

Abstract
Precise secondary and tertiary structure formation is critically important for the cellular functionality of ribonucleic acids (RNAs). RNA folding studies were mainly conducted in vitro, without the possibility of validating these experiments inside cells. Here, we directly resolve the folding stability of a hairpin‐structured RNA inside live mammalian cells. We find that the stability inside the cell is comparable to that in dilute physiological buffer. On the contrary, the addition of in vitro artificial crowding agents, with the exception of high‐molecular‐weight PEG, leads to a destabilization of the hairpin structure through surface interactions and reduction in water activity. We further show that RNA stability is highly variable within cell populations as well as within subcellular regions of the cytosol and nucleus. We conclude that inside cells the RNA is subject to (localized) stabilizing and destabilizing effects that lead to an on average only marginal modulation compared to diluted buffer.
Keywords: biophysics, folding stability, in-cell spectroscopy, macromolecular crowding, RNA
The process of RNA folding has been studied in great detail in the past years.1 However, most experiments were performed in dilute aqueous solution, commonly in the presence of particular ions to shield the negative charges of the phosphodiester backbone and promote folding.2 However, such in vitro studies are not sufficient to understand how RNAs fold and function in the native environment of the cell, which is densely crowded with up to 40 % of the volume taken up by differently sized biopolymers, lipids, osmolytes, and salts.3 To understand how cellular environments modulate RNA folding, various attempts have been made to mimic the cellular milieu in vitro by the addition of artificial crowding agents such as polyethylene glycol and Ficoll.4 Interestingly, the effects of such crowding agents on nucleic acid folding are diverse and depend on the complexity of the folding process. The crowding effect on tertiary and quaternary nucleic acid structures can be generalized to be stabilizing,5 whereas its impact on secondary hairpin structure is mostly destabilizing.5e, 6 To rationalize these findings three major contributions were discussed by which crowding modulates RNA stability. First, crowding reduces the available volume for RNA unfolding.4 In this way, the so‐called excluded volume effect stabilizes the more compact folded native state relative to the unfolded more extended state. The magnitude of this effect depends on the relative size of the RNA and the crowder.4b, 6a, 7 Thus, the most stabilizing effects of macromolecular crowding agents were found for tertiary and quaternary RNA structures. Second, increased crowding leads to a reduction in water activity and polarity.8 As RNA duplex and hairpin structures are assumed to be more hydrated than their unfolded states, most destabilizing effects were observed for RNA secondary structures.6 Third, specific surface interactions between the nucleobases and the crowder were observed.5e, 9 Such interactions stabilize the unfolded state of the RNA leading to a destabilization of the secondary structure.
Although such studies yielded mechanistic insights into how artificial crowding agents modulate RNA folding, it remains unclear whether they adequately mimic the cellular environment and report on in‐cell RNA structure and functionality. It was recently reported that different crowding agents lead to different results for RNA tertiary structure formation10 which could lead to a misinterpretation of the in‐cell functionality. Further insight comes from a growing line of experimental evidence in the field of protein folding where crowding agents do not reflect the folding behavior observed in the living cell.11
Here, we study RNA hairpin folding directly in living cells and interpret the results by comparative studies with artificial crowding agents. As a model system we utilize a well‐studied temperature‐sensitive RNA structure. The Salmonella fourU RNA thermometer (4U) is located in the 5′‐untranslated region (5′‐UTR) of the agsA (aggregation‐suppression protein) gene and functions as a temperature‐sensitive control element of gene expression of a small heat shock protein at the mRNA level.12 Free‐living microorganisms commonly use such built‐in biosensors to surveil the environmental temperature and to adapt to the environmental change.13 Here, we explore how the temperature sensitivity of the 4U RNA thermometer is modulated within cellular environments. We determine changes of the folding stability with subcellular resolution and compare the results to those obtained with commonly used synthetic crowding agents. Therefore, we used Fast Relaxation Imaging (FReI), a recently developed technique to study biomolecular kinetics and thermodynamics in single living cells.14 FReI combines fast laser‐induced temperature jumps with Förster resonance energy transfer (FRET) microscopy. To study the 4U RNA thermometer we implemented a tailored temperature jump protocol that consisted of 2.4 °C consecutive temperature jumps. Thereby, we were able to study the thermal melting curve of the 4U RNA in the cell within 300 s avoiding cytotoxic effects (Figure 1 A).14b
Figure 1.
