

Abstract
Aims: Parkinson's disease (PD) heavily affects humans and little is known about its cause and pathogenesis. Sirtuin 3 (Sirt3) plays a key role in regulating mitochondrial dysfunction, which is the main cause of DAergic neuronal loss in PD. We investigated the mechanisms of neuroprotective role of Sirt3 in DAergic neuronal survival. Results: Sirt3 was reduced in 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP)-treated neurons with its overexpression being neuroprotective. We identified that Sirt3 interacted with manganese superoxide dismutase (SOD2) and adenosine triphosphate (ATP) synthase β and modulated their activities by deacetylating SOD2 (K130) and ATP synthase β (K485) to prevent reactive oxygen species accumulation and ATP depletion, and to alleviate DAergic neuronal death upon MPTP treatment. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) interacted with estrogen-related receptor alpha (ERRα) that bound to the Sirt3 promoter as its transcription factor to regulate Sirt3 expression and DAergic neuronal death. In the mouse midbrain, MPTP administration led to the loss of PGC-1α and Sirt3, high acetylation level of SOD2 and ATP synthase β, and the specific loss of DAergic neurons, while Sirt3 overexpression could protect against DAergic neuronal loss. Sirt3 knockout mice exhibited more sensitive and more DAergic neuronal loss to MPTP treatment. Innovation: The study provides new insights into a critical PGC-1α/ERRα-Sirt3 pathway, linking regulation of mitochondrial protein acetylation and DAergic neuronal death in PD pathogenesis, which provide a potential therapeutic strategy and target in PD treatment. Conclusion: These results provide a vital PGC-1α/ERRα-Sirt3 pathway that protects against DAergic neuronal death by directly deacetylating SOD2 (K130) and ATP synthase β (K485) in PD. Antioxid. Redox Signal. 24, 312–328.
Introduction
Parkinson's disease (PD) is a severe neurodegenerative disorder characterized by the selective and progressive loss of DAergic neurons in the substantia nigra pars compacta (SNc) (8). Mutated genes, including Parkin, LRRK2, PINK1, DJ-1, and α-Synuclein, have been identified in some cases of inherited PD (13, 37). However, the vast majority of PD cases are idiopathic, which are far from understood. Experimentally, the administration of mitochondrial complex I toxin 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) can mimic key features of PD in humans and nonhuman primates, including the marked loss of DAergic neurons (4, 29). 1-Methyl-4-phenylpyridinium iodide (MPP+) as a toxic metabolite of MPTP is taken up by DAergic neurons and subsequently inhibits complex I activity, which can seriously damage mitochondrial function and lead to DAergic neuronal death (4).
Innovation.
This study is the first detailed report providing new insights into a critical PGC-1α/ERRα-Sirt3 pathway, linking regulation of mitochondrial protein acetylation and DAergic neuronal death in Parkinson's disease (PD) pathogenesis, which provide a potential therapeutic strategy and target in PD treatment.
Oxidative stress as the cellular outcome of mitochondrial dysfunction is strongly implicated as a contributing factor to PD pathogenesis (22, 28). Some evidence suggests that DAergic neurons are highly vulnerable to oxidative stress because of high levels of reactive oxygen species (ROS) (6). As an accumulation of ROS could activate mitochondrial apoptotic pathways in DAergic neurons (22, 42), antioxidants are of particular importance in the treatment of oxidative stress in DAergic neurons. Mitochondrial manganese superoxide dismutase (SOD2) is the main enzyme responsible for converting harmful superoxide radicals to hydrogen peroxide in mitochondria. However, the way in which SOD2 is regulated in PD is yet to be elucidated.
Energy status is a known determinant of neuronal death (36). A reduction in intracellular adenosine triphosphate (ATP) level has been associated with neuronal damage in the brains of PD patients, as well as in cell and mouse models of PD (31, 35). ATP synthase is the key enzyme in ATP synthesis, which provides energy for neurons. Thus, how ATP synthase is regulated in DAergic neuronal loss of PD should be investigated.
Accumulating evidence suggests that protein acetylation is a key regulatory mechanism in mitochondrial function. Sirtuins are a family of nicotinamide (NAM) adenine dinucleotide (NAD+)-dependent protein deacetylases that regulate numerous cellular processes (7, 19, 23). As the main mitochondrial deacetylase, sirtuin 3 (Sirt3) plays a key role in energy metabolism and oxidative stress (5, 60). Many mitochondrial enzymes involved in energy metabolism are substrates of Sirt3, by which Sirt3 can affect ATP production (44, 53), tricarboxylic acid (TCA) cycle (61), electron transport chain (1, 15), amino acid metabolism (20), fatty acid β-oxidation (24), and ketone body production (49).
Previous studies have also shown that Sirt3 can regulate mitochondrial ROS levels by deacetylating SOD2 (9, 26, 41). Recent evidence suggests that Sirt3 has a positive role in several neurological disorders, including Alzheimer's disease (21, 56), PD (33), Huntington's disease (16), and Amyotrophic lateral sclerosis (51), but the detailed mechanism is still unknown. Mitochondrial protein acetylation plays a key role in mitochondrial dysfunction, which is a key feature of DAergic neuronal loss in PD. However, little is known about the role of Sirt3-mediated deacetylation in the loss of DAergic neurons of PD pathogenesis.
In this study, we define a vital pathway in which Sirt3 reduction mediates DAergic neuronal loss with Sirt3 overexpression being neuroprotective both in vitro and in vivo. We demonstrate that Sirt3 deacetylates SOD2 (K130) and ATP synthase β (K485) to decrease ROS and elevated ATP levels. The reduction of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α), interacting with transcription factor estrogen-related receptor alpha (ERRα), is first connected with the decreased Sirt3 in MPP+-treated neurons. The results provide new insights into a key PGC-1α/ERRα-Sirt3 pathway, linking the regulation of mitochondrial protein acetylation and DAergic neuronal death in PD pathogenesis.
Results
Sirt3 protects against MPP+-induced neuronal death
MPP+, a neurotoxic product of MPTP, has been extensively used in a variety of cell injury and PD models (12, 27, 54). In this study, MPP+ induced a series of mitochondrial dysfunction in Neuro-2a (N-2a) mouse neuroblastoma cells and mouse primary midbrain neurons, resulting in cell viability reduction (Supplementary Fig. S1A–D; Supplementary Data are available online at www.liebertpub.com/ars), ROS accumulation (Supplementary Fig. S1E–I), ATP depletion (Supplementary Fig. S1J), mitochondrial membrane potential reduction (Supplementary Fig. S2A–D), decreased mitochondrial fusion process (Supplementary Fig. S3A–H), and neuronal apoptosis (Supplementary Fig. S1K, L).
Sirt3 as the main mitochondrial deacetylase plays a key role in regulating mitochondrial metabolic processes. Consistent with previous reports (38, 48), exogenous Sirt3 colocalized with mitochondria when EGFP-C1-Sirt3 or Sirt3-EGFP-N3 was cotransfected with pDsRed-mito into N-2a cells (Fig. 1A, B). Moreover, endogenous Sirt3 was exclusively localized to mitochondria in N-2a cells and mouse primary midbrain neurons following MPP+ treatment (Fig. 1C, D). To determine whether Sirt3 expression is regulated by MPTP administration, we used Western blotting and found a dose- and time-dependent Sirt3 reduction in N-2a cells, SH-SY5Y cells, and mouse and rat primary midbrain neurons under MPP+ treatment (Fig. 1E–L).
FIG. 1.

