

 This work is licensed under a
This work is licensed under a Summary
Acromegaly due to ectopic GHRH secretion from a neuroendocrine tumor (NET) is rare and comprises <1% of all acromegaly cases. Herein we present a 57-year-old woman with clinical and biochemical features of acromegaly and a 6 cm pancreatic NET (pNET), secreting GHRH and calcitonin. Following surgical resection of the pancreatic tumor, IGF1, GH and calcitonin normalized, and the clinical features of acromegaly improved. In vitro studies confirmed that the tumor secreted large amounts of both GHRH and calcitonin, and incubation of pNET culture-derived conditioned media stimulated GH release from a cultured human pituitary adenoma. This is a unique case of pNET secreting both GHRH and calcitonin. The ability of the pNET-derived medium to stimulate in vitro GH release from a human pituitary-cell culture, combined with the clinical and hormonal remission following tumor resection, confirmed the ectopic source of acromegaly in this patient.
Learning points
Signs, symptoms and initial work-up of acromegaly due to ectopic GHRH secretion are similar to pituitary-dependent acromegaly. However, if no identifiable pituitary lesion is found, somatostatin receptor scan and further imaging (CT, MRI) should be performed.
Detection of GHRH in the blood and in the tumor-derived medium supports the diagnosis of ectopic GHRH secretion.
Functional bioactivity of pNET-secreted GHRH can be proved in vitro by releasing GH from human pituitary cells.
Background
Acromegaly due to ectopic growth hormone (GH) releasing hormone (GHRH) secretion from a neuroendocrine tumor (NET) is rare, and up to 100 cases have been reported in the literature (1, 2, 3). Half of the tumors were lung carcinoids and most others were pancreatic neuroendocrine tumors (pNET) (1, 2, 3). We hereby report a unique case of acromegaly due to pNET co-secreting GHRH and calcitonin with an extensive in vitro evaluation confirming the ability of the tumor to stimulate GH release from human pituitary cells in culture.
Case presentation
A 57-year-old woman was referred for assessment of a pancreatic mass. Her medical history included type 2 diabetes mellitus, hypertension, hyperlipidemia, hypothyroidism, hysterectomy and oophorectomy. Two months prior to the endocrine evaluation, she developed sub-acute endocarditis after sclerotherapy for varicose veins, had aortic valve insufficiency and underwent aortic valve replacement.
After surgery, a chest and an abdominal computed tomography (CT) scan was performed due to persistent fever that demonstrated a 6 cm mass in the pancreatic head blocking the main pancreatic duct (Fig. 1A), with an additional 2.2 cm incidental mass in the left adrenal, compatible with adrenal adenoma (Fig. 2A). The findings were confirmed by an abdominal MRI.
Figure 1.
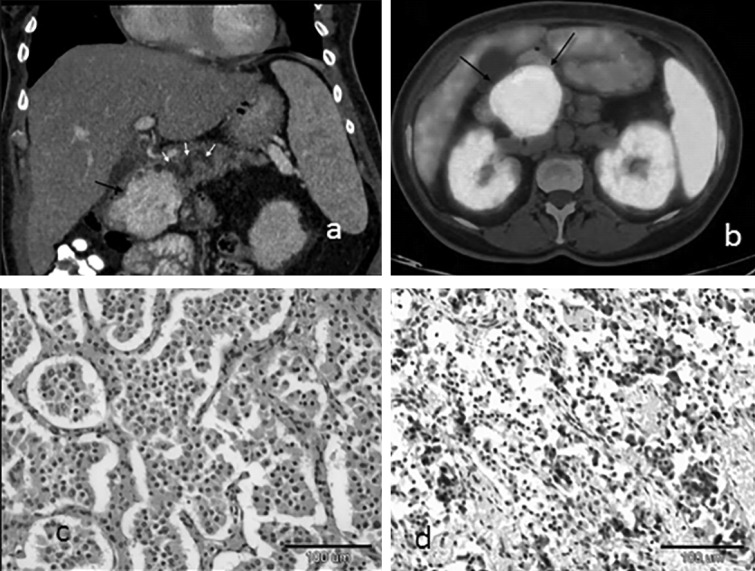
(A) Coronal contrast enhanced CT of the abdomen shows a hypervascular 6.0 cm mass in the head of the pancreas (black arrow) obstructing the main pancreatic duct which is dilated (white arrows). (B) Axial Ga-68 DOTATATE PET/CT image shows uptake in the pancreatic mass (black arrow). (C) Pancreatic neuroendocrine tumor (hematoxylin–eosin stain, original magnification X 150. (D) Immunohistochemistry staining positive for calcitonin.
