

Abstract
Reactive oxygen species (ROS) are believed to be important mediators of muscle atrophy and weakness in aging and many degenerative conditions. However, the mechanisms and physiological processes specifically affected by elevated ROS in neuromuscular units that contribute to muscle weakness during aging are not well defined. Here we investigate the effects of chronic oxidative stress on neurotransmission and excitation-contraction (EC) coupling mechanisms in the levator auris longus (LAL) muscle from young (4–8 months) and old (22–28 months) wild type mice and young adult Sod1−/− mice. The frequency of spontaneous neurotransmitter release and the amplitude of evoked neurotransmitter release in young Sod1−/− and old wild type LAL neuromuscular junctions were significantly reduced from the young wild type values, and those declines were mirrored by decreases in synaptic vesicle pool size. Presynaptic cytosolic calcium concentration and mitochondrial calcium uptake amplitudes showed substantial increases in stimulated young Sod1−/− and old axon terminals. Surprisingly, LAL muscle fibers from old mice showed a greater excitability than fibers from either young wild type or young Sod1−/− LAL muscle fibers. Both evoked excitatory junction potential (EJP) and spontaneous mini EJP amplitudes were considerably higher in LAL muscles from old mice than in fibers from young Sod1−/− LAL muscle. Despite a greater excitability, sarcoplasmic calcium influx in both old wild type and young Sod1−/− LAL muscle fibers was significantly smaller. Sarcoplasmic reticulum calcium levels were also smaller in both mice, but the difference was not statistically significant in muscle fibers from old wild type mice. The protein ratio of triad calcium channels – RyR1/DHPR was not different in all groups. However, fibers from both Sod1−/− and old mice had substantially elevated levels of protein carbonylation and S-nitrosylation modifications. Overall, our results suggest that young Sod1−/− recapitulate many neuromuscular and muscle fiber changes seen in old mice. We also conclude that muscle weakness in old mice might in part be driven by ROS mediated EC uncoupling, while both EC uncoupling and reduced neurotransmitter release contribute to muscle weakness in Sod1−/− mice.
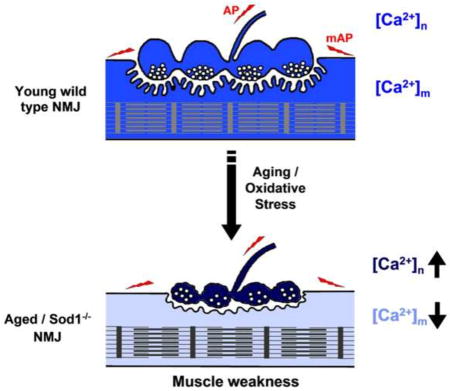
Graphical abstract
INTRODUCTION
Aging is associated with a progressive loss in muscle mass and strength, which causes a substantial reduction in quality of life and mobility. Deterioration in skeletal muscle structure and function with age is thought to be driven by multiple processes, such as loss of trophic factor support, hormonal changes, inadequate nutrition and activity, and reduced muscle regenerative capacity; elevated cytosolic reactive oxygen species (ROS) and resultant macromolecular damage is believed to be one of their major downstream effectors [1,2]. In support of the ROS role in muscle atrophy, a number of studies have shown that caloric restriction [3,4] and boosting of cellular stress responses [5,6] result in reduced oxidative stress and preservation of muscle function in older animals. Conversely, elevation of cellular cytosolic oxidative stress either by the deletion of Cu-Zn superoxide dismutase (Sod1−/−) or by introduction of mutations in CuZnSOD (e.g., the SOD1-G93A ALS miutant mouse model) increases ROS and causes premature muscle atrophy and weakness in mice [7,8,9], which is phenotypically similar to muscle changes seen in older animals. Both Sod1−/− and SOD1-G93A mice have dysfunctional mitochondria and a substantial elevation in the biomarkers of protein and nucleic acid oxidative damage in skeletal muscles. It has also been shown that oxidative stress levels correlate strongly with the extent of muscle mass reduction, neuromuscular junction (NMJ) denervation and muscle fiber type switching in Sod1−/−, SOD1-G93A and old wild type mice. Those changes can be detected as early as 4–5 months of age in Sod1−/− animals [9]. However, despite these similarities in old and Sod1 mutant mice, the use of SOD1-G93A and other SOD1 mutant models to decipher the mechanistic aspects of oxidative stress in muscle atrophy and weakness is confounded by the toxic gain of function that results in the formation of CuZnSOD protein aggregates [10], an issue that is absent in Sod1−/− mice.
More recent studies have attempted to evaluate the contributions of elevated oxidative stress in distinct cell populations to muscle atrophy and weakness. Neuronal rescue of Sod1 expression in Sod1−/− mice prevented early declines in muscle mass and isometric force [11], whereas muscle specific deletion of Sod1 led to the decline in muscle force without muscle atrophy [12]. This suggests that muscle changes observed in Sod1−/− mice might be driven by neuronal oxidative stress, which is similar to normal aging wherein motor neuronal dysfunction is believed to precede muscular changes [13]. Overall, the Sod1−/− mouse is a promising model of sarcopenia in the fact that both Sod1−/− and old mice have quite similar neuromuscular phenotypes; however, it still remains to be shown that physiological mechanisms underlying those phenotypes are also the same.
In the current study, we used the levator auris longus (LAL) muscle to characterize physiological changes in the neuromuscular unit in response to chronic oxidative stress in old and young Sod1−/− mice, and also to explore potential links between those changes and muscle weakness. We found that presynaptic terminals in LAL muscles from young Sod1−/− and old wild type mice had comparable changes in neurotransmitter release and presynaptic calcium signaling. Young Sod1−/− and old wild type mice showed ROS-dependent decreases in both spontaneous and evoked neurotransmitter release (quantal content) at LAL NMJs. Presynaptic cytosolic calcium concentration and mitochondrial calcium uptake were significantly elevated in stimulated young Sod1−/− and old LAL axon terminals. Surprisingly, LAL muscle fibers from old wild type mice showed increased excitability and higher evoked EJPs. LAL sarcoplasmic calcium influx during both twitch and titanic stimuli in old wild type and young Sod1−/− mice was significantly smaller than in young animals, which was potentially the result of oxidative modification to muscle calcium channels, as the protein levels of RyR1 and DHPR were unchanged. Therefore, LAL muscles from young Sod1−/− and old wild type mice have reduced EC coupling and muscle fiber recruitment compared to young wild type LAL muscles, which suggests that ROS induced EC uncoupling might be one of the causes of muscle weakness in old mice.
