntyre R.S. Diabetes mellitus and disturbances in brain connectivity: a bidirectional relationship? Neuromolecular Med. 2014;16(4):658–668. doi: 10.1007/s12017-014-8316-8. [DOI] [PubMed] [Google Scholar]
- 95.Rotte M., Baerecke C., Pottag G., Klose S., Kanneberg E., Heinze H.J., Lehnert H. Insulin affects the neuronal response in the medial temporal lobe in humans. Neuroendocrinology. 2005;81(1):49–55. doi: 10.1159/000084874. [DOI] [PubMed] [Google Scholar]
- 96.Strachan M.W. Insulin and cognitive function in humans: experimental data and therapeutic considerations. Biochem. Soc. Trans. 2005;33(Pt 5):1037–1040. doi: 10.1042/BST0331037. [DOI] [PubMed] [Google Scholar]
- 97.Benedict C., Hallschmid M., Schultes B., Born J., Kern W. Intranasal insulin to improve memory function in humans. 2007. [DOI] [PubMed]
- 98.Benedict L., Nelson C.A., Schunk E., Sullwold K., Seaquist E.R. Effect of insulin on the brain activity obtained during visual and memory tasks in healthy human subjects. Neuroendocrinology. 2006;83(1):20–26. doi: 10.1159/000093338. [DOI] [PubMed] [Google Scholar]
- 99.Shemesh E., Rudich A., Harman-Boehm I., Cukierman-Yaffe T. Effect of intranasal insulin on cognitive function: a systematic review. J. Clin. Endocrinol. Metab. 2012;97(2):366–376. doi: 10.1210/jc.2011-1802. [DOI] [PubMed] [Google Scholar]
- 100.McIntyre R.S., Soczynska J.K., Woldeyohannes H.O., Lewis G.F., Leiter L.A., MacQueen G.M., Miranda A., Fulgosi D., Konarski J.Z., Kennedy S.H. Thiazolidinediones: novel treatments for cognitive deficits in mood disorders? Expert Opin. Pharmacother. 2007;8(11):1615–1628. doi: 10.1517/14656566.8.11.1615. [DOI] [PubMed] [Google Scholar]
- 101.Salehi-Sadaghiani M., Javadi-Paydar M., Gharedaghi M.H., Zandieh A., Heydarpour P., Yousefzadeh-Fard Y., Dehpour A.R. NMDA receptor involvement in antidepressant-like effect of pioglitazone in the forced swimming test in mice. Psychopharmacology (Berl.) 2012;223(3):345–355. doi: 10.1007/s00213-012-2722-0. [DOI] [PubMed] [Google Scholar]
- 102.Gold P.W., Licinio J., Pavlatou M.G. Pathological parainflammation and endoplasmic reticulum stress in depression: potential translational targets through the CNS insulin, klotho and PPAR-γ systems. Mol. Psychiatry. 2013;18(2):154–165. doi: 10.1038/mp.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 104.Hotamisligil G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kemp D.E., Ismail-Beigi F., Ganocy S.J., Conroy C., Gao K., Obral S., Fein E., Findling R.L., Calabrese J.R. Use of insulin sensitizers for the treatment of major depressive disorder: a pilot study of pioglitazone for major depression accompanied by abdominal obesity. J. Affect. Disord. 2012;136(3):1164–1173. doi: 10.1016/j.jad.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sepanjnia K., Modabbernia A., Ashrafi M., Modabbernia M.J., Akhondzadeh S. Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: randomized double-blind placebo-controlled trial. Neuropsychopharmacology. 2012;37(9):2093–2100. doi: 10.1038/npp.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kashani L., Omidvar T., Farazmand B., Modabbernia A., Ramzanzadeh F., Tehraninejad E.S., Ashrafi M., Tabrizi M., Akhondzadeh S. Does pioglitazone improve depression through insulin-sensitization? Results of a randomized double-blind metformin-controlled trial in patients with polycystic ovarian syndrome and comorbid depression. 2013. [DOI] [PubMed]