Sirt3 protects against MPP+-induced neuronal death. Exogenous Sirt3 localization was observed in N-2a cells with 500 μM MPP+ for 24 h by live cell imaging. EGFP-C1-Sirt3 (A) or Sirt3-EGFP-N3 (B) was cotransfected with pDsRed-mito into N-2a cells. The nucleus was stained with Hoechst 33342 (blue). Scale bar is 10 μm. (C, D) Endogenous Sirt3 localization was observed by confocal microscopy in N-2a cells and mouse primary midbrain neurons with MPP+ for 24 h. N-2a cells were treated with 500 μM MPP+ (C), while the mouse primary midbrain neurons were treated with 50 μM MPP+ (D). Mitochondria were indicated with COX IV antibody (red), while the nucleus was stained with DAPI (blue). Scale bar is 10 μm. Sirt3 expression was detected by Western blotting in N-2a cells under different concentrations of MPP+ for 24 h (E) and with 500 μM MPP+ for different times (F), in mouse primary midbrain neurons under different concentrations of MPP+ for 24 h (G), and with 50 μM MPP+ for different times (H), in SY5Y cells under different concentrations of MPP+ for 24 h (I) and with 500 μM MPP+ for different times (J), in rat primary midbrain neurons under different concentrations of MPP+ for 24 h (K), and with 50 μM MPP+ for different times (L). Cell viability was measured by MTT in N-2a cells and mouse primary midbrain neurons with Sirt3 knockdown (M) or overexpression (N). Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01; ***p < 0.001 or ###p < 0.001, * represents significance between N-2a cells while # represents significance between mouse primary midbrain neurons, compared with control group, paired two-tailed Student's t-test. The complex I activity was measured in N-2a cells and mouse primary midbrain neurons with Sirt3 knockdown (O) or in the midbrains of Sirt3 knockout mice (P). Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01; ***p < 0.001 or ###p < 0.001, * represents significance between N-2a cells while # represents significance between mouse primary midbrain neurons, paired two-tailed Student's t-test. (Q–T) Sirt3-3×FLAG, 3×FLAG vector, Sirt3 siRNA or control siRNA was transfected into mouse primary midbrain neurons treated with 50 μM MPP+. PARP-1 and caspase 3 were measured by Western blotting with Sirt3 overexpression (Q) or knockdown (R). Neuronal apoptosis was measured by TUNEL assay with Sirt3 overexpression (S) or knockdown (T). Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA with Newman–Keuls test. MPP+, 1-methyl-4-phenylpyridinium iodide; N-2a, Neuro-2a; PARP-1, poly (ADP-ribose) polymerase-1; Sirt3, sirtuin 3. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Furthermore, we examined whether Sirt3 reduction leads to neuronal damage and found that Sirt3 knockdown decreased cell viability (Fig. 1M) and complex I activity (Fig. 1O, P). We also found that Sirt3 could protect against MPP+-induced mitochondrial dysfunction, including membrane potential reduction (Supplementary Fig. S2E, F) and mitochondrial fusion process inhibition (Supplementary Fig. S3M, N). These results indicate that Sirt3 has positive roles in maintaining mitochondrial membrane potential and fusion/fission process, which is consistent with the previous studies (3, 39, 45).
Then we examined whether Sirt3 is required for neuronal survival in PD and found that Sirt3 overexpression alleviated neuronal death induced by MPP+ treatment (Fig. 1K, M), whereas Sirt3 knockdown induced cell death of mouse primary midbrain neurons in the absence of MPP+ (Fig. 1L, N). The deacetylase-deficient Sirt3 mutant (H248Y) completely eliminated the neuroprotective effects of Sirt3 (Supplementary Fig. S4), indicating that Sirt3 deacetylase activity is required for neuroprotection. Together, these results indicate that Sirt3 is reduced under MPP+ treatment and Sirt3 overexpression protects against MPP+-induced neuronal death.
Sirt3 interacts with SOD2 and ATP synthase β in vitro and in vivo
As the main mitochondrial deacetylase, Sirt3 is known to regulate mitochondrial function through interacting with and deacetylating the substrates to modulate their activities. Increased ROS and decreased ATP levels are closely related to the mitochondrial dysfunction, which is the main cause of DAergic neuronal loss in PD. To find Sirt3 targets participating in Sirt3 neuroprotection of PD through ROS and ATP regulation, we used immunoprecipitation and liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) to identify the interacting proteins of Sirt3. We had identified 102 proteins from the immunopurified Sirt3-3× FLAG complex, among which ATP synthase β, the catalytic subunit of ATP synthase, was highly enriched (Fig. 2A). Previous studies have shown that Sirt3 deacetylates and activates SOD2 to modulate cellular ROS levels in the liver (41).
FIG. 2.

Sirt3 interacts with SOD2 and ATP synthase β in vitro and in vivo. (A) The immunopurified complex of Sirt3 was separated by SDS-PAGE, followed by silver staining. pCold-TF-Sirt3 and its control pCold-TF were purified and incubated with purified SOD2-3×FLAG (B) or ATP synthase β-3×FLAG (C). The interaction was observed by Western blotting with SOD2 or ATP synthase β antibody after nickel beads purification. (D) Sirt3-3×FLAG was transfected into N-2a cells and immunopurified, followed by Western blotting with SOD2 and ATP synthase β antibodies. (E) SOD2-3×FLAG or ATP synthase β-3×FLAG was transfected into N-2a cells and immunopuriifed, followed by Western blotting with Sirt3 antibody. (F) Endogenous Sirt3 was immunopurified from N-2a cells with Sirt3 antibody, followed by Western blotting with SOD2 and ATP synthase β antibodies. (G) Endogenous SOD2 and ATP synthase β were immunopurified from N-2a cells with SOD2 and ATP synthase β antibodies, followed by Western blotting with Sirt3 antibody. ATP, adenosine triphosphate; SOD2, manganese superoxide dismutase.
Then, we examined whether Sirt3 can interact with SOD2 and ATP synthase β in vitro and in vivo. We found that Sirt3 could interact with SOD2 and ATP synthase β in vitro (Fig. 2B, C). Co-IP results indicated that exogenous Sirt3 interacts with SOD2 and ATP synthase β, respectively (Fig. 2D, E). We also examined the endogenous interaction of Sirt3 with SOD2 and ATP synthase β and found that MPP+ treatment reduced these interactions owing to a reduction in Sirt3 (Fig. 2F, G). These results indicated that Sirt3 interacts with SOD2 and ATP synthase β both in vitro and in vivo.
Sirt3 deacetylates and activates SOD2 and ATP synthase β to protect against MPP+-induced ROS accumulation and ATP depletion
The acetylation levels of mitochondrial proteins are closely linked with the activities of many important metabolic enzymes (5). Therefore, we determine whether Sirt3 reduction induced by MPP+ leads to mitochondrial protein hyperacetylation. We found a notable increase in mitochondrial protein acetylation because of decreased Sirt3 expression in MPP+-induced N-2a cells and mouse primary midbrain neurons (Supplementary Fig. S5B, C).
Thus, we determined whether SOD2 and ATP synthase β are regulated by Sirt3-mediated deacetylation. We demonstrated that Sirt3 decreased the acetylation levels of SOD2 and ATP synthase β in vitro (Fig. 3A, B). MPP+ treatment increased the total acetylation levels of immunoprecipitated SOD2 and ATP synthase β with the reduction of Sirt3, whereas no significant protein expression changes were observed in SOD2 and ATP synthase β (Fig. 3C). Moreover, Sirt3, but not catalytically inactive SIRT3-H248Y, decreased the acetylation levels of SOD2 and ATP synthase β, which indicated Sirt3 deacetylation of SOD2 and ATP synthase β in vivo (Fig. 3D).
FIG. 3.

Sirt3 deacetylates and activates SOD2 and ATP synthase β to protect against ROS accumulation and ATP depletion. pCold-TF-Sirt3 and its control pCold-TF were purified and incubated with purified SOD2-3×FLAG (A) or ATP synthase β-3×FLAG (B). Then, deacetylation of SOD2 and ATP synthase β was observed by Western blotting with acetyl-lysine antibody. (C–F) The acetylation levels of SOD2 and ATP synthase β were measured by Western blotting with SOD2 and ATP synthase β antibodies after immunopurification with acetyl-lysine antibody in N-2a cells. N-2a cells were treated with 500 μM MPP+ for 24 h (C). Sirt3-3×FLAG, 3×FLAG vector, or enzymatically inactive Sirt3-H248Y-3×FLAG was transfected into N-2a cells (D). N-2a cells were transfected with Sirt3-3×FLAG or 3×FLAG vector and then treated with 500 μM MPP+ for 24 h (E). N-2a cells were transfected with Sirt3 siRNA or control siRNA and then treated with 500 μM MPP+ for 24 h (F). (G) ROS were measured in mouse primary midbrain neurons transfected with Sirt3-3×FLAG or 3×FLAG vector and treated with 50 μM MPP+ for 24 h. (H) ATP was measured in mouse primary midbrain neurons transfected with Sirt3-3×FLAG or 3×FLAG vector and treated with 50 μM MPP+ for 24 h. (I) ROS were measured in mouse primary midbrain neurons transfected with Sirt3 siRNA or control siRNA and treated with 50 μM MPP+ for 24 h. (J) ATP was measured in mouse primary midbrain neurons transfected with Sirt3 siRNA or control siRNA and treated with 50 μM MPP+ for 24 h. Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA with Newman–Keuls test. The mRNA (K), protein (L) level, and activities of SOD2 (M) and ATP synthase β (N) were measured after transfection of Sirt3-3×FLAG, 3×FLAG vector, or enzymatically inactive Sirt3-H248Y-3×FLAG into N-2a cells. Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01, ANOVA with Newman–Keuls test. ROS, reactive oxygen species.