MATERIALS AND METHODS
Animals
All animal procedures and manipulations were performed in accordance with the Guide for the Care and Use of Laboratory Animals by the NIH and approved by the Institutional Animal Care and Use Committees at UTHSCSA and Audie L. Murphy Memorial Veterans Hospital. Sod1−/− (a gift from Dr. Charles Epstein, UCSF) mice were generated by crossing breeder Sod1−/+ animals previously backcrossed to C57/BL6 mice. Thy1-EYFP+/tg mice (YFP-16; Jackson Laboratory, Bar Harbor, ME) were crossed with Sod1−/− mice to generate Thy1-EYFP+/tg;Sod1−/− mice. All genotypes were confirmed by PCR of DNA isolated from mouse tail tips and performed according specific genotyping protocols described at www.jax.org. C57/BL6, Sod1−/−, Thy1-EYFP+/tg, and Thy1-EYFP+/tg;Sod1−/− mice were maintained until ~ 22–28 month old under specific pathogen-free conditions to generate the “old” experimental group. Mice of 4–8 months old were assigned to the “young” group.
Chemical and antibodies
Unless otherwise mentioned chemical reagents were purchased from Sigma-Aldrich (Saint Louis, MO). Primary DHPR α1 (Cav1.1), Mouse RyR1 (clone 34C) and G3PD (GAPDH) antibodies and secondary goat anti-mouse IgG HRP-conjugated antibody were bought from Thermo Scientific (Rockford, IL). anti-MHCIIa-FITC antibody was from Biorbyt (San Francisco, CA). β-actin antibody was bought from GenScript (Piscataway, NJ). Synaptobrevin (VAMP1) mouse antibody was bought from Invitrogen (Carlsbad, CA).
Muscle electrophysiology
Mice were euthanized by an intraperitoneal injection of pentobarbital (200 mg/kg of body weight). The levator auris longus (LAL) muscle together with the nerve was surgically dissected from the mouse neck by following the procedure described in ref. 14. LAL was pinned to the bottom of a 35 mm Sylgard covered Petri dish and perfused with oxygenated (95% O2/5% CO2) Ringer’s solution (mM: 120 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 0.4 NaH2PO4, 23.8 NaHCO3 and 5.6 D-glucose) at room temperature. After ~ 1 hour of recovery, the nerve was drawn into a glass stimulating pipette filled with Kreb’s solution and 1 μM μ-conotoxin GIIIB (Peptide International, Louisville, Kentucky, USA) was added to the bathing solution to block muscle action potentials. Recordings began after the muscle stopped producing twitches in response to single stimuli (3V, 300 μs), and were done at room temperature. Nerve stimuli were delivered via a Master-8 stimulator (AMPI, Israel) connected to a digitimer - DS2A Mk.II (Digitimer, Hertfordshire, UK). The electrophysiological recording setup consisted of an upright BX51WI Olympus microscope equipped with 20X and 40X Olympus water immersion lenses, and a 1.0 gain head stage connected to an Axoclamp 900A amplifier (Molecular Devices). Microelectrodes used for recordings were filled with a mixture of 1.5M KCl and 1.5M KAc solution to give the final resistance of ~ 50 MΩ. Data were digitized (4/30, PowerLab) and recorded with Chart 5.5.6 software package (AD Instruments). Data analysis was performed using both Chart and AxoGraph software. EJP amplitude, miniature EJP frequency and amplitude, and quantal content were measured and calculated in the same way as described in our earlier study ref. 15.
Fluorescent measurements
LAL for fluorescent imaging of both presynaptic and muscle variables was prepared in the same way as for electrophysiological recordings. Muscle contractions were inhibited either by applying 10 μM tubocurarine (Sigma-Aldrich, Saint Louis, MO) to the bathing solution for presynaptic pH and mitochondrial calcium imaging or by stretching the muscle at ~1.5x of its resting length for muscle fiber calcium imaging. Fluorescent images were captured using an Andor iXon DU-860D back illuminated EMCCD camera (Andor Technology, South Windsor, CT) controlled by Andor IQ software (ver 1.8) and wide-field microscopy on an upright BX51WI Olympus microscope fitted with a 60X and 100X Olympus water immersion lenses. A xenon arc lamp in a Lambda DG-4 illumination system (Novato, CA) was used as the excitation light source. Emitted light was filtered by using a Lambda 10-B optical filter changer. Optical filters and dichroic mirrors were purchased from Chroma Technology (Rockingham, VT) and Semrock (Rochester, NY). Image processing was performed with either Andor iQ or ImageJ software package. ROS production by presynaptic mitochondria was imaged using MitoSox Red (Invitrogen, Carlsbad, CA). 5 mM MitoSox stock solution in DMSO was added to LAL in Ringer’s solution to the final concentration of 10 μM and incubated for 30 min in the dark at room temperature. After a brief rinse, basal MitoSox fluorescence (emission 580 nm) was collected from visually identified NMJs. LAL was next stimulated with a 10 Hz train of stimuli for 30 min, and imaged again. To obtain maximal values of MitoSox fluorescence, LAL was incubated with 50 μM antimycin A (Sigma-Aldrich, Saint Louis, MO) for 30 min. MitoSox fluorescent intestines were background subtracted, averaged and normalized to background subtracted fluoresce obtained with antimycin A. Presynaptic pH changes were imaged at LAL NMJs by monitoring EYFP fluorescence (emission 525 nm, 120 ms/frame) changes in response to 2s of 30 Hz and 100 Hz stimulus trains. EYFP fluorescence was background subtracted and normalized to the resting fluorescence. Presynaptic mitochondrial calcium uptake was measured with rhod-2AM (Invitrogen, Carlsbad, CA). 1 mM rhod-2AM dissolved in DMSO was added to LAL in Ringer’s solution to the final concentration of 1 μM and incubated for 30 min in the dark at room temperature. The preparation was then distained for 30 min in dye free solution. Rhod-2 fluorescence was imaged (emission 580 nm, 5 Hz acquisition rate) during LAL stimulations with 2s, 30 Hz and 100 Hz stimulus trains. Rhod-2 fluorescence values 2s after termination of the stimulus for both 30 Hz and 100 Hz were background subtracted and averaged. Muscle fiber sarcoplasmic calcium transients were imaged with fluo-4AM (Invitrogen, Carlsbad, CA). LAL was incubated with 1 μM of fluo-4AM for ~ 15 min and distained in dye free solution for additional 30 min at room temperature. Fluo-4 fluorescent responses (emission 510 nm) were recorded in response to 3 single pulses (twitches) and a 2s 30 Hz stimulus train at an image acquisition rate of 55 Hz. Raw fluo-4 fluorescence values were background subtracted and normalized to the resting fluorescence. Sarcoplasmic reticulum calcium content was estimated by measuring the peak intensity of fluo-4 fluorescent response and normalizing it to the resting fluo-4 intensity in muscles exposed to 10 mM caffeine (Sigma-Aldrich, Saint Louis, MO).
