

Abstract
The elucidation of factors that activate the regeneration of the adult mammalian heart is of major scientific and therapeutic importance. Here we found that epicardial cells contain a potent cardiogenic activity identified as follistatin-like 1 (Fstl1). Epicardial Fstl1 declines following myocardial infarction and is replaced by myocardial expression. Myocardial Fstl1 does not promote regeneration, either basally or upon transgenic overexpression. Application of the human Fstl1 protein (FSTL1) via an epicardial patch stimulates cell cycle entry and division of pre-existing cardiomyocytes, improving cardiac function and survival in mouse and swine models of myocardial infarction. The data suggest that the loss of epicardial FSTL1 is a maladaptive response to injury, and that its restoration would be an effective way to reverse myocardial death and remodelling following myocardial infarction in humans.
The epicardium of the heart is an external epithelial layer that contributes to myocardial growth during development by providing progenitor cells1,2 as well as mitogens, including FGFs, IGF2, and PDGFs3–5. Recent studies suggest that the epicardium might also preserve function of the adult myocardium following injury, possibly as a source of myogenic progenitors6,7. To our knowledge no epicardial-secreted factors have been shown to support adult myocardial regeneration in mammals to date.
Epicardial signal activates cardiomyocyte division
We co-cultured an epicardial mesothelial cell (EMC) line with Myh6+ mouse embryonic stem cell-derived cardiomyocytes (referred to as mCMsESC; Extended Data Fig. 1, Supplementary Videos 1 and 2, and Methods). Co-cultures consistently increased the number of cardio-myocytes (α-actinin+ cells, Fig. 1a–c) and the expression of cardio-myocyte markers (Fig. 1d). Conditioned media from EMC cultures recapitulated this effect (Fig. 1e–h). The number of α-actinin+ cells exhibiting rhythmic Ca2+ transients also increased with the addition of EMC media (8.6-fold) (Fig. 1i), as quantified automatically by kinetic imaging cytometry. Similarly, conditioned media prepared from adult epicardial-derived cells8 increased proliferation and nearly doubled the incidence of aurora B kinase in the cleavage furrow connecting adjacent embryonic cardiomyocytes (Tnnt2+ cells; 0.19 to 0.33%, P < 0.05, Fig. 1j–m), indicating a secreted activity in the adult epicardium that promotes cytokinesis of embryonic cardiomyocytes.
Figure 1. Epicardial secretome has cardiogenic activity, and improves cardiac function after MI via embryonic epicardium-like patches.
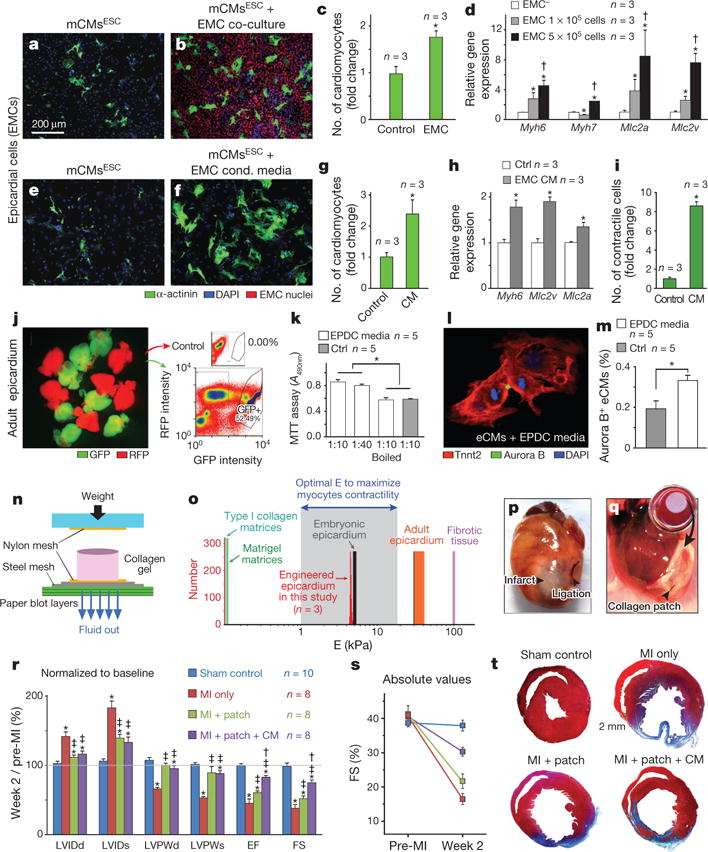
a–d, Co-culture of mCMsESC cardiomyocytes with epicardial EMC cells. a, b, Representative micrographs. c, d, Quantification of myocyte number (c) and cardiac gene expression (d). *P < 0.05 compared to acellular (EMC−) control; †P < 0.05 compared to 1 × 105 cells condition. e–i, Culture of mCMsESC cardiomyocytes with EMC-conditioned media. Representative micrographs (e, f). Quantification of myocyte number (g), cardiac gene expression (h), and cardiomyocytes with rhythmic calcium transients (i). *P < 0.05 compared to control. j–m, Effect of adult epicardial media on embryonic cardiomyocytes from E12.5 GFP+ cells (Tnnt2-cre;Rosa26mTmG/+) (j). Conditioned media obtained from adult epicardial-derived cells (EPDCs) promotes cardiomyocyte proliferation that can be heat-inactivated (k) and cytokinesis analysed by double immunostaining for aurora B and Tnnt2 (cardiomyocytes) (l, m). *P < 0.05. n, Schematic of collagen patch generation (reconstructed from ref. 26). o, Evaluation of mechanical properties of engineered patch, measured by atomic force microscopy. p, q, Suture procedure of patch over ischaemic myocardium. r, Echocardiography analysis normalized to individual pre-surgery baseline values. s, Absolute values of fractional shortening (FS%). t, Masson’s trichrome staining of the animal cohorts: sham (control, n = 10), infarcted mice without treatment (MI only, n = 8), MI treated with patch only (MI plus patch, n = 8), and infarcted animals treated with patch laden with epicardial conditioned media (MI plus patch plus CM, n = 8), 2 weeks after MI. *P < 0.05 compared to Sham control, ‡P < 0.05 compared to MI-only, and †P < 0.05 compared to MI plus patch (see Methods for details.)
Engineered epicardium improves function after injury
We next evaluated the effect of epicardial-secreted factors in the adult injured heart by delivering conditioned media in three-dimensional collagen nano-fibrillar patches9. Patches were designed with an elastic modulus emulating the embryonic epicardium (E ~12 kPa)9, lower than the mature epicardium (E > 30–40 kPa) and fibrotic cardiac tissue (E > 100 kPa), but higher than those for the most currently used scaffolding biomaterials (E ≤ 1 kPa) (Fig. 1n, o). Patches seeded with EMC-media (33% of total volume) were sutured onto the heart immediately following surgical-induced myocardial infarction (MI, permanent ligation of the left anterior descending LAD coronary artery, Fig. 1p, q). Two weeks later, patch-treated hearts (both with or without EMC-media) showed improved morphometric parameters (Fig. 1r–t and Extended Data Table 1), consistent with collagen patch providing a mechanical support that inhibits remodelling9. Notably, only patch with EMC media treatment improved cardiac function (Fig. 1r, s and Extended Data Table 1).
Fstl1 is an epicardial cardiomyogenic factor
To identify bioactive proteins, we analysed EMC-conditioned media by mass spectrometry. Comparison of spectra to the IPI rat database identified 1,596 peptide reads corresponding to 311 unique proteins. Ten secreted proteins with the highest spectral counts were selected for testing in the mCMsESC assay. Of these, cardiogenic activity was noted only with follistatin-like 1 (also known as Fstl1, FRP or TSC36, accession number: NP_077345.1) (Fig. 2a). Unlike follistatin, Fstl1 does not block activin and its biochemical and biological functions are poorly characterized10. Fstl1 levels increase in the blood stream following acute MI and, for this reason, it has been considered a biomarker for acute coronary syndrome11.
Figure 2. Fstl1 is an epicardial cardiogenic factor with dynamic expression after ischaemic injury.

a, MS/MS spectrum of Fstl1. b–g, Fstl1 treatment of mCMsESC cardiomyocytes measured by immunostaining (α-actinin, green) (b, c), quantification of myocyte number (d), expression of cardiac-specific markers (e), cardiomyocytes with rhythmic calcium transient (f), and individual cardiomyocyte cell size (g). *P < 0.05 indicates statistically significantly different from control. h, Fstl1 immunostaining in the mouse embryonic heart (days E12.5, E15.5 and E17.5). Fstl1 (red), Wt1 (epicardial marker), α-actinin (myocardial marker), DAPI (nuclei). Fstl1 is expressed in epicardium (white arrowheads), no myocardium (yellow arrowhead). i) Expression shift of Fstl1 in the mouse heart after MI. Trichrome staining (upper), labels fibrosis (blue) Fstl1 immunohistochemistry (lower panels, brown). In injured hearts Fstl1 expression is depleted from the epicardium (brown) and upregulated in the myocardium. j–l, High resolution images of Fstl1 expression-shift after MI (see Methods for details).
Treating mCMsESC for 8 days with bacterially synthesized recombinant human FSTL1 (10 ng ml−1) increased the number of cardiomyocytes (Fig. 2b–d), the transcription levels of myocardial-specific proteins (Myh6, Mlc2v, and Mlc2a, Fig. 2e), and the number of α-actinin+ cells with rhythmic Ca2+ transients (Fig. 2f). FSTL1 treatment did not induce hypertrophy. Indeed FSTL1 decreased myocyte cell size in a dose-dependent manner (Fig. 2g) in 48 h. Thus, FSTL1 recapitulates the cardiomyogenic activity of the epicardial conditioned medium.
Dynamic expression of Fstl1 after ischaemic injury
During fetal development, endogenous Fstl1 is expressed throughout the myocardium of the primitive heart tube12, but becomes restricted to the epicardium by mid-gestation (Fig. 2h). Epicardial expression persists throughout adulthood (Fig. 2i–k). Remarkably, Fstl1 localization shifts strikingly following ischaemic injury, such that it becomes abundant in the myocardium (Fig. 2i–l) and absent in the epicardium and infarcted area (Fig. 2i, l).
Epicardial FSTL1 promotes regeneration
Prior studies showed that transient overexpression of Fstl1, either by myocardial transgenic expression (Fstl1-TG13, Extended Data Fig. 2a, b) or systemic infusion, is anti-apoptotic following acute ischaemia-reperfusion (I/R)14,15. In the context of permanent myocardial infarction, myocardial Fstl1-TG mice did not recapitulate the effect of the patch containing EMC-media (Extended Data Fig. 2a–j). We assessed epicardial-hFSTL1 delivery by collagen patches loaded with 10 μg of recombinant bacterial-synthetized hFSTL1 per patch. Patches retained immune-detectable hFSTL1 up to 21 days in vitro, and 28 days in vivo, the longest lengths of times tested (Extended Data Fig. 3a–f). Application of hFSTL1-loaded patches simultaneously with MI significantly improved survival (Fig. 3a) and sustained long-term recovery of cardiac function (Fig. 3b and Extended Data Table 2). Epicardial patch with FSTL1 also improved cardiac function when applied onto infarcted hearts of Fstl1-TG mice (Fig. 3c); thus, myocardial overexpression of FSTL1 is insufficient for long-term recovery but epicardial reconstitution of recombinant FSTL1 is necessary to induce the beneficial effects.
Figure 3. FSTL1 recapitulates the in vivo restorative effect of epicardial conditioned media in the engineered epicardial patch, and promotes cardiomyocyte proliferation.