- 108.Kemp D.E., Ismail-Beigi F., Calabrese J.R. Antidepressant response associated with pioglitazone: support for an overlapping pathophysiology between major depression and metabolic syndrome. Am. J. Psychiatry. 2009;166(5):619. doi: 10.1176/appi.ajp.2008.08081195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.de Sousa R.T., Machado-Vieira R., Zarate C.A., Jr, Manji H.K. Targeting mitochondrially mediated plasticity to develop improved therapeutics for bipolar disorder. 2014. [DOI] [PMC free article] [PubMed]
- 110.Allen P.J. Creatine metabolism and psychiatric disorders: Does creatine supplementation have therapeutic value? Neurosci. Biobehav. Rev. 2012;36(5):1442–1462. doi: 10.1016/j.neubiorev.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Iosifescu D.V., Bolo N.R., Nierenberg A.A., Jensen J.E., Fava M., Renshaw P.F. Brain bioenergetics and response to triiodothyronine augmentation in major depressive disorder. Biol. Psychiatry. 2008;63(12):1127–1134. doi: 10.1016/j.biopsych.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 112.Nierenberg A.A., Kansky C., Brennan B.P., Shelton R.C., Perlis R., Iosifescu D.V. Mitochondrial modulators for bipolar disorder: a pathophysiologically informed paradigm for new drug development. Aust. N. Z. J. Psychiatry. 2013;47(1):26–42. doi: 10.1177/0004867412449303. [DOI] [PubMed] [Google Scholar]
- 113.Roitman S., Green T., Osher Y., Karni N., Levine J. 2007. [DOI] [PubMed]
- 114.Kondo D.G., Sung Y.H., Hellem T.L., Fiedler K.K., Shi X., Jeong E.K., Renshaw P.F. 2011. [DOI] [PMC free article] [PubMed]
- 115.Papakostas G.I., Alpert J.E., Fava M. S-adenosyl-methionine in depression: a comprehensive review of the literature. Curr. Psychiatry Rep. 2003;5(6):460–466. doi: 10.1007/s11920-003-0085-2. [DOI] [PubMed] [Google Scholar]
- 116.Papakostas G.I., Cassiello C.F., Iovieno N. Folates and S-adenosylmethionine for major depressive disorder. Can. J. Psychiatry. 2012;57(7):406–413. doi: 10.1177/070674371205700703. [DOI] [PubMed] [Google Scholar]
- 117.Papakostas G.I., Fava M., Thase M.E. Treatment of SSRI-resistant depression: a meta-analysis comparing within- versus across-class switches. Biol. Psychiatry. 2008;63(7):699–704. doi: 10.1016/j.biopsych.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 118.Alpert J.E., Papakostas G., Mischoulon D., Worthington J.J., III, Petersen T., Mahal Y., Burns A., Bottiglieri T., Nierenberg A.A., Fava M. S-adenosyl-L-methionine (SAMe) as an adjunct for resistant major depressive disorder: an open trial following partial or nonresponse to selective serotonin reuptake inhibitors or venlafaxine. J. Clin. Psychopharmacol. 2004;24(6):661–664. doi: 10.1097/01.jcp.0000145339.45794.cd. [DOI] [PubMed] [Google Scholar]
- 119.Papakostas G.I., Mischoulon D., Shyu I., Alpert J.E., Fava M. 2010.