Western immunoblot
LAL was isolated and homogenized in 5 volumes of ice cold homogenation solution (50 mM Tris, 10 mM EDTA, pH 8.3). An equal volume of extracting solution (120 mM Tris, 4% SDS, 20% glycerol, pH 6.8) was added to the homogenate. The sample was mixed thoroughly and heated in a 50 °C waterbath for 15 min and then, centrifuged at 13,400 rpm for 20 min in an Eppendorf minispin centrifuge (Eppendorf, Hamburg, Germany). The supernatant was collected and aliquoted for further use in protein assays. Protein concentrations were determined spectrophotometrically using a Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL) and an Ultrospec 3000 spectrophotometer (Pharmacia Biotech, Pittsburgh, PA). For a regular immunoblot, protein samples were diluted with an equal volume of Laemmli sample buffer plus β-mercaptoethanol (Bio-Rad, Hercules, CA) and heated at 95 °C for 5 min. After cooling, the samples together with a PAGE-MASTER protein standard (GenScript, Piscataway, NJ) were loaded 10 μg/lane onto a pre-cast 4–15% Mini-PROTEAN TGX polyacrylamide gel (Bio-Rad, Hercules, CA) and separated at 200V for 60 min. Separated proteins were then transferred to PVDF membrane (Bio-Rad, Hercules, CA) and blocked in 5% non-fat dry milk TBS (20 mM Tris, 150 mM NaCl, 0.3% Tween 80, pH 7.5) for 1 hour. The membrane was then incubated for 3 hours with a primary antibody diluted in TBS followed by 3×5 min washes with TBS, and then an HRP conjugated secondary antibody. After the final wash, the membrane was incubated with Pierce ECL western blotting substrate (Thermo Scientific, Rockford, IL) and exposed to an X-ray film (Thermo Scientific, Rockford, IL). Proteins samples for carbonyl (Oxidized Protein Western Blot Detection Kit, Abcam, Cambridge, MA) and S-nitrosothiol (Pierce S-Nitrosylation Western Blot Kit, Thermo Scientific, Rockford, IL) group detection were derivatized before loading onto a gel according to the manufacturer instructions.
Statistical analysis
Statistical analysis was performed either in Microsoft Excel or SigmaPlot. Fluorescence values and electrophysiological data in the text are shown as group mean ± standard error of mean. Statistical significance was tested using paired Student’s t-test (two tails) unless otherwise mentioned. P values as shown in figures: * p<0.05, **p<0.01, ***p<0.001.
RESULTS
We investigated the effects of age-related oxidative stress and experimentally induced reactive oxygen species (ROS) overproduction by the deletion of the Cu-Zn superoxide dismutase gene (Sod1−/−) on muscle-nerve communication at neuromuscular junctions (NMJs) in the levator auris longus muscle (LAL) in mice. LAL is an exceptionally thin and flat fast-twitch skeletal muscle, which makes it an ideal preparation for both imaging and electrophysiological assays. Moreover, its relatively invariant daily activity unlike in other skeletal muscles provides for a greater control over NMJ response variability with age. LAL was isolated from young (4–8 months) and old (22–28 months) mice of mixed sex. The caudal (smallest) segment of the LAL has been reported to undergo a substantial denervation and atrophy with age and also, in ALS and spinal muscular atrophy [16]. Morphologically, LAL from old wild type mice had more split muscle fibers than LAL from young mice, but overall LAL muscle fibers from old mice did not appear to be significantly atrophied. Muscle fiber typing analysis performed using wild type mice expressing DsRed fluorescent protein under the control of Myh2 promoter or with anti-MHCIIa antibody revealed that LAL from old mice had significantly fewer pure type IIa fibers (the predominant muscle fiber type in young LAL muscle, [17]) than LAL from young mice (Fig. 1A, Young 50.7±4.1%, n=5 (muscles); Old 35.1±3.3%, n=5 (muscles), p=0.019). In addition, a number of weakly stained fibers could be detected in old but not young LAL, which were categorized as mixed type and were not used for type IIa fiber counting. The average muscle fiber diameter (Ferret’s diameter) measured in bright-field muscle images was not significantly different between the groups (Young 17.8±1.1 μm, n=72 (fibers); Old 18.3±1.1 μm, n=100 (fibers)).
Figure 1. Age-related changes in muscle fiber composition and ROS production in the levator auris longus (LAL) muscle.

(A) Bar charts showing the abundance of type IIa muscle fibers (right) and the average Ferret’s diameter for all muscle fibers in old and young LAL (left). (B) Quantification of ROS production in young and old LAL NMJs during rest and nerve electrical stimulation. The values shown are MitoSOX fluorescence intensities normalized to the maximal MitoSOX responses obtained with antimycin A. Error bars are SEMs.
We used MitoSOX to quantify the relative rates of ROS production by presynaptic mitochondria in young and old LAL NMJs. Both basal (unstimulated) and stimulated rates (facial nerve electrical stimulation) of ROS production normalized to the maximal rate obtained with antimycin A were substantially elevated in presynaptic mitochondria in LAL muscles from old mice (Fig. 1B, F/Fantimycin, basal: Young 4.4±1.4%, n=14 (NMJs), Old 15.0±3.4%, n=12 (NMJs); 10Hz 30 min stimulation: Young 15.3±1.7%, n=14 (NMJs), Old 56.7±8.6%, n=12 (NMJs)). This suggests that dysfunctional mitochondria and elevated rates of ROS production are important markers of old LAL.