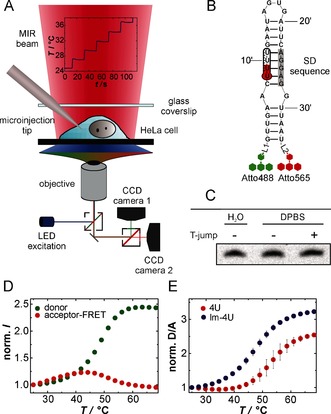
4U RNA thermal unfolding studied by Fast Relaxation Imaging (FReI). A) Representation of the FReI setup. B) 4U structure labeled with FRET‐capable dyes Atto488 and Atto565. SD=Shine‐Dalgarno. For detailed structure of linker regions L1 and L2 see Figure S1. C) Gel electrophoresis and EtBr staining reveal the intactness of the hairpin after temperature stepping in DPBS buffer. D) Normalized donor‐ and acceptor‐FRET fluorescence intensities for 4U RNA in DPBS buffer studied by FReI. E) Normalized D/A FRET signal as a function of temperature for 4U RNA and its low‐melting variant G12A‐C23U (lm‐4U) in vitro. n=4. Error bars represent mean ±s.d.
To study the 4U RNA unfolding equilibrium by FRET, the 4U RNA was functionalized by end‐group labeling using Atto488 and Atto565 via orthogonal N‐hydroxysuccinimide (NHS) and click chemistry (Figure 1 B, see the Supporting Information for details). Thermal unfolding of the RNA led to a decrease in FRET between the two dyes, with an increase in donor (D, Atto488) and a decrease in the acceptor fluorescence intensity (A, Atto565) resulting in an increase of the D/A FRET signal (Figure 1 D,E). The intrinsic response of the D/A FRET signal to temperature was marginal (Figure S2). Each temperature step was maintained for 20 s allowing for a complete equilibration of the sample (Figure S3).
The thermal unfolding process of 4U was fully reversible and left the RNA intact (Figure 1 C, Figure S4A). Owing to the cooperativity of 4U RNA unfolding,12b the sigmoidal curves were fitted to a two‐state folding model (see the Supporting Information for details). The melting temperature was in agreement with that from previous in vitro measurements on 4U (Table S1).12b This shows that the dyes do not modify the thermal unfolding. In the physiological buffer, Dulbecco's phosphate‐buffered saline (DPBS), we measured a melting temperature of T m=52.7±1.6 °C. However, for an accurate thermodynamic analysis the applied temperatures of FReI would be too high for cells. We therefore studied the low‐melting (G12A‐C23U) variant of 4U RNA,15 which we refer to as lm‐4U RNA. In DPBS lm‐4U shows a melting temperature of T m=47.4±0.6 °C and an unfolding free energy of ΔG°u (37 °C)=5.9±0.8 kJ mol−1 (Figure S4B, Table S1).
Next, we probed the lm‐4U RNA in differently crowded environments. First, we investigated the size dependence of the crowding effect and tested the commonly used crowding agent polyethylene glycol (PEG) with different degrees of polymerization (molecular weight (MW): 200 Da, 400 Da, 6 kDa, and 20 kDa) and its monomer, ethylene glycol (EG, 62 Da), at a concentration of 300 g L−1. The lm‐4U hairpin was destabilized in the presence of (P)EG with MW<6 kDa as indicated by the lowered melting temperatures and free energies of unfolding (Table S1, Figure 2 A).
Figure 2.
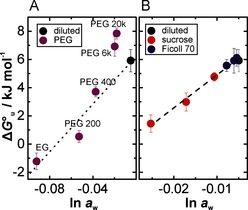
lm‐4U RNA folding stability in different crowded solutions in vitro. The free energy for unfolding at 37 °C, ΔG°u, is plotted against the activity of water, ln a w. The DPBS buffer is used as diluted reference buffer. A) Size‐dependent effect of PEG (300 g L−1) on the RNA folding stability. The dotted line indicates the dependence of the folding stability on ln a w for PEG<6 kDa. B) Concentration‐dependent effect of sucrose and Ficoll 70 (100, 200, and 300 g L−1). The dashed line represents a globally linear fit for sucrose and Ficoll 70, as an analysis of covariance reveals no significant differences for the individual fits. n=4. Error bars represent mean ±s.d.