- 120.Papakostas G.I., Shelton R.C., Zajecka J.M., Etemad B., Rickels K., Clain A., Baer L., Dalton E.D., Sacco G.R., Schoenfeld D., Pencina M., Meisner A., Bottiglieri T., Nelson E., Mischoulon D., Alpert J.E., Barbee J.G., Zisook S., Fava M. L-methylfolate as adjunctive therapy for SSRI-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am. J. Psychiatry. 2012;169(12):1267–1274. doi: 10.1176/appi.ajp.2012.11071114. [DOI] [PubMed] [Google Scholar]
- 121.Farah A. The role of L-methylfolate in depressive disorders. CNS Spectr. 2009;14(1) Suppl. 2:2–7. doi: 10.1017/s1092852900003473. [DOI] [PubMed] [Google Scholar]
- 122.Miller A.L. The methylation, neurotransmitter, and antioxidant connections between folate and depression. Altern. Med. Rev. 2008;13(3):216–226. [PubMed] [Google Scholar]
- 123.Owen R.T. Folate augmentation of antidepressant response. 2013. [DOI] [PubMed]
- 124.Fava M., Mischoulon D. Folate in depression: efficacy, safety, differences in formulations, and clinical issues. J. Clin. Psychiatry. 2009;70(70) Suppl. 5:12–17. doi: 10.4088/JCP.8157su1c.03. [DOI] [PubMed] [Google Scholar]
- 125.Reichardt L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006;361(1473):1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Duman R.S., Aghajanian G.K. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338(6103):68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ota K.T., Duman R.S. Environmental and pharmacological modulations of cellular plasticity: role in the pathophysiology and treatment of depression. Neurobiol. Dis. 2013;57:28–37. doi: 10.1016/j.nbd.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Duman R S., Li N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. 2012. [DOI] [PMC free article] [PubMed]
- 129.Ogunshola O.O., Bogdanova A.Y. Epo and non-hematopoietic cells: what do we know? Methods Mol. Biol. 2013;982:13–41. doi: 10.1007/978-1-62703-308-4_2. [DOI] [PubMed] [Google Scholar]
- 130.Miskowiak K.W., Vinberg M., Harmer C.J., Ehrenreich H., Kessing L.V. Erythropoietin: a candidate treatment for mood symptoms and memory dysfunction in depression. 2012. [DOI] [PubMed]
- 131.Miskowiak K.W., Vinberg M., Christensen E.M., Bukh J.D., Harmer C.J., Ehrenreich H., Kessing L.V. Recombinant human erythropoietin for treating treatment-resistant depression: a double-blind, randomized, placebo-controlled phase 2 trial. Neuropsychopharmacology. 2014;39(6):1399–1408. doi: 10.1038/npp.2013.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Abelaira H.M., Réus G.Z., Neotti M.V., Quevedo J. The role of mTOR in depression and antidepressant responses. Life Sci. 2014;101(1-2):10–14. doi: 10.1016/j.lfs.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 133.Niciu M.J., Henter I.D., Luckenbaugh D.A., Zarate C.A., Jr, Charney D.S. Glutamate receptor antagonists as fast-acting therapeutic alternatives for the treatment of depression: ketamine and other compounds. Annu. Rev. Pharmacol. Toxicol. 2014;54:119–139. doi: 10.1146/annurev-pharmtox-011613-135950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Browne C.A., Lucki I. Antidepressant effects of ketamine: mechanisms underlying fast-acting novel antidepressants. 2013. [DOI] [PMC free article] [PubMed]
- 135.McGirr A., Berlim M.T., Bond D.J., Fleck M.P., Yatham L.N., Lam R.W. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol. Med. 2014;•••:1–12. doi: 10.1017/S0033291714001603. [DOI] [PubMed] [Google Scholar]
- 136.Zarate C.A., Jr, Singh J.B., Carlson P.J., Brutsche N.E., Ameli R., Luckenbaugh D.A., Charney D.S., Manji H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry. 2006;63(8):856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 137.Murrough J.W., Iosifescu D.V., Chang L.C., Al Jurdi R.K., Green C.E., Perez A.M., Iqbal S., Pillemer S., Foulkes A., Shah A., Charney D.S., Mathew S.J. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am. J. Psychiatry. 2013;170(10):1134–1142. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Torjesen I. Ketamine helps a third of patients with treatment resistant depression, finds small UK study. BMJ. 2014;348:g2576. doi: 10.1136/bmj.g2576. [DOI] [PubMed] [Google Scholar]
- 139.Murrough J.W., Perez A.M., Pillemer S., Stern J., Parides M.K. 2013.