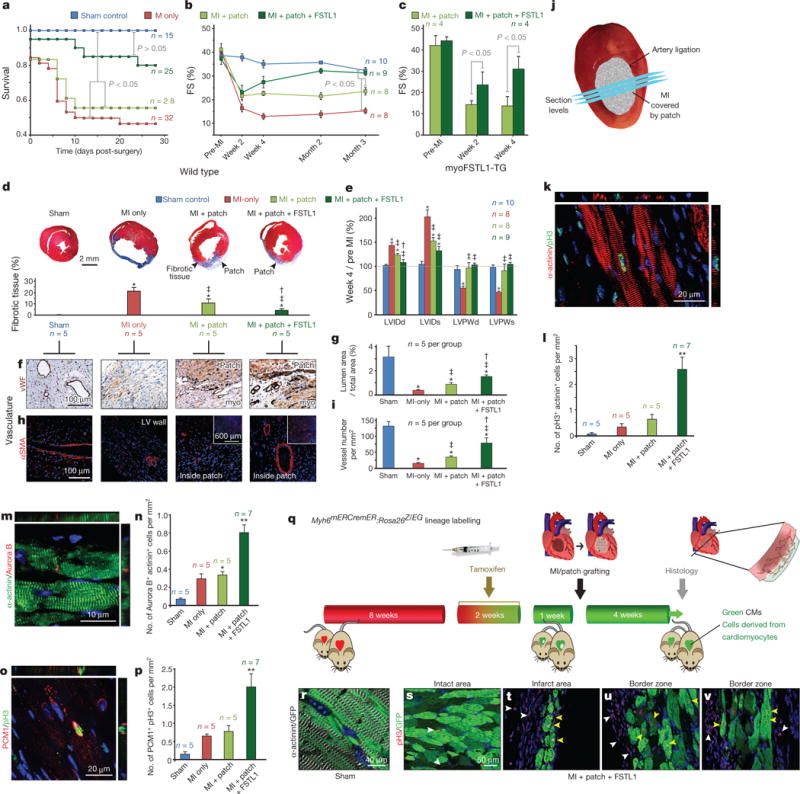
a, b, Survival (a) and kinetics of FS(%) (b) analyses after MI in the indicated treatments. c, Effect of epicardial hFSTL1 patches on FS% in Fstl1-TG mice. d–i, Masson’s trichrome staining (d), morphometric analysis by echocardiography (e), and vascularization analysis (f–i) 4 weeks after MI. *P < 0.05 compared to sham, ‡P < 0.05 vs MI only, and †P < 0.05 vs MI plus patch. j, Cross-sections covering infarct/patch area separated 250 μm, 1–2 mm from apex used for cardiomyocytes proliferation analysis (k–p), 4 weeks after MI. k, m, o, Co-staining of pH3 and α-actinin (k), midbody-localized aurora B kinase between α-actinin+ cells (m), and double-positive cells for pH3 and PCM1 (cardiomyocyte nuclei16) (o) 4 weeks post-MI, quantified in l, n, p, normalized to myocardium area quantified by trichrome staining of immediately adjacent section. *P < 0.05 from sham. **P < 0.05 from all other groups. q–v, Lineage tracing of FSTL1-responsive cells in 4-OH-tamoxifen treated Myh6mERcremER:Rosa26Z/EG mice; patch with FSTL1 applied simultaneously to MI, and hearts were collected 4 weeks post-MI (q) with efficient labelling of cardiomyocytes (r). Infarcted hearts showing eGFP+ (pre-existing, green) cardiomyocytes positive for pH3 (yellow arrowheads) (white arrowheads: pH3+ eGFP− cells) (s–v). (length of treatment (a–c) and n for each experiment indicated in graph, see Methods for details).
The improved cardiac function and survival following patch with FSTL1 treatment was accompanied by attenuated fibrosis (Fig. 3d, e, Extended Data Fig. 3g–j, Extended Data Table 2, and Supplementary Videos 3–5), increased vascularization of the patch and underlying myocardium at the border of the infarcted region (Fig. 3f–i), as reflected by the increased area occupied by vessels (Fig. 3f, g), and the increased number of vessels (of any size) per area unit (Fig. 3h, i). Masson’s trichrome staining showed contiguous engraftment of the patch with FSTL1 onto the host myocardium and demonstrated migration of host cells into the patch including evidence of striated cells (green arrows, last two columns in Extended Data Fig. 3k).
A similar recovery was found when FSTL1-loaded patches were grafted one week after I/R injury in the mouse, when cardiac function had substantially decreased (about 15% reduction in fractional shortening, FS%). As is typical, cardiac function of untreated animals progressively declined (22%, 20% and 16% fractional shortening at 1, 3 and 5 weeks post-I/R). In contrast, the patch with FSTL1 cohort showed a nearly complete and stable recovery of fractional shortening (to 34% three weeks post-I/R) (Extended Data Fig. 4a–d and Extended Data Table 3), suggesting that epicardial-delivered FSTL1 is sufficient to revert the loss of cardiac function after experimental MI.
FSTL1 induces cardiomyocyte proliferation in vivo
Four weeks following MI, the patch with FSTL1 cohort showed evidence of striated myocytes (α-actinin+ cells) within the patch (Extended Data Fig. 5a–d). Cardiomyocytes in the border zone had undergone cell division (Extended Data Fig. 5e–h) by several independent criteria, including an increased number of double-positive α-actinin+, phospho-histone H3 (pH3)+ cells (Fig. 3j–l; and Extended Data Fig. 5e–k), increased incidence of aurora B kinase localized to the midbody between α-actinin+ cells (Fig. 3m, n and Supplementary Video 6), and increased incidence of cells that were double-positive for pH3 and the nuclear cardiomyocyte maker PCM1 (ref. 16) (Fig. 3o, p) relative to MI and patch-only cohorts (Extended Data Fig. 5l–r). Thus, epicardial FSTL1 delivery activates cardiomyocyte cell cycle entry and cytokinesis in vivo reminiscent of the in vitro results above. Proliferating cardiomyocytes were found only in the border zone and, to a lesser extent, the infarcted area (Extended Data Fig. 5s, t). Increased cardiomyocyte proliferation was also observed in the I/R injury model with delayed patch implantation (Extended Data Fig. 4e, f). Notably, FSTL1 did not diminish cardiomyocyte apoptosis, the extent of the infarcted area or the area at risk (hypoperfused area) acutely after MI; nor did it affect apoptosis or inflammation at day 4 and day 8 post-MI (Extended Data Fig. 6).
Figure 5. Epicardial FSTL1 delivery activates cardiac regeneration in preclinical model of ischaemic heart injury.
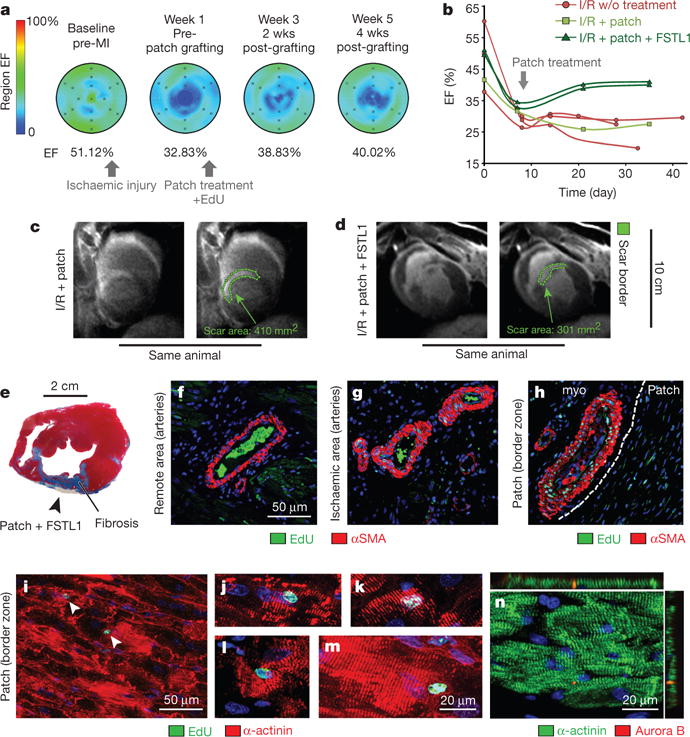
a–d, Time course MRI analysis of cardiac function in pigs. Functional analysis by measurement of ejection fraction (EF%) (a, b). Scar size at week 4 post-grafting (c, d). Green lines highlight scar perimeter. e–n, Analysis at week 4 post-grafting. Masson’s trichrome staining (e). EdU incorporation (newly synthesized DNA) in the vascular smooth muscle cells (f–h). White line demarcates patch and host tissue. i–n, EdU (i–m) incorporation and aurora B kinase positivity (n) in cardiomyocytes at week-4 post-grafting (see Methods for details).
Figure 4. FSTL1 proliferative activity on early cardiomyocytes depends on the cells’ selective post-transcriptional FSTL1 modifications.
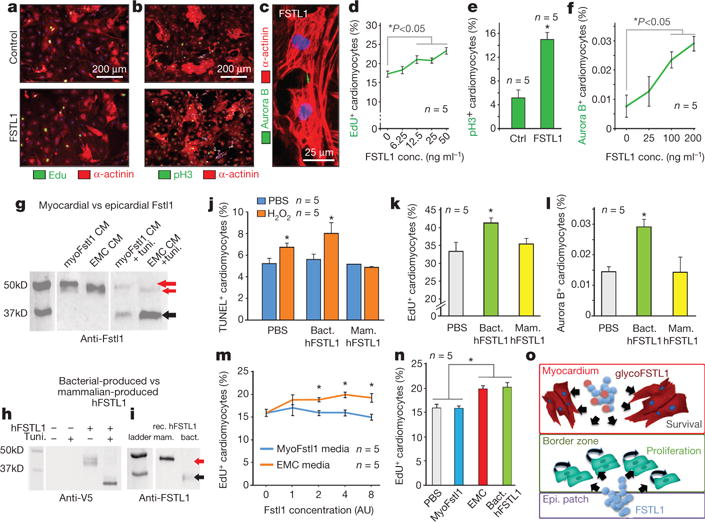
a–f, FSTL1 promotes proliferation of mCMsESC, measured by EdU incorporation (a), pH3 (b), and aurora B immunostaining (c), and quantified in d–f. g–i, Western blot analysis of Fstl1 secreted in cultured cardiomyocytes (myoFSTL1 CM) infected with Adeno-Fstl1 and in EMC (EMC CM) in the presence of tunicamycin (glycosylation inhibitor) (g), hFSTL1-V5 tagged expressed in AD-293 cells (h), and mammalian and bacterial-produced FSTL1 (i). Red arrows, glycosylated; black arrows, hypoglycosylated. j, Mammalian-produced FSTL1 attenuates H2O2 induced apoptosis, while bacterial-produced FSTL1 cannot. k, l, Bacterially-produced FSTL1 promotes mCMsESC EdU incorporation and aurora B positivity whereas mammalian-produced FSTL1 does not. m, n, Quantification of EdU incorporation in mCMsESC treated with conditioned media of EMC and Fstl1-overexpressing NRVC (concentration normalized to Fstl1 content). *P < 0.05 indicates statistically different from control (see Methods for details). o, Working model of FSTL1 in distinct cardiac compartments.
In contrast to patch with FSTL1 delivery, transgenic overexpression of Fstl1 (Fstl1-TG mice) did not show any evidence of cardiomyocyte proliferation after MI (Extended Data Fig. 2k, l), despite increased vascularization described previously13 (Extended Data Fig. 2m, n), indicating that epicardial-delivered FSTL1 might function differently than myocardial-expressed Fstl1.
To distinguish whether the FSTL1-responsive cells arise from preexisting myocytes (Myh6+ cells) or de novo from a progenitor population, we heritably labelled Myh6+ cardiomyocytes using a 4-OH-tamoxifen-inducible cre17 before injury (Fig. 3q). 4-OH tamoxifen injected into Myh6mERcremER: Rosa26Z/EG mice17 efficiently labelled pre-existing cardiomyocytes with eGFP before MI (Fig. 3r). Four weeks after patch engraftment, eGFP+, pH3+ double-positive cells were visible in the infarct area and border zone (Fig. 3s–v), indicating that the proliferating cardiomyocytes expressed Myh6 before MI. We treated cardiomyocytes at different stages of differentiation with FSTL1 in order to determine which stage(s) can respond by proliferating. Neither adult mouse cardiomyocytes (Extended Data Fig. 7a–f), neonatal rat cardiomyocytes (Extended Data Fig. 7g–j) nor cardiomyogenic progenitor cells (Lin−, Sca1+, SP+)18 responded (Extended Data Fig. 7k–m). Of the cells tested, only immature cardiomyocytes (mCMsESC) proliferated in response to FSTL1 (Fig. 4a–f).
It remained paradoxical that neither the endogenous Fstl1 induced by MI nor myocardially overexpressed Fstl1 could induce a regenerative response (Fig. 3 and Extended Data Fig. 2). Western blot analysis indicated that myocardially overexpressed Fstl1 (in neonatal rat ventricular myocytes, NRVC) migrates substantially slower in SDS–polyacryamide gels than does epicardially synthesized Fstl1 (EMC), and that tunicamycin treatment eliminates the difference (Fig. 4g), suggesting cell-type specific glycosylation. The bacterially produced recombinant human FSTL1 (as used in the patch) showed a faster migration consistent with less extensive glycosylation (Fig. 4h, i). Direct comparison of recombinant human FSTL1 produced in bacterial versus mammalian cells (NS0-derived mouse myeloma cell line) revealed that mammalian-expressed FSTL1, but not bacterial FSTL1, protects mCMsESC from H2O2-induced apoptosis (Fig. 4j), consistent with evidence of cardioprotection14 but not regeneration (Extended Data Fig. 2a–j) in Fstl1 TG mice. In contrast, bacterially synthesized human FSTL1 promotes mCMsESC proliferation, whereas human FSTL1 produced in NS0-derived cells or in NRVCs cannot stimulate proliferation of mCMsESC (Fig. 4k–n). Thus, whether FSTL1 induces cardioprotection versus proliferation correlates with cell source and might reflect post-translational modification.