We found a monotonic decrease of ΔG°u with decreasing water activity (ln a w) caused by EG, PEG 200, and PEG 400, indicating a relationship between water activity and folding stability. Similarly, such water sensitivity was observed for other nucleic acid secondary structures.6 However, the addition of 300 g L−1 PEG 6 kDa or 20 kDa caused a significant stabilization of the folded state despite a reduction in ln a w (see the Supporting Information for details). This stabilization can be explained by the excluded volume effect, where the shift of the folding equilibrium towards the more compact native state increases the hairpin stability.4b, 6b, 7
Next, we tested the hairpin stability of lm‐4U in the presence of Ficoll 70 and its monomeric building block sucrose by varying their concentration from 0 to 300 g L−1. Ficoll 70 and PEG 20 kDa have comparable particle sizes. However, we found that the hairpin stability decreased linearly with ln aw for sucrose and Ficoll 70 following the same trend (Figure 2 B). The comparison of PEG and Ficoll further showed that chemically different crowding agents act differently on RNA stability, as shown by the different slopes of ΔG°u versus ln a w. This discrepancy could be explained by different interactions between the cosolute and the nucleobases upon unfolding. Small PEG molecules can interact with the RNA via hydrophobic effects to the nucleobases in a PEG size‐dependent manner.9b, 16 In contrast, sucrose features more available hydroxyl groups than EG at same mass concentration enabling preferential and hydrophilic interaction with the nucleobases which are exposed to the solvent upon unfolding.6a Recent MD simulations and experimental data suggest hydrogen‐bond interactions between cosolute and nucleic acid base.6a, 9a Thus, our findings show that the stabilizing effect of volume exclusion is counteracted by a reduction of water activity and the attractive interactions between the cosolute and the nucleobases, both with destabilizing contributions.
We then microinjected the lm‐4U RNA into living HeLa cells and used FReI to measure its folding stability. Surprisingly, we found that the T m and ΔG°u values measured in the cell are similar to the results obtained from the dilute DPBS buffer solution (Figure 3 A,B). This is surprising as the different contributions to RNA folding stability observed in vitro revealed significant net effects at high concentrations of 300 g L−1, which is comparable to the cellular crowding density. In fact, this means that any destabilization of lm‐4U RNA by the reduced intracellular water activity8a,8c or the interaction of nucleobases5e, 9 must be offset by stabilizing contributions, for example, from excluded volume effects, yielding an overall marginal stability modulation.
Figure 3.

Folding stability of lm‐4U in single HeLa cells. Box plots for A) melting temperature, T m, and B) free energy of unfolding at 37 °C, ΔG°u. Error bars indicate mean ±s.d. and statistical significance was tested by an unpaired t‐test.
However, we observed a broad distribution of the melting temperature and the unfolding free energy between different cells (Table S1, Figure 3 A,B). We estimated its width by two standard deviations which describe 95 % of the distribution of unfolding free energies. The free energy spanned a range from ≈2.9 to 3.5 k B T (7.6 and 9.0 kJ mol−1) in the nucleus and cytosol, respectively. Although on average no significant thermodynamic difference between cytosol and nucleus was observed, analysis on a single‐cell level revealed that for certain cells the RNA is more stable in the nucleus than in the cytosol or vice versa (Figure S5). Such variations of the crowding effect at the subcellular level could be rationalized by multiple cell physiological processes, for example, the cell cycle or genetic noise.17 Specifically, cell cycle progression may lead to a change in intracellular crowding densities. It was shown that cell volume and cell dry mass increase at different rates during the cell cycle leading to different crowding densities.18 It was previously suggested that this effect could also modulate protein folding stability during the cell cycle.19
Further, we show that the folding stability of lm‐4U RNA is heterogeneously distributed both within the cytosol and the nucleus as indicated by the color‐coded cell map and histogram (Figure 4). Subcellular heterogeneity led to variations of ΔG°u by ≈1.7 to 2.9 k B T (4.4 and 7.6 kJ mol−1 compared to 0.4 kJ mol−1 for comparative in vitro experiments) in the cytosol and nucleus. Although such energetic modulations are small, they may have a significant impact on the cooperative folding process of RNA. As the cell is highly structured and compartmentalized on many length scales,20 one may speculate that subcellular variations can be utilized by the cell to locally adjust the energy landscape for RNA folding.
Figure 4.

A) Single‐pixel‐based titration curves for cytosolic and nucleus regions of a single cell. Insets: False‐colored images of the free energy of unfolding at 37 °C, ΔG°u, calculated for each pixel. B) ΔG°u (37 °C) and C) T m single‐pixel histograms of the cytosol and nucleus of the same cell as shown in (A).