- 140.Diamond P.R., Farmery A.D., Atkinson S., Haldar J., Williams N., Cowen P.J., Geddes J.R., McShane R. Ketamine infusions for treatment resistant depression: a series of 28 patients treated weekly or twice weekly in an ECT clinic. J. Psychopharmacol. (Oxford) 2014;28(6):536–544. doi: 10.1177/0269881114527361. [DOI] [PubMed] [Google Scholar]
- 141.Price R.B., Iosifescu D.V., Murrough J.W., Chang L.C., Al Jurdi R.K., Iqbal S.Z., Soleimani L., Charney D.S., Foulkes A.L., Mathew S.J. Effects of ketamine on explicit and implicit suicidal cognition: a randomized controlled trial in treatment-resistant depression. Depress. Anxiety. 2014;31(4):335–343. doi: 10.1002/da.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ballard E.D., Ionescu D.F., Vande Voort J.L., Niciu M.J., Richards E.M., Luckenbaugh D.A., Brutsché N.E., Ameli R., Furey M.L., Zarate C.A., Jr Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J. Psychiatr. Res. 2014;58:161–166. doi: 10.1016/j.jpsychires.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lapidus K.A., Levitch C.F., Perez A.M., Brallier J.W., Parides M.K., Soleimani L., Feder A., Iosifescu D.V., Charney D.S., Murrough J.W. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol. Psychiatry. 2014;76(12):970–976. doi: 10.1016/j.biopsych.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Owen R.T. Glutamatergic approaches in major depressive disorder: focus on ketamine, memantine and riluzole. Drugs Today (Barc) 2012;48(7):469–478. doi: 10.1358/dot.2012.48.7.1832873. [DOI] [PubMed] [Google Scholar]
- 145.Zarate C.A., Jr, Payne J.L., Quiroz J., Sporn J., Denicoff K.K., Luckenbaugh D., Charney D.S., Manji H.K. An open-label trial of riluzole in patients with treatment-resistant major depression. Am. J. Psychiatry. 2004;161(1):171–174. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- 146.Sanacora G., Kendell S.F., Levin Y., Simen A.A., Fenton L.R., Coric V., Krystal J.H. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol. Psychiatry. 2007;61(6):822–825. doi: 10.1016/j.biopsych.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Preskorn S.H., Baker B., Kolluri S., Menniti F.S., Krams M., Landen J.W. 2008. [DOI] [PubMed]
- 148.Moghaddam B., Adams B., Verma A., Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997;17(8):2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nations K.R., Bursi R., Dogterom P., Ereshefsky L., Gertsik L., Mant T., Schipper J. Maximum tolerated dose evaluation of the AMPA modulator Org 26576 in healthy volunteers and depressed patients: a summary and method analysis of bridging research in support of phase II dose selection. Drugs R D. 2012;12(3):127–139. doi: 10.2165/11634360-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tokita K., Yamaji T., Hashimoto K. Roles of glutamate signaling in preclinical and/or mechanistic models of depression. Pharmacol. Biochem. Behav. 2012;100(4):688–704. doi: 10.1016/j.pbb.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 151.Bauer M., Adli M., Bschor T., Pilhatsch M., Pfennig A., Sasse J., Schmid R., Lewitzka U. Lithium’s emerging role in the treatment of refractory major depressive episodes: augmentation of antidepressants. Neuropsychobiology. 2010;62(1):36–42. doi: 10.1159/000314308. [DOI] [PubMed] [Google Scholar]
- 152.Du J., Gray N.A., Falke C.A., Chen W., Yuan P., Szabo S.T., Einat H., Manji H.K. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J. Neurosci. 2004;24(29):6578–6589. doi: 10.1523/JNEUROSCI.1258-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nonaka S., Chuang D.M. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9(9):2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- 154.Hashimoto R., Hough C., Nakazawa T., Yamamoto T., Chuang D.M. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J. Neurochem. 2002;80(4):589–597. doi: 10.1046/j.0022-3042.2001.00728.x. [DOI] [PubMed] [Google Scholar]