We examined whether Sirt3 deacetylase activity is required for neuroprotection in PD. We found that Sirt3 overexpression decreased, while Sirt3 knockdown further increased acetylation levels of SOD2 and ATP synthase β in MPP+-induced neuronal injury models (Fig. 3E, F). These results indicate that Sirt3 deacetylates SOD2 and ATP synthase β both in vitro and in vivo, and Sirt3 overexpression can alleviate the increased acetylation levels of SOD2 and ATP synthase β in MPP+-induced neuron injury models.
Furthermore, we measured ROS and ATP levels to determine whether the acetylation state of SOD2 and ATP synthase β affected their enzymatic activities. We found that Sirt3 overexpression alleviated ROS accumulation and ATP depletion (Fig. 3G, H), while Sirt3 knockdown had the opposite effects (Fig. 3I, J) in mouse primary midbrain neurons. The same results were found in N-2a cells (Supplementary Fig. S6A–D). We also found that Sirt3 overexpression could increase SOD2 and ATP synthase β activity without changing their expression, while inactive Sirt3-H248Y did not have this effect (Fig. 3K–N).
These results indicated that Sirt3 modulated intracellular ROS and ATP levels by deacetylating and activating SOD2 and ATP synthase β. Sirt3 reduction was responsible for the increased acetylation levels of SOD2 and ATP synthase β, resulting in reduced enzymatic activities associated with ROS accumulation and ATP depletion under MPP+ treatment.
Sirt3 deacetylates SOD2 at K130 and ATP synthase β at K485 to alleviate ROS accumulation and ATP depletion
To identify the functional lysine acetylation residues of SOD2 and ATP synthase β regulated by Sirt3, we mutated lysine (K53, K68, K89, K130) in SOD2 and lysine (K133, K198, K256, K426, K485, K522) in ATP synthase β based on previously reported acetylation sites in mass spectrometry-based surveys (11, 55, 62). Compared with wild-type Flag-SOD2 and Flag-ATP synthase β, only the K130R mutant of SOD2 and the K485R mutant of ATP synthase β did not significantly decrease acetylation levels following cotransfection of Sirt3-EGFP (Fig. 4A, B). Moreover, only the K130R mutant of SOD2 and the K485R mutant of ATP synthase β did not significantly increase following MPP+ treatment (Fig. 4C, D). In addition, we found that only the K130R mutant of SOD2 did not increase ROS level (Fig. 4E), and the K485R mutant of ATP synthase β did not exhibit the same decrease in ATP levels following MPP+ treatment (Fig. 4F).
FIG. 4.

Sirt3 deacetylates SOD2 at K130 and ATP synthase β at K485 to alleviate ROS accumulation and ATP depletion. (A, B) WT-SOD2, WT-ATP synthase β, and their mutants were cotransfected with Sirt3-EGFP into N-2a cells. The acetylation levels of SOD2, ATP synthase β, and their mutants were measured by Western blotting after immunoprecipitation with ANTI-FLAG M2 Affinity Gel. (C, D) WT-SOD2, WT-ATP synthase β, and their mutants were transfected into N-2a cells with 500 μM MPP+ for 24 h. The acetylation levels of SOD2, ATP synthase β, and their mutants were measured by Western blotting after immunoprecipitation with ANTI-FLAG M2 Affinity Gel. (E) ROS level was measured in N-2a cells transfected with WT-SOD2 and its mutants with 500 μM MPP+ for 24 h. Quantitative data = mean ± SEM, n = 3, **p < 0.01; ***p < 0.001, comparison against untreated, paired two-tailed Student's t-test. (F) ATP was measured in N-2a cells transfected with WT-ATP synthase β and its mutants with 500 μM MPP+ for 24 h. Quantitative data = mean ± SEM, n = 3, *p < 0.05, comparison against WT, ANOVA with Dunnett test. WT-SOD2, WT-ATP synthase β, and their mutants were transfected into N-2a cells with 5 mM NAM (G, H) or Sirt3 siRNA (I, J). Quantitative data = mean ± SEM, n = 3, **p < 0.01, comparison against control group, paired two-tailed Student's t test. The acetylation levels of SOD2, ATP synthase β, and their mutants were measured by Western blotting after immunoprecipitation with ANTI-FLAG M2 Affinity Gel. (K) Protein sequence alignment of SOD2 and ATP synthase β cross species was performed by UniProt. (L) The acetylation levels of SOD2 and ATP synthase β were measured by Western blotting with SOD-K130-Ac and synthase β-K485-Ac antibodies. N-2a cells were transfected with Sirt3-3×FLAG or Sirt3 siRNA, or treated with 500 μM MPP+. (M) The acetylation levels of SOD2 ATP synthase β in mouse primary midbrain neurons, rat primary midbrain neurons, and SH-SY5Y cells were measured by Western blotting under MPP+ treatment for 24 h. SH-SY5Y cells were treated with 500 μM MPP+, while the other two neurons were 50 μM MPP+. Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01, comparison against control group, paired two-tailed Student's t test. NAM, nicotinamide.
To further examine whether these two acetylation sites were regulated by Sirt3, we observed the effects of Sirt3 inhibition on the two lysine acetylation sites on SOD2 and ATP synthase β. Sirt3 inhibition by NAM (a widely used sirtuins inhibitor) or siRNA did not increase acetylation levels in SOD2-K130R mutant or the ATP synthase β-K485R mutant, but did significantly increase acetylation levels in the corresponding wild types (Fig. 4G–J). These results demonstrate that K130 and K485 are the functional acetylation sites of SOD2 and ATP synthase β that can be regulated by Sirt3.
Protein sequence alignment showed that K130 on SOD2 and K485 on ATP synthase β are highly conserved across species (Fig. 4K). To verify that K130 of SOD2 and K485 of ATP synthase β were the accurate deacetylation sites by Sirt3, we made SOD-K130-Ac and ATP synthase β-K485-Ac antibodies. SOD2 (K130) and ATP synthase β (K485) acetylation levels increased under MPP+ treatment and Sirt3 knockdown, but decreased with Sirt3 overexpression (Fig. 4L). Moreover, we found that the acetylation levels of SOD2 (K130) and ATP synthase β (K485) also increased following Sirt3 reduction in mouse and rat primary midbrain neurons, and in SH-SY5Y cells under MPP+ treatment (Fig. 4M). These results indicate that Sirt3 deacetylates K130 on SOD2 and K485 on ATP synthase β cross species.
PGC-1α interacts with transcription factor ERRα to regulate Sirt3 expression and neuronal death
Next, we sought to determine which factor regulates Sirt3 expression. We measured mRNA levels of Sirt3 and found a significant decrease in Sirt3 mRNA in both N-2a cells and mouse primary midbrain neurons under MPP+ treatment (Fig. 5A, B), suggesting that Sirt3 reduction was regulated at the transcription level following MPP+ treatment.
FIG. 5.