To further elucidate the role of oxidative stress in aging, and more importantly, the impact of oxidative stress on physiological mechanisms involved in motor neuron – muscle fiber communication, we employed a mouse model lacking the antioxidant enzyme Cu-Zn superoxide dismutase (Sod1−/− mice), which has been used previously as a model of accelerated aging and sarcopenia [4,8]. In this study we used 4–8 month old Sod1−/− mice to avoid any overlap with NMJ/muscle early developmental changes. Previous studies showed that young Sod1−/− mice have substantial muscle atrophy and weakness typical of much older mice. These atrophic changes were accompanied by elevated rates of mitochondrial O2·− and hydrogen peroxide production, similarly to the increase in ROS generation measured in muscle mitochondria from old wild type mice [9]. Immunochemistry for MHCIIa revealed that the number of type IIa fibers in Sod1−/− LAL was reduced 44.3±3.1% n=35 (fibers) compared to young LAL (difference was not significant), and the Ferret’s fiber diameter was unchanged 17.1±1.5 μm n=35 (fibers). Electrophysiological recordings in LAL showed that EJP and mini EJPs amplitudes were increased in old and decreased in Sod1−/− mice when compared to young controls Fig. 2A. Membrane input resistance was considerably higher in LAL muscle from old mice compared to muscles from either young wild type or Sod1−/− mice, see Table 1. Together, these results and LAL morphological findings suggest that changes in muscle fiber electrical properties rather than muscle fiber diameter are likely responsible for the augmented EJP/mini EJP amplitudes in old mice. There was no significant difference in the resting membrane potential between the groups. Conversely, the quantum content (QC), a number of synaptic vesicles released per action potential, was reduced at old and Sod1−/− LAL NMJs. Also, both old and Sod1−/− LAL muscles had substantially reduced mini EJP frequencies than younger controls, Fig. 2A. Decreased synaptobrevin (VAMP1) protein levels in both old and Sod1−/− LAL muscle extracts, Fig. 2B, indicate that synaptic vesicle pool sizes are substantially smaller at old and Sod1−/− LAL NMJs, which is in agreement with the calculated QC values. These findings suggest that elevated oxidative stress at old and Sod1−/− NMJs likely has a common mechanism of action, which results in reduced neurotransmitter release.
Figure 2. Evoked and spontaneous neurotransmitter release properties and synaptic vesicle pool size are changed with increased oxidative stress.

(A) Representative traces of evoked EJPs (top row) and spontaneous miniature EJPs (bottom row) recorded in LAL obtained from young and old wild type and Sod1−/− mice. (B) Representative western blot of young, old and Sod1−/− LAL muscle protein extracts (left) incubated with anti-VAMP1 antibodies. Quantification of VAMP1 protein levels (integrated band density normalized to β-actin levels) in young (n=5) and old BL6 (n=5) and Sod1−/− (n=3) LAL muscles (right). Error bars are SEM.
Table 1.
Summary of electrophysiological parameters measured in LAL from young and old wild type mice and Sod1−/− mice.
| YOUNG n=20(4) |
OLD n=15(3) |
Sod1−/− n=18(3) |
|
|---|---|---|---|
| EJP (mV) | 31.1±1.1*** | 37.5±1.6 | 24.5±0.9 |
| mini EJP (mV) | 0.69±0.03 | 1.86±0.22*** | 0.63±0.09 |
| RMP (mV) | 70.8±0.6 | 70.7±1.1 | 70.8±1.2 |
| Quantal Content | 76.1±5.3*** | 42.5±4.8 | 33.1±4.7 |
| Input Res. (MOhm) | 1.8±0.3 | 2.9±0.4* | 1.6±0.2 |
| Freq. mini EJP (Hz) | 2.5±0.3* | 1.5±0.3 | 0.7±0.4 |
t-test p values:
p<0.001
p<0.05
We next examined presynaptic terminal morphology and cytosolic calcium/pH kinetics in Thy1-EYFP+/tg and Thy1-EYFP+/tg; Sod1−/− transgenic mice. Old wild type and Sod1−/− mice appeared to have very similar LAL NMJ morphologies, both were smaller in size and more fragmented than young control NMJs, but without the signs of possible denervation (Fig. 3A). Calcium influx into the nerve terminal could be assessed indirectly by monitoring cytosolic pH changes with EYFP. Cytosolic acidification is produced through fast calcium extrusion by the plasma membrane calcium ATPase, and it can be used as an indirect and crude measure of calcium influx. It has been previously demonstrated that NMJ electrical stimulation results in calcium-dependent cytosolic acidification, which reversibly decreases EYFP fluorescence in presynaptic terminals, Fig. 3B. On the other hand, the rate of following re-alkalization and recovery in EYFP fluorescence is proportional to the extent of exocytosis (the number of synaptic vesicle fused with the membrane) [18]. 2s, 30 and 100 Hz stimuli produced cytosolic acidifications in nerve terminals from old wild type and Sod1−/− LAL muscles that were substantially larger than in young controls (Fig. 3C; 100Hz, −dF/Fo: Young 1.9±0.2%, n=10 (NMJs); Old 3.3±0.2%, n=12 (NMJs); Sod1−/− 4.2±0.6%, n=9 (NMJs)). Acidification amplitudes at 30Hz, however, were larger but not statistically significant, which is likely the result of fast pH buffering by the Na+/H+ exchanger at lower stimulation magnitudes [18]. Moreover, realkalization rates at 100Hz were also slower in NMJs from old wild type and Sod1−/− mice, suggesting that fewer synaptic vesicles were released from those junctions despite higher calcium influx/cytosolic acidifications.
Figure 3. Oxidative stress leads to large acidification and slow re-alkalization in stimulated presynaptic terminals of old and Sod1−/− LAL muscles.

(A) Fluorescent images showing morphology of LAL axon terminals in young, old, and Thy1-EYFP+/tg;Sod1−/− mice. (B) EYFP fluorescence changes in LAL nerve terminals from young, old, and Thy1-EYFP+/tg;Sod1−/− mice in response to 2s trains of 30Hz and 100Hz stimuli. (C) Average absolute decrease in EYFP fluorescence recorded from LAL nerve terminals from young, old, and Thy1-EYFP+/tg;Sod1−/− mice in response to 2s, 100Hz stimulus train. Error bars are SEMs.
Increased cytosolic calcium levels might in part be the result of lost mitochondrial cytosolic calcium sequestration. Mitochondrial cytosolic calcium uptake is necessary for the stimulation of oxidative phosphorylation and synchronization of mitochondrial ATP synthesis with synaptic activity [19,20]. However, excessive mitochondrial calcium levels are known to cause ROS production and trigger mitochondrial dependent apoptosis. We measured cytosolic calcium uptake by presynaptic mitochondria at LAL presynaptic terminal loaded with rhod-2, a calcium sensitive fluorescent dye. Mitochondrial calcium concentration was expressed as the background subtracted increase in rhod-2 fluorescence intensity after 2s, 30 and 100Hz stimuli. Fig. 4. Presynaptic mitochondrial calcium in old and Sod1−/− NMJs rose to considerably higher levels at 100Hz than in young controls (100Hz, F−Fo: Young 323±22, n=9 (NMJs); Old 443±41, n=9 (NMJs); Sod1−/− 662±52, n=11 (NMJs)). Even after a 30Hz stimulation, mitochondrial calcium uptake in Sod1−/− NMJs was also higher than in either young or old NMJs (30Hz, F−Fo: Young 279±30, n=9 (NMJs); Old 264±34, n=9 (NMJs); Sod1−/− 412±43, n=11 (NMJs)), which indicates a potentially heightened vulnerability of Sod1−/− presynaptic mitochondria for calcium overload. Overall, these results are in agreement with our findings of higher cytosolic Ca2+ levels and elevated ROS production rates seen in presynaptic terminals from old and Sod1−/− LAL muscles.