- 155.Berrocoso E., Sánchez-Blázquez P., Garzón J., Mico J.A. Opiates as antidepressants. Curr. Pharm. Des. 2009;15(14):1612–1622. doi: 10.2174/138161209788168100. [DOI] [PubMed] [Google Scholar]
- 156.Tenore P.L. Psychotherapeutic benefits of opioid agonist therapy. J. Addict. Dis. 2008;27(3):49–65. doi: 10.1080/10550880802122646. [DOI] [PubMed] [Google Scholar]
- 157.Lutz P.E., Kieffer B.L. Opioid receptors: distinct roles in mood disorders. Trends Neurosci. 2013;36(3):195–206. doi: 10.1016/j.tins.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Torregrossa M.M., Isgor C., Folk J.E., Rice K.C. ; Torregrossa M.M., Isgor C., Folk J.E., Rice K.C., Watson S.J., Woods J.H. The delta-opioid receptor agonist (+)BW373U86 regulates BDNF mRNA expression in rats. Neuropsychopharmacology. 2004;29(4):649–659. doi: 10.1038/sj.npp.1300345. [DOI] [PubMed] [Google Scholar]
- 159.Zhang H., Shi Y.G., Woods J.H., Watson S.J., Ko M.C. Central kappa-opioid receptor-mediated antidepressant-like effects of nor-Binaltorphimine: behavioral and BDNF mRNA expression studies. 2007. [DOI] [PMC free article] [PubMed]
- 160.Gerra G., Borella F., Zaimovic A., Moi G., Bussandri M., Bubici C., Bertacca S. Buprenorphine versus methadone for opioid dependence: predictor variables for treatment outcome. 2004. [DOI] [PubMed]
- 161.Bodkin J.A., Zornberg G.L., Lukas S.E., Cole J.O. Buprenorphine treatment of refractory depression. J. Clin. Psychopharmacol. 1995;15(1):49–57. doi: 10.1097/00004714-199502000-00008. [DOI] [PubMed] [Google Scholar]
- 162.Janowsky D.S., el-Yousef M.K., Davis J.M., Sekerke H.J. A cholinergic-adrenergic hypothesis of mania and depression. 1972. [DOI] [PubMed]
- 163.Dilsaver S.C. Pharmacologic induction of cholinergic system up-regulation and supersensitivity in affective disorders research. 1986. [PubMed]
- 164.Kasper S., Moises H.W., Beckmann H. The anticholinergic biperiden in depressive disorders. Pharmacopsychiatria. 1981;14(6):195–198. doi: 10.1055/s-2007-1019597. [DOI] [PubMed] [Google Scholar]
- 165.Drevets W.C., Zarate C.A., Jr, Furey M.L. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol. Psychiatry. 2013;73(12):1156–1163. doi: 10.1016/j.biopsych.2012.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rami A., Krieglstein J. Muscarinic-receptor antagonist scopolamine rescues hippocampal neurons from death induced by glutamate. Brain Res. 1998;788(1-2):323–326. doi: 10.1016/S0006-8993(98)00041-9. [DOI] [PubMed] [Google Scholar]
- 167.Furey M.L., Drevets W.C. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry. 2006;63(10):1121–1129. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Drevets W.C., Furey M.L. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol. Psychiatry. 2010;67(5):432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Khajavi D., Farokhnia M., Modabbernia A., Ashrafi M., Abbasi S.H., Tabrizi M., Akhondzadeh S. Oral scopolamine augmentation in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled study. J. Clin. Psychiatry. 2012;73(11):1428–1433. doi: 10.4088/JCP.12m07706. [DOI] [PubMed] [Google Scholar]
- 170.Philip N.S., Carpenter L.L., Tyrka A.R., Price L.H. 2010.
- 171.George T.P., Sacco K.A., Vessicchio J.C., Weinberger A.H., Shytle R.D. Nicotinic antagonist augmentation of selective serotonin reuptake inhibitor-refractory major depressive disorder: a preliminary study. J. Clin. Psychopharmacol. 2008;28(3):340–344. doi: 10.1097/JCP.0b013e318172b49e. [DOI] [PubMed] [Google Scholar]
- 172.Philip N.S., Carpenter L.L., Tyrka A.R., Whiteley L.B., Price L.H. Varenicline augmentation in depressed smokers: an 8-week, open-label study. J. Clin. Psychiatry. 2009;70(7):1026–1031. doi: 10.4088/JCP.08m04441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zarate C.A., Jr, Mathews D., Ibrahim L., Chaves J.F., Marquardt C., Ukoh I., Jolkovsky L., Brutsche N.E., Smith M.A., Luckenbaugh D.A. A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol. Psychiatry. 2013;74(4):257–264. doi: 10.1016/j.biopsych.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]