PGC-1α interacts with Sirt3 transcription factor ERRα to regulate Sirt3 expression and neuronal death. Sirt3 mRNA level was measured by qPCR in N-2a cells treated with 500 μM MPP+ for different times (A) and mouse primary midbrain neurons treated with 50 μM MPP+ for different times (B). Quantitative data = mean ± SEM, n = 3, *p < 0.05; ***p < 0.001, comparison against 0 h, ANOVA with Dunnett test. ChIP-qPCR was performed with ERRα (C) and PGC-1α (D) antibodies in N-2a cells. Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01, ANOVA with Newman–Keuls test. The mRNA of Sirt3 was measured in N-2a cells with ERRα and PGC-1α overexpression (E) or knockdown (F). The expression of PGC-1α and ERRα was measured by Western blotting in N-2a cells (G) and mouse primary midbrain neurons (H). ChIP-qPCR was performed with ERRα (I) and PGC-1α (J) antibodies in N-2a cells under MPP+ treatment. (K) The expression of Sirt3 and the acetylation levels of SOD2 and ATP synthase β were measured by Western blotting after transfection with PGC-1α siRNA. (L) The acetylation level of mitochondrial proteins was measured by Western blotting after mitochondrial isolation from mouse primary midbrain neurons following PGC-1α knockdown. The protein gel was a loading control. ROS (M) and ATP (N) were measured with DCFH-DA staining and ATP Assay Kit in N-2a cells transfected with Sirt3-3×FLAG and PGC-1α siRNA, respectively. Quantitative data = mean ± SEM, n = 3, *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA with Newman–Keuls test. PARP-1 and caspase 3 were measured by Western blotting in N-2a cells (O) and mouse primary midbrain neurons (P) transfected with Sirt3-3×FLAG and PGC-1α siRNA, respectively. The protein (Q) and mRNA (R) levels of PGC-1α were measured in mouse primary midbrain neurons treated with 50 μM MPP+, 2 mM NAC, or 50 μM MPP+ and 2 mM NAC for 2 and 24 h. Quantitative data = mean ± SEM, n = 3, ***p < 0.001, ANOVA with Newman–Keuls test. ChIP, chromatin immunoprecipitation; ERRα, estrogen-related receptor alpha; NAC, N-acetyl-L-cysteine; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator 1α; qPCR, quantitative real-time polymerase chain reaction.
Transcriptional coactivator PGC-1α is a crucial regulator in mitochondrial function. Recent work indicates that PGC-1α plays a key role in DAergic neuronal survival and PD pathogenesis (50). PGC-1α was reported to interact with nuclear respiratory factor 1 (NRF1) and nuclear factor erythroid 2 (NF-E2)-related factor 2 (NRF2) to regulate mitochondrial biogenesis, oxidative stress response, and energy generation (10, 14, 18, 58). Sirt3 was reported to be regulated by PGC-1α and ERRα in brown adipocytes (17). Furthermore, we found that PGC-1α interacted with ERRα and NRF1 (Supplementary Fig. S7A).
To find the transcription factor of Sirt3, we performed chromatin immunoprecipitation (ChIP) assays and found that ERRα had a strong binding in the Sirt3 promoter (Fig. 5C), compared with NRF1 and NRF2 (Supplementary Fig. S7B, C). Moreover, because of the interaction between PGC-1α and ERRα, we found that PGC-1α had a similar binding pattern with ERRα in the Sirt3 promoter (Fig. 5D). We also found that ERRα or PGC-1α overexpression increased Sirt3 expression and cotransfection of ERRα and PGC-1α could increase more expression of Sirt3 (Fig. 5E), while ERRα or PGC-1α knockdown decreased Sirt3 expression and cotransfection of ERRα siRNA and PGC-1α siRNA could decrease more expression of Sirt3 (Fig. 5F). These results indicate that ERRα and PGC-1α coordinately regulate Sirt3 expression.
To find which factor was responsible for Sirt3 reduction in MPP+ treatment, we examined PGC-1α and ERRα expression with Western blotting and found that PGC-1α, but not ERRα, was reduced with a similar change pattern of Sirt3 expression under MPP+ treatment in both N-2a cells and mouse primary midbrain neurons (Fig. 5G, H). The ChIP assays also revealed that ERRα did not decrease its binding with the Sirt3 promoter under MPP+ treatment (Fig. 5I), while PGC-1α decreased its binding to the Sirt3 promoter owing to PGC-1α reduction (Fig. 5J).
Then, we examined whether PGC-1α reduction could affect Sirt3 downstream effects. We found that PGC-1α knockdown reduced Sirt3 expression, and increased the acetylation levels of SOD2 (K130) and ATP synthase β (K485) with SOD2-K130-Ac and ATP synthase β-K485-Ac antibodies (Fig. 5K). Moreover, PGC-1α knockdown increased mitochondrial protein acetylation levels (Fig. 5L). These results suggest that PGC-1α is a positive activator of Sirt3 transcription and involved in the regulation of mitochondrial protein acetylation.
We examined whether Sirt3 overexpression could rescue the effects of PGC-1α knockdown and found that Sirt3 overexpression alleviated ROS increase, ATP reduction, and neuronal apoptosis induced by PGC-1α knockdown (Fig. 5M–P). PGC-1α expression was suggested to be affected by ROS in DAergic neuronal survival and PD pathogenesis (50). We examined PGC-1α expression following N-acetyl-L-cysteine (NAC, a widely used antioxidant) treatment and found that the PGC-1α reduction induced by MPP+ treatment could be alleviated by NAC treatment in the early stage (Fig. 5Q, R). These results suggest that PGC-1α interacts with Sirt3 transcription factor ERRα as a transcriptional coactivator and is responsible for the expression and downstream effects of Sirt3 in MPP+-induced neuronal injury models.
Sirt3 protects against DAergic neuronal loss in an MPTP mouse model of PD
Given the PGC-1α-Sirt3 signaling in cell models of PD, we carried out in vivo experiments to examine the pathological relevance of our findings. PGC-1α and Sirt3 were reduced, and the acetylation levels of SOD2 (K130) and ATP synthase β (K485) were increased in MPTP-treated mouse midbrains (Fig. 6A).
FIG. 6.
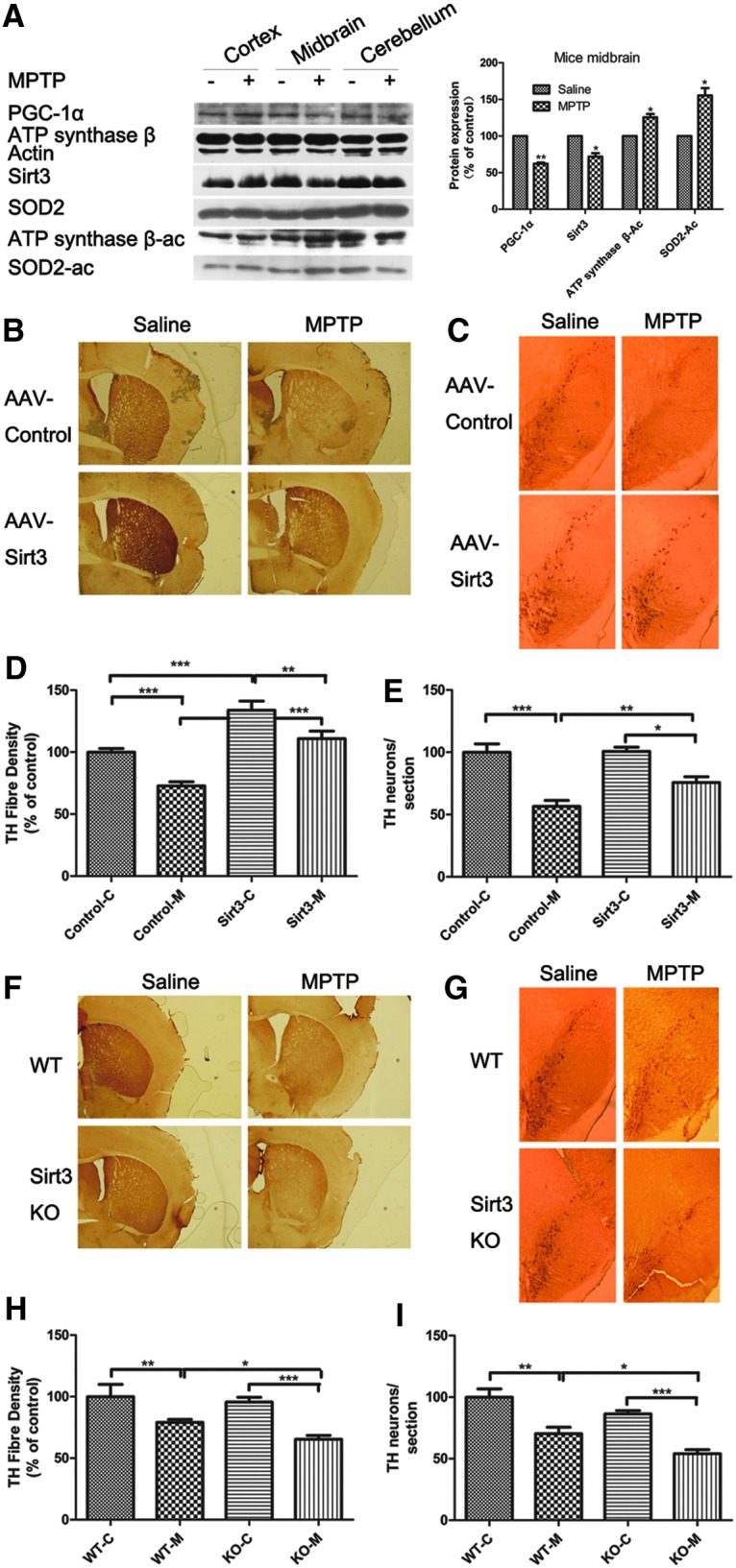
Sirt3 protects against DAergic neuronal loss in an MPTP mouse model of Parkinson's disease. (A) The expression of PGC-1α and Sirt3, and the acetylation levels of SOD-K130 and ATP synthase β-K485 in an MPTP mouse model were measured by Western blotting. (B, C) Brain sections from the striatum and SNc showing TH immunoreactivity after intraperitoneal injection of saline or MPTP preceded by brain injection of either control vector or AAV-Sirt3. (D, E) The quantification of TH fiber density and TH-positive neurons according to (B) and (C). (F, G) Brain sections from the striatum and SNc showing TH immunoreactivity after intraperitoneal injection of saline or MPTP in WT or Sirt3 KO mice. (H, I) The quantification of TH fiber density and TH-positive neurons according to (F) and (G). Quantitative data = mean ± SEM, n = 6, *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA with Newman–Keuls test (D, E, H, I). MPTP, 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine; SNc, substantia nigra pars compacta; TH, tryosine hydroxylase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
To substantiate the in vitro findings regarding the neuroprotective effects of Sirt3, we injected an AAV-Sirt3 vector into the dorsolateral striatum of mice. Immunohistochemical staining demonstrated that the MPTP-induced loss of tryosine hydroxylase (TH)-positive neurons in the striatum and SNc was remarkably attenuated by Sirt3 overexpression (Fig. 6B, C). Densitometric analysis of TH staining demonstrated that Sirt3-overexpressed animals had more TH-positive DA fiber in the striatum than control vector-treated animals (Fig. 6D). Quantification of SNc TH-positive neurons showed that Sirt3-overexpressed animals had a greater number of immunopositive neurons after MPTP lesioning than control vector-treated animals (Fig. 6E).