Figure 4. Mitochondrial calcium uptake is increased with age and oxidative stress.

Representative fluorescent images of young, old and Sod1−/− LAL NMJs loaded with rhod-2 and stimulated for 2s with 30Hz and 100Hz stimulus trains (left). LAL presynaptic mitochondrial calcium levels were quantified for young, old, and Sod1−/− mice (right) and plotted as the background subtracted increases in rhod-2 fluorescence intensities at 30Hz and 100Hz stimuli. Error bars are SEMs.
Reduction in neurotransmitter release may potentially cause a failure in muscle action potential generation and inhibition of muscle contraction. It is especially critical for old muscles as they typically show a reduction in neurotransmission safety factor [21]. Moreover, elevated ROS may also affect muscle contractility directly [22]. Here, we employed single muscle fiber sarcoplasmic calcium ([Ca2+]i) kinetics measurements in LAL loaded with fluo-4 (Fig. 5A) to assess both muscle action potential failures and excitation-contraction coupling. Titanic stimulation of the facial nerve innervating the LAL muscle revealed that 20±2% of young, 37±4% of old and 49±2% of Sod1−/− muscle fibers (n=70, each) failed to elevate their [Ca2+]i. In the muscle fibers that responded, single pulse stimulation (twitch) of the nerve led to an increase in [Ca2+]i which was substantially smaller in old and Sod1−/− (Fig. 5 B,C) than in young muscle fibers (F/Fo: Young 2.52±0.10, n=20 (fibers); Old 1.85±0.09, n=24 (fibers); Sod1−/−1.94±0.16, n=22 (fibers)). 30Hz, 2s stimulation similarly resulted in a lower [Ca2+]i in old and Sod1−/− LAL compared to young controls (F/Fo: Young 3.34±0.20, n=20 (fibers); Old 2.34±0.14, n=24 (fibers); Sod1−/− 2.52±0.25, n=22 (fibers)). However, twitch [Ca2+]i half-clearance times (τ1/2) appeared to be unchanged in all muscles (Fig. 5D; ms: Young 61.0±1.4, n=20 (fibers); Old 62.6±2.7, n=20 (fibers); Sod1−/− 62.0±3.2, n=22 (fibers)). Furthermore, sarcoplasmic reticulum calcium ([Ca2+]s) contents of LAL muscles was also significantly lower in Sod1−/− compared to young mice, as revealed by the activation of ryanodine receptors (RyR) with 10 mM caffeine. Caffeine induced [Ca2+]i peak was significantly lower in Sod1−/− than in young LAL, Fig. 5E. However, [Ca2+]s in old LAL was lower but not significantly different than young controls (F/Fo: Young 3.12±0.37, n=12 (fibers); Old 2.79±0.19, n=10 (fibers); Sod1−/− 2.04±0.22 n=14 (fibers)).
Figure 5. Old and Sod1−/− LAL muscles have reduced sarcoplasmic calcium levels upon stimulation.

(A) Bright-field and fluorescent images of a young LAL muscle fiber loaded with fluo-4 and stimulated with single stimulus (twitch) and 2s, 30Hz stimulus train. (B) Representative traces showing kinetics of fluo-4 fluorescent responses to twitches and a 2s, 30Hz stimulus train in young, old and Sod1−/− LAL muscle fibers. The inset is a magnified single fluo-4 twitch response from young LAL. (C) Average fluo-4 fluorescent response amplitudes to twitch (left) and 2s, 30Hz stimulus train (right) in young, old and Sod1−/− LAL muscle fibers. (D) Twitch calcium clearance time constants (time to 50% of the peak) calculated for young, old and Sod1−/− LAL muscle fibers. (E) Average fluo-4 fluorescent responses in muscle fibers induced by application of 2 mM caffeine to young, old and Sod1−/− LAL. Error bars are SEMs.
The observed declines in [Ca2+]i and [Ca2+]s could potentially be the result of changes in muscle calcium channel protein levels such as ryanodine receptor (RyR) and dihydropyridine receptor (DHPR) or their oxidative posttranslational modification, both of which have been reported to occur in old skeletal muscles [23,24]. We evaluated RyR1 and DHRP α1S protein levels in whole LAL muscle lysates by Western blot. No substantial differences in either RyR1 or DHRP α1S protein levels were observed between young, old and Sod1−/− LALs Fig 6A. The ratio of RyR1/DHRP α1S proteins appeared to be slightly but not significantly elevated in old and Sod1−/− LALs (RyR1/DHRP α1S band density: Young 1.25±0.11, n=5 (muscles); Old 1.29±0.14, n=5 (muscles); Sod1−/− 1.37±0.13, n=3 (muscles); p=0.79). Oxidized protein and S-nitrosylation western blots of the same muscle lysates showed that old and Sod1−/− LALs had elevated levels of protein carbonylation and S-nytrosylation, Fig 6B,C. Notably, these protein modifications were most prominent in heavy proteins, which are typically structural and integral membrane proteins that have slow turnover rates.
Figure 6. Old and Sod1−/− LAL muscles show increased levels of protein oxidative modifications without significant changes in protein levels of key calcium receptors.

(A) Western blot of whole LAL muscle extracts from young, old and Sod1−/− mice probed with anti-RyR1, DHPR α1S and β-actin antibodies. (B) Representative western blot with anti-DNP antibodies to detect protein carbonyls in young, old and Sod1−/− LAL muscle protein extracts. (C) Representative western blot with anti-TMT antibodies to detect cysteine nitrosylation in young, old and Sod1−/− LAL muscle protein extracts.
DISCUSSION
In this work, we evaluate the contributions of increased reactive oxygen species (ROS) to the physiological changes seen in old skeletal muscles by looking at both neuronal and muscle physiological properties in young Sod1−/−, and young and old wild type levator auris longus (LAL) muscles. One of the advantages for using the LAL, apart from its great accessibility for both electrophysiological and optical assays, is the decreased rate of muscle fiber denervation compared to other old skeletal muscles. This, in turn, allows for the analysis of elevated oxidative stress influence on neuromuscular unit properties without interference from the already well known denervation effects in old skeletal muscles. As a result of slow muscle denervation, LAL undergoes a delayed atrophy with age [17], Fig 1A. Nevertheless, like many limb skeletal muscles, LAL exhibits many common early aging changes, such as the fast to slow muscle fiber type switch, and an increase in presynaptic mitochondrial dysfunction accompanied by elevated ROS levels Fig 1B, which suggests some overlap between the mechanisms for the age-dependent neuromuscular unit dysfunction in LAL and other skeletal muscles.
The results of our previous [15] and present study show that elevated oxidative stress leads to the inhibition of neurotransmitter release at NMJs in Sod1−/− mice. Similarly, old LAL NMJs had also decreased levels of both spontaneous (mini EJP frequency) and evoked (quantal content) neurotransmitter release Table 1. Decreased synaptic vesicle pool sizes seen at old and Sod1−/− LAL NMJs, Fig. 2B, are likely responsible in part for the reduced neurotransmission. Elevated ROS have been shown to interfere with synaptic vesicle axonal transport [25] and formation of new vesicles in the trans-Golgi network [26]. This, in turn, may lead to the accumulation of fewer synaptic vesicles at NMJs resulting in reduced neurotransmitter release. However, ROS may also directly modulate synaptic vesicle release in neurons. External application of low concentration H2O2 led to LTP potentiation, whereas high concentrations (>20 μM) inhibited both LTP and synaptic release in rat hippocampal slices [27]. Similarly, frog and mouse NMJs showed an acute inhibition of both spontaneous and evoked neurotransmitter release after a brief treatment with H2O2 [28]. These effects are believed due to the direct ROS action on the synaptic fusion proteins, particularly, SNAP-25, a member of SNARE proteins that has a number of redox sensitive cysteine residues [29]. ROS-dependent inhibition of evoked neurotransmitter release might be in part responsible for muscle weakness typical of old and Sod1−/− mice. As the amount of neurotransmitter released per action potential falls, it gets more difficult for each incoming stimulus to reach the threshold to set off muscle fiber contraction. It is particularly true for some old muscles wherein a lower neurotransmission safety factor have been reported even without denervation [21]. However, our electrophysiological measurements found that LAL muscle input resistance was significantly higher in older than in either young or Sod1−/− samples, Fig 2A. It means that even with fewer vesicles released, muscle fiber plasma membrane in old LAL is sensitive enough to reach the threshold. The observed differences in input resistances without a substantial change in muscle fiber diameter might stem from the differential expression of ion channels in old and young muscles, and are the result of an adaptation to reduced neurotransmitter release in old muscles. For instance, muscle BK channel conductance slightly increases with age [30], but its activity is inhibited by elevated ROS [31]. Additionally, it has been shown that the expression of the major CIC-1 chloride channel and also, macroscopic chloride conductance in rat fast twitch muscles substantially decrease with age [32], thereby increasing muscle excitability. Therefore, ROS effects on neurotransmitter release alone cannot fully explain denervation independent muscle weakness in old muscles, suggesting that muscle specific ROS mechanisms might possibly be involved.
Substantial declines in neurotransmitter release observed at both Sod1−/− and old LAL NMJs might be the result of a decrease in cytosolic calcium influx. Surprisingly, cytosolic calcium levels at 100 Hz trains measured indirectly by monitoring cytosolic pH changes with EYFP were significantly elevated in Sod1−/− and old LAL presynaptic terminals, Fig 3B,C. Our estimates of cytosolic calcium levels are also quite likely underestimated, since the plasma membrane calcium ATPase activity in neurons is reduced by oxidative stress and during aging (reduced proton influx [33]) while cytosolic pH buffering capacity is only modestly affected with age [34]. Several different mechanisms may cause a rise in the cytosolic calcium level such as an increase in the activity or expression of calcium channels, action potential prolongation and reduced cytosolic calcium buffering. P/Q voltage-gated calcium channels are quite similar to N-type calcium channel, the major neuronal type calcium channel, have been shown to increase their activity after oxidation. H2O2 treatment of Xenopus oocytes heterologously expressing P/Q voltage-gated calcium channels greatly increased the opening probability and ion conductance of the channels [35]. Neuronal cytosolic calcium buffering capacity is also decreased with age. The expression of major cytosolic calcium buffering proteins such parvalbumin and calbindin D-28k has been shown to decline in neurons during elevated oxidative stress and aging [36]. Conversely, upregulation of the A-type current potassium channel activity has been reported with elevated ROS in many neurodegenerative disorders and aging, which leads to the reduced neuronal excitability [37]. Nevertheless, despite these opposing forces, the net effect is still increased calcium levels in motor neurons seen during aging and oxidative stress.
Mitochondrial calcium uptake serves to synchronize neuronal activity and mitochondrial ATP production [20] by stimulating the TCA cycle enzymes, but presynaptic mitochondria may also buffer cytosolic calcium [38]. Mitochondrial calcium concentration in old and Sod1−/− LAL presynaptic mitochondria rose to substantially higher levels than in young NMJs during stimulation, Fig 4, which suggests that indeed cytosolic calcium influx is increased at old and Sod1−/− LAL NMJs. The increases in cytosolic and mitochondrial calcium levels are likely a compensatory response to decreased mitochondrial ATP output by already dysfunctional mitochondria, which is necessary for powering both synaptic vesicle exocytosis and cytosolic calcium removal. We have previously shown that presynaptic mitochondrial density and mitochondrial ATP production correlate with synaptic activity in hippocampal synaptosomes [39], and thus, a reduction in mitochondrial ATP production will also likely reduce neurotransmitter release at NMJs. However, substantial elevations in mitochondrial and cytosolic calcium levels, in return, increase energy metabolic demands and also, sensitize mitochondria to calcium overload, which might cause axonal degeneration [40].