Figure 2.
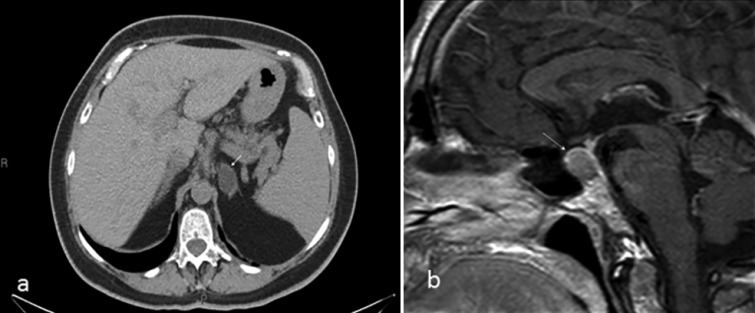
(A) Non-contrast abdominal CT scan showing a hypodense left adrenal mass measuring (−16) HU, consistent with an adrenal adenoma. (B) Sagittal T1-weighted sellar MRI with gadolinium enhancement showing slight enlargement of the pituitary gland, without a focal lesion.
The patient had acromegalic features with multiple skin tags, coarse facial features and soft tissue swelling of hands and feet. Serum insulin-like growth factor 1 (IGF1) was elevated, 116.1 nmol/l (normal: 12.2–29.8 nmol/l). The diagnosis of acromegaly was confirmed by a 75 g glucose tolerance test (OGTT) with a nadir growth hormone (GH) level of 13.5 ng/ml. Other pituitary hormones were normal. Adrenal hormones and chromogranin A were all within the normal range, whereas calcitonin, measured as part of a pancreatic tumor work-up, was markedly elevated – 978 pg/ml (normal <5 pg/ml) (Table 1). An ultrasound-guided fine needle aspiration of the pancreatic mass revealed uniform cells with centrally located round oval nuclei, eosinophilic cytoplasm and no mitoses. Immunohistochemical staining was positive for pankeratin, chromogranin A and synaptophysin. KI-67 index was 5%. Ga-68 DOTATATE PET scan depicted strong Ga-68 uptake in the pancreatic mass (Fig. 1B) and physiological uptake in the pituitary gland. No uptake was seen in the left adrenal.
Table 1.
Hormonal evaluation of pNET. pNET medium was collected after 48 h incubation for GHRH and calcitonin measurements. The pituitary cell culture was incubated for 4 h with the pNET-conditioned medium which then was collected for GH measurement
| Test | Patient | Control/normal range |
|---|---|---|
| Blood | ||
| GHRH (ng/l) | 1273 | <60 |
| Calcitonin (pg/ml) | 978 | <5 |
| In vitro | ||
| Incubation of pNET with medium | ||
| pNET-derived GHRH release (ng/l) | 57 404 | Undetected |
| pNET-derived calcitonin release (pg/ml) | 3798 | Undetected |
| Effect of pNET-derived medium on GH release from human pituitary culture | ||
| Change in GH release | 36% increase | No change in the presence of control medium |
The MRI of the sella turcica showed an enlarged pituitary, 11×14 mm, with a suprasellar extension but without any visible adenoma (Fig. 2B). The plasma GH releasing hormone (GHRH) level was markedly elevated 1273 ng/l (normal <60 ng/l) (Table 1) consistent with ectopic acromegaly.
Investigation
Hormonal evaluation of pancreatic neuroendocrine tumor (pNET) and pNETs' effect on pituitary culture
Methods
Hormonal tests
Serum and tumor medium GHRH concentrations were measured using an in-house double-antibody RIA (4).
Serum and tumor medium calcitonin levels were determined using a solid phase, enzyme-labeled, two-site chemiluminescent immunoenzymatic assay (Immulite 2000 Calcitonin, Siemens Medical Solutions Diagnostic Ltd., Lanberis, Gwynedd, UK).