Sirt3 KO mice were more sensitive to MPTP treatment and exhibited fewer TH-positive neurons and less TH fiber density following MPTP treatment (Fig. 6F–I). These results indicate that Sirt3 protects against DAergic neuronal loss in an MPTP mouse model of PD.
Discussion
In this study, we found that the major mitochondrial deacetylase Sirt3 acts as a key regulator of mitochondrial dysfunction in DAergic neuronal damage and demonstrated that Sirt3 protects against DAergic neuronal damage both in vivo and in vitro. Sirt3 reduction leads to increased mitochondrial protein acetylation and dysregulation of two critical acetylation substrates: SOD2 and ATP synthase β, which are implicated in the regulation of ROS elimination and ATP production. Moreover, we found that two functional acetylation sites (SOD2-K130 and ATP synthase β-K485) are regulated by Sirt3 and essential for the modulation of ROS and ATP levels. Our study further demonstrates that the PGC-1α is responsible for the regulation of Sirt3 by interacting with ERRα and is implicated in mitochondrial dysfunction characterized by ROS elevation and ATP reduction in DAergic neuronal injury (Fig. 7).
FIG. 7.
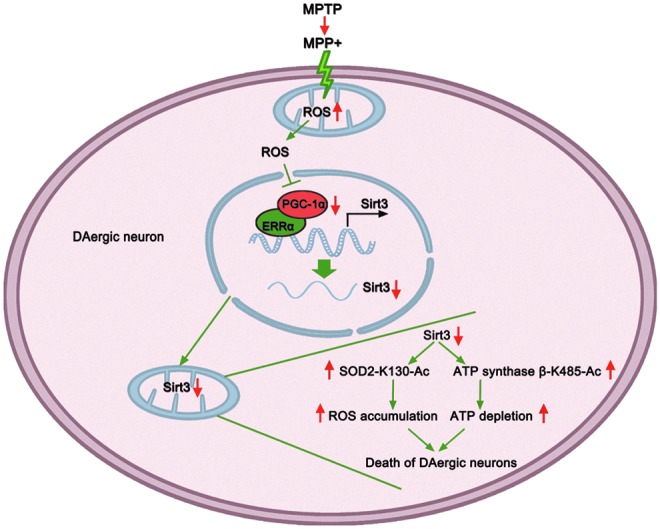
Model of PGC-1α/ERRα-Sirt3 pathway in protecting against DAergic neuronal death. Extrinsic neurotoxic chemicals such as MPTP or intrinsic factors target mitochondria to stimulate ROS production at the early stage, which reduces PGC-1α expression. PGC-1α interacts with ERRα as a transcriptional coactivator and its reduction decreases Sirt3 transcription. A reduction in Sirt3 decreases the deacetylation and activation of SOD2 and ATP synthase β, resulting in ROS accumulation and ATP depletion. Increased ROS and reduced ATP affect a variety of metabolic activities, resulting in DAergic neuronal loss. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Mitochondrial dysfunction is closely linked to the pathogenesis of aging-associated neurodegenerative disorders, including PD (2, 13, 40). Sirt3 plays a critical role in regulating various key mitochondrial metabolic pathways, such as the TCA cycle (15, 27), the electron transport chain (1, 15), amino acid metabolism (20), fatty acid β-oxidation (24), and ketone body production (49). Sirt3 also has a positive role in maintaining mitochondrial membrane potential (39) and mitochondrial fusion/fission homeostasis (45).
In addition to these metabolic functions, Sirt3 controls ROS elimination and ATP production, the dysregulation of which are two key phenotypes of neuronal injury in neurodegenerative disorders. Accumulating evidence suggests that Sirt3 plays a role in various neurodegenerative diseases (16, 21, 33, 51, 56), but whether and how Sirt3 mediates DAergic neuronal death in PD remains to be elucidated. The present results demonstrate that Sirt3 is reduced in a series of MPP+-induced neuronal injury and mouse models, while Sirt3 overexpression prevents neuronal loss in cell and mouse models of PD induced by MPTP/MPP+.
ROS accumulation resulting from mitochondrial dysfunction is the main characteristic of DAergic neuronal death. ROS levels are very high in the brains of PD patients (6). The antioxidant system is essential for neuroprotection in neurons. Previously, Qu et al. reported that Prx2 phosphorylation mediated by CDK5 is essential for the regulation of cytoplasmic ROS levels (42). However, it remains unclear how mitochondrial ROS are regulated in DAergic neurons.
Mitochondrial antioxidant enzyme SOD2 plays an important role in maintaining redox homeostasis. Previous studies have shown that ROS are scavenged in the liver during calorie restriction because of the deacetylation and activation of SOD2 by Sirt3 (9, 41). However, the mechanism by which Sirt3 regulates ROS accumulation in DAergic neuronal loss is not well understood. Our study revealed for the first time that functional K130 acetylation in SOD2 is regulated by Sirt3, the loss of which results in the deacetylation and inactivation of SOD2, which is implicated in ROS accumulation in DAergic neuronal damage induced by MPP+.
Intracellular ATP level is critical for neuronal survival. A lack of Sirt3 is associated with a marked reduction of intracellular ATP level (22), which suggests that Sirt3 has positive roles in neuronal survival through regulating ATP levels. As the core catalytic unit of ATP synthase, ATP synthase β plays essential roles in regulating intracellular ATP levels. Also, we have identified that ATP synthase β is the interacting protein of Sirt3 from the MS results (Supplementary Table S1). The MS results showed that the identified proteins were mainly located in mitochondria and nucleus (Supplementary Fig. S8A) and enriched in regulating ATP levels, including the ATP biosynthetic process (Supplementary Fig. S8B), which suggested to us that Sirt3 might interact with and deacetylate ATP synthase β to regulate ATP production directly.
Among the MS results, another protein related to ATP production is ATP synthase α. However, we have not detected the endogenous interaction between ATP synthase α and Sirt3 (Supplementary Fig. S8C). Recently, Rahman et al. revealed that Sirt3 regulates mitochondrial complex V activity by deacetylating ATP synthase β in Drosophila (44). In our study, Sirt3 reduction mediated the elevated acetylation of ATP synthase β, which eventually resulted in intracellular ATP depletion in MPP+-induced neuronal injury models. Moreover, we showed that Sirt3 regulates the functional acetylation site (K485) of ATP synthase β. Given this, our results indicated that Sirt3 reduced ATP depletion by deacetylating and activating ATP synthase β, which attenuates neuronal loss in PD.