Elevated oxidative stress may also produce muscle weakness through a direct impact on muscle contraction by modifying muscle contractile machinery and excitation-contraction (EC) coupling. Skinned muscle fibers from old rodents show a reduction in specific muscle force, indicating a probable impairment in the muscle contractile machinery [41]. It has been shown that old muscle fibers show only a slight decrease in the actin-myosin filament ratio [42], however, the activity of myosin ATPase is substantially reduced [43]. Myosin but not actin oxidative modification levels are also increased in old muscles [22]. However, while both protein carbonylation and S-nitrosylation levels were increased in Sod1−/− and old LAL, Fig 6B,C, Sod1−/− skinned muscle fibers did not show a reduction in specific muscle force [44]. Sarcoplasmic calcium amplitude ([Ca2+]i) in stimulated Sod1−/− and old LAL muscle fibers was substantially lower than in wild-type LAL, Fig 5, which is a potential sign of weak EC coupling in Sod1−/− and old muscle fibers. The direct relationship has been shown between [Ca2+]i and specific isometric muscle force [45], suggesting that lower [Ca2+]i in Sod1−/− and old LAL muscle fibers contribute to muscle weakness. A number of mechanisms may cause a decrease in [Ca2+]i, such as a reduction in sarcoplasmic reticulum calcium level ([Ca2+]s), decreased ryanodine receptor 1 (RyR1) and dihydropyridine receptor (DHRP) coupling, and calcium channel oxidative modification. [Ca2+]s in old LAL was lower, but not significantly reduced from the young LAL [Ca2+]s levels. However, Sod1−/− [Ca2+]s was notably reduced. Elevated RyR1 nitrosylation during aging has been demonstrated to cause [Ca2+]s leak and muscle weakness [24]. Muscle aging also leads to the decline in both DHRP and RyR1 binding sites, which is thought to reduce their coupling [46]. However, there was no significant change in either DHRP and RyR1 protein levels or their ratio in Sod1−/− and old LAL compared to young LAL, Fig 6A. Nevertheless, this does not exclude the possibility of a disrupted physical interaction between DHRP and RyR1 at the triad junctions, even if their ratio remains unchanged.
In summary, we show that Sod1−/− and old mice share similarities and some distinctions in how their neuromuscular units adapt to chronic oxidative stress. For instance, young Sod1−/− and old wild type mice show ROS-dependent decrease in synaptic vesicle pool size and neurotransmitter release at NMJs, however, muscle fibers in old LAL muscles show increases in their excitability to compensate for that decrease. Although more excitable than Sod1−/− LAL, both young Sod1−/− and old LALs have defective EC coupling. This suggests that muscle weakness in old mice might in part be caused by ROS mediated EC uncoupling, while both EC uncoupling and reduced neurotransmitter release contribute to muscle weakness in Sod1−/− mice. Furthermore, our results are also suggestive of a likely sequence of the events that lead to muscle weakness in aging animals, in which neuronal ROS induced dysfunction likely occurs before the decline in muscle EC coupling in aging animals. However, future studies with neuron specific Sod1 ablation models would be needed to elucidate the mechanistic aspects of those transitions in the neuromuscular unit.
Research highlights.
Induced and age-related oxidative stress produce similar changes in neuromuscular transmission.
Chronic ROS reduce synaptic vesicle size pool and evoked neurotransmitter release.
Neuronal cytosolic and mitochondrial calcium levels show compensatory increases.
Old wild type LAL muscles exhibit higher excitability than young Sod1−/− LAL muscles.
Old wild type and young Sod1−/− LAL muscles have reduced sarcoplasmic calcium influx.
Acknowledgments
We would like to thank Kavithalakshmi Sataranatarajan for preparing muscle samples used in this study. We are also grateful to Dr Gregory Macleod and Dr Rodolfo Llinas for their valuable suggestions and comments. This work was supported by NIH grants AG020591 and AG021890.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Demontis F, Piccirillo R, Goldberg AL, Perrimon N. Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis Model Mech. 2013;6:1339–1352. doi: 10.1242/dmm.012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez-Freire M, de Cabo R, Studenski SA, Ferrucci L. The neuromuscular junction: aging at the crossroad between nerves and muscle. Front Aging Neurosci. 2014;6:208. doi: 10.3389/fnagi.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colman RJ, Beasley TM, Allison DB, Weindruch R. Attenuation of sarcopenia by dietary restriction in rhesus monkeys. J Gerontol Ser A-Biol Sci Med Sci. 2008;63:556–559. doi: 10.1093/gerona/63.6.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jang YC, Liu Y, Hayworth CR, Bhattacharya A, Lustgarten MS, Muller FL, Chaudhuri A, Qi W, Li Y, Huang JY, Verdin E, Richardson A, Van Remmen H. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging cell. 2013;11:770–782. doi: 10.1111/j.1474-9726.2012.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broome CS, Kayani AC, Palomero J, Dillmann WH, Mestril R, Jackson MJ, McArdle A. Effect of lifelong overexpression of HSP70 in skeletal muscle on age-related oxidative stress and adaptation after non damaging contractile activity. FASEB. 2006;20:1549–1551. doi: 10.1096/fj.05-4935fje. [DOI] [PubMed] [Google Scholar]
- 6.Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, Huang TT, Epstein CJ, Roberts LJ, Csete M, Faulkner JA, Van Remmen H. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Phys-Reg Int Comp Ph. 2007;293:R1159–R1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe M, Dykes-Hoberg M, Cizewski Culotta V, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- 11.Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A, Brooks SV. Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice. FASEB. 2014;28:1666–1681. doi: 10.1096/fj.13-240390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, Bhattacharya A, Richardson A, Jackson MJ, McArdle A, Brooks SV, Van Remmen H. CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB. 2013;27:3536–3548. doi: 10.1096/fj.13-228130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deschenes MR, Roby MA, Eason MK, Harris MB. Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp Gerontol. 2010;45:389–393. doi: 10.1016/j.exger.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright M, Kim A, Son YJ. Subcutaneous administration of muscarinic antagonists and triple-immunostaining of the levator auris longus muscle in mice. J Vis Exp. 2011;55:3124. doi: 10.3791/3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Y, Ivannikov MV, Walsh ME, Liu Y, Zhang Y, Jaramillo CA, Macleod GT, Van Remmen H. The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PLOS ONE. 2014;9:e100834. doi: 10.1371/journal.pone.0100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomson SR, Nahon JE, Mutsaers CA, Thomson D, Hamilton G, Parson SH, Gillingwater TH. Morphological characteristics of motor neurons do not determine their relative susceptibility to degeneration in a mouse model of severe spinal muscular atrophy. PLOS ONE. 2012;7:e52605. doi: 10.1371/journal.pone.0052605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLOS ONE. 2012;7:e34640. doi: 10.1371/journal.pone.0034640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Nguyen KT, Barrett EF, David G. Vesicular ATPase inserted into the plasma membrane of motor terminals by exocytosis alkalinizes cytosolic pH and facilitates endocytosis. Neuron. 2010;68:1097–1108. doi: 10.1016/j.neuron.2010.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chouhan AK, Ivannikov MV, Lu Z, Sugimori M, Llinas RR, Macleod GT. Cytosolic calcium coordinates mitochondrial energy metabolism with presynaptic activity. J Neurosci. 2012;32:1233–1243. doi: 10.1523/JNEUROSCI.1301-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivannikov MV, Macleod GT. Mitochondrial Free Ca2+ Levels and Their Effects on Energy Metabolism in Drosophila Motor Nerve Terminals. Biophys J. 2013;104:2353–2361. doi: 10.1016/j.bpj.2013.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacob JM, Robbins N. Age differences in morphology of reinnervation of partially denervated mouse muscle. J Neurosci. 1990;10:1530–1540. doi: 10.1523/JNEUROSCI.10-05-01530.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prochniewicz E, Thomas DD, Thompson LV. Age-related decline in actomyosin function. J Gerontol Ser A-Biol Sci Med Sci. 2005;60:425–431. doi: 10.1093/gerona/60.4.425. [DOI] [PubMed] [Google Scholar]
- 23.Carmeli E, Coleman R, Reznick AZ. The biochemistry of aging muscle. Exp Gerontol. 2002;37:477–489. doi: 10.1016/s0531-5565(01)00220-0. [DOI] [PubMed] [Google Scholar]
- 24.Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14:196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang C, Bourdette D, Banker G. Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases. Mol Neurodegener. 2012;7:29. doi: 10.1186/1750-1326-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Chang A. Heat shock response relieves ER stress. EMBO. 2008;27:1049–1059. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamsler A, Segal M. Hydrogen peroxide modulation of synaptic plasticity. J Neurosci. 2003;23:269–276. doi: 10.1523/JNEUROSCI.23-01-00269.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giniatullin AR, Darios F, Shakirzyanova A, Davletov B, Giniatullin R. SNAP25 is a presynaptic target for the depressant action of reactive oxygen species on transmitter release. J Neurochem. 2006;98:1789–1797. doi: 10.1111/j.1471-4159.2006.03997.x. [DOI] [PubMed] [Google Scholar]
- 29.Washbourne P, Cansino V, Mathews J, Graham M, Burgoyne R, Wilson M. Cysteine residues of SNAP-25 are required for SNARE disassembly and exocytosis, but not for membrane targeting. Biochem J. 2001;357:625–634. doi: 10.1042/0264-6021:3570625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tricarico D, Petruzzi R, Camerino DC. Changes of the biophysical properties of calcium-activated potassium channels of rat skeletal muscle fibres during aging. Pflugers Arch. 1997;434:822–829. doi: 10.1007/s004240050471. [DOI] [PubMed] [Google Scholar]
- 31.Tang XD, Garcia ML, Heinemann SH, Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol. 2004;11:171–178. doi: 10.1038/nsmb725. [DOI] [PubMed] [Google Scholar]
- 32.Pierno S, De Luca A, Beck CL, George AL, Jr, Conte Camerino D. Aging-associated down-regulation of ClC-1 expression in skeletal muscle: phenotypic-independent relation to the decrease of chloride conductance. FEBS Lett. 1999;449:12–16. doi: 10.1016/s0014-5793(99)00202-1. [DOI] [PubMed] [Google Scholar]
- 33.Zaidi A, Michaelis ML. Effects of reactive oxygen species on brain synaptic plasma membrane Ca 2+-ATPase. Free Radic Biol Med. 1999;27:810–821. doi: 10.1016/s0891-5849(99)00128-8. [DOI] [PubMed] [Google Scholar]
- 34.Roberts EL, Jr, Chih CP. The pH buffering capacity of hippocampal slices from young adult and aged rats. Brain Res. 1998;779:271–275. doi: 10.1016/s0006-8993(97)01120-7. [DOI] [PubMed] [Google Scholar]
- 35.Li A, Ségui J, Heinemann SH, Hoshi T. Oxidation Regulates Cloned Neuronal Voltage-Dependent Ca2+ Channels Expressed in Xenopus Oocytes. J Neurosci. 1998;18:6740–6747. doi: 10.1523/JNEUROSCI.18-17-06740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kishimoto J, Tsuchiya T, Cox H, Emson PC, Nakayama Y. Age-related changes of calbindin-D28k, calretinin, and parvalbumin mRNAs in the hamster brain. Neurobiol Aging. 1998;19:77–82. doi: 10.1016/s0197-4580(97)00166-8. [DOI] [PubMed] [Google Scholar]
- 37.Hsieh CP. Redox modulation of A-type K+ currents in pain-sensing dorsal root ganglion neurons. Biochem Biophys Res Commun. 2008;370:445–449. doi: 10.1016/j.bbrc.2008.03.097. [DOI] [PubMed] [Google Scholar]
- 38.Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2008;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ivannikov MV, Sugimori M, Llinás RR. Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J Mol Neurosci. 2013;49:223–230. doi: 10.1007/s12031-012-9848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, Hetz C, Twiss JL, Alvarez J, Court FA. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowe DA, Thomas DD, Thompson LV. Force generation, but not myosin ATPase activity, declines with age in rat muscle fibers. Am J Physiol-Cell Ph. 2002;283:C187–C192. doi: 10.1152/ajpcell.00008.2002. [DOI] [PubMed] [Google Scholar]
- 42.Thompson LV, Durand D, Fugere NA, Ferrington DA. Myosin and actin expression and oxidation in aging muscle. J Appl Physiol. 2006;101:1581–1587. doi: 10.1152/japplphysiol.00426.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowe DA, Husom AD, Ferrington DA, Thompson LV. Myofibrillar myosin ATPase activity in hindlimb muscles from young and aged rats. Mech Ageing Dev. 2004;125:619–627. doi: 10.1016/j.mad.2004.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larkin LM, Hanes MC, Kayupov E, Claflin DR, Faulkner JA, Brooks SV. Weakness of whole muscles in mice deficient in Cu, Zn superoxide dismutase is not explained by defects at the level of the contractile apparatus. Age. 2013;35:1173–1181. doi: 10.1007/s11357-012-9441-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.González E, Messi ML, Zheng Z, Delbono O. Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J Physiol. 2003;552:833–844. doi: 10.1113/jphysiol.2003.048165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payne AM, Delbono O. Neurogenesis of excitation-contraction uncoupling in aging skeletal muscle. Exerc Sport Sci Rev. 2004;32:36–40. doi: 10.1097/00003677-200401000-00008. [DOI] [PubMed] [Google Scholar]