Human GH concentrations in the pituitary cell culture were measured by RIA (hGH-IRMA kit; DIA source ImmunoAssays, Louvain-la-Neuve, Belgium).
pNET medium and pituitary tissue preparation
A fresh tissue sample of the pNET was obtained during surgery and was cultured. Pituitary tissue culture was derived from a pituitary tumor sample obtained from a man with GH-secreting adenoma who underwent curative transsphenoidal surgical resection. These procedures were approved by local IRB, and both patients gave informed consent. Both pNET and pituitary-resected specimens were prepared, as described previously (5). After 48 h, the pNET medium was collected and stored at −20 °C for further evaluation. The pituitary cell culture was incubated with the pNET-conditioned medium, or with control medium (low glucose DMEM; five wells each), for 4 h and then the medium was collected for GH measurement.
Results
As shown in Table 1, when the pNET was incubated with a medium, markedly high levels of GHRH and calcitonin were found to be secreted from the tumor.
Furthermore, when the pNET-derived medium was incubated with human pituitary cell culture, it led to a 36% increase in the secretion of GH from the pituitary cells, confirming the functional bioactivity of the pNET-derived GHRH.
Treatment
The patient underwent radical pancreatectomy–duodenectomy.
Outcome and follow-up
Histological examination revealed a partially encapsulated NET of 6.0 cm confined to the pancreatic head (Fig. 1C). Immunohistochemical staining was positive for calcitonin (Fig. 1D), pankeratin, chromogranin A, synaptophysin, CD-56 and Ki-67 of 3%. Dissection margins were free of tumor, and peripancreatic lymph nodes were normal. These findings were compatible with pNET, Grade 2.
In total, 3 months after surgery, there was an improvement in the clinical features of acromegaly, and the IGF1 level was 30.4 nmol/l (normal: 12.2–29.8 nmol/l). Calcitonin level was undetectable now. Repeated Ga-68 DOTATATE PET did not demonstrate any pathological Ga-68 uptake.
Discussion
Acromegaly secondary to ectopic GHRH secretion is rare. Signs and symptoms of ectopic acromegaly are similar to pituitary-dependent acromegaly. Diagnosis is usually delayed and is based on elevated GH and IGF1 levels in the presence of a normal or diffusely enlarged pituitary and an extra-pituitary NET. Acromegaly remission may be achieved by the extra-pituitary tumor resection.
We describe a case of ectopic GHRH secretion caused by pNET in which in addition to detection of GHRH in the blood and in the tumor-derived medium, we could confirm for the first time the functional bioactivity of GHRH secreted from the pNET by its effect of releasing GH from cultured human pituitary tumor cells, known to express the GHRH receptor. A previous two studies have shown similar results in acromegalic patients with foregut carcinoid tumor (6) and bronchial carcinoid tumor (7); however, in both cases, the effect was studied in rat pituitary cells.
Another unique feature of the presented case was that the tumor also expressed and secreted calcitonin (Fig. 1D). Calcitonin is typically secreted by thyroid C-cells and is considered a tumor marker for medullary thyroid carcinoma. Functional pNETs ectopically secreting calcitonin were rarely reported in the literature (8, 9). However, most recently ten cases of pNET-secreting calcitonin were reported (10). To the best of our knowledge, only one case of pNET secreting both GHRH and calcitonin has been described previously (11).
Distinction between pituitary GH adenoma and ectopic GHRH secretion is important, as pituitary hyperplasia secondary to GHRH over-secretion may be misdiagnosed as a pituitary tumor, leading to unnecessary pituitary surgery. However, an experienced radiologist should distinguish between a diffuse, symmetrical pituitary enlargement secondary to hyperplasia vs an adenoma, usually a macroadenoma. The finding of increased serum GHRH levels in acromegaly caused by an ectopic tumor compared with the suppressed and undetectable GHRH levels in cases of GH-secreting pituitary adenomas may be helpful when ectopic GHRH secretion is suspected. Yet, GHRH measurement is not widely available; thus, distinction of other than pituitary adenoma causes of acromegaly is primarily based on radiological findings. Functional assay confirming the bioactivity of GHRH secreted from the ectopic source by its in vitro effect of releasing GH from human pituitary cells could be of substantial assistance in establishing the correct diagnosis.
Patient consent
Written informed consent has been obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
T Zornitzki researched the data, wrote the manuscript, contributed to discussion and reviewed/edited the manuscript. H Rubinfeld made pNET medium and pituitary tissue preparation, wrote the manuscript, contributed to discussion and reviewed/edited manuscript. L Lysyy researched the data, wrote the manuscript and contributed to discussion. T Schiller researched the data and wrote the manuscript. V Raverot did hormonal (GHRH) evaluation in blood and medium. I Shimon researched the data, wrote the manuscript, contributed to discussion and reviewed/edited manuscript. H Knobler researched the data, wrote the manuscript, contributed to discussion and reviewed/edited manuscript.