Epicardial FSTL1 in a preclinical swine model
Epicardial delivery of FSTL1 was evaluated in the swine model of I/R injury.I/R decreased left ventricular ejection fraction (EF %) from ~50% before MI, as determined by magnetic resonance imaging (MRI), to ~30% at 1 week after injury. Application of the patch with FSTL1 to the epicardium over the injured tissue at this time (1 week post-MI I/R) stimulated recovery of contractile function (to ~40% EF) in 2 weeks (3 weeks post-MI I/R) (Fig. 5a, b). The recovery remained stable for an additional 2 weeks, the longest time analysed, and was in contrast to the steady decline seen without treatment or following treatment with patch alone (Fig. 5b). FSTL1-treated pigs demonstrated the least scar size of all treatments, including the patch-only condition (see representative MRI images (Fig. 5c, d)). Examination of histological sections of tissues 4 weeks after patch implantation confirmed the limited fibrosis and showed integration of the patch into the host tissue (Fig. 5e). Cardiomyocytes in the border zone and ischaemic area of the patch with FSTL1 treated hearts also had evident EdU labelling (Fig. 5i–m) and midbody-localized aurora B kinase (indicative of cytokinesis) (Fig. 5n). Vascular smooth muscle cells were also EdU+ suggestive of arteriogenesis (Fig. 5g, h). Thus, the patch with FSTL1 appears therapeutic in the swine MI I/R model.
Discussion
Heart regeneration studies in zebrafish suggested that the epicardium is activated by injury to produce factors and cells that sustain cardiac function19. Unlike lower vertebrate hearts, which are robustly regenerative, the mammalian heart retains negligible regenerative potency in adulthood and, instead, sustains cardiomyocyte death and scarring following injury. Very little is known of the endogenous mechanisms that limit regeneration and the topic remains a subject of intense therapeutic interest and scientific debate20. Our data suggest a new view of epicardial function after injury in the mammalian heart. Rather than activation to support cardiac function, the loss of epicardial FSTL1 expression after injury, and the functional and anatomical recovery by reconstitution in an engineered biomaterial, indicate that ischaemic injury induces a maladaptive loss of FSTL1 in the epicardium.
We sought to identify the cell population that proliferates in response to FSTL1. FSTL1 could not stimulate mature adult ventricular cardiomyocytes to synthesize DNA or divide, nor did it induce hypertrophy (as can occur in response to mitogens) either at 48 h (Fig. 2g) or 4 weeks post-MI (Extended Data Fig. 5o). In contrast, FSTL1 stimulated replication of newly emerging cardiomyocytes from mouse ESC cultures (Fig. 4a–f). FSTL1 did not enhance replication of either ESC-derived progenitors before the appearance of Myh6 (not shown) or a population of cardiac progenitors isolated from the adult murine heart (Extended Data Fig. 7k–m), suggesting that competence to respond to FSTL1 occurs transiently. Although at least some mono-nuclear adult cardiomyocytes can be induced to divide21, the FSTL1-responsive cardiomyocytes in our experiments have even less mature sarcomeric and electrophysiological properties22 (for example, automaticity, relatively high maximum diastolic potential, and slow action potential peak Vmax) (Extended Data Fig. 1). The cells that respond to FSTL1 might overlap cells identified in an earlier analysis of infarcted hearts labelled with Myh6-cre, in which a minor population of cre-labelled cells were reported to divide and give rise new cardiomyocytes upon ischaemic injury23. Whether the FSTL1-responsive cells reflect resident Myh6+ recruited upon injury for example23, or derive from de-differentiation24 (thus recapitulating the zebrafish model25) is an interesting question whose resolution will depend on improved method to identify and/or isolate such cells.
Myocardial Fstl1 induced by MI cannot promote a regenerative response, either basally or when abundantly overexpressed transgenically in cardiomyocytes (Extended Data Fig. 2). However, transgenic myocardial Fstl1 is cardioprotective post-MI14. Direct comparison of FSTL1 overexpressed in cardiomyocytes versus the epicardial (EMC) protein revealed tunicamycin-sensitive differences in SDS–PAGE mobility (Fig. 4), consistent with the possibility of differential glycosylation (or other post-translational modification) depending on the cell in which it is expressed. We infer from these data that native epicardial and myocardial FSTL1 have analogous differences in glycan structure that affect their function. It will be important to determine the structure of the glycans, as well as elucidate how post-translation modifications dictate whether FSTL1 promotes anti-apoptosis (myocardial) or cardiomyocyte proliferation (EMC and bacterially produced).
These studies identified FSTL1 as a regenerative factor that is normally present in healthy epicardium, but lost upon MI, suggesting a mechanism whereby injury maladaptively diminishes the regenerative potency of the mammalian heart. Reconstitution of FSTL1 by an engineered epicardial biomaterial improved cardiac function in the mouse MI, mouse MI I/R and preclinical swine MI I/R models with evidence of cardiomyocyte regeneration amenable to clinical translation.
METHODS
Data reporting
No statistical methods were used to predetermine sample size.
Cell preparation
Progenitor cells
Sca1+, Myh6− cardiomyocyte progenitors were obtained by the Schneider laboratory as described18 (Extended Data Fig. 7k–m).
Epicardial mesothelial cells (EMCs)
These were maintained in DMEM with 10% FBS and antibiotics/antimycotic as described27. EMCs are stably transduced with H2B–mCherry lentivirus for nuclei labelling (Figs 1 and 4).
Mouse embryonic stem cell-derived cardiomyocytes (mCMsESC)
A stable mouse ESC line for drug resistance selection of cardiomyocytes (Myh6-Puror;Rex-Blastr) was generated by lentiviral transduction and blasticidin selection, similarly to our previously reported human line28
mCMsESC
These were obtained by differentiation of Myh6-Puror;Rex-Blastr mESCs in a differentiation media containing: Iscove’s Modified Dulbecco Media (IMDM) supplemented with 10% FBS, 2 mM glutamine, 4.5 × 10−4 M monothioglycerol, 0.5 mM ascorbic acid, 200 μg ml−1 transferrin (Roche), 5% protein-free hybridoma media (PFHM-II, Invitrogen) and antibiotics/antimycotic as embryoid bodies (EBs) until day 4 and plated onto adherent cell culture plate until 9, one day after the onset of spontaneous beating. To purify Myh6+ cardiomyocytes, puromycin was added at differentiation day 9 for 24 h. Subsequently cells were trypsinized and plated as monolayer cardiomyocytes. Conditioned media and FSTL1 treatments were typically performed 24 h after monolayer plating. The length of the treatments is indicated in each figure legends (Figs 1, 2, 4 and Extended Data Fig. 1).
AD-293
These cells were directly purchased from Stratagene avoiding misidentification, and cultured in DMEM media with 10% FBS and with pen/strep. It’s used for its high transfection efficiency and yield of recombinant proteins (Fig. 4h).
EMCs and Myh6-Puror;Rex-Blastr mESCs, and AD-293 cells are quarterly tested for mycoplasma contamination when in use.
Embryonic cardiomyocytes
We used fluorescence activated cell sorting (FACS) to purify cardiomyocytes from Tnt-cre;Rosa26mTmG/+ (C57BL/6J and ICR mixed background) hearts from e12.5 embryos. Hearts were dissociated collagenase IV digestion and GFP+ cells for FACS purification. The GFP+ cells were cultured and confirmed to be cardiomyocytes by their expression of the cardiomyocyte specific markers alpha actinin (ACTN2) and cardiac troponin T (TNNT2). They were rhythmically beating when cultured in vitro (Fig. 1j–m).
Neonatal rat ventricular cardiomyocytes (NRVCs)
These cells were isolated with the neonatal rat cardiomyocyte isolation kit (Cellutron) and cultured at 37 °C with 5% CO2. In brief, ventricles were dissected from 1–2-day-old Hsd:s.d. rats (Sprague Dawley), then digested five times for 15 min each with the enzyme cocktail at 37 °C. Cells were pooled, pre-plated for 90 min on an uncoated cell culture dish to remove fibroblasts, and plated on 1% gelatin-coated cell culture plastic dishes in high-serum media (DME/F12 [1:1], 0.2% BSA, 3 mM sodium-pyruvate, 0.1 mM ascorbic acid, 4 mg l−1 transferrin, 2 mM L-glutamine, and 5 mg l−1 ciprofloxacin supplemented with 10% horse serum and 5% fetal bovine serum (FBS)) at 3 × 105 cells per cm2. After 24 h, media was changed to low-serum medium (same but with 0.25% FCS) and cells cultured until use (Fig. 4g, m, n and Extended Data Fig. 7g–j).
Adult mouse cardiomyocytes
These were isolated from 3 month old Myh6mERcremER:Rosa26Z/EG. C57BL/6J mice as previously published29. Briefly, mice were anesthetized with pentobarbital sodium (100 mg per kg IP). The heart was removed and retrograde perfused at 37 °C with a Ca2+-free solution (in mM, 120 NaCl, 14.7 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4-7H2O, 4.6 NaHCO3, 10 Na-HEPES, 30 taurine, 10 BDM, 5.5 glucose) followed by enzymatic digestion with collagenase. Ventricles were cut into small pieces and further digested. Stop buffer (Ca2+-free solution, 12.5 μM CaCl2, 10% bovine calf serum) was added and the cell suspension was centrifuged at 40 g for 3 min. Myocytes were resuspended in stop buffer in increasing CaCl2 concentrations until 1 mM was achieved. Cells were then resuspended in MEM, 5% bovine calf serum, 10 mM BDM, 2 mM L-glutamine and added to the collagen solution, pre-polymerization (250,000 cells per ml or per patch). Following collagen gelation and plastic compression, cellular patches were cultured in aforementioned (plating) media overnight and then transferred into culture media: MEM, 1 mg ml−1 bovine serum albumin, 25 μM blebbistatin, 2 mM L-glutamine, in presence or absence of recombinant FSTL1 (AVISCERA BIOSCIENCE, 10 ng ml−1). At day 7, fluorescent ubiquitination-based cell-cycle indicator (FUCCI, Premo FUCCI Cell Cycle Sensor, Life Technologies, US) assay was conducted on the 3D culture specimens as previously described30. In this assay, G1 and S/G2/M cells emit red and green fluorescence, respectively. The volume of Premo geminin-GFP and Premo Cdt1–RFP reagent were calculated using the equation below:
where the number of cells is the estimated total number of cells at the time of cell labelling (equal to CM seeding density, PPC (particles per cell) is the number of viral particles per cell (40 in this assay), and 1 × 108 is the number of viral particles per ml of the reagent. The volumes of reagents calculated above were directly added to the cellular patches in complete cell medium, mixed gently, and incubated overnight in the culture incubator (≥ 16 h). Patch samples were imaged using a conventional fluorescence microscope, using GFP and RFP filter sets (Extended Data Fig. 7a–f).
Co-culture experiments
mCMsESC are co-cultured with H2B–mCherry EMCs for 4 days and visualized by α-actinin immunofluorescent staining and H2B–mCherry fluorescence (Fig. 1a, b), cardiomyocyte counting (Fig. 1c, n = 3), and cardiogenic gene expression normalized to Gapdh gene expression (Fig. 1d n = 3)
Epicardial conditioned media
Rat epicardial mesothelial cells (EMC) conditioned media
EMC27 cells were cultured in 10% FBS DMEM with penicillin/streptromycin until confluent (~1 × 106 cm−2), then washed with PBS three times and media is changed to serum free DMEM with penicillin/streptromycin without phenol red and cultured for 2 additional days before the media was collected as conditioned media (20 ml of media is added for conditioning and 18 ml is collected after 2 days). Collected media was filtered through 0.22 μm pore membrane (Millipore). Control conditioned media were prepared the same way but without EMC cells (Fig. 1e–i, n–t).
Neonatal rat ventricular cardiomyocytes (NRVCs) conditioned media. NRVC were infected with adenovirus expressing un-tagged mouse Fstl1 at MOI 50. 24 h post-infection culture media was replaced by serum free media (DMEM/F12 with penicillin/streptromycin). The media was conditioned with the infected NRVC and EMC cells for 24 h (Fig. 4m, n).
mCMsESC were treated with control and EMC-conditioned media for 8 days before α-actinin immunofluorescent staining (Fig. 1e, f), cardiomyocyte counting (Fig. 1g, n = 3), analysis of cardiogenic gene expression normalized to Gapdh gene expression (Fig. 1h. n = 3) and quantification of the number of cardiomyocytes with rhythmic calcium transient measured automatically using a Kinetic Imaging Cytometer (Vala Sciences) (Fig. 1i, n = 3).
mCMsESC were treated with serial dilutions of conditioned media of EMC and Fstl1-overexpressing NRVC for 24 h with 10 μg ml−1 EdU, and stained for α-actinin and EdU (Fig. 4m, n, n = 5). The concentrations of the conditioned media are normalized to amount of Fstl1 expression by western blot.
Adult mouse EPDC conditioned media
This was generated in the Zhou laboratory8. Briefly, eight-week old adult Wt1creERT2/+;Rosa26mTmG/+ hearts mice in C57BL/6J and ICR mixed background were injected orally with 4 mg tamoxifen by gavage, four to five oral injections were administered during a two-week period. Myocardial infarction was then induced by ligation of left anterior descending coronary artery on (11 weeks old) adult mice. One week after injury, we collected Wt1creERT2/+;Rosa26mTmG/+ hearts, which were then digested with collagenase IV into single cells. Digestion solution was made by adding 4 ml 1% collagenase IV and 1 ml 2.5% trypsin into 44.5 ml Hanks’ balanced salt solution, and supplemented with 0.5 ml chicken serum and 0.5 ml horse serum. Cells were resuspended in Hank’s balanced salt solution, 4 ml digestion solution was added to each tube and rocked gently in 37 °C shaker for 6 min. After removing the supernatant containing dissociated cells, we added another 4 ml digestion solution to repeat the digestion 6 times. After final digestion, we filtrated the cells through 70 μm filter and pellet cells by centrifuging at 200 g for 5 min at 4 °C. Cells were then re-suspended by Hanks’ balanced salt solution for FACS isolation. Dissociated cells from GFP- hearts were used as a control for gate setting in FACS. GFP+ cells (epicardium-derived cells, EPDCs) were isolated from GFP+ Wt1creERT2/+;Rosa26mTmG/+ hearts by FACS and these GFP+ purified populations were confirmed to be GFP+ cells under fluorescence microscope (Fig. 1j). FSTL1 expression (determined by PCR) was restored in cultured GFP+ EDPCs. Complete conditioned media from EPDCs was then added to embryonic cardiomyocytes culture for 48 h before assay for proliferation (Fig. 1k–m, n = 5).