In summary, we uncover a vital mechanism by which PGC-1α/ERRα-mediated Sirt3 function regulates DAergic neuronal loss in an MPTP model of PD via deacetylating and activating SOD2 and ATP synthase β. As such, the present findings hold important implications for PD treatment.
Materials and Methods
Cell culture
The N-2a, SH-SY5Y, and 293T cell lines were cultured in Dulbecco's modified Eagle's medium (Hyclone) containing 10% (v/v) fetal bovine serum (Gibco BRL), penicillin (50 U/ml), and streptomycin sulfate (50 mg/ml), incubating at 37°C in a humidified atmosphere with 5% CO2.
The primary neuron cultures were derived from the midbrain or cortex of E16 ICR mice or Sprague-Dawley rats (Vitalriver) as described previously (25, 57, 59). The neurons were isolated and plated in the Dulbecco's modified Eagle's medium: nutrient mixture F-12 containing 2% (v/v) B27 supplements (Invitrogen), penicillin (50 U/ml), and streptomycin sulfate (50 mg/ml), incubating at 37°C in a humidified atmosphere with 5% CO2. After 4 h, the medium was changed. Then, half of the medium was changed every 3 days.
Plasmid constructs and transfection
Sirt3 was subcloned into p3×FLAG-CMV-14, pEGFP-C1, and pEGFP-N3, respectively. SOD2, PGC-1α, and ERRa were subcloned into p3×FLAG-CMV-14. ATP synthase β was cloned into p3×FLAG-CMV-14 from the C57BL mouse brain cDNA. The pDsRed-mito was obtained from Professor Yi Rao. Sirt3 mutant (H248Y), SOD2 mutants (K53R, K68R, K89R, K130R), and ATP synthase β mutants (K133R, K189R, K256R, K426R, K485R, K522R) were mutated from Sirt3-p3×FLAG-CMV-14, SOD2-p3×FLAG-CMV-14, and ATP synthase β-p3×FLAG-CMV-14. The plasmids were transfected with Lipofectamine 3000 (Invitrogen). The cells were harvested 24 h later for the following experiments.
Lentivirus infection
Sirt3 was subcloned into Fugw3 vector to overexpress Sirt3 in mouse primary midbrain neurons. Transfection mixture containing RRE, REV, VsVg, and Fugw3 was transfected into 293T cells with Lipofectamine LTX (Invitrogen), followed by changing the medium 12 h later. Then, the supernatant was collected and the neurons were infected for 24 h. Then, neurons were harvested 72–96 h later for the following experiments.
RNA interference
Cells were transfected with siRNAs against target genes (Sirt3 siRNA: sc-61556; PGC-1 siRNA: sc-38885; ERRa siRNA: sc-44707; Santa Cruzc Biotechnology) with Lipofectamine RNAiMAX (Invitrogen) for 48 h.
Generation of Sirt3 knockout mice
Sirt3-knockout mice were generated by a CRISPR-Cas system. In brief, a fusion of the target-specific crRNA and tracrRNA expression vector was generated as described by Li et al. (30). The vector was linearized and purified by the DNA clear-up kit and then transcribed using the Transcription T7 Kit (TaKaRa) following the manufacturer's instructions. Zygotes derived from superovulation of C57BL-6J females mating with the males were cultured in the KSOM embryo culture medium. The customizable synthetic (sgRNA) with target-specific sequence and Cas-9-encoding mRNA was injected into the cytoplasm of one-cell stage embryos using an Eppendorf transferMan NK2 micromanipulator.
Injected zygotes were transferred into pseudopregnant ICR female mice. The extracted DNA from newborn mice was amplified via polymerase chain reaction (PCR) and then sequenced to find appropriate deficiency on Sirt3 genes. Mice with 50-nucleotides-deleted Sirt3 (open reading disrupted) genotype were selected for breeding and crossed with C57BL-6J genetic background. PCR was performed to distinguish mice genotypes using AGACTTGGGTCCTCTGAAAC and GTG ACTGTAGTCTCCGACGCTT primers.
Intrastriatal administration of adenoviruses
The adeno-associated viral (AAV) expressing Sirt3 were engineered using the pAOV-Syn-EGFP system. Each AAV was injected directly into the striatum of animals for 1 month and then we carried out the MPTP treatment. A single unilateral injection of each virus (2 μl, 1 × 1013 particles per μl) was delivered to the right striatum (1.1 mm rostral, 2.2 mm right of bregma, and 3.7 mm below the skull surface) at a constant rate (0.1 μl/min) by using a syringe pump system.
MPTP treatment
MPTP (Sigma) was dissolved in 0.9% saline to a final concentration of 2.5 mg/ml. The mice (six animals per group) were given intraperitoneal injections of either MPTP (15 mg/kg) or 0.9% saline alone for four administrations at 2-h intervals. The mice were sacrificed 2 days after the first injection. All animal protocols used in this investigation were approved by the Institutional Animal Care and Use Committee at the Peking University.
Measurement of ROS and mitochondrial superoxide levels
ROS or mitochondrial superoxide production was determined by incubating the cells in a medium containing 10 μM DCFH-DA (Sigma) or 5 mM Mito-SOX (Invitrogen) at 37°C for 30 min, followed by flow-cytometric analysis (BD Biosciences). The results were analyzed by WinMDI 2.9 software.
COX I activity assay
The COX I activity assay was carried out with the COX I Activity Kit (Genmed Scientifics, Inc.) (32, 34, 52). In brief, the mitochondria of cells and tissues were isolated by Dounce tissue grinders as described previously (47, 52). The mitochondrial protein concentration was measured by the 2-D Quantitative Kit (GE Healthcare). The mitochondrial protein was incubated with reagent A/B/D of the kit according to the manufacturer's instructions. After incubation, the decrease in absorption was measured at 340 nm by Smart Spec™ Plus spectrophotometer (Bio-Rad).
Measurement of ATP level
The ATP level was determined using the ATP Assay Kit (Beyotime). In brief, cells were harvested and lysed with the lysis buffer, followed by centrifugation at 16,000 g for 5 min at 4°C. Finally, the level of ATP was determined by mixing 100 μl of the supernatant with 100 μl of luciferase reagent and measured by Varioskan Flash (ThermoFisher Scientific).
RNA isolation and quantitative real-time PCR
Cells were harvested for RNA extraction using TRIzol (Invitrogen). One microgram of total RNA was reverse transcribed to cDNA using a HiFi-MMLV cDNA First Strand Synthesis Kit (CW Bio). Quantitative real-time PCR (qPCR) was performed on the CFX96™ Real-Time System (Bio-Rad) using the GoTaq® qPCR Master Mix (Promega). The primer sequences of qPCR are listed in the Supplementary Table S2.
Western blotting analysis
Cells or tissues were lysed, followed by measuring the protein concentration with the 2-D Quantitative Kit. Equal amount of protein was resolved by 12.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride membranes using a semidry blotting apparatus. After blocking with 5% nonfat milk solution, the membranes were incubated with primary antibodies overnight at 4°C and then horseradish peroxidase (HRP)-conjugated secondary antibodies (1031-05 or 4050-05; Southern Biotech) for 2 h at room temperature. Protein bands were detected using the Immobilon Western Chemiluminescent HRP substrate (Millipore) followed by X-ray films.
The following primary antibodies were used in this study: β-actin antibody (ab3280), PGC-1 antibody (ab54481), SOD2 antibody (ab13533), and COX IV antibody (ab33985) from Abcam; ATPB antibody (sc-166443) and SOD2 antibody (sc-133134) from Santa Cruz; Sirt3 antibody (5490), acetylated-lysine antibody (9814), NRF1 antibody (12381), NRF2 antibody (12721), and ERRα antibody (13826) from Cell Signaling Technology; PGC-1α antibody (4C1.3) from Calbiochem.
