

Abstract
Objectives
‘Idiopathic’ cardiac conditions such as dilated cardiomyopathy (DCM) and resuscitated sudden cardiac death (RSCD) may be familial. We suspected that inpatient cardiology services fail to recognise this. Our objective was to compare diagnostic value of family histories recorded by inpatient cardiology teams with a multigenerational family tree obtained by specially trained allied professionals.
Methods
2 experienced cardiology nurses working in 2 tertiary adult cardiac units were trained in cardiac-inherited diseases and family history (FHx) taking, and established as regional coordinators for a National Cardiac Inherited Disease Registry. Over 6 months they sought ‘idiopathic’ cardiology inpatients with conditions with a possible familial basis, reviewed the FHx in the clinical records and pursued a minimum 3-generation family tree for syncope, young sudden death and cardiac disease (full FHx).
Results
37 patients (22 males) were selected: mean age 51 years (range 15–79). Admission presentations included (idiopathic) RSCD (14), dyspnoea or heart failure (11), ventricular tachycardia (2), other (10). 3 patients had already volunteered their familial diagnosis to the admitting team. FHx was incompletely elicited in 17 (46%) and absent in 20 (54%). 29 patients (78%) provided a full FHx to the coordinator; 12 of which (41%) were strongly consistent with a diagnosis of a cardiac-inherited disease (DCM 7, hypertrophic cardiomyopathy 3, long QT 1, left ventricular non-compaction 1). Overall, a familial diagnostic rate rose from 3/37(8%) to 12/37 (32%).
Conclusions
Adult cardiology inpatient teams are poor at recording FHx and need to be reminded of its powerful diagnostic value.
Keywords: GENETICS
Key questions.
What is already known about this subject?
Trained cardiac genetic healthcare professionals in cardiology outpatient services have been shown to increase familial detection rate of cardiac-inherited diseases, such as hypertrophic cardiomyopathy and long QT syndrome, in a cost-effective manner and increase patient satisfaction. No such information exists for inpatient cardiac services.
What does this study add?
Family histories taken by cardiology inpatient teams from patients potentially affected by cardiac-inherited diseases, are usually insufficient to diagnose these conditions or to identify at-risk family members. However, when trained cardiac genetic healthcare professionals elicit at least a three-generation family pedigree, detection of cardiac-inherited diseases increases. This facilitates appropriate use of cardiac and genetic testing and allows risk reduction not only in the proband, but also their presymptomatic relatives.
How might this impact on clinical practice?
Cardiology inpatient staff should be advised to obtain multigenerational pedigrees during the evaluation of cardiology inpatients, particularly in cases of non-ischaemic cardiomyopathy and resuscitated cardiac death. Training and time is required for this which can be facilitated and led by an allied professional within cardiology services and with links to a cardiac genetic service.
Introduction
Cardiac-inherited diseases such as hypertrophic cardiomyopathy or long QT syndrome (LQTS) are individually relatively rare but collectively common.1 Presenting symptoms include dyspnoea, syncope, arrhythmias and even sudden unexpected cardiac arrest.
Timely identification of cardiac-inherited diseases is paramount because there is extensive evidence that the risk of sudden death can be significantly reduced with a variety of management options, including β-blockers,2 3 implantable cardiac defibrillators (ICDs)4 5 and lifestyle modification.6 Studies of such autosomal-dominant conditions have shown that it is possible to identify up to 8–9 affected family members per proband.7 8 Non-ischaemic dilated cardiomyopathy (DCM) may be familial in up to 40%,9–11 and thorough investigation of non-coronary disease-related resuscitated sudden cardiac death (RSCD), including pharmacological provocative testing reveals an inherited cause in up to 60%.12
Therefore, consideration of familial cardiac-inherited diseases, even in seemingly isolated cases of structural or electrical disorders, and prompt diagnosis of a potential new proband is of vital importance regardless of their presentation, as many other unidentified family members may also be at risk.8 13 14
As a result of these combined observations, a proactive, integrated approach from cardiology and genetics professionals is recommended.15 16 Central to this is an in-depth, multigenerational family history (FHx). This has the potential to establish a diagnosis, avoid unnecessary, extensive and potentially expensive testing for underlying conditions, and can assist with devising a genetic testing strategy, interpreting genetic test results, and providing ongoing risk assessments for sudden cardiac death (SCD).14 17 However, to our knowledge, there are no other studies of the everyday practice of inpatient cardiology units to see if these guidelines are being adhered to, nor any to demonstrate the overall effect of their enforced introduction in these institutions.