Acknowledgements
We thank Meital Adi for imaging evaluation and Nadya Ziv-Sokolovskaja for pathology work-up.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- 1. Borson-Chazot F, Garby L, Raverot G, Claustrat F, Raverot V & Sassolas G. 2012. Acromegaly induced by ectopic secretion of GHRH: a review 30 years after GHRH discovery. Annales d'Endocrinologie 73 497–502. 10.1016/j.ando.2012.09.004 [DOI] [PubMed] [Google Scholar]
- 2. Garby L, Caron P, Claustrat F, Chanson P, Tabarin A, Rohmer V, Arnault G, Bonnet F, Chabre O, Christin-Maitre S et al. 2012. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of growth hormone-releasing hormone (GHRH): A French nationwide series of 21 cases. Journal of Clinical Endocrinology and Metabolism 97 2093–2104. 10.1210/jc.2011-2930 [DOI] [PubMed] [Google Scholar]
- 3. Ghazi AA, Amirbaigloo A, Dezfooli AA, Saadat N, Ghazi S, Pourafkari M, Tirgari F, Dhall D, Bannykh S, Melmed S et al. 2013. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine 43 293–302. 10.1007/s12020-012-9790-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Girard P, Cohen R, Sassolas G, Harthe C, Cabrera P & Boissel JP. 1987. Pharmacokinetics of human growth hormone releasing factor (hgrf-44 nh2) in normal men after intravenous administration of a large range of doses. European Journal of Clinical Pharmacology 32 507–513. 10.1007/BF00637679 [DOI] [PubMed] [Google Scholar]
- 5. Rubinfeld H, Hadani M, Barkai G, Taylor JE, Culler MD & Shimon I. 2006. Cortistatin inhibits growth hormone release from human fetal and adenoma pituitary cells and prolactin secretion from cultured prolactinomas. Journal of Clinical Endocrinology and Metabolism 91 2257–2263. 10.1210/jc.2005-2148 [DOI] [PubMed] [Google Scholar]
- 6. Leveston SA, McKeel DW Jr, Buckley PJ, Deschryver K, Greider MH, Jaffe BM & Daughaday WH. 1981. Acromegaly and Cushing's syndrome associated with a foregut carcinoid tumor. Journal of Clinical Endocrinology and Metabolism 53 682–689. 10.1210/jcem-53-4-682 [DOI] [PubMed] [Google Scholar]
- 7. Shalet SM, Beardwell CG, MacFarlane IA, Ellison ML, Norman CM, Rees LH & Hughes M. 1979. Acromegaly due to production of a growth hormone releasing factor by a bronchial carcinoid tumor. Clinical Endocrinology 10 61–67. 10.1111/j.1365-2265.1979.tb03034.x [DOI] [PubMed] [Google Scholar]
- 8. Cronin MJ, Rogol AD, Dabney LG & Thorner MO. 1982. Selective growth hormone and cyclic amp stimulating activity is present in human pancreatic islet cell tumor. Journal of Clinical Endocrinology and Metabolism 55 381–383. 10.1210/jcem-55-2-381 [DOI] [PubMed] [Google Scholar]
- 9. Kon T, Wada R, Suzuki R, Nakayama Y, Ebina Y & Yagihashi S. 2012. Vip and calcitonin-producing pancreatic neuroendocrine tumor with watery diarrhea: clinicopathological features and the effect of somatostatin analogue. Journal of the Pancreas 13 226–230. 10.6092/1590-8577/567 [DOI] [PubMed] [Google Scholar]
- 10. Kovacova M, Filkova M, Potocarova M, Kinova S & Pajvani UB. 2014. Calcitonin-secreting pancreatic neuroendocrine tumors: A case report and review of the literature. Endocrine Practice 20 e140–e144. 10.4158/EP13505.CR [DOI] [PubMed] [Google Scholar]
- 11. Nozieres C, Chardon L, Goichot B, Borson-Chazot F, Hervieu V, Chikh K, Lombard-Bohas C & Walter T. 2015. Neuroendocrine tumors producing calcitonin: characteristics, prognosis and potential interest of calcitonin monitoring during follow-up. European Journal of Endocrinology 174 335–341. 10.1530/EJE-15-0917 [DOI] [PubMed] [Google Scholar]