Immunohistochemistry
Mice were transcardially perfused with 0.9% saline followed by an equal volume of buffered 4% paraformaldehyde (PFA). Brains were removed, postfixed in 4% PFA, and then cryoprotected in sucrose solution before cryosectioning. Brains were sectioned in the coronal plane at 30 μm thickness. Immunohistochemical analysis was performed as follows: coronal sections were incubated overnight at 4°C in the TH primary antibody (22941; ImmunoStar), washed in phosphate-buffered saline (PBS), incubated in a biotinylated secondary antibody (E030310-01; Southern Biotech) overnight at 4°C, washed in PBS, and incubated in a streptavidin horseradish peroxidase tertiary antibody (E030100; Southern Biotech). Immunohistochemical staining was visualized using a diaminobenzidine/glucose oxidase reaction. Photomicrographs were taken by Leica MZ APO and Leica DMRE microscope (Leica).
Immunocytochemistry
Cells were washed with PBS, fixed with 4% polyoxymethylene for 20 min, permeabilized with 1% Triton X-100 for 20 min, blocked with 10% bovine serum albumin (BSA) for 1 h at 37°C, and then incubated with a primary antibody in 1% BSA overnight at 4°C, after which TRITC- or FITC-conjugated secondary antibodies (1031-03 or 4050-02; Southern Biotech) in 1% BSA were added for 1 h at room temperature in the dark. Coverslips were fixed onto microslides using the Antifade Mounting Medium (Solarbio Science Technology Co., Ltd.), and images were acquired with the LSM 710 NLO and DuoScan System confocal laser-scanning microscope (Zeiss).
Immunoprecipitation
Cells were harvested and lysed in five volumes of lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 3 mM MgCl2, 5% glycerol, 0.5% Nonidet P-40, 1 mM dithiothreitol, 1% protein inhibitor (PI), pH 7.4) for 2 h with rotation at 4°C. Cell lysates were cleared by centrifugation at 21,000 g for 30 min, followed by measuring the supernatant with 2-D Quantitative Kit. Equal amount of protein was incubated with the ANTI-FLAG M2 affinity gel (Sigma) at 4°C overnight, washed with a wash buffer (25 mM Tris-HCl, 150 mM NaCl, 3 mM MgCl2, 0.2 mM EDTA, 0.1% Tween-20, 5% glycerol, 1% PI, pH 7.4) for 10 min for three times, and proteins were eluted with 20 μl elution buffer containing 400 μg/ml 3×FLAG peptides (ChinaPeptides Co., Ltd.).
LC-MS/MS analysis
The elution proteins of immunoprecipitation were separated by 12.5% SDS-PAGE gel and visualized by a Silver Staining Kit (Beyotime). The gels were destained and dehydrated and the proteins were digested with sequencing-grade trypsin (Promega). The peptides were extracted from gel pieces with 0.1% formic acid (FA) and 50% acetonitrile, and dried in a vacuum centrifuge (ThermoFisher Scientific).
The peptides were dissolved in 10 μl of 0.2% FA and separated by an online Nano-LC system (Microtech Scientific), which was equipped with a C18 reverse phase column. The column was eluted with linear gradient of 5–30% acetonitrile in 0.2% FA at the rate of 500 nl/min for 100 min.
The mass spectra were acquired by LTQ-Orbitrap mass spectrometer (ThermoFisher) equipped with a nano-ES ion source (Proxeon Biosystems). Full scan spectra (from m/z 300–1600) were acquired in the Orbitrap analyzer with a resolution of 60,000 at 400 m/z after the accumulation of 1,000,000 ions. The five most-intense ions per scan were selected for collision-induced dissociation fragmentation in the linear ion trap after the accumulation of 3000 ions. We set the maximal filling times at 500 ms for the full scans and 150 ms for the MS/MS scans. The dynamic exclusion list was restricted to a maximum of 500 entries with a maximum retention period of 60 s and a relative mass window of 10 ppm.
Data analysis
All raw files were processed with the MaxQuant software (Version 1.3.0.5). The generated peak list files were searched against the UniProt mouse protein sequence database (released 2013.08). The searching parameters were set as follows: enzyme was trypsin; up to two missed cleavages; carbamidomethyl cysteine as fixed modification; and oxidation methionine and protein N-terminal acetylation as variable modifications. MS tolerance was 6 ppm, while MS/MS tolerance was 0.5 Da. The required false discovery rate was set to 1% at peptide and protein levels, and the minimum required peptide length was seven amino acids. At least one unique or razor peptide per protein group was required for protein identification.
In vitro interaction and deacetylation
Sirt3 was subcloned into pColdTM TF DNA (Code. 3365) and was expressed in BL21 (CD601; Transgen) on the Isopropyl β-d-1-thiogalactopyranoside induction. pCold-TF-Sirt3 and the vector were purified with nickel beads. For in vitro interaction assay, 5 μg of SOD2-Flag or ATP synthase β proteins was incubated with 15 μg pCold-TF-Sirt3 protein in tris buffer at 4°C overnight. The nickel beads were added and washed for three times with 50 mM imidazole. Finally, the proteins were elluted by 200 mM imidazole. For in vitro deacetylation assay, 5 μg of SOD2-Flag or ATP synthase β proteins was incubated with 15 μg pCold-TF-Sirt3 proteins in HEPES buffer at 30°C for 6 h.
ChIP assay
ChIP assay was performed following the manufacturer's instruction of Chromatin Immunoprecipitation Kit (9002; Cell Signaling Technology). qPCR was performed to find out the binding sites in the Sirt3 promoter by PGC-1α. Primer sequences of Sirt3 for amplifying purified DNA are listed in the Supplementary Table S3.
Statistical analysis
All functional experiments were repeated at least three times. The protein bands in Western blotting were quantified by ImageJ. Data were analyzed by GraphPad Prism 5.0 software and shown by the mean ± SEM. Multiple sets of data were analyzed by one-way or two-way ANOVA, and Student's t-test was used to analyze two sets of data, considering statistically significant when p < 0.05.
Supplementary Material
Abbreviations Used
- ATP
adenosine triphosphate
- ChIP
chromatin immunoprecipitation
- ERRα
estrogen-related receptor alpha
- FA
formic acid
- HRP
horseradish peroxidase
- LC-MS/MS
liquid chromatography-mass spectrometry/mass spectrometry
- MPP+
1-methyl-4-phenylpyridinium iodide
- MPTP
1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine
- MTT
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- N-2a
Neuro-2a
- NAC
N-acetyl-L-cysteine
- NAM
nicotinamide
- NRF1
nuclear respiratory factor 1
- NRF2
nuclear factor erythroid 2 (NF-E2)-related factor 2
- PARP-1
poly (ADP-ribose) polymerase-1
- PBS
phosphate-buffered saline
- PD
Parkinson's diseases
- PFA
paraformaldehyde
- PGC-1α
peroxisome proliferator-activated receptor-γ coactivator 1α
- PI
protein inhibitor
- qPCR
quantitative real-time polymerase chain reaction
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- Sirt3
sirtuin 3
- SNc
substantia nigra pars compacta
- SOD2
manganese superoxide dismutase
- TCA
tricarboxylic acid
Acknowledgments
We are grateful to Professor Toren Finkel of the National Institutes of Health for providing helpful discussion. Additionally, we would like to thank Professor David T. Dexter of Imperial College London, for providing discussion and sample preparation. This work was supported by grants from the National Natural Science Foundation of China (Nos. 31270872, 31470807, 31200610), the National Key Basic Research Program of China (Nos. 2010CB912203, 2011CB915504), and funds from the State Key Laboratory of Protein and Plant Gene Research, College of Life Sciences, Peking University.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, and Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A 105: 14447–14452, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreux PA, Houtkooper RH, and Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 12: 465–483, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, and Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic Biol Med 49: 1230–1237, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beal MF. Experimental models of Parkinson's disease. Nat Rev Neurosci 2: 325–334, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Bell Eric L. and Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell 42: 561–568, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butterfield DA, Perluigi M, Reed T, Muharib T, Hughes CP, Robinson RA, and Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal 17: 1610–1655, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalkiadaki A. and Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol 8: 287–296, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Chao J, Leung Y, Wang M, and Chang RC. Nutraceuticals and their preventive or potential therapeutic value in Parkinson's disease. Nutr Rev 70: 373–386, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Zhang J, Lin Y, Lei Q, Guan K-L, Zhao S, and Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 12: 534–541, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherry AD, Suliman HB, Bartz RR, and Piantadosi CA. Peroxisome proliferator-activated receptor gamma co-activator 1-alpha as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J Biol Chem 289: 41–52, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325: 834–840, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Dauer W. and Przedborski S. Parkinson's disease: mechanisms and models. Neuron 39: 889–909, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Exner N, Lutz AK, Haass C, and Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J 31: 3038–3062, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finck BN. and Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 116: 615–622, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finley L, Haas W, Desquiret-Dumas V, Wallace D, Procaccio V, Gygi S, and Haigis M. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One 6: 6, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu J, Jin J, Cichewicz RH, Hageman SA, Ellis TK, Xiang L, Peng Q, Jiang M, Arbez N, Hotaling K, Ross CA, and Duan W. trans-(-)- -Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington disease. J Biol Chem 287: 24460–24472, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giralt A, Hondares E, Villena JA, Ribas F, Diaz-Delfin J, Giralt M, Iglesias R, and Villarroya F. Peroxisome proliferator-activated receptor- coactivator-1 controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J Biol Chem 286: 16958–16966, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gleyzer N, Vercauteren K, and Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol 25: 1354–1366, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guarente L. Calorie restriction and sirtuins revisited. Genes Dev 27: 2072–2085, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallows WC, Yu W, Smith BC, Devires MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, Zhao S, Guan K-L, and Denu JM. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol Cell 41: 139–149, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han P, Tang Z, Yin J, Maalouf M, Beach TG, Reiman EM, and Shi J. Pituitary adenylate cyclase-activating polypeptide protects against β-amyloid toxicity. Neurobiol Aging 35: 2064–2071, 2014 [DOI] [PubMed] [Google Scholar]
- 22.Hauser DN. and Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis 51: 35–42, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herskovits AZ. and Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res 23: 746–758, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, and Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464: 121–125, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karaki S, Becamel C, Murat S, Mannoury la Cour C, Millan MJ, Prezeau L, Bockaert J, Marin P, and Vandermoere F. Quantitative phosphoproteomics unravels biased phosphorylation of serotonin 2A receptor at Ser280 by hallucinogenic versus nonhallucinogenic agonists. Mol Cell Proteomics 13: 1273–1285, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim H-S, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park S-H, Singh KK, Abdulkadir SA, Spitz DR, Deng C-X, and Gius D. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17: 41–52, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim-Han JS, Antenor-Dorsey JA, and O'Malley KL. The parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J Neurosci 31: 7212–7221, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang-Rollin IC, Rideout HJ, Noticewala M, and Stefanis L. Mechanisms of caspase-independent neuronal death: energy depletion and free radical generation. J Neurosci 23: 11015–11025, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langston JW, Ballard P, Tetrud JW, and Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219: 979–980, 1983 [DOI] [PubMed] [Google Scholar]
- 30.Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y, and Liu M. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 31: 681–683, 2013 [DOI] [PubMed] [Google Scholar]
- 31.Lim KM, Kim HH, Bae ON, Noh JY, Kim KY, Kim SH, Chung SM, Shin S, Kim HY, and Chung JH. Inhibition of platelet aggregation by 1-methyl-4-phenyl pyridinium ion (MPP+) through ATP depletion: evidence for the reduced platelet activities in Parkinson's disease. Platelets 20: 163–170, 2009 [DOI] [PubMed] [Google Scholar]
- 32.Liu G, Tian H, Huang YQ, Hu J, Ji YX, Li SQ, Feng YQ, Guo L, and Zhu YG. Alterations of mitochondrial protein assembly and jasmonic acid biosynthesis pathway in Honglian (HL)-type cytoplasmic male sterility rice. J Biol Chem 287: 40051–40060, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Peritore C, Ginsberg J, Kayhan M, and Donmez G. SIRT3 attenuates MPTP-induced nigrostriatal degeneration via enhancing mitochondrial antioxidant capacity. Neurochem Res 40: 600–608, 2015 [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Yang N, Hao W, Zhao Q, Ying T, Liu S, Li Q, Liang Y, Wang T, Dong Y, Ji C, and Zuo P. Dynamic proteomic analysis of protein expression profiles in whole brain of Balb/C mice subjected to unpredictable chronic mild stress: implications for depressive disorders and future therapies. Neurochem Int 58: 904–913, 2011 [DOI] [PubMed] [Google Scholar]
- 35.McNaught KS, Jackson T, JnoBaptiste R, Kapustin A, and Olanow CW. Proteasomal dysfunction in sporadic Parkinson's disease. Neurology 66: S37–S49, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Nicotera P. and Leist M. Energy supply and the shape of death in neurons and lymphoid cells. Cell Death Differ 4: 435–442, 1997 [DOI] [PubMed] [Google Scholar]
- 37.Olanow CW. and Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci 22: 123–144, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Onyango P, Celic I, McCaffery JM, Boeke JD, and Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A 99: 13653–13658, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pellegrini L, Pucci B, Villanova L, Marino ML, Marfe G, Sansone L, Vernucci E, Bellizzi D, Reali V, Fini M, Russo MA, and Tafani M. SIRT3 protects from hypoxia and staurosporine-mediated cell death by maintaining mitochondrial membrane potential and intracellular pH. Cell Death Differ 19: 1815–1825, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perier C, Bové J, and Vila M. Mitochondria and programmed cell death in Parkinson's disease: apoptosis and beyond. Antioxid Redox Signal 16: 883–895, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Qiu X, Brown K, Hirschey MD, Verdin E, and Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 12: 662–667, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Qu D, Rashidian J, Mount MP, Aleyasin H, Parsanejad M, Lira A, Haque E, Zhang Y, Callaghan S, Daigle M, Rousseaux MW, Slack RS, Albert PR, Vincent I, Woulfe JM, and Park DS. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson's disease. Neuron 55: 37–52, 2007 [DOI] [PubMed] [Google Scholar]
- 43.This reference has been deleted.
- 44.Rahman M, Nirala NK, Singh A, Zhu LJ, Taguchi K, Bamba T, Fukusaki E, Shaw LM, Lambright DG, Acharya JK, and Acharya UR. Drosophila Sirt2/mammalian SIRT3 deacetylates ATP synthase beta and regulates complex V activity. J Cell Biol 206: 289–305, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC, and Gupta MP. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol 34: 807–819, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.This reference has been deleted.
- 47.Scher MB, Vaquero A, and Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev 21: 920–928, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwer B. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol 158: 647–657, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, Jacobson MP, and Verdin E. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab 12: 654–661, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shin J-H, Ko Han S, Kang H, Lee Y, Lee Y-I, Pletinkova O, Troconso Juan C, Dawson Valina L, and Dawson Ted M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell 144: 689–702, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song W, Song Y, Kincaid B, Bossy B, and Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1α. Neurobiol Dis 51: 72–81, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su Y, Sun H, Fang J, Hu G, and Xiao M. Brain mitochondrial dysfunction in ovariectomized mice injected with D-galactose. Neurochem Res 35: 399–404, 2010 [DOI] [PubMed] [Google Scholar]
- 53.Vassilopoulos A, Pennington JD, Andresson T, Rees DM, Bosley AD, Fearnley IM, Ham A, Flynn CR, Hill S, Rose KL, Kim H-S, Deng C-X, Walker JE, and Gius D. SIRT3 deacetylates ATP synthase F1complex proteins in response to nutrient- and exercise-induced stress. Antioxid Redox Signal 21: 551–564, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venderova K. and Park DS. Programmed cell death in Parkinson's disease. Cold Spring Harb Perspect Med 2: a009365, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, and Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327: 1004–1007, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weir H, Murray T, Kehoe P, Love S, Verdin E, O'Neill M, Lane JD, and Balthasar B. CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer's disease. PLoS One 7: 7, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiss S, Pin JP, Sebben M, Kemp DE, Sladeczek F, Gabrion J, and Bockaert J. Synaptogenesis of cultured striatal neurons in serum-free medium: a morphological and biochemical study. Proc Natl Acad Sci U S A 83: 2238–2242, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, and Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999 [DOI] [PubMed] [Google Scholar]
- 59.Xu S-Y, Wu Y-M, Ji Z, Gao X-Y, and Pan S-Y. A modified technique for culturing primary fetal rat cortical neurons. J Biomed Biotechnol 2012: 1–7, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshino J. and Imai S. Mitochondrial SIRT3: a new potential therapeutic target for metabolic syndrome. Mol Cell 44: 170–171, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu W, Dittenhafer-Reed KE, and Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 287: 14078–14086, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, and Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science 327: 1000–1004, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.