New Zealand has a National Cardiac Inherited Disease Registry run by a multidisciplinary clinical and scientific network called the Cardiac Inherited Disease Group (CIDG).13 Two new part-time coordinators (both experienced cardiac nurses) in two large district hospitals each with a regional cardiology referral base noted FHx of people at high risk of having a cardiac-inherited disease was not always performed by the cardiology inpatient team, and that patients could be discharged without identification of the underlying aetiology of their symptoms. Since cardiac-inherited diseases remain a differential diagnosis in presentations such as syncope, heart failure or RSCD, the coordinators were aware that FHx could potentially improve accuracy of diagnosis and improve quality of care for the patient and their family. Therefore, we wanted to test the following hypotheses:
That documentation of family histories in inpatient hospital records was inadequate, and
That a multigenerational family tree obtained by a trained allied professional could lead to the increased detection of hitherto unrecognised familial cardiac disease.
Methods
Two experienced cardiac nurses were educated in cardiac-inherited diseases and employed as part time regional coordinators13 within their tertiary adult cardiology services.
Patients were admitted, as usual, to the tertiary adult cardiac referral centre directly, or via transfer, from their local hospital. Over a 6-month period, through liaising with the relevant cardiology staff, they identified hospital inpatients who were definitely or potentially affected by cardiac-inherited disease. This included those who were already diagnosed, newly diagnosed through inpatient cardiac investigation, or had a FHx of cardiac-inherited disease, or those who presented with heart failure, sudden death including motor vehicle accidents, events during swimming and ‘heart attacks’ with unknown aetiology. Presence of permanent pacemakers and ICDs was elicited and other symptoms consistent with undiagnosed cardiac conditions were documented. The aim was to offer family cascade screening.
Coordinators took a ‘full’ FHx, including drawing at least a three-generation family tree, with syncope, young sudden death and cardiac disease documented. Additional baseline demographic data were also obtained. Admission presentations were recorded as documented in the clinical notes by the admitting physician and those with clinically apparent vasovagal syncope were not included.
The original clinical notes taken by the admitting staff were retrospectively reviewed and qualitatively compared for quality and content of FHx documentation.
Ethical approval
All patients were enrolled prospectively with the Cardiac Inherited Diseases Registry New Zealand. At this time, they provided informed consent for their de-identified clinical data to be used in research publications. The Cardiac Inherited Diseases Registry New Zealand, and the consent forms used were given multiregional (national) ethical approval by the New Zealand Health and Disability Ethics Committees AKX/02/00/107/AM02.13 Consent forms can be reviewed at http://www.cidg.org.nz. Patients with a proven cardiac-inherited disease were subsequently offered genetic counselling, genetic testing and clinical family screening as recommended by international guidelines.15 16 18–21
Results
Thirty-seven patients were recruited: 22 males and 15 females with a mean age of 51 (range 15–79; table 1). Clinical presentation included RSCD,14 dyspnoea or heart failure,11 and ventricular tachycardia2 (figure 1A). Their diagnoses, after inpatient cardiology investigation, are documented in figure 1B.
Table 1.