MTT Assays
Proliferation of cardiomyocytes treated with conditional medium was measured by MTT assay using Celltiter 96 Aqueous One solution (Promega) as previously described8. After adding the Celltiter 96 Aqueous One reagent into the cell culture medium, we incubate the plate at 37 °C for 3–4 h, and then record the absorbance at 490 nm using a 96-well plate reader. Absorbance at 490 nm is tightly correlated with cell number. The MTT readout on the y-axis, labelled MTT assay (A490), thus reflects the relative number of cells from each well between groups of treatment (Fig. 1k). Boiling of conditioned media abolished the growth-promoting effects (Fig. 1k), suggesting a proteinaceous nature of the effective components.
Recombinant FSTL1 was purchased from AVISCERA BIOSCIENCE (00347-02-100, produced in E. coli) and R&D system (1694-FN-050, produced in mouse myeloma cell line, NS0derived).
mCMsESC were treated with bacteria-synthetized recombinant human FSTL1 (10 ng ml−1) for 8 days with media change every 2 days, before α-actinin immunofluorescent staining (Fig. 2b, c), cardiomyocyte counting (Fig. 2d, n = 8), analysis of cardiogenic gene expression normalized to Gapdh gene expression (Fig. 2e, n = 3) and quantification of the number of cardiomyocytes with rhythmic calcium transient measured automatically using a Kinetic Imaging Cytometer (Vala Sciences) (Fig. 2f, n = 6). mCMsESC were treated with bacteria-synthetized recombinant human FSTL1 (6.25–50 ng ml−1) for 2 days before measurement of individual cardiomyocyte cell size (in pixels) (Fig. 2g, n = 5).
mCMsESC were stimulated with 6.25, 12.5, 25 and 50 ng ml−1 of bacteria produced FSTL1 for 24 h with 10 μg ml−1 EdU, and stained for α-actinin and EdU (Fig. 4a, d, n = 5). mCMsESC were stimulated with 10 ng ml−1 FSTL1 for 48 h and stained for α-actinin and pH3 (Fig. 4b, e, n = 5). mCMsESC were stimulated with 25, 100, 200 ng ml−1 FSTL1 for 48 h, and stained for α-actinin (red) and aurora B (Fig. 4c, f, n = 5)
mCMsESC were stimulated with 10 nM H2O2, and 10 ng ml−1 bacteria and mammalian produced FSTL1 for 24 h, and staining for α-actinin and TUNEL for cell death (Fig. 4j, n = 5).
mCMsESC were stimulated with 10 ng ml−1 of bacteria and mammalian produced FSTL1 for 24 h with 10 μg ml−1 EdU, and stained for α-actinin and EdU (Fig. 4k, n = 5), and α-actinin and aurora B (Fig. 4l, n = 5)
FSTL1 overexpression and western blot
AD-293 cells were transiently transfected with human FSTL1 plasmid (GE Dharmacon, ID: ccsbBroad304_02639 pLX304-Blast-V5-FSTL1) using lipofectamine 2000 (mocked transfection was done with lipofectamine and no plasmid). 48 h post-transfection serum containing media was replaced by serum free DMEM and incubated with the cells for 24 h. Tunicamycin was used at 2 μg ml−1. Conditioned media from tunicamycin samples was collected during 16 h (cells looked healthy). Conditioned media was spun at 400 g for 7 min and then concentrated approximately 20 times using Microcon-10 kDa cut off columns (Millipore). Samples were combined 1 to 1 ratio with 2 × SDS sample buffer containing protease inhibitor, DTT and 5 mM EDTA, boiled for 10 min at 95 °C and run in a 4–15% acrylamide Mini-Protean TGX gel, transferred to nitrocellulose membrane and incubated with anti-V5 primary antibody MAB 15253 (Pierce) 1:1,000 dilution and anti-mouse 800 nm conjugated secondary antibody at 1:10,000 dilution (Odyssey), and scanned using the Odyssey Clx Imager (Fig. 4h).
Neonatal rat ventricular cardiomyocytes were infected with adenovirus expressing un-tagged mouse Fstl1 at MOI 50. 24 h post-infection culture media was replaced by serum-free media. Serum free DMEM/F12 pen/strep media was conditioned with the infected NRVC and EMC cells for 24 h. Tunicamycin was used at 1 μg ml−1 and media was conditioned for 16 h. Conditioned media was spun at 400 g 7 min and then concentrated using Microcon-10 kDa cut off columns (Millipore). Samples were combined 1 to 1 ratio with 2 × SDS sample buffer containing protease inhibitor, DTT and 5 mM EDTA, boiled 10 min at 95 °C and run in Any KD Mini-Protean TGX gel, transferred to nitrocellulose membrane and incubated with anti-FSTL1 MAB1694 (R&D) primary antibody 1:500 dilution and anti-rat 800 nm conjugated secondary antibody at 1:10,000 dilution (Odyssey), and scanned using Odyssey Clx Imager. Blocking and antibody incubation was done in Odyssey blocker. The western blot for recombinant FSTL1 (100 ng each) was performed the same way (Fig. 4g, i).
RNA extraction and qRT–PCR
Total RNA was extracted with TRIzol (Invitrogen) and reverse transcribed to cDNA with QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions. cDNA samples synthesized from 100 ng of total RNA were subjected to RT–qPCR with LightCycler 480 SYBR Green I Master kit (Roche) performed with LightCycler 480 Real-Time PCR System (Roche) (Figs 1d, h and 2e and Extended Data Fig. 7b–d). Primer sequences are listed in Supplementary Table 1.
LC-MS/MS analysis of conditioned-media
First, Tris(2-carboxyethyl)phosphine (TCEP) was added into 1 ml of conditional media to 10 mM and the protein sample was reduced at 37 °C for 30 min. Then iodoacetamide was added to 20 mM and the solution was alkylated at 37 °C for 40 min in the dark. Mass spectrometry grade of trypsin (Promega) was then added to the solution as 1:100 ratio. After overnight digestion at 37 °C, the sample was then desalted using a SepPack cartridge, dried using a SpeedVac and re-suspended in 100 μl of 5% formic acid. The resulting peptides were analysed on-line by an LC-MS/MS system, which consisted of a Michrom HPLC, a 15 cm Michrom Magic C18 column, a low flow ADVANCED Michrom MS source, and a LTQ-Orbitrap XL (Thermo Scientific, Waltham, MA). A 120-min gradient of 0–30% B (0.1% formic acid, 100% acetonitrile) was used to separate the peptides, and the total LC time was 141 min. The LTQ-Orbitrap XL was set to scan the precursors in the Orbitrap at a resolution of 60,000, followed by data-dependent MS/MS of the top 4 precursors.
The raw LC-MS/MS data was then submitted to Sorcerer Enterprise (Sage-N Research Inc.) for protein identification against the IPI rat protein database, which contains semi-tryptic peptide sequences with the allowance of up to 2 missed cleavages and precursor mass tolerance of 50.0 p.p.m. A molecular mass of 57 Da was added to all cysteines to account for carboxyamidomethylation. Differential search includes 16 Da for methionine oxidation. The search results were viewed, sorted, filtered, and statically analysed using PeptideProphet and ProteinProphet (ISB). The minimum trans-proteomic pipeline (TPP) probability score for proteins and peptides was set to 0.95, respectively, to assure a TPP error rate of lower than 0.01. The example MS/MS spectrum R.GLCVDALIELSDENADWK.L was identified as Fstl1 (Fig. 2a). Peptide probability = 1.0, Xcorr = 6.276, delta Cn = 0.471.
Automated in vitro cell proliferation and cell death assay
Cells (mCMsESC and NRVC) were incubated with EdU (details of dosage and length of exposure are specified in figure legends) in a 384-well plate format, and were fixed for 2 h in 4% PFA, washed in PBS and stained for EdU using Click-it EdU assay kit (Life Technologies). The cells were then washed in PBS, immunostained with an α-actinin antibody (Sigma, A7811, 1:500) to identify cardiomyocytes and stained with DAPI (4′,6-diamidino-2-phenylindole, 1:10,000) to identify nuclei. The plates were then imaged using InCell 1000 system (GE Healthcare) and automatically analysed in Developer Toolbox (GE Healthcare) as described31. Ratios of EdU+/α-actinin+ nuclei and α-actinin+ nuclei were generated for the percentage of cardiomyocyte incorporated EdU in the chromosomal DNA.
Similarly, cells (mCMsESC and NRVC) in 384-well plate format were fixed for 2 h in 4% PFA, washed in PBS, and were immunostained with pH3 antibody (Millipore 06-570, 1:200) for nuclei in mitosis, or aurora B (Millipore 04-1036, 1:200) for cytokinesis, or TUNEL (Roche) for cell death, and α-actinin antibody (Sigma, A7811, 1:500) for cardiomyocytes and DAPI (1:10,000) for nuclei. The same imaging and analysis were done for pH3 staining as the EdU assays, and the aurora B+, α-actinin+ double positive cells were manually counted. The percentages of pH3+, α-actinin+ double positive nuclei, aurora B+, α-actinin+ double positive cells, and TUNEL+, α-actinin+ double positive nuclei relative to the total number of α-actinin+ cell nuclei were calculated to determine the percentages of cardiomyocytes undergoing mitosis, cytokinesis and apoptosis, respectively.Calcium Imaging. Contractile calcium transients were recorded using a Kinetic Image Cytometer (KIC, Vala Sciences) using Fluo4 NW calcium indicator (Life Science). Data was processed using Cyteseer software containing the KIC analysis package (Vala Sciences) as described32.
Compressed collagen gel for use as an engineered epicardial patch
Highly hydrated collagen gels, used as cardiac patch in this study, were produced by adding 1.1 ml 1 × DMEM (Sigma, MO, US) to 0.9 ml of sterile rat tail type I collagen solution in acetic acid (3.84 mg ml−1, Millipore, MA, US). The resulting 2 ml collagen-DMEM mixture was mixed well and neutralized with 0.1 M NaOH (~50 μl). The entire process was conducted on ice to avoid premature gelation of collagen. In the case of patches containing epicardial factors, the EMC culture media was collected as above and 0.6 ml of that was mixed with 0.5 ml DMEM. The collagen solution (0.9 ml) was then distributed into the wells of 24-well plates (15.6 mm in diameter) and placed in a tissue culture incubator for 30 min at 37 °C for polymerization. For pig studies, 6.8 ml of collagen was mixed with 8.2 ml DMEM to obtain a 15 ml solution that was then cast into a 6-cm Petri dish (area = 28.3 cm2). Plastic compression was performed as described previously33,34. Briefly, as cast, highly hydrated collagen gels (at ~0.9 and 15 ml volumes for the mice and swine study, respectively) underwent unconfined compression via application of a static compressive stress of ~1,400 Pa for 5 min (see refs 33, 35 for details), resulting in ~98–99% volume reduction (Fig. 1n). The elastic modulus of the compressed collagen, aimed to approximate that of the embryonic epicardium which is optimal for contractility of immature cardiomyocytes (see ref. 36), was assessed by atomic force microscopy (AFM) in nano-indentation mode, using a force trigger that resulted in a minimal local strain of less than 10% (indentation of ~100 nm) to minimize the effect of substrate-related artefacts. A custom-made flat AFM tip was manufactured using focused ion beam milling and used to probe the stiffness of the gels by scanning areas of 90 μm × 90 μm. Histogram of the distribution of measured micro stiffness of the patch is compared with the range of elasticity reported for common scaffolding biomaterials37, and previously described36 optimal range of elasticity to maximize myocyte contractility (Fig. 1o, n = 3).
Myocardial infarction and application of the epicardial patch
Permanent LAD occlusion (MI)
Male 10–12-week-old C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbour, ME, USA). Fstl1-TG mice used in MI experiments are C57BL6 background, female and male mice aged 12–15 weeks old. Mice were anaesthetized using an isoflurane inhalational chamber, endotracheally intubated using a 22-gauge angiocatheter (Becton, Dickinson Inc., Sandy, Utah) and connected to a small animal volume-control ventilator (Harvard Apparatus, Holliston, MA). A left thoracotomy was performed via the fourth intercostal space and the lungs retracted to expose the heart. After opening the pericardium, a 7-0 suture was placed to occlude the left anterior descending artery (LAD) ~2 mm below the edge of the left atrium. Ligation was considered successful when the LV wall turned pale (Fig. 1p). In the case of experimental groups treated with patch, immediately after the ligation, prepared collagen patch was sutured (at two points) onto the surface of ischaemic myocardium (Fig. 1g). The patch size used was ~ one-third of the 15.6 mm-diameter collagen gel. Animals were kept on a heating pad until they recovered. Another group of mice underwent sham ligation; they had a similar surgical procedure without LAD ligation. A minimum number of n = 8 was used in each study group.