In conclusion, we have presented the first thermodynamic insights into the folding stability of a RNA hairpin in living cells with subcellular resolution. On average, the stability of the lm‐4U hairpin in cells is similar to that in physiological buffer. This results from neutralization of the counteracting contributions from excluded volume versus nonspecific interactions and changes in water activity. However, spatial and temporal fluctuation of all three factors can cause folding heterogeneities of up to ≈3 k B T as indicated by the observed cell‐to‐cell variability and subcellular differences. This is in line with previous studies demonstrating the existence of cell‐to‐cell and subcellular heterogeneities of crowding as well as of protein folding landscapes.21 Further, we have demonstrated that individual synthetic polymers fail to mimic the cellular milieu in its entirety. Such crowding agents lead to an unbalanced view of the cellular crowding effect, overrepresenting the destabilizing changes in water activity and nonspecific interaction contributions. This demands the development and application of experimental techniques such as in‐cell chemical modification,10a,10b in‐cell NMR spectroscopy,10c single‐molecule FRET,22 and temperature modulation techniques23 to probe RNA folding directly in the cell. Such studies will make it possible to decipher individual contributions of the cellular crowding effect and understand how RNA structure and function evolved within living cells.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge funding from the Ministry of Innovation, Science and Research of the State of NRW (Rückkehrerprogramm) and the Cluster of Excellence RESOLV (EXC 1069) funded by the German Research Foundation (DFG). M.G. is supported by Fonds der Chemischen Industrie and D.G. by the International Graduate School of Neuroscience (RUB). We thank J. Drenckhan for assistance with RNA synthesis and labeling.
M. Gao, D. Gnutt, A. Orban, B. Appel, F. Righetti, R. Winter, F. Narberhaus, S. Müller, S. Ebbinghaus, Angew. Chem. Int. Ed. 2016, 55, 3224.
References
- 1.
- 1a. Schroeder R., Barta A., Semrad K., Nat. Rev. Mol. Cell Biol. 2004, 5, 908–919; [DOI] [PubMed] [Google Scholar]
- 1b. Chen S.-J., Annu. Rev. Biophys. 2008, 37, 197–214; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Woodson S. A., Annu. Rev. Biophys. 2010, 39, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Draper D. E., Grilley D., Soto A. M., Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 221–243. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Zimmerman S. B., Trach S. O., J. Mol. Biol. 1991, 222, 599–620; [DOI] [PubMed] [Google Scholar]
- 3b. Ellis R. J., Minton A. P., Nature 2003, 425, 27–28. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Nakano S. I., Miyoshi D., Sugimoto N., Chem. Rev. 2014, 114, 2733–2758; [DOI] [PubMed] [Google Scholar]
- 4b. Zhou H.-X., Rivas G., Minton A. P., Annu. Rev. Biophys. 2008, 37, 375–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Zimmerman S. B., Trach S. O., Nucleic Acids Res. 1988, 16, 6309–6326; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Nakano S., Karimata H. T., Kitagawa Y., J. Am. Chem. Soc. 2009, 131, 16881–16888; [DOI] [PubMed] [Google Scholar]
- 5c. Kilburn D., Roh J. H., Guo L., Briber R. M., Woodson S. A., J. Am. Chem. Soc. 2010, 132, 8690–8696; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Dupuis N. F., Holmstrom E. D., Nesbitt D. J., Proc. Natl. Acad. Sci. USA 2014, 111, 8464–8469; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Lambert D., Draper D. E., J. Mol. Biol. 2007, 370, 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Nakano S., Karimata H., Ohmichi T., Kawakami J., Sugimoto N., J. Am. Chem. Soc. 2004, 126, 14330–14331; [DOI] [PubMed] [Google Scholar]
- 6b. Spink C. H., Chaires J. B., Biochemistry 1999, 38, 496–508; [DOI] [PubMed] [Google Scholar]
- 6c. Nakano S., Yamaguchi D., Tateishi-Karimata H., Miyoshi D., Sugimoto N., Biophys. J. 2012, 102, 2808–2817; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Rozners E., Moulder J., Nucleic Acids Res. 2004, 32, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kilburn D., Roh J. H., Behrouzi R., Briber R. M., Woodson S. A., J. Am. Chem. Soc. 2013, 135, 10055–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Garlid K. D., Int. Rev. Cytol. 2000, 192, 281–302; [DOI] [PubMed] [Google Scholar]