Details of cohort showing the initial presentations of 37 cardiology inpatients with presentations of unknown aetiology, their discharge diagnoses after inpatient assessment according to the CIDG registry coordinators, and whether a family history was used by the cardiology inpatient team or the CIDG registry coordinators to determine whether patients were potentially affected by a CID
| Patient | Age | Clinical presentation | Final diagnosis | Familial condition identified by cardiology team | Familial condition identified by coordinators |
|---|---|---|---|---|---|
| 1 | 15 | RSCD | LQTS | N | N |
| 2 | 39 | Syncope | Atrioventricular block | N | N |
| 3 | 58 | RSCD | DCM | N | N |
| 4 | 48 | Dyspnoea | DCM | N | N |
| 5 | 26 | Chest pain | Infiltrative cardiomyopathy | N | N |
| 6 | 47 | RSCD | Unknown | N | N |
| 7 | 21 | RSCD | Unknown | N | N |
| 8 | 51 | RSCD | Unknown | N | N |
| 9 | 57 | Chest pain | NSTEMI, known HCM | N | Y |
| 10 | 64 | RSCD | Other—viral myocarditis | N | N |
| 11 | 58 | Syncope | DCM | N | Y |
| 12 | 72 | RSCD | DCM | N | Y |
| 13 | 66 | Chest pain | NSTEMI, obligate LQTS carrier* | N | Y |
| 14 | 44 | RSCD | Unknown | N | N |
| 15 | 57 | Chest pain | ARVC | N | N |
| 16 | 49 | Presyncope | BrS | N | N |
| 17 | 62 | Dyspnoea | DCM | N | Y |
| 18 | 58 | Dyspnoea | DCM | N | N |
| 19 | 62 | Dyspnoea, known DCM | DCM | N | N |
| 20 | 60 | Dyspnoea, known DCM | DCM | N | Y |
| 21 | 49 | VT | DCM | N | N |
| 22 | 20 | Abdominal pain | DCM | N | Y |
| 23 | 62 | Dyspnoea | DCM | N | Y |
| 24 | 20 | RSCD | DCM | N | N |
| 25 | 68 | Heart failure | DCM | N | N |
| 26 | 70 | Presyncope | DCM | N | N |
| 27 | 43 | Dyspnoea | DCM | N | Y |
| 28 | 54 | Dyspnoea | DCM | N | N |
| 29 | 50 | VT | DCM | N | N |
| 30 | 25 | RSCD | Other—electrocution | N | N |
| 31 | 19 | RSCD | HCM | N | N |
| 32 | 79 | Heart failure | HCM | N | N |
| 33 | 48 | RSCD | LVNC | N | Y |
| 34 | 55 | RSCD | Unknown | N | N |
| 35 | 34 | RSCD | Unknown | N | N |
| 36 | 45 | Chest pain | HCM | N | Y |
| 37 | 57 | Dyspnoea | HCM | N | Y |
*This patient was diagnosed with a NSTEMI, the cardiology inpatient assessment included FHx documenting ‘LQTS’; however, FHx obtained by coordinator revealed he was an obligate LQTS carrier.
ARVC, arrhythmogenic right ventricular cardiomyopathy; BrS, Brugada syndrome; CID, cardiac-inherited disease; CIDG, Cardiac Inherited Disease Group; DCM, dilated cardiomyopathy; FHx, family history; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; LVNC, left ventricular non-compaction; NSTEMI, non-ST elevation myocardial infarction; RSCD, resuscitated sudden cardiac death; VT, ventricular tachycardia.
Figure 1.
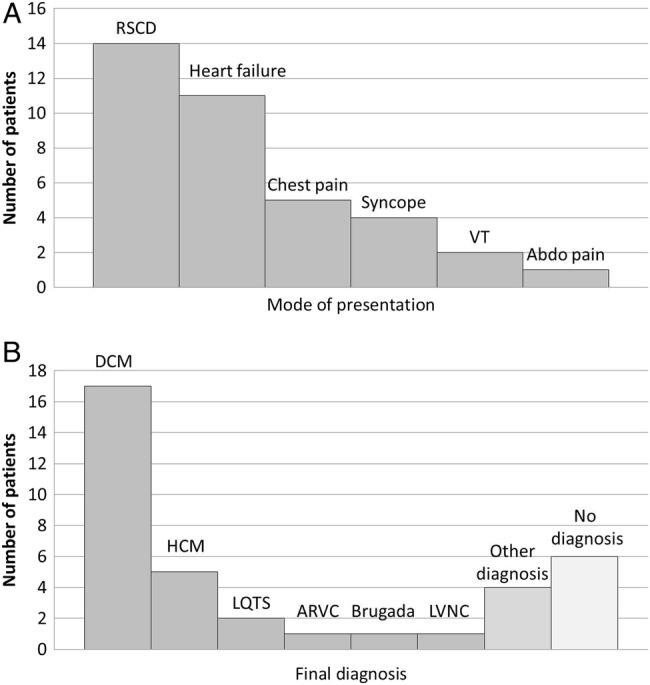
Diagnosis at presentation and diagnoses finally reached at the end of the inpatient admission. (A) Mode of clinical presentation of 37 cardiology inpatients identified by the CIDG coordinators as definitely or possibly having inherited heart conditions. (B) Diagnoses of 37 patients after full assessment by both the inpatient cardiology team and CIDG registry coordinators. CIDG, Cardiac Inherited Disease Group; RSCD, resuscitated sudden cardiac death; VT, ventricular tachycardia; abdo pain, abdominal pain; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; ARVC, arrhythmogenic right ventricular cardiomyopathy; Brugada, Brugada syndrome; LVNC, left ventricular non-compaction.
Documentation of FHx by inpatient team
Twenty participants (54%) had no documentation of FHx during their admission (see figure 2). Of the 17 patients (46%) who did have some FHx documented, 14 were insufficient to either identify or exclude a familial basis for their presentation. In three cases, where a familial condition was documented, the patients knew their diagnosis, its heritable nature and volunteered this to the treating physician (however, no FHx was documented by the cardiology team even after this occurred). Six patients had significant factual errors: one was an obligate LQTS carrier, two had unrecognized immediate family members with ICDs, and in the remaining three, clear diagnoses in first degree relatives was incorrectly reported (such as documenting a SCD which did not occur).
Figure 2.

Flow diagram of assessment of 37 cardiology inpatients. This compares the diagnostic success rate for detecting familial conditions between the regular inpatient cardiology team and the specialist CID nurses. CIDG, Cardiac Inherited Disease Group; CID, cardiac-inherited disease.
In comparison, when pedigrees were obtained by the coordinators, 29 (78%) patients were able to provide three full generations of FHx with sufficiently detailed clinical knowledge to enable appropriate assessment of heritability patterns (see figure 2). All remaining patients were able to recall details for at least some of their family members. Twelve of the 29 pedigrees (41%) were strongly consistent with a heritable pathology for the patient's condition. Of these 12 patients, 5 had no FHx documented by the inpatient team, and 4 had inaccurate FHx documented. The remaining three had already been identified by the admitting team. Therefore, the link with an inherited disease had not been made in 9 of the 12 familial cases (see table 1 and figure 2 for details of entire cohort).
Full pedigrees took between 30 and 90 min to complete. Identification of unrelated familial conditions arose and through follow-up, a familial breast and ovarian cancer (BRCA1) mutation was exposed in one such case. Various factors impeded taking a thorough FHx, including the patient's physical condition (such as severe heart failure symptoms), emotional state and social or cultural values about health privacy. Family dynamics and structure such as adoption (n=2) also played an important part, particularly in some Ma¯ori patients where multiple adoptions in and out of the family is common cultural practice.22 Low health literacy of the patient was also a common limiting factor as some patients did not comprehend the unfamiliar conditions of family members. In most cases, assistance from relatives was necessary in eliciting details of the family's demographics and health history which occasionally required a second consultation from the coordinator.
Discussion
This simple study demonstrates that detailed family histories are usually not obtained by inpatient general medical and cardiology staff, and when they are attempted, often contain significant errors and misleading information. In comparison, detailed family pedigrees can be effectively obtained by trained allied professionals, which can lead to the identification of inherited cardiac conditions.
To our knowledge, there are no other studies examining the impact of a cardiogenetics team in an inpatient cardiology service. By comparing inpatient hospital records with details obtained by the coordinators, we have seen an increased detection rate of cardiac-inherited diseases when such diseases are properly considered. While most patients seen in cardiology wards are discharged with a definitive diagnosis and underlying aetiology, such as coronary artery disease, this sample demonstrates that in the remainder of cases, the simple act of obtaining a FHx can reveal an inherited disease: ‘idiopathic’ becomes ‘familial’ in a striking number of cases. In many cases, the history was not subtle—containing multiple members with a cardiac transplant or death from DCM, for example. This study reveals the significant proportion of probands who would remain unrecognised without a supplemental assessment. With respect to the detection of presymptomatic individuals in the community,13 for autosomal dominant conditions, it is possible to detect up to 8–9 affected relatives with extensive family cascade screening,7 although 2–4 is more common.13 Therefore, this simple diagnostic test, the FHx, also has important positive implications for the rest of the family and the community at large.13 Cardiac genetic screening generally identifies 25–40% of asymptomatic relatives to be at some increased risk.18 23
We document that a detailed three-generation pedigree provides valuable clinical information and increases detection rate of cardiac-inherited disease. However, we also found that it takes considerable time, and that input from family members is highly beneficial. In busy general cardiology practice, these factors are likely to significantly impede uptake of the latest consensus guidelines recommending cardiogenetics team involvement in the assessment and management of relevant patients.15 Previous studies demonstrate the importance of specialised cardiac-inherited disease team presence in the outpatient setting, particularly in the investigation of sudden unexplained death and cardiac arrest.7 12 24 25 Our study identifies for the first time, the importance of a cardiac-inherited disease team presence in the inpatient setting. The diagnostic value of provocative pharmacological tests to detect mostly inherited arrhythmic substrates in cardiac arrest survivors has been established beyond doubt.26 The present study reminds us that an essential part of that investigative algorithm is a thorough FHx,24 and individual centres may wish to devise their own solution using skilled/trained cardiac nurses such as this study, genetic counsellors or to facilitate more time and training for their admitting physicians to perform this activity.
With the subsequent passage of time, our coordinators have noticed anecdotally an increased awareness of cardiac-inherited diseases among cardiology staff, and an increased rate of referrals. This has been facilitated by a specialised cardiac-inherited disease team inpatient presence, scheduled lectures for nursing, physiology staff and clinicians, as well as bedside clinical learning. Another positive effect is the increased awareness in general of the importance of the FHx with each patient.
A potential limitation of this paper is its performance in tertiary referral centres, and as such may not represent the general population. Furthermore, the referral to the CIDG coordinators was not systematic and it is possible that some other patients with similar conditions will have escaped the attention of the CIDG coordinator. While we consider it is likely that the cohort is representative of this section of cardiology inpatients, it may not be. Replication of this study in a larger cohort, with further assessment to determine effects on community and familial detection rate, cost-effectiveness (including diagnostic tests avoided) and patient satisfaction would be contributory.
When patient and families are seen in a specialised cardiac-inherited disease clinic, patient satisfaction has been consistently high.27–29 It is imperative now to bring an element of that specialised knowledge into the inpatient cardiology wards. This study has shown that the addition of a standard, three-generation (minimum) family tree to an inpatient assessment is of itself a powerful diagnostic investigation which results in an increased rate of identification of cardiac-inherited diseases in probands and their relatives.
Conclusion
Family histories taken during inpatient cardiology admissions are usually either absent or inaccurate. Admitting cardiology staff needs to be reminded of the diagnostic value of a thorough FHx for the patient, family and potentially the community. The appointment of dedicated trained staff to obtain a full FHx results in a remarkably increased detection of cardiac-inherited diseases. Therefore, we suggest that protocols are developed and appropriate staff be trained and employed in all cardiology inpatient services.
Acknowledgments
The authors gratefully acknowledge the secretarial assistance of Charlene Nell for manuscript preparation from the Department of Cardiovascular Services, Green Lane Cardiovascular Services, Auckland City Hospital, Auckland, New Zealand.
Footnotes
Collaborators: On behalf of the Cardiac Inherited Disease Group New Zealand.
Contributors: All authors fulfil the International Committee of Medical Journal Editors (ICMJE) uniform requirements for manuscript submission. JRS as corresponding author acts as guarantor. All authors were involved in the study conception and design; analysis and interpretation of data; drafted the article; and final approval of the manuscript. All authors were involved in the acquisition and analysis of data; revising the article; and final approval of the manuscript. All authors were involved in the study conception and design; interpretation of data; drafting and revising the article; and final approval of the manuscript.
Funding: JRS, TD and AG receive salary support from Cure Kids (grant 4507) and KEW-S is supported by Fellowship funding from the National Heart Foundation (New Zealand) (grant 1516) and the Green Lane Research and Education Fund (grant 14/45/4105).
Competing interests: None declared.
Ethics approval: All patients were enrolled prospectively with the Cardiac Inherited Diseases Registry New Zealand. At this time, they provided informed consent for their clinical data to be used in de-identified research publications. The Cardiac Inherited Diseases Registry New Zealand, including consent form, was given ethical approval by the New Zealand Health and Disability Ethics Committees AKX/02/00/107/AM02.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Burton H, Alberg C, Stewart A. Heart to heart: inherited cardiovascular conditions services—a needs assessment and service review. Cambridge, UK: PHG Foundation, 2009. http://www.phgfoundation.org/reports/4986/ [Google Scholar]
- 2.Goldenberg I, Bradley J, Moss A et al. . Beta-blocker efficacy in high-risk patients with the congenital long-QT syndrome types 1 and 2: implications for patient management. J Cardiovasc Electrophysiol 2010;21:893–901. 10.1111/j.1540-8167.2010.01737.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vincent GM, Schwartz PJ, Denjoy I et al. . High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation 2009;119: 215–21. 10.1161/CIRCULATIONAHA.108.772533 [DOI] [PubMed] [Google Scholar]
- 4.Maron BJ, Spirito P, Shen WK et al. . Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA 2007;298:405–12. [Erratum appears in JAMA 2007;298:1516], [Reprint in Nat Clin Pract Cardiovasc Med 2008;5:76–7] 10.1001/jama.298.4.405 [DOI] [PubMed] [Google Scholar]
- 5.Gersh BJ, Maron BJ, Bonow RO et al. . 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 2011;58: e212–60. 10.1016/j.jacc.2011.06.011 [DOI] [PubMed] [Google Scholar]
- 6.Schwartz PJ, Priori SG, Spazzolini C et al. . Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001;103:89–95. 10.1161/01.CIR.103.1.89 [DOI] [PubMed] [Google Scholar]
- 7.Tan HL, Hofman N, van Langen IM et al. . Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 2005;112:207–13. 10.1161/CIRCULATIONAHA.104.522581 [DOI] [PubMed] [Google Scholar]
- 8.Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ et al. . Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 2001;357:165–8. 10.1016/S0140-6736(00)03587-X [DOI] [PubMed] [Google Scholar]
- 9.Gregori D, Rocco C, Miocic S et al. . Estimating the frequency of familial dilated cardiomyopathy in the presence of misclassification errors. J Appl Statistics 2001;28:53–62. 10.1080/02664760120011590 [DOI] [Google Scholar]
- 10.Mestroni L, Maisch B, McKenna WJ et al. . Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J 1999;20:93–102. 10.1053/euhj.1998.1145 [DOI] [PubMed] [Google Scholar]
- 11.Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2011;57:1641–9. 10.1016/j.jacc.2011.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Werf C, Hofman N, Tan HL et al. . Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: the experience of a tertiary referral center in The Netherlands. Heart Rhythm 2010;7:1383–9. 10.1016/j.hrthm.2010.05.036 [DOI] [PubMed] [Google Scholar]
- 13.Earle N, Crawford J, Smith W et al. . Community detection of long QT syndrome with a clinical registry: an alternative to ECG screening programs? Heart Rhythm 2013;10:233–8. 10.1016/j.hrthm.2012.10.043 [DOI] [PubMed] [Google Scholar]
- 14.Dunn KE, Caleshu C, Cirino AL et al. . A clinical approach to inherited hypertrophy: the use of family history in diagnosis, risk assessment, and management. Circ Cardiovasc Genet 2013;6:118–31. 10.1161/CIRCGENETICS.110.959387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Priori SG, Wilde AA, Horie M et al. . HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013;10:1932–63. 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 16.Vohra J, Skinner J, Semsarian C. Cardiac genetic investigation of young sudden unexplained death and resuscitated out of hospital cardiac arrest. Heart Lung Circ 2011;20:746–50. 10.1016/j.hlc.2011.07.015 [DOI] [PubMed] [Google Scholar]
- 17.Hershberger RE, Lindenfeld J, Mestroni L et al. . Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail 2009;15:83–97. 10.1016/j.cardfail.2009.01.006 [DOI] [PubMed] [Google Scholar]
- 18.Ackerman MJ, Priori SG, Willems S et al. . HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011;13:1077–109. 10.1093/europace/eur245 [DOI] [PubMed] [Google Scholar]
- 19.Skinner JR. Guidelines for the diagnosis and management of familial long QT syndrome. Heart Lung Circ 2007;16:22–4. 10.1016/j.hlc.2006.10.021 [DOI] [PubMed] [Google Scholar]
- 20.Fatkin D, members of the CCGDCWG. Guidelines for the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ 2011;20:691–3. 10.1016/j.hlc.2011.07.008 [DOI] [PubMed] [Google Scholar]
- 21.Semsarian C, for the CSANZ Cardiac Genetics Diseases Council Writing Group. Guidelines for the diagnosis and management of hypertrophic cardiomyopathy. Heart Lung Circ 2011;20:688–90. 10.1016/j.hlc.2011.07.017 [DOI] [PubMed] [Google Scholar]
- 22.Newman E. History of transracial adoption: a New Zealand perspective. Am Indian Q 2013;37:237–57. 10.1353/aiq.2013.0010 [DOI] [Google Scholar]
- 23.Miller EM, Wang Y, Ware SM. Uptake of cardiac screening and genetic testing among hypertrophic and dilated cardiomyopathy families. J Genet Couns 2013;22:258–67. 10.1007/s10897-012-9544-4 [DOI] [PubMed] [Google Scholar]
- 24.Skinner JR. Investigation following resuscitated cardiac arrest. Arch Dis Child 2013;98:66–71. 10.1136/archdischild-2011-301515 [DOI] [PubMed] [Google Scholar]
- 25.Behr ER, Dalageorgou C, Christiansen M et al. . Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J 2008;29:1670–80. 10.1093/eurheartj/ehn219 [DOI] [PubMed] [Google Scholar]
- 26.Krahn AD, Healey JS, Chauhan VS et al. . Epinephrine infusion in the evaluation of unexplained cardiac arrest and familial sudden death: from the cardiac arrest survivors with preserved Ejection Fraction Registry. Circ Arrhythm Electrophysiol 2012;5:933–40. 10.1161/CIRCEP.112.973230 [DOI] [PubMed] [Google Scholar]
- 27.Ingles J, Lind JM, Phongsavan P et al. . Psychosocial impact of specialized cardiac genetic clinics for hypertrophic cardiomyopathy. Genet Med 2008;10:117–20. 10.1097/GIM.0b013e3181612cc7 [DOI] [PubMed] [Google Scholar]
- 28.Christiaans I, van Langen IM, Birnie E et al. . Genetic counseling and cardiac care in predictively tested hypertrophic cardiomyopathy mutation carriers: the patients’ perspective. Am J Med Genet A 2009;149A:1444–51. 10.1002/ajmg.a.32915 [DOI] [PubMed] [Google Scholar]
- 29.Ormondroyd E, Oates S, Parker M et al. . Pre-symptomatic genetic testing for inherited cardiac conditions: a qualitative exploration of psychosocial and ethical implications. Eur J Hum Genet 2014;22:88–93. 10.1038/ejhg.2013.81 [DOI] [PMC free article] [PubMed] [Google Scholar]