Ischaemia reperfusion (I/R)
Male C57/BL6, aged 10 to 11 weeks, were anaesthetized and intubated as described above. A left lateral thoracotomy was then performed. Pericardium was gently pulled off and an 8-0 Nylon suture (Ethicon, Inc. Johnson & Johnson Co., USA) was used to ligate the left anterior descending coronary artery against a PE10 tubing, which was removed after 30 min occlusion. Successful performance of coronary artery occlusion was verified by visual inspection (by noting the development of a pale colour in the distal myocardium upon ligation). The chest was then closed using 7–0 sutures around adjacent ribs, and the skin was closed with 6–0 suture. Buprenorphine was administered subcutaneously for a minimum of 1 day at BID dosing. For the animal group treated with patch, a second thoracotomy was performed one week post the incidence of I/R and the prepared collagen patch was sutured (at two points) onto the surface of ischaemic myocardium. Sham-operated controls consisted of age-matched mice that underwent identical surgical procedures (two thoracotomies) with the exception of LAD ligation (Extended Data Fig. 4).
Echocardiography
In vivo heart function was evaluated by echocardiography at 2 weeks (Figs 1r, s and 3b, c and Extended Data Fig. 2 h–j), 4 weeks (Fig. 3b, c, e and Extended Data Fig. 2h–j), and 2 and 3 months (Fig. 3b) after LAD ligation. Two-dimensional (2D) analysis was performed on mice using a GE Vivid 7 ultrasound platform (GE Health Care, Milwaukee, WI) equipped with 13 MHz transducer. The mice were sedated with isoflurane (100 mg per kg, inhalation), and the chest was shaved. The mice were placed on a heated platform in the supine or left lateral decubitus position to facilitate echocardiography. 2D clips and M-mode images were recorded in a short axis view from the mid-left ventricle at the tips of the papillary muscles. LV internal diameter (LVID) and posterior wall thickness (LVPW) were measured both at end diastolic and systolic. Fractional shortening (FS, %) and ejection fraction (EF, %, via extrapolation of 2D data) were calculated from LV dimensions in the 2D short axis view. A minimum number (n) of 8 mice per experimental group was used for the echo evaluations. Measurements were performed by two independent groups in a blind manner. In ischaemia reperfusion study, in vivo heart function was evaluated pre-surgery (baseline), 1 week after the incidence of I/R, and two and four weeks post-implantation (Extended Data Fig. 4a–d).
In vivo delayed-enhanced magnetic resonance imaging (DEMRI)
To prepare for scanning, induction of anaesthesia was accomplished with 2% and maintained with 1.25–1.5% isoflurane with monitoring of the respiratory rate. ECG leads were inserted subcutaneously to monitor the heart rate while the body temperature was maintained at 37 °C. Using 3T GE Signa Excite clinical scanner with a dedicated mouse coil (Rapid MR International, Germany), functional parameters were recorded on weeks 1 and 4 after treatment. The following sequences were performed for MRI acquisitions: (1) DEMRI was performed following i.p. injection of 0.2 mmol per kg gadopentetate dimeglumine (Magnevist, Berlex Laboratories) using gated fGRE-IR sequences with FOV 3.4 cm, slice thickness 0.9 mm, matrix 128 × 128, TE 5 ms, TI 150–240 ms, and FA 60°; and (2) cardiac MRI of volumes were performed using fSPGR with FOV 7 cm, slice thickness 0.9 mm, matrix 256 × 256, TE 5.5 ms, and FA 30. Coronal and axial scout images were used to position a 2-dimensional imaging plane along the short axis of the left ventricular (LV) cavity (Extended Data Fig. 3h–j).
Histology, immunohistochemistry and immunofluorescent staining
Histological analysis (Mason’s trichrome staining) was performed according to standard protocols for paraffin embedded samples. For immunohistochemistry and immunofluorescent staining, embedded hearts were sectioned at a thickness of 7 μm, unless described otherwise. Antibodies used were as follows: 1:200 α-actinin (Sigma, A7811), 1:300 α-smooth muscle actin (Sigma A2547) 1:100 phospho-Histone3 (rabbit Millipore 06-570), 1:300 phospho-Histone3 (mouse Abcam ab14955) 1:100 WT1 (Abcam, ab15249), 1:100 (Fig. 1l–m) and 1:250 (Figs 3m, n and 5n) aurora B (Millipore 04-1036), 1:100 Tnnt2 (DSHB, Ct3), 1:100 Tnni3 (Abcam, ab56357), 1:200 PCM1 (Sigma-Aldrich HPA023370), 1:200 FSTL1 (R&D MAB17381). At least 5 sections per heart were used per staining for Mason’s Trichrome staining and 3 sections per heart per staining for immunohistochemistry and immunofluorescent staining, respectively. HRP Anti-rat secondary antibody (Jackson ImmunoResearch 712-036-153, 1:500) was used for immunohistochemistry, and respective fluorescent secondary antibodies (Life Technologies 1:200) were used for immunofluorescent staining. The Trichrome staining and immunohistochemistry images were taken using an upright Zeiss microscope and dissection scopes. The fluorescent images were taken using Apotome Optical Sectioning (Zeiss). An inclusion criterion for the patch engraftment was that the patch covered > 70% of the infarct (controlled by histology). TUNEL assay (Roche 11684795910).and EdU assay (Life Technologies C10337) were performed as instructed.
Lineage tracing experiments
Epicardial lineage labelling was achieved by oral delivery of tamoxifen (4 mg) in eight-week old Wt1creERT2/+;Rosa26RFP/+ mice with C57BL/6J and ICR mixed background (delivered 6 times for duration of 3 weeks and stopped 1 week before MI). Hearts were collected at 2 weeks after MI. Immunostaining of RFP for Wt1 linage cells, Fstl1 and Tnni3 shows that Fstl1 is absent in epicardial cells and their derivatives, but abundant in the myocardium after MI (Fig. 2l).
Cardiomyocyte lineage labelling was achieved by injecting 4-OH tamoxifen intraperitoneally into eight-week old Myh6mERcremER:Rosa26Z/EG mice17 of C57BL6 background at a dose of 20 mg per kg per day for 2 weeks, and stopped 1 week before havesting cardiomyocytes (Extended Data Fig. 7a–f), or MI operation and patch grafting. 4 weeks after MI, the animals were collect for immunostaining (Fig. 3q–v).
TTC staining
At day 2 post MI/patch treatment, the mouse hearts from all four groups were harvested and sectioned perpendicularly to the long axis into four sections (approximately 2 mm thick). The sections were placed in the wells of a 12-well cell culture plate and incubated with 1% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich) solution for 15 min at 37 °C. Subsequently section were washed with PBS and visualized using a stereomicroscope and photographed with a digital camera (Extended Data Fig. 6a, b).
Vessel counting
Blood vessel density parameters were measured from histological sections of heart samples stained for von Willebrand factor (vWF) as a marker of endothelial cells in the vessel wall. Up to 60 sections were analysed for each treatment group (4 mice in each group). Analysis was performed using ImageJ to calculate: (1) the total luminal area of blood vessels, and (2) the number of vessels that stained + for the vWF. In each case, a histogram of the vessel parameters as a fraction of total surface area analysed was obtained and the mid-values plotted for each treatment group. Statistical significance (P < 0.05) of the differences from sham group was determined by one-tailed ANOVA (Fig. 3f–i).
Cardiomyocyte proliferation quantification in vivo
Data collected from 5–7 hearts in each group (7 for MI plus patch with FSTL1, 5 for Sham, MI-only and MI plus patch) with 3 different cross-sections (each section covered the infarct, patch, and separated by 250 μm, between 1–2 mm from the apex) counted exhaustively for total pH3+/α-actinin+, aurora B+/α-actinin+, and pH3+/PCM1+ cells in each section, and normalized to myocardium area quantified by trichrome staining of immediate adjacent section (Fig. 5j–p, and Extended Data Fig. 5).
Enzyme-linked immunosorbent assay
In order to assess the FSTL1 retention within the engineered patch system in vitro, collagen scaffolds laden with FSTL1 (5 μg ml−1) were immersed in PBS and shaken for various times (0, 12 h, 1 day, and 21 days) at 4 °C and the FSTL1 concentration was determined using Enzyme-linked Immunosorbent Assay kit (USCN Life Science, Inc., Houston, USA). The detection limit for this technique was 0.50 ng ml−1. Scaffolds were pretreated with 1 mg ml−1 collagenase type I (Sigma Aldrich, MO, US) and 5 mg ml−1 hyaluronidase (Sigma Aldrich, MO, US) dissolved in phosphate buffered saline for 5 min followed by centrifugation at 5,000 g for 20 min.
Aliquots of 100 μl of the collected samples were added to the 96-well plates and incubated for 2 h at 37 °C. Then, 100 μl of the prepared detection reagent A were added to the wells followed by 1 h incubation at same temperature. After aspiration and washing 3 times, 100 μl of the prepared detection reagent B was added to the wells and incubated for 30 min at 37 °C. After aspiration and washing 5 times, 90 μl of substrate solution was added to the wells following by incubation for 25 min at 37 °C. 50 μL of stopping solution was added to the wells and the absorbance of each well was read at 450 nm, immediately. The concentration of FSTL1 was defined using standard curve of the standard solutions. The test was performed 4 times (Extended Data Fig. 3a).
Application of the patch in a swine model of ischaemia-reperfusion
The swine study was performed by inflation of a percutaneous coronary angioplasty dilation catheter to occlude the LAD in Yorkshire pigs (45 days old). Occlusion time of 90 min was followed by fully reperfusion to mimic the clinical MI disease model. One week after MI, a left thoracotomy was performed and the patch (6-cm diameter) was sutured onto the infarct. Animal groups included: sham controls, I/R with no treatment (n = 3), I/R treated with patch alone (I/R plus patch, n = 1), and I/R treated with patch laden with FSTL1 (I/Rplus patch with FSTL1, n = 2). EdU delivery: 250 mg per week EdU was infused into circulation during the 4-week time course of study (week 1 to week 5 post I/R), using osmotic mini pumps (Fig. 5).
Animal compliance
The procedures involving animal use and surgeries were approved by the Stanford Institutional Animal Care and Use Committee (IACUC). Animal care and interventions were provided in accordance with the Laboratory Animal Welfare Act (C57BL/6J wildtype mice (Figs 1p–t, 2h–k, 3a–p, 3–6) Myh6mERcremER:Rosa26Z/EG. C57BL/6J mice (Fig. 3q–v, Extended Data Fig. 7a–f) Yorkshire pigs (Fig. 5)).
The study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Boston University (wild-type and Fstl1-TG C57BL/6J mice, (Fig. 3c and Extended Data Fig. 2)).
Mice were used in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Tnt-cre;Rosa26mTmG/+, Wt1creERT2/+;Rosa26mTmG/+.mice (C57BL/6J and ICR mixed background) (Fig. 1j–m) Wt1creERT2/+;Rosa26RFP/+ mice (C57BL/6J and ICR mixed background) (Fig. 2l)
All animal study was approved by the Institutional Animal Care and Use Committee (IACUC) of Sanford-Burnham-Prebys Medical Discovery Institute. All animal procedures performed conform the NIH guidelines (Hsd:s.d. rats (Fig. 4g, m, n and Extended Data Fig. 7g–j)).
Statistical analysis
The number of samples (n) used in each experiment is recorded in the text and shown in figures. All in vitro experiments have been done at least twice independently. Gene expression experiments have been done 3 times independently and EdU proliferation assays and cell size measurement have been done more than 10 times independently. Sample size was not predetermined, with retrospective analysis of significantly different results in most in vitro studies using Gpower 3.1 produces power > 0.8. Sample sizes for animal studies were estimated. Animals which did not survive up to 4 weeks after surgery were excluded from functional and histological studies. Randomization was not applied. Blinding to group allocation was practiced between animals surgery and results analysis of mouse myocardial infarction experiments. The values presented are expressed as means ± s.e.m. The rationale to use means ± s.e.m. instead of s.d. is that s.e.m. quantifies uncertainty in an estimate of the mean whereas s.d. indicates dispersion of the data from mean. In other words, the SEM provides an estimate of the reported mean value, while the s.d. gives an idea of the variability of single observations. Normal distribution were tested and confirmed in automatic analysis of mCMsESC (Figs 1c, g, i, 2d, f, g and 4d, e, j,–n and Extended Data Figs 1 and 7h, j). We did not estimate variations in the data. The variances are similar between the groups that are being statistically compared. One-way ANOVA with multiple comparisons (Fig. 1r, 3 and Extended Data Figs 2b, d and 4, 5, 6) and Student’s t-test (Fig. 1a–m, 2 and 4 and Extended Data Fig. 2e–n and 7) were used to test for statistical significance (P < 0.05). Survival curve were generated by Kaplan–Meier method using PRISM (GraphPad) and Log-rank (Mantel–Cox) test was used to test the significant differences between the survival of mice in different conditions (Fig. 3a).
Extended Data
Extended Data Figure 1. Characterization of mCMsESC cells used in this study.

a, Schematic time-line of cell preparation and treatment. b–d, Immunostaining of α-actinin of mCMsESC, showing that the majority of the cells are α-actinin+ (b), and the α-actinin lacks striation structures (c). d, Immunostaining of α-smooth muscle actin (aSMA) of mCMsESC, showing the majority of the cells are αSMA+, unlike mature cardiomyocytes with no SMA expression38. e, f, Automatic detection of EdU incorporation in mCMsESC. Captured image of mCMsESC treated with 10 μg ml−1 EdU for 24 h, stained with EdU, α-actinin and DAPI using InCell 1000 (General Electric) (e). Overlay of masks of EdU, α -actinin and DAPI channels with automatic detection software (f). g, EdU incorporation profile of mCMsESC over time. mCMsESC are treated with 10 μg ml−1 EdU for 24 h at time 0 h, 24 h, 48 h, and 144 h. The percentage of EdU+/α-actinin+ cardiomyocytes of all α-actinin+ cardiomyocytes is calculated for each time period. Note the decrease of EdU incorporation rate over time. h, i, Fluo 4 calcium images of mCMsESC, with baseline background image (h) and peak image (i). j, Comparison of representative calcium transients of mCMsESC (red) and neonatal rat ventricular cardiomyocytes (NRVC, blue). Note the reduced amplitude, slower rate of up and down strokes, and elongated duration of the calcium transient in mCMsESC compared to NRVC, suggesting immature calcium handling in mCMsESC. In all experiments, FSTL1 was added one day after plating of the mCMsESC (time 0–24 in this figure).
Extended Data Figure 2. Myocardial overexpression of Fstl1 (Fstl1-TG) mice after permanent LAD ligation.

a–d, Fstl1 protein expression kinetics after myocardial infarction. Fstl1-TG mice (C57/Bl6 background) and littermate wild-type (WT) mice underwent LAD ligation. Heart tissue and serum were collected at baseline, day 1, day 3, day 7 and day 28 after surgery. Fstl1 protein levels in ischaemic area (IA) and remote area (RM) of heart were analysed by western blotting (a). Fstl1 expression expressed relative to tubulin levels is reported (b). Fstl1 serum levels were analysed by western blotting (c). Also shown in Ponceau-S staining to indicate equal loading of serum. Quantification of serum Fstl1 level is shown in (d). n > 3 in all groups. *P < 0.05 compared to WT baseline, #P < 0.05 compared to Fstl1-TG baseline. ANOVA was used for statistical significance (P < 0.05). e–j, Morphometric and functional response of Fstl1-TG mice to permanent LAD ligation at long-term. Representative Masson’s trichrome staining of WT (e) and Fstl1-TG (f) 4 weeks after MI. Quantification of content in fibrotic tissue at week 4 after MI (g). Echocardiographic measurement of left ventricular internal dimension in systole (LVIDs) (h), and left ventricular internal diameter in diastole (LIVDd) (i) at weeks 2 and 4 after MI. Echocardiographic determination of fractional shortening (FS%) in the indicated genotypes at 2 and 4 weeks after MI (j). k–n, Double immunofluorescent staining of α-actinin (cardiomyocytes) and pH3 (mitosis) (k) and α-actinin (cardiomyocytes) and von Willebrand factor (vascular endothelial cells) (m) in the Fstl1-TG and WT mice, quantified in (i, n). n = 5, *P < 0.05 indicates significantly different from WT.
Extended Data Figure 3. Patch with FSTL1 attenuated fibrosis after MI.
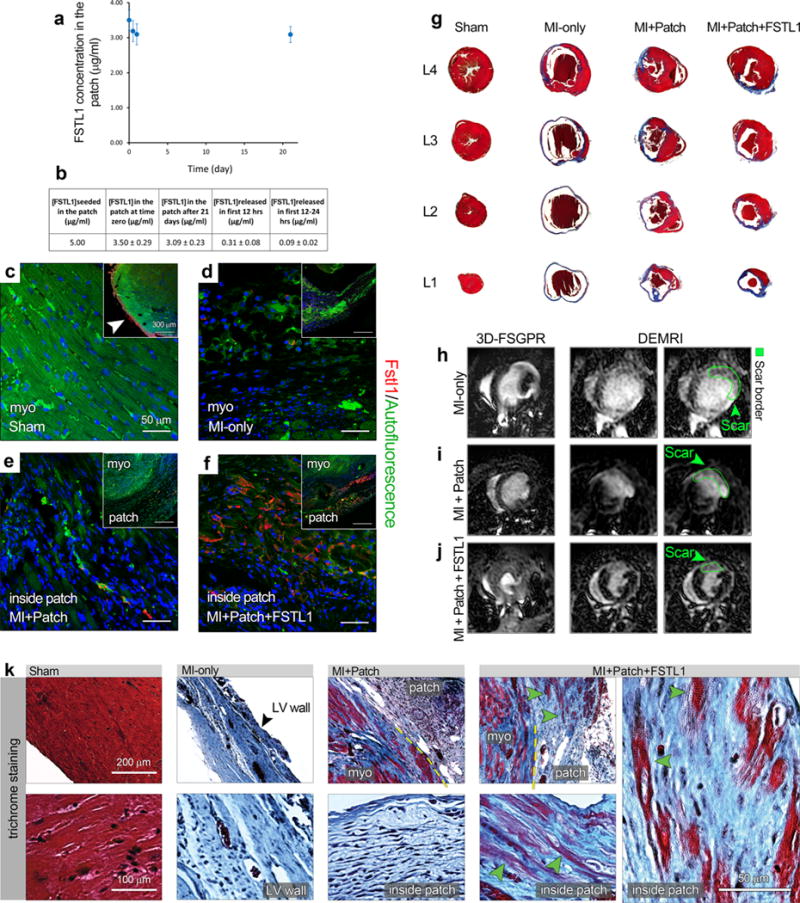
a–f, FSTL1 retention in the patch in vitro and in vivo. a, b, Enzyme-linked immunosorbent assay used to measure the amount of FSTL1 retained within collagen scaffolds exposed to PBS in vitro for different time intervals (0–21 days) (a). The Table lists the initial and final FSTL1 concentration, as well as the release values within the first 24 h (b). c–f, FSTL1 retention in the patch in vivo. Representative images of Fstl1 immunostaining in the indicated animal treatment groups, week 4 after surgery. Note that, while Fstl1 is expressed in the uninjured epicardium (arrow in the inset in c), its expression became undetectable within the infarct area after MI (d). Similarly, no FSTL1 was detected in the MI plus patch animals (e), while it still persists (red staining) in the patch area of the MI plus patch with FSTL1 group (f). g, Representative Masson’s Trichrome staining on serial cross sections of hearts under 4 conditions (sham, MI only, MI with patch and MI plus patch with FSTL1) 4 weeks after MI. Note the severe fibrosis in MI only condition, and reduced fibrosis in MI plus patch condition, and further reduction in MI plus patch with FSTL1 condition, quantified in Fig. 3d. h–j, Representative MRI images from the mouse MI only, MI plus patch and MI plus patch with FSTL1 treatment groups showing the 3D-FSPGR (fast spoiled gradient-echo) images and the delayed enhancement images using gadolinium contrasting agents, confirming a reduction in infarct area (demarcated in green) and preserved contractility (Supplementary Videos 3–5). k, Trichrome staining of infarct and border zone of the indicated treatments demonstrates the integration of the patch with the host tissue and massive patch cellularization by the native cardiac cells. Observe the abundant muscle (red) inside the patch and in the border zone of the patch with FSTL1 treated animals (three right panels, green arrowheads).
Extended Data Figure 4. Analysis of patch with FSTL1 function in the mouse model of ischaemia/reperfusion (I/R) with delayed patch grafting.
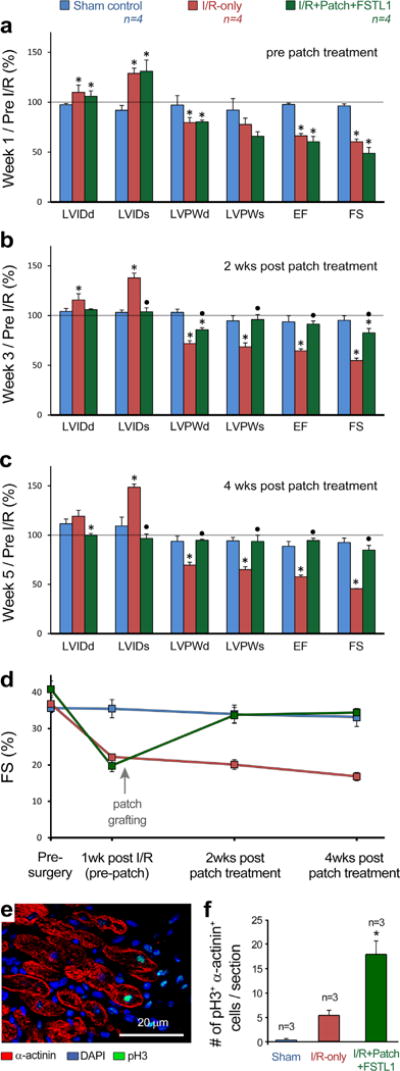
a–c, Heart function evaluation for sham, I/R, and I/R treated with patch with FSTL1, at end- diastolic and systolic, pre-grafting (a, 1 week post-injury), 2 weeks post patch implantation (b), and 4 weeks post grafting (c). Values were normalized by dividing to pre-surgery baseline values for each individual animal. d, Absolute values of fractional shortening (FS, %) at different times pre and post I/R as evaluated by echocardiography of mice from a–c. Abbreviations same as in Fig. 3. *P < 0.05 compared to sham and black circle P < 0.05 compared to I/R. e, Co-immunofluorescence staining of DNA duplication marker phospho-Histone3 Ser10 (pH3, green) and α-actinin (red) in the border zone of patch with FSTL1 treated heart 4 weeks after MI. f, Quantification of incidence of pH3+, α-actinin+ double positive cells in the 3 experimental groups. Data collected from 3 hearts in each group with 3 different cross sections counted for total pH3+, α-actinin+ cells in each heart. *P < 0.05 indicates statistically different from all other groups.
Extended Data Figure 5. Representative images and quantification of cardiomyocyte proliferation in vivo after patch with FSTL1 treatment.

a–h, Immunostaining of the cardiomyocyte marker α-actinin (red) in the infarct area (b–d) and co-immunofluorescence staining of DNA duplication marker phospho-Histone3 Ser10 (pH3, green) and α-actinin (red) in the border zone (f–h), in the 4 treatment groups analysed 4 weeks post-MI, compared to sham-operated animals (a, e). Insets in (a–d) show lower magnification images with broken lines demarcating the border between the patch and host tissues. Arrowheads in g, h, indicate α-actinin+ cardiomyocytes with pH3+ nuclei. i–k, Representative images of pH3+ cardiomyocytes in a patch with FSTL1 treated heart. Masson’s Trichrome staining of a heart after MI 4 weeks treated with patch with FSTL1 (i). The adjacent slide was stained for α-actinin in j, corresponding to the black box area with infarction and the patch in i. The spotted line in j indicates the boundary between the heart and the patch. The adjacent slide was stained for α-actinin and pH3, and all α-actinin+, pH3+ double positive cardiomyocytes found were shown in k (white arrowhead), with each image corresponding to the area in numbered white boxes in j. l–n, Quantification of cardiomyocyte proliferation measured in 3 cross sections covering the infarct, patch, and separated by 250 μm, between 1–2 mm from the apex in each heart (Fig. 3j). Data collected from 5–7 hearts in each group with the 3 cross-sections counted exhaustively for incidence of α-actinin+ cells positive for pH3 (l), midbody-localized aurora B kinase between α-actinin+ cells (m), and double-positive cells for pH3 and the nuclear cardiomyocyte maker PCM1 (n), and normalized to myocardium area quantified by trichrome staining of immediate adjacent section. *P < 0.05 statistically different from sham. **P < 0.05, statistically different from all other groups. o, Quantification of hypertrophy in all experimental groups, measured by counting cardiomyocytes in areas of intraventricular wall with perpendicular cross-sections of cardiomyocytes in all hearts analysed for cardiomyocyte proliferation. No significance were found between samples. p–r, Quantification of incidence of α-actinin+ cells positive for pH3 (p), midbody-localized aurora B kinase between α-actinin+ cells (q), and double-positive cells for pH3 and the nuclear cardiomyocyte maker PCM1 (r) measured in l, m, to total number of cardiomyocytes, calculated using hypertrophic analysis results in o. *P < 0.05, statistically different from sham, **P < 0.05,: statistically different from all other groups. s, t, Quantification of incidence of α-actinin+ cells positive for pH3 (s) and midbody-localized aurora B kinase between α-actinin+ cells (t), separated by their localization in the border zone or infarcted area. Note the majority of proliferation quantified by both methods are located in the border zone, *P < 0.05, statistically different from all other groups.
Extended Data Figure 6. Effect of implantation of patch with FSTL1 on apoptosis and inflammation.
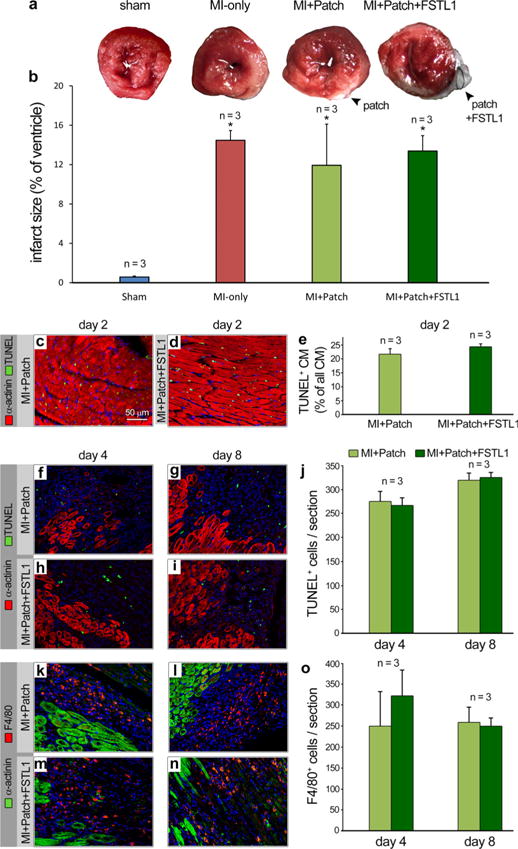
a, Representative TTC staining of day 2 post MI/patch treatment of all four groups (sham, MI, MI plus patch, MI plus patch with FSTL1). b, Quantification of area at risk comparing all 4 groups. Data collected from 4 hearts in each group, with 4 cross-sections, approximately 2 mm thick each, encompassing each heart. *P < 0.05, statistically different from the sham. c, d, Representative image of TUNEL assays (TUNEL, green, α-actinin, red) comparing hearts 2 days after MI with patch alone and patch with FSTL1. e, Quantification of TUNEL+, α-actinin+ in infarcted area, as percentage of total number of cardiomyocyte. No difference is observed between MI plus patch and MI plus patch with FSTL1 conditions. Data collected from 3 hearts in each group with 3 different cross-sections (same as in Fig. 3j) Ten 0.09 mm2 images were taken from infarcted area of each section and counted for TUNEL+, α-actinin+ and total α-actinin+ cells. f–j, TUNEL staining for cell death and α-actinin staining for cardiomyocytes were performed on hearts treated with patch-only and patch with FSTL1 at day 4 and day 8 after MI (f–i). Minimal TUNEL+, α-actinin+ cells are detected while there are signification amount of TUNEL+, α-actinin− cells. Quantification of all TUNEL+ nuclei showed no significant differences between patch and patch with FSTL1 treated hearts at both time points (j). k–o, Immunostaining of F4/80 for macrophages and α-actinin for cardiomyocyte were performed on the same hearts as in panels a–d (k–n). Quantification of F4/80+ cells showed no significant differences between patch and patch with FSTL1 treated hearts at both time points (o).
Extended Data Figure 7. FSTL1 does not induce proliferation in adult and neonatal cardiomyocytes, or cardiac progenitor cells.

a–f, Adult cardiomyocytes derived from mouse primary isolation. a, Visualization of GFP+ cardiomyocytes isolated from Myh6mERcremER:Rosa26Z/EG mice treated with 4-OH-tamoxifen (OH-Tam) in 3D-collagen patches. b–d) Gene expression changes in adult cardiomyocyte treated with FSTL1, including proliferation (b), cardiac-specific (c), and hypertrophy (d) markers. Note no changes in expression of cardiac specific genes, no increase in cell cycle markers (consistent with undetectable Ki67 immunostaining), and decreased hypertrophy markers (n = 3). Cardiomyocytes were embedded within 3D patch were treated with FSTL1 (10 ng ml−1) for duration of 7 days with media change every 2 days. e, f, FUCCI assay in 3D-cultured adult cardiomyocytes, conducted 1 week after the 3D culture. e, Treatment with FSTL1 was performed for 7 days with media change every 2 days. f, Adult cardiomyocytes 3D-cultured control in absence of FSTL1. Note no detectable sign of cardiomyocytes in S/G2/M phases (GFP+) in either condition. Purple arrows point to purple-colored nuclei resulting from co-localization of Hoechst (blue) and G1 phase FUCCI (red) labelling. g–j, Primary neonatal rat ventricular cardiomyocytes (NRVC). g, h, Freshly isolated NRVCs stimulated with FSTL1 for 48 h with 10 μg ml−1 EdU, and stained for α-actinin (red) and EdU (green). Percentages of EdU+/α-actinin+ cardiomyocytes of all α-actinin+ cardiomyocytes are quantified (h). i, j, NRVCs stimulated with FSTL1 for 48 h, and stained for α-actinin (red) and pH3 (green). Percentages of pH3+/α-actinin+ cardiomyocytes of all α-actinin+ cardiomyocytes are quantified (j). No increase of proliferation is found upon FSTL1 treatment. (n = 4) *P < 0.05, statistically different from control,. k–m, Sca1+ progenitor cells18 were starvation-synchronized for 48 h and stimulated with FSTL1 or control growth medium for 72 h in presence of EdU. Clone 3 was obtained by clonal growth from the Lin-Sca1+SP fraction. Sca1 pool was obtained from lin-Sca1+ without clonal growth. k, EdU and DAPI staining of Sca1+ cells after 72 h treatment. l, Percentage of EdU+ Sca1+ cells after 72 h treatment. FSTL1 concentration: 0, 1, 10, 100 ng ml−1. Abbreviation s: SS, serum starvation; CGM, control growth medium. m, Number of Sca1+ cells after 72 h FSTL1 treatment (n = 5). No significant change is found upon FSTL1 treatment.
Extended Data Table 1.
Raw echocardiography values (average ± s.e.m.) obtained at days 0 (baseline), 14, and 28 post treatment in a mouse model of permanent LAD ligation.
| LVIDd (mm) | LVIDs (mm) | LVPWd (mm) | LVPWs (mm) | EF (%) | FS (%) | |
|---|---|---|---|---|---|---|
|
Sham Baseline, n=10 |
3.89 ± 0.07 | 2.44 ± 0.07 | 1.10 ± 0.06 | 1.44 ± 0.04 | 75.40 ± 1.33 | 37.76 ± 1.48 |
|
MI-only Baseline, n=10 |
3.94 ± 0.06 | 2.28 ± 0.09 | 1.29 ± 0.09 | 1.68 ± 0.11 | 78.53 ± 1.68 | 43.50 ± 1.82 |
|
MI+Patch Baseline, n=10 |
4.08 ± 0.10 | 2.50 ± 0.16 | 1.07 ± 0.03 | 1.49 ± 0.12 | 77.75 ± 2.67 | 41.54 ± 1.98 |
|
MI+Patch+CM Baseline, n=10 |
3.84 ± 0.09 | 2.28 ± 0.07 | 1.19 ± 0.04 | 1.60 ± 0.05 | 77.63 ± 0.71 | 40.72 ± 0.66 |
|
MI+Patch+FSTL1 Baseline, n=10 |
4.04 ± 0.14 | 2.42 ± 0.11 | 1.19 ± 0.05 | 1.47 ± 0.08 | 73.12 ± 2.52 | 39.12 ± 2.04 |
|
Sham Week 2, n=10 |
3.96 ± 0.17 | 2.53 ± 0.11 | 1.14 ± 0.04 | 1.43 ± 0.07 | 73.33 ± 2.21 | 38.01 ± 1.58 |
|
MI-only Week 2 post injury, n=8 |
5.31 ± 0.23 | 4.08 ± 0.22* | 0.85 ± 0.05 | 0.91 ± 0.09 | 35.58 ± 3.57 | 16.48 ± 1.67 |
|
MI+Patch Week 2 post injury, n=8 |
4.63 ± 0.08*● | 3.56 ± 0.16*● | 1.11 ± 0.05● | 1.36 ± 0.09● | 48.17 ± 3.52*● | 21.69 ± 2.17*● |
|
MI+Patch+CM Week 2 post injury, n=8 |
4.45 ± 0.16*● | 3.10 ± 0.15*● | 1.12 ± 0.04*● | 1.40 ± 0.06*● | 64.18 ± 1.90*●■ | 30.35 ± 1.30*●■ |
|
MI+Patch+FSTL1 Week 2 post injury, n=9 |
4.77 ± 0.23*● | 3.72 ± 0.33*● | 1.02 ± 0.07*● | 1.14 ± 0.08*● | 50.31 ± 5.64*●▲ | 23.17 ± 3.05*●▲ |
|
Sham Week 4, n=10 |
3.98 ± 0.09 | 2.55 ± 0.12 | 1.02 ± 0.07 | 1.41 ± 0.07 | 71.32 ± 2.63 | 35.17 ± 1.60 |
|
MI-only Week 4 post injury, n=8 |
5.27 ± 0.20* | 4.55 ± 0.24* | 0.70 ± 0.04* | 0.78 ± 0.06* | 35.32 ± 2.64* | 12.71 ± 1.28* |
|
MI+Patch Week 4 post injury, n=8 |
5.04 ± 0.12*● | 3.77 ± 0.11*● | 1.02 ± 0.08● | 1.28 ± 0.08● | 51.43± 1.62*● | 22.71 ± 0.92*● |
|
MI+Patch+CM Week 4 post injury, n=8 |
4.35 ± 0.18●■ | 3.20 ± 0.24*●■ | 1.23 ± 0.05● | 1.53 ± 0.06● | 62.15 ± 4.36●■ | 27.52 ± 2.50*●■ |
|
MI+Patch+FSTL1 Week 4 post injury, n=9 |
4.24 ± 0.12●■ | 2.86 ± 0.13*■ | 1.15 ± 0.06● | 1.50 ± 0.08● | 67.73 ± 1.79●■ | 32.87 ± 1.26*●■ |
The patch was implanted simultaneously with injury.
P<0.05 statistically significant difference in comparison with Sham. Black circle (P<0.05), statistically significant difference in comparison with MI-only. black square (P<0.05) statistically significant difference in comparison with MI plus patch; black triangle statistically significant difference (P<0.05) in comparison with MI plus patch plus CM.
Extended Data Table 2.
Raw echocardiography values (average ± s.e.m.) in a long term (months 2 and 3) post treatment, in a mouse model of permanent LAD ligation.
| LVIDd (mm) | LVIDs (mm) | LVPWd (mm) | LVPWs (mm) | EF (%) | FS (%) | |
|---|---|---|---|---|---|---|
|
Sham Month 2, n=4 |
4.26 ± 0.05 | 2.68 ± 0.03 | 1.20 ± 0.03 | 1.41 ± 0.07 | 72.00 ± 0.41 | 35.75 ± 0.25 |
|
MI Month 2, n=4 |
5.53 ± 0.17* | 4.76 ± 0.19* | 0.73 ± 0.04* | 0.91 ± 0.09* | 33.98 ± 3.24* | 13.88 ± 1.56* |
|
MI+Patch Month 2, n=4 |
5.05 ± 0.09*● | 3.92 ± 0.04*● | 0.92 ± 0.09* | 1.27 ± 0.08* | 51.13 ± 2.70*● | 21.50 ± 1.44*● |
|
MI+Patch+FSTL1 Month 2, n=4 |
3.96 ± 0.16●■ | 2.58 ± 0.09●■ | 1.27 ± 0.04●■ | 1.50 ± 0.09●■ | 68.25 ± 0.75●■ | 33.19 ± 0.70●■ |
|
Sham Month 3, n=4 |
4.41 ± 0.13 | 3.01 ± 0.13 | 1.12 ± 0.02 | 1.34 ± 0.01 | 67.00 ± 1.74 | 32.38 ± 1.28 |
|
MI Month 3, n=4 |
4.93 ± 0.27* | 4.14 ± 0.16* | 0.90 ± 0.03* | 0.99 ± 0.09* | 38.00 ± 2.65* | 15.33 ± 1.20* |
|
MI+Patch Month 3, n=4 |
4.94 ± 0.23 | 3.68 ± 0.22* | 0.90 ± 0.10 | 1.18 ± 0.02* | 53.50 ± 2.63*● | 23.58 ± 1.57*● |
|
MI+Patch+FSTL1 Month 3, n=4 |
4.65 ± 0.04● | 3.19 ± 0.06●■ | 0.93 ± 0.09 | 1.17 ± 0.07●■ | 66.19 ± 1.12●■ | 31.38 ± 1.46●■ |
The patch was implanted simultaneously with injury.
P<0.05, statistically significant difference in comparison with sham. Black circle, statistically significant difference (P<0.05) in comparison with MI-only, black square, statistically significant difference (P<0.05) in comparison with MI plus patch; black triangle, statistically significant difference (P<0.05) in comparison with MI plus patch plus CM.
Extended Data Table 3.
Raw echocardiography values (average ± s.e.m) of delayed grafting in a mouse model of ischaemia/reperfusion.
| LVIDd (mm) | LVIDs (mm) | LVPWd (mm) | LVPWs (mm) | EF (%) | FS (%) | |
|---|---|---|---|---|---|---|
|
Sham Baseline, n=4 |
3.62 ± 0.15 | 2.39 ± 0.14 | 1.13 ± 0.09 | 1.44 ± 0.10 | 74.98 ± 2.57 | 35.67 ± 1.03 |
|
IR Baseline, n=4 |
3.90 ± 0.10 | 2.59 ± 0.03 | 1.26 ± 0.05 | 1.45 ± 0.11 | 73.06 ± 2.98 | 36.81 ± 2.36 |
|
IR+Patch+FSTL1 Baseline, n=4 |
4.09 ± 0.09 | 2.67 ± 0.06 | 1.27 ± 0.03 | 1.50 ± 0.05 | 77.50 ± 2.57 | 40.83 ± 2.24 |
|
Sham Week 1 post injury Pre-graft, n=4 |
3.62 ± 0.07 | 2.27 ± 0.05 | 1.16 ± 0.05 | 1.43 ± 0.03 | 71.67 ± 3.33 | 35.50 ± 2.50 |
|
IR Week 1 post injury Pre-graft, n=4 |
4.27 ± 0.16* | 3.33 ± 0.16* | 1.00 ± 0.03* | 1.12 ± 0.03 | 48.33 ± 0.51* | 22.13 ± 0.84* |
|
IR+Patch+FSTL1 Week 1 post injury Pre-graft, n=4 |
4.34 ± 0.32* | 3.49 ± 0.32* | 1.02 ± 0.03* | 0.99 ± 0.05 | 46.69 ± 3.04* | 19.74 ± 1.57* |
|
Sham Week 3 post injury Week 2 post graft, n=4 |
3.78 ± 0.24 | 2.46 ± 0.15 | 1.15 ± 0.06 | 1.35 ± 0.06 | 69.95 ± 3.59 | 34.04 ± 2.44 |
|
IR Week 3 post injury Week 2 post graft, n=3 |
4.49 ± 0.16* | 3.56 ± 0.14* | 0.91 ± 0.07* | 0.99 ± 0.04* | 47.22 ± 2.54* | 20.12 ± 1.36* |
|
IR+Patch+FSTL1 Week 3 post injury Week 2 post graft, n=4 |
4.34 ± 0.12 | 2.77 ± 0.14● | 1.08 ± 0.00*● | 1.44 ± 0.04● | 70.78 ± 2.42● | 33.67 ± 2.19*● |
|
Sham Week 5 post injury Week 4 post graft, n=4 |
4.03 ± 0.16 | 2.75 ± 0.21 | 1.05 ± 0.09 | 1.34 ± 0.10 | 66.46 ± 3.92 | 33.08 ± 2.47 |
|
IR Week 5 post injury Week 4 post graft, n=3 |
4.63 ± 0.16 | 3.84 ± 0.10* | 0.87 ± 0.05* | 0.95 ± 0.09* | 42.08 ± 0.96* | 16.81 ± 1.13* |
|
IR+Patch+FSTL1 Week 5 post injury Week 4 post graft, n=4 |
4.09 ± 0.15* | 2.57 ± 0.12● | 1.20 ± 0.04● | 1.41 ± 0.10● | 73.16 ± 1.13● | 34.44 ± 0.87● |
Data obtained at baseline (pre-injury, pre-grafting), weeks 1 (post injury, pre grafting) and weeks 3, 5 (post-injury, post-grafting). The patch was implanted 1week after injury.
P<0.05, statistically significant difference in comparison with sham; black circle indicates statistically significant difference (P<0.05) in comparison with MI-only; black square indicates statistically significant difference (P<0.05) in comparison with MI plus patch; black triangle statistically significant difference (P<0.05) in comparison with MI plus patch plus CM.
Supplementary Material
Acknowledgments
We thank L. M. Brill for spectrometry, F. Cerignolli for providing valuable cells, S. Metzler and P. Kim for help with imaging, and P. Shah for assistance in mouse and pig experiments. This work was supported by NIH grants to P.R.-L. (HL065484 and R01 HL086879); M.Me. (HL113601, HL108176, P01 HL098053); P30 AR061303 and P30 CA030199 for shared services and by the California Institute for Regenerative Medicine (CIRM, RC1-00132) to M.Me. K.We. and C.H. were SBMRI CIRM postdoctoral fellows (TG2-0116). V.S. was an Oak Foundation postdoctoral fellow. Support was also provided by NIH/NHLBI 5UM1 HL113456 to P.C.Y; HL116591 to K.Wa., M.J.B. was supported by the NIH (K08 AI079268) and the Stanford BioX Interdisciplinary Initiatives Program and NSF NSEC(PHY-0830228). B.Z. was supported by the National Basic Research Program of China (2013CB945302 and 2012CB945102) and the National Natural Science Foundation of China (91339104, 31271552, and 31222038). A Seed Grant to P.R.-L. from the Stanford Cardiovascular Institute supported the swine study.
Footnotes
Author Contributions K.We. and W.C. performed experiments on EMCs and mESCs. K.We. generated mCMsESC, and performed cardiomyogenic and proliferation assays on mCMsESC, proliferation assays on NRVC. K.We. and W.Z. performed mass spectrometry experiments. M.D.-C. performed immunostaining of Fstl1. K.We. and A.S. performed calcium transient experiment. V.S. generated cardiac patch. V.S., A.W., and M.J.B. performed biomechanical analysis of cardiac patch biomaterial. M.Z. and V.S. performed mouse MI experiments and echocardiography. Y.M. and P.C.Y. performed MRI analysis. K.We. and W.C. performed immunostaining of α-actinin, pH3 and aurora B. K.We. performed GFP, TUNEL and f4/80 staining. K.We. and P.B. performed analysis of high throughput imaging results. M.Mh. analysed the release of Fstl1 from the patch in vitro. V.S. and G.F. performed assays of adult mouse cardiomyocytes. K.Wa., K.N. and S.M. performed experiments with Fstl1-TG mice. B.Z. performed experiments with adult mouse epicardium-conditioned media. X.T., Q.L. and B.Z. performed Wt1CreERT2 lineage tracing studies and experiments with adult mouse epicardium conditioned medium. K.Wa. and S.M. provided data on systemic delivery of FSTL1. M.N. and M.D.S. provided data on myocyte progenitors. M.J.B. supervised AFM studies. D.B. supervised and coordinated in vivo mouse physiology experiments. V.S., Y.M., and P.C.Y. conducted the preclinical swine study. C.H. performed FSTL1 overexpression and western blot experiments. K.We., V.S., M.Me. and P.R.-L. designed experiments and prepared the manuscript. M.J.B.v.d.H. provided materials.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
References
- 1.van Wijk B, Gunst QD, Moorman AF, van den Hoff MJ. Cardiac regeneration from activated epicardium. PLoS ONE. 2012;7:e44692. doi: 10.1371/journal.pone.0044692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai CL, et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454:104–108. doi: 10.1038/nature06969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavine KJ, Ornitz DM. Rebuilding the coronary vasculature: hedgehog as a new candidate for pharmacologic revascularization. Trends Cardiovasc Med. 2007;17:77–83. doi: 10.1016/j.tcm.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brade T, et al. Retinoic acid stimulates myocardial expansion by induction of hepatic erythropoietin which activates epicardial Igf2. Development. 2011;138:139–148. doi: 10.1242/dev.054239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mellgren AM, et al. Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ Res. 2008;103:1393–1401. doi: 10.1161/CIRCRESAHA.108.176768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smart N, et al. Myocardial regeneration: expanding the repertoire of thymosin beta4 in the ischemic heart. Ann NY Acad Sci. 2012;1269:92–101. doi: 10.1111/j.1749-6632.2012.06708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kikuchi K, et al. tcf21+ epicardial cells adoptnon-myocardial fates during zebrafish heart development and regeneration. Development. 2011;138:2895–2902. doi: 10.1242/dev.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou B, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011;121:1894–1904. doi: 10.1172/JCI45529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serpooshan V, et al. The effect of bioengineered acellular collagen patch on cardiac remodeling and ventricular function post myocardial infarction. Biomaterials. 2013;34:9048–9055. doi: 10.1016/j.biomaterials.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka M, et al. DIP2 disco-interacting protein 2 homolog A (Drosophila) is a candidate receptor for follistatin-related protein/follistatin-like 1–analysis of their binding with TGF-β superfamily proteins. FEBS J. 2010;277:4278–4289. doi: 10.1111/j.1742-4658.2010.07816.x. [DOI] [PubMed] [Google Scholar]
- 11.Widera C, et al. Identification of Follistatin-Like 1 by expression cloning as an activator of the growth differentiation factor 15 gene and a prognostic biomarker in acute coronary syndrome. Clin Chem. 2012;58:1233–1241. doi: 10.1373/clinchem.2012.182816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams D, Larman B, Oxburgh L. Developmental expression of mouse Follistatin-like 1 (Fstl1): Dynamic regulation during organogenesis of the kidney and lung. Gene Expr Patterns. 2007;7:491–500. doi: 10.1016/j.modgep.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimano M, et al. Cardiac myocyte follistatin-like 1 functions to attenuate hypertrophy following pressure overload. Proc Natl Acad Sci USA. 2011;108:E899–E906. doi: 10.1073/pnas.1108559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshima Y, et al. Follistatin-Like 1 Is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation. 2008;117:3099–3108. doi: 10.1161/CIRCULATIONAHA.108.767673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogura Y, et al. Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models. Circulation. 2012;126:1728–1738. doi: 10.1161/CIRCULATIONAHA.112.115089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergmann O, et al. Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover. Exp Cell Res. 2011;317:188–194. doi: 10.1016/j.yexcr.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 17.Sohal DS, et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 18.Oh H, et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lepilina A, et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127:607–619. doi: 10.1016/j.cell.2006.08.052. [DOI] [PubMed] [Google Scholar]
- 20.Mercola M, Ruiz-Lozano P, Schneider MD. Cardiac muscle regeneration: lessons from development. Genes Dev. 2011;25:299–309. doi: 10.1101/gad.2018411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 22.Chen HS, Kim C, Mercola M. Electrophysiological challenges of cell-based myocardial repair. Circulation. 2010;120:2496–2508. doi: 10.1161/CIRCULATIONAHA.107.751412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senyo SE, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, et al. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS ONE. 2010;5:e12559. doi: 10.1371/journal.pone.0012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jopling C, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–609. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown RA, Wiseman M, Chuo CB, Cheema U, Nazhat SN. Ultrarapid engineering of biomimetic materials and tissues: fabrication of nano- and microstructures by plastic compression. Adv Funct Mater. 2005;15:1762–1770. [Google Scholar]
- 27.Eid H, et al. Role of epicardial mesothelial cells in the modification of phenotype and function of adult rat ventricular myocytes in primary coculture. Circ Res. 1992;71:40–50. doi: 10.1161/01.res.71.1.40. [DOI] [PubMed] [Google Scholar]
- 28.Kita-Matsuo H, et al. Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PLoS ONE. 2009;4:e5046. doi: 10.1371/journal.pone.0005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fajardo G, et al. Deletion of the β2-adrenergic receptor prevents the development of cardiomyopathy in mice. J Mol Cell Cardiol. 2013;63:155–164. doi: 10.1016/j.yjmcc.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakaue-Sawano A, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 31.Bushway PJ, Mercola M. High-throughput screening for modulators of stem cell differentiation. Methods Enzymol. 2006;414:300–316. doi: 10.1016/S0076-6879(06)14017-3. [DOI] [PubMed] [Google Scholar]
- 32.Cerignoli F, et al. High throughput drug risk assessment in human cardiomyocytes by kinetic image cytometry. J Pharm Toxicol Methods. 2012;66:246–256. doi: 10.1016/j.vascn.2012.08.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serpooshan V, et al. Reduced hydraulic permeability of three-dimensional collagen scaffolds attenuates gel contraction and promotes the growth and differentiation of mesenchymal stem cells. Acta Biomater. 2010;6:3978–3987. doi: 10.1016/j.actbio.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 34.Serpooshan V, Muja N, Marelli B, Nazhat SN. Fibroblast contractility and growth in plastic compressed collagen gel scaffolds with microstructures correlated with hydraulic permeability. J Biomed Mater Res A. 2011;96:609–620. doi: 10.1002/jbm.a.33008. [DOI] [PubMed] [Google Scholar]
- 35.Abou Neel EA, Cheema U, Knowles JC, Brown RA, Nazhat SN. Use of multiple unconfined compression for control of collagen gel scaffold density and mechanical properties. Soft Matter. 2006;2:986–992. doi: 10.1039/b609784g. [DOI] [PubMed] [Google Scholar]
- 36.Engler AJ, et al. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J Cell Sci. 2008;121:3794–3802. doi: 10.1242/jcs.029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venugopal JR, et al. Biomaterial strategies for alleviation of myocardial infarction. J Royal Soc Interface. 2012;9:1–19. doi: 10.1098/rsif.2011.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clement S, et al. Expression and function of alpha-smooth muscle actin during embryonic-stem-cell-derived cardiomyocyte differentiation. J Cell Sci. 2007;120:229–238. doi: 10.1242/jcs.03340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.