- 8b. Sasmal D. K., Ghosh S., Das A. K., Bhattacharyya K., Langmuir 2013, 29, 2289–2298; [DOI] [PubMed] [Google Scholar]
- 8c. Harada R., Sugita Y., Feig M., J. Am. Chem. Soc. 2012, 134, 4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Pincus D. L., Hyeon C., Thirumalai D., J. Am. Chem. Soc. 2008, 130, 7364–7372; [DOI] [PubMed] [Google Scholar]
- 9b. Knowles D. B., Lacroix A. S., Deines N. F., Shkel I., Record M. T., Proc. Natl. Acad. Sci. USA 2011, 108, 12699–12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Tyrrell J., McGinnis J. L., Weeks K. M., Pielak G. J., Biochemistry 2013, 52, 8777–8785; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Tyrrell J., Weeks K. M., Pielak G. J., Biochemistry 2015, 54, 6447–6453; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Hänsel R., Löhr F., Foldynová-Trantírková S., Bamberg E., Trantírek L., Dötsch V., Nucleic Acids Res. 2011, 39, 5768–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Guzman I., Gelman H., Tai J., Gruebele M., J. Mol. Biol. 2014, 426, 11–20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Danielsson J., Mu X., Lang L., Wang H., Binolfi A., Theillet F., Bekei B., Logan D. T., Selenko P., Wennerström H., Oliveberg M., Proc. Natl. Acad. Sci. USA 2015, 112, 12402–12407; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Monteith W. B., Cohen R. D., Smith A. E., Guzman-Cisneros E., Pielak G. J., Proc. Natl. Acad. Sci. USA 2015, 112, 1739–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Waldminghaus T., Heidrich N., Brantl S., Narberhaus F., Mol. Microbiol. 2007, 65, 413–424; [DOI] [PubMed] [Google Scholar]
- 12b. Rinnenthal J., Klinkert B., Narberhaus F., Schwalbe H., Nucleic Acids Res. 2010, 38, 3834–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kortmann J., Narberhaus F., Nat. Rev. Microbiol. 2012, 10, 255–265. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Ebbinghaus S., Dhar A., McDonald J. D., Gruebele M., Nat. Methods 2010, 7, 319–323; [DOI] [PubMed] [Google Scholar]
- 14b. Guo M., Xu Y., Gruebele M., Proc. Natl. Acad. Sci. USA 2012, 109, 17863–17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rinnenthal J., Klinkert B., Narberhaus F., Schwalbe H., Nucleic Acids Res. 2011, 39, 8258–8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buscaglia R., Miller M. C., Dean W. L., Gray R. D., Lane A. N., Trent J. O., Chaires J. B., Graham J., Cancer B., Hancock S., Nucleic Acids Res. 2013, 41, 7934–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Pardee A. B., Dubrow R., Hamlin J. L., Kletzien R. F., Animal Cell Cycle. Annu. Rev. Biochem. 1978, 47, 715–750; [DOI] [PubMed] [Google Scholar]
- 17b. Elowitz M., Levine A., Siggia E., Swain P., Science 2002, 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Tzur A., Kafri R., Lebleu V. S., Lahav G., Kirschner M. W., Science 2009, 325, 167–171; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Mir M., Wang Z., Shen Z., Bednarz M., Bashir R., Golding I., Prasanth S. G., Popescu G., Proc. Natl. Acad. Sci. USA 2011, 108, 13124–13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wirth A. J., Platkov M., Gruebele M., J. Am. Chem. Soc. 2013, 135, 19215–19221. [DOI] [PubMed] [Google Scholar]
- 20. Weiss M., Elsner M., Kartberg F., Nilsson T., Biophys. J. 2004, 87, 3518–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Gnutt D., Gao M., Brylski O., Heyden M., Ebbinghaus S., Angew. Chem. Int. Ed. 2015, 54, 2548–2551; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2578–2581; [Google Scholar]
- 21b. Ebbinghaus S., Gruebele M., J. Phys. Chem. Lett. 2011, 2, 314–319; [Google Scholar]
- 21c. Dhar A., Girdhar K., Singh D., Gelman H., Ebbinghaus S., Gruebele M., Biophys. J. 2011, 101, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. König I., Zarrine-Afsar A., Aznauryan M., Soranno A., Wunderlich B., Dingfelder F., Stüber J. C., Plückthun A., Nettels D., Schuler B., Nat. Methods 2015, 12, 773–779. [DOI] [PubMed] [Google Scholar]
- 23. Schoen I., Krammer H., Braun D., Proc. Natl. Acad. Sci. USA 2009, 106, 21649–21654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary