

Fish et al. identified muscleblind-like 1 (MBNL1) as a robust suppressor of multiorgan breast cancer metastasis. MBNL1 binds the 3′ UTRs of DBNL and TACC1 and, by enhancing the stability of these genes’ transcripts, suppresses cell invasiveness.
Keywords: MBNL1, RNA-binding proteins, metastasis
Abstract
Post-transcriptional deregulation is a defining feature of metastatic cancer. While many microRNAs have been implicated as regulators of metastatic progression, less is known about the roles and mechanisms of RNA-binding proteins in this process. We identified muscleblind-like 1 (MBNL1), a gene implicated in myotonic dystrophy, as a robust suppressor of multiorgan breast cancer metastasis. MBNL1 binds the 3′ untranslated regions (UTRs) of DBNL (drebrin-like protein) and TACC1 (transforming acidic coiled-coil containing protein 1)—two genes that we implicate as metastasis suppressors. By enhancing the stability of these genes’ transcripts, MBNL1 suppresses cell invasiveness. Consistent with these findings, elevated MBNL1 expression in human breast tumors is associated with reduced metastatic relapse likelihood. Our findings delineate a post-transcriptional network that governs breast cancer metastasis through RNA-binding protein-mediated transcript stabilization.
For metastasis to occur, cancer cells must acquire cellular phenotypes that enable migration into distant organs, where their outgrowth gives rise to end-organ failure (Chaffer and Weinberg 2011). Enhanced invasiveness and migratory capacity may be generated through the coordinated expression of specific gene sets (Chiang and Massague 2008). Gene expression levels are established not only by transcriptional induction but also through cotranscriptional processing and post-transcriptional regulation of mRNA stability and expression. Two important classes of molecules that act as post-transcriptional regulators are microRNAs (miRNAs) and RNA-binding proteins (RBPs). miRNAs have well-documented roles in post-transcriptional regulation of mRNA expression both in normal development and during cancer progression (Tavazoie et al. 2008; Zhang and Ma 2012; Gurtan and Sharp 2013; Pencheva and Tavazoie 2013; Llobet-Navas et al. 2014). RBPs can act similarly, where a single RBP can interact with and modulate the expression of many transcripts (Keene 2007). Through their binding to specific sets of transcripts, RBPs regulate numerous processes, including pre-mRNA processing, RNA stability, and translation. Together, these processes contribute to a cell's gene expression state, which drives biological phenotypes such as those required for metastatic progression (Licatalosi and Darnell 2010; Mitchell and Parker 2014).
In this study, we identify the RBP muscleblind-like protein 1 (MBNL1) as a metastasis suppressor in human breast cancer and elucidate a mechanism through which it acts. The MBNL1 gene encodes a protein containing two pairs of conserved RNA-binding CCCH zinc finger domains (Pascual et al. 2006) and has a well-characterized role in pre-mRNA splicing regulation (Ho et al. 2004). Loss of MBNL1 function results in a shift to fetal and embryonic stem cell-like splicing expression patterns (Lin et al. 2006; Han et al. 2013) and impairs erythroid terminal differentiation (Cheng et al. 2014). The Drosophila ortholog of human MBNL1 is required for normal development of photoreceptor cells and normal muscle differentiation and attachment (Begemann et al. 1997; Artero et al. 1998). MBNL1 is also required for normal mammalian development. Mice with genetic knockout of the MBNL1 gene display muscle weakness and develop cataracts (Kanadia et al. 2003a). MBNL2 and MBNL3, the remaining members of the MBNL family, are highly homologous to MBNL1, yet individual MBNL family members have distinct and tissue-specific expression patterns during development and in somatic tissues (Kanadia et al. 2003b).
In addition to its role in splicing, MBNL1 has been implicated as a regulator of other steps of RNA processing and expression. MBNL1 has been shown to influence mRNA localization and translation in mouse myoblasts (Wang et al. 2012) and promote mRNA decay in this same system (Masuda et al. 2012). MBNL1 also affects developmentally regulated alternative polyadenylation in mice (Batra et al. 2014) and regulates the processing of pre-miR-1 (Rau et al. 2011). Taken together, these studies highlight the wide range of tissue-specific RNA processing steps mediated by MBNL1.
By integrating high-throughput sequence analysis of transcripts bound to MBNL1 with genome-wide transcriptomic measurements of mRNA stability, we uncovered a metastasis regulatory network governed by MBNL1 in breast cancer. We found that endogenous MBNL1 can suppress metastasis by human breast cancer cells. MBNL1 directly binds to the 3′ untranslated regions (UTRs) of two genes that suppress metastatic invasion. MBNL1 promotes the stability of transcripts encoding these genes. MBNL1 depletion reduces the abundance of these transcripts, increasing metastatic colonization. In support of these experimental findings, analysis of MBNL1 expression in breast tumor samples reveals a significant association between the expression of MBNL1 and metastasis-free survival.
Results
MBNL1 is a suppressor of breast cancer metastasis, and its expression is associated with reduced clinical metastatic relapse outcomes
Loss of MBNL1 activity is associated with the pathogenesis of myotonic dystrophy (Miller et al. 2000) and MBNL1 has been found to regulate developmental phenotypes (Kanadia et al. 2003a). A recent study, for example, implicated MBNL1 as a negative regulator of endocardial cell invasion during chick heart development (Vajda et al. 2009). Given the impact of MBNL1 on gene expression, its implicated regulatory role in developmental cell invasion, and its expression in breast cancer cells (Supplemental Fig. S1A), we investigated its potential role in cancer metastasis—a process that requires cell invasiveness. Depletion of MBNL1 using two independent shRNAs robustly enhanced the metastatic colonization capacity of MDA-231 human breast cancer cells in tail vein metastatic colonization assays (Fig. 1A). Moreover, MBNL1 depletion also significantly enhanced (∼15-fold) metastatic colonization by LM2 cells, a metastatic in vivo selected subline derived from the MDA-231 cell parental population (Fig. 1B; Minn et al. 2005). We next examined the effect of MBNL1 knockdown on metastasis by an independent and minimally passaged human breast cancer cell line, CN34 (Gupta et al. 2007). Again, we found that MBNL1 knockdown in this line significantly increased (∼100-fold) lung colonization (Fig. 2A). These findings reveal that endogenous MBNL1 can act as a suppressor of breast cancer metastatic colonization in these human breast cancer cells.
Figure 1.

MBNL1 suppresses breast cancer metastasis in a xenograft model, and MBNL1 levels are associated with metastasis-free survival. (A) Western blot for MBNL1 in whole-cell lysate of MDA-231 cells expressing either of two independent shRNAs targeting MBNL1 or a control shRNA. Of these cells, 1 × 105 were intravenously injected into NOD-Scid mice. Lung colonization was assessed by bioluminescence imaging and histology at the indicated time points. Representative H&E-stained lungs correspond to day 83 after injection. n = 10–11. (B) LM2 cells (2 × 104) expressing an shRNA targeting MBNL1 or a control shRNA were intravenously injected into NOD-Scid mice, and lung colonization was monitored by bioluminescence imaging. Representative H&E- and vimentin-stained lungs correspond to day 64 after injection. n = 5. (C) Kaplan-Meier survival curves of distant metastasis-free survival (DMFS) for breast cancer patients with tumors expressing high (red) or low (black) levels of the MBNL1 transcript. n = 1609. (D) Dot plot representation of MBNL1 expression in a set of 117 primary breast cancers and 36 distal metastases from previously published data sets. Cancer samples were transcriptomically profiled using a common platform and normalized to allow for intra- and intercohort comparisons. n = 153.
Figure 2.

MBNL1 is sufficient to suppress breast cancer metastasis in a xenograft model. (A) Western blot for MBNL1 in whole-cell lysate of CN34 cells expressing either of two independent shRNAs targeting MBNL1 or a control shRNA. Of these cells, 7.5 × 104 were intravenously injected into NOD-Scid mice, and lung colonization was assessed by bioluminescence imaging at day 78 after injection. Representative vimentin-stained lungs correspond to day 78 after injection. n = 6. (B) After intracardiac injection of 5 × 104 MDA-231 cells expressing a control hairpin or an shRNA targeting MBNL1 into athymic nude mice, systemic metastasis was monitored over time by bioluminescence imaging. n = 10. (C) Bioluminescence signal quantification of bone and representative H&E- stained gross bone histology 35 d after intracardiac injection of 5 × 105 MDA-231 cells expressing a control hairpin or an shRNA targeting MBNL1 into athymic nude mice. n = 10. (D) Western blot for MBNL1 in whole-cell lysate of LM2 cells stably expressing exogenous MBNL1 or an empty vector. Of these cells, 1 × 105 were injected intravenously into NOD-Scid mice. Lung colonization was monitored by bioluminescence imaging. Representative H&E- and vimentin-stained lungs correspond to day 37 after injection. n = 10.
Clinical data coupled with microarray analysis of primary tumors from three independent cohorts of breast cancer patients (n = 1609) revealed a significant association between MBNL1 transcript levels in tumors and increased metastasis-free survival (Fig. 1C). Furthermore, we observed that breast cancer metastases expressed significantly lower levels of MBNL1 relative to primary tumors (Fig. 1D). The two other human MBNL family proteins, MBNL2 and MBNL3, share sequence similarity with MBNL1. However, MBNL1 is the most abundant MBNL transcript in breast cancer tumors, suggesting that, of the MBNL proteins, MBNL1 plays the major regulatory role in this tissue (Supplemental Fig. S1A). Furthermore, in the cell lines used in this study, MBNL1 depletion by RNAi did not down-regulate MBNL2 transcript levels (Supplemental Fig. S1B,C). Together, these data reveal a clinical association between MBNL1 expression and metastasis-free survival in breast cancer that is consistent with the functional metastasis-suppressive role that we identified for this RBP.
We next assessed the impact of MBNL1 loss of function on systemic metastasis to multiple organs through intracardiac injection of breast cancer cells into the arterial systemic circulation. This revealed a significant increase in colonization of multiple distal organs, including the brain, lungs, and bone, by MBNL1-depleted cells relative to control cells (Fig. 2B,C). We next sought to determine whether MBNL1 overexpression could, conversely, suppress metastatic colonization in vivo. Overexpression of MBNL1 in highly metastatic LM2 cells significantly reduced their lung colonization capacity (Fig. 2D). These experiments reveal MBNL1 to be a robust suppressor of metastasis to multiple distal organs in these breast cancer models.
MBNL1 suppresses invasion and transendothelial migration in breast cancer cells
We next sought to define the cellular phenotypes altered in cells depleted of MBNL1. shRNA-mediated knockdown of MBNL1 did not enhance in vitro proliferation rates. In fact, MBNL1 depletion actually reduced in vitro proliferation (Fig. 3A). Furthermore, cells depleted of MBNL1 did not exhibit enhanced primary tumor growth rates in vivo (Supplemental Fig. S1D). Therefore, the enhanced in vivo metastasis phenotype exhibited by MBNL1-depleted cells is unlikely to be caused by increased proliferative or growth rates.
Figure 3.

MBNL1 suppresses in vitro invasion and transendothelial migration. (A) Cell proliferation of MDA-231 cells expressing shRNAs targeting MBNL1 or a control shRNA was assessed by seeding 2.5 × 104 cells and, after 5 d, quantifying the viable number of cells using Trypan blue exclusion. n = 3. (B) Cell migration capacity of MDA-231 MBNL1 knockdown cells compared with control cells was assessed by seeding 5 × 104 cells in a Boyden chamber with 3.0-μm pores. After 12 h, the number of cells that migrated to the basal side of each insert was quantified. n = 6–7. (C) The transendothelial migration capacity of MDA-231 cells with MBNL1 knockdown compared with control cells was assessed by seeding 5 × 104 cells on a transwell insert with 3.0-μm pores coated with a monolayer of human umbilical vein endothelial cells (HUVECs). After 20 h, the number of cancer cells that migrated through the HUVEC monolayer was quantified by counting the number of cells on the bottom side of each insert. n = 6. (D) Transwell invasion capacity of MDA-231 cells with MBNL1 knockdown compared with control cells was assessed by seeding 5 × 104 cells on a Matrigel-coated transwell insert with 8.0-μm pores. After 20 h, the number of cells invaded onto the bottom side of each insert was quantified. n = 12–13. (E) Transwell invasion assays were performed using MDA-231 cells with MBNL1 knockdown and stable overexpression of MBNL1. n = 5. (F) Transwell invasion assays were performed using CN34 cells with MBNL1 knockdown or control cells. n = 5–6. Data are shown as mean ± SEM.
To identify potential phenotypes displayed by MBNL1-depleted cells that could enhance metastatic activity, we analyzed the ability of breast cancer cells depleted of MBNL1 to invade through Matrigel, a model of cancer cell invasion through the basement membrane. MBNL1 depletion significantly enhanced invasion of both MDA-231 and CN34 breast cancer cells through Matrigel (Fig. 3D,F). Furthermore, this increase in invasiveness was abrogated upon stable overexpression of MBNL1 in cells depleted of MBNL1 (Fig. 3E; Supplemental Fig. S1E). These effects appeared to be cell type-specific, as knockdown of MBNL1 in a HER2-positive breast cancer cell line (SKBR3) did not enhance cell invasiveness. Another phenotype required for efficient metastasis to organs such as the lungs or brain, which contain endothelial barriers, is enhanced transendothelial migration capacity. We found that cancer cells depleted of MBNL1 displayed enhanced transendothelial migration capacity relative to control cells (Fig. 3C). Importantly, MBNL1 depletion did not enhance general migratory capacity in the absence of Matrigel or endothelial cells as a barrier (Fig. 3B). These results reveal that MBNL1 impedes both the invasion and transendothelial migration capacity of MDA-231 breast cancer cells and suggest that these processes, fundamental to metastatic progression, may contribute to the in vivo metastasis-suppressive role of MBNL1.
MBNL1 binds and stabilizes transcripts in breast cancer cells
We next investigated the molecular mechanism through which MBNL1 could mediate metastasis suppression. Given that MBNL1 is an RBP, we sought to identify its direct targets as potential mediators of its effects. To identify endogenous RNA targets of MBNL1 in breast cancer cells, we performed high-throughput sequencing–cross-linking immunoprecipitation (HITS-CLIP), as previously described (Fig. 4A; Supplemental Fig. S2A; Jensen and Darnell 2008). Statistically significant MBNL1-bound regions were then identified using the CIMS (cross-link-induced mutation site) algorithm, which incorporates information provided by the cross-link-induced nucleotide deletions present at the site of RNA–protein interactions, a mutation type that is a consequence of the UV radiation used to cross-link RNA and proteins in the HITS-CLIP procedure (Zhang and Darnell 2011). MBNL1-bound CLIP tag clusters were significantly enriched in the GCUU motif (Fig. 4A), which is in agreement with the YGCY motif previously determined as the optimal MBNL1-binding motif by SELEX (systematic evolution of ligands by exponential enrichment) (Goers et al. 2010). This finding was also in agreement with HITS-CLIP data for MBNL1 in mouse myoblasts, where MBNL1-binding sites were enriched for UGC and GCU containing 4 mers (Wang et al. 2012). We also found that, as expected from its known role as a regulator of alternative splicing, the majority of MBNL1-binding sites occurred in introns. However, MBNL1 was also bound to many sites in coding regions and UTRs (Fig. 4A), consistent with recent findings from HITS-CLIP studies of MBNL1 in mouse myoblasts (Masuda et al. 2012; Wang et al. 2012).
Figure 4.
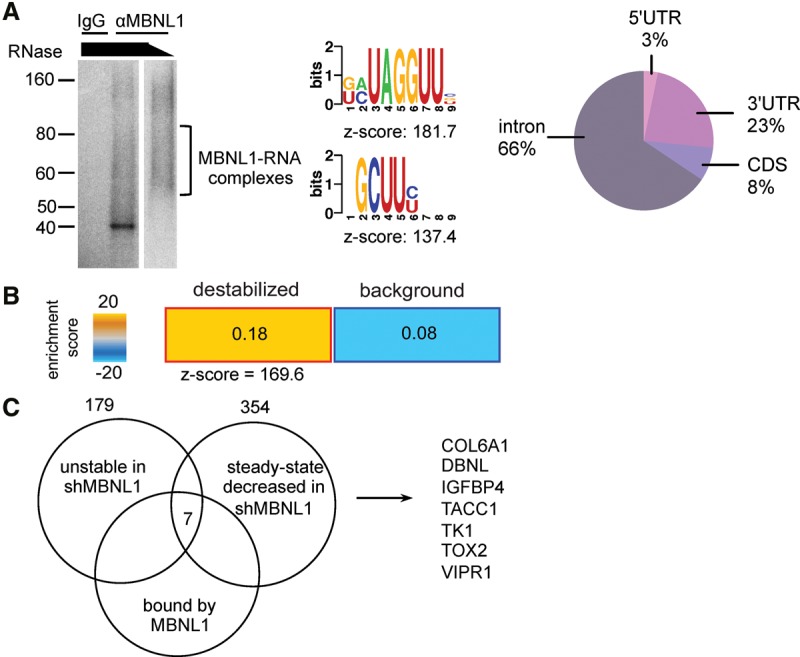
MBNL1 depletion affects the stability of transcripts directly bound by MBNL1. (A) 32P-labeled RNA–MBNL1 complexes were visualized by autoradiography after RNase treatment, immunoprecipitation with anti-MBNL1 or IgG, and separation by SDS-PAGE. The nucleotide motifs shown are significantly enriched in the MBNL1-bound HITS-CLIP CIMS clusters. Pie chart depicting the percentage of MBNL1 HITS-CLIP clusters mapping to the indicated transcript regions. (B) Heat map depicting the relative enrichment of MBNL1-bound transcripts in the set of transcripts destabilized by MBNL1 depletion compared with transcripts with no MBNL1-dependent changes in stability. Transcript stability was measured by transcriptomic profiling of MDA-231 sh1MBNL1 and shCTRL cells at 0 and 9 h after α-amanitin treatment. MBNL1-bound transcript enrichment in the stability data set was assessed using TEISER. (C) Venn diagram of transcripts stabilized and bound by MBNL1. Numbers are transcripts bound by MBNL1 in the HITS-CLIP data, transcripts with steady state levels down-regulated in shMBNL1 versus control cells (fold change down >1.3 in shMBNL1 vs. shCTRL, q < 0.05), and transcripts destabilized upon MBNL1 knockdown (fold change down >1.1 after amanitin treatment in shMBNL1 vs. control cells).
MBNL1 was originally characterized as a regulator of alternative splicing, and recent studies have implicated it in the regulation of RNA stability. Given its known role in alternative splicing regulation, we used MISO (mixture of isoforms) (Katz et al. 2010) to analyze RNA sequencing (RNA-seq) data from MDA-231 breast cancer cells with MBNL1 knockdown and control cells to identify MBNL1-dependent alternative splicing events. Through this analysis, we identified 21 MBNL1-dependent alternatively spliced exons that were in transcripts also bound by MBNL1 (Supplemental Table S1). We validated the MBNL1-dependent changes in exon inclusion for these transcripts using semiquantitative RT–PCR and arrived at four genes that exhibited large MBNL1-dependent changes in exon inclusion (Supplemental Fig. S2B). The MBNL1-dependent changes in relative transcript splice variant abundance were then tested to see whether they could affect the invasion phenotype observed in MBNL1-depleted cells. To do this, we specifically depleted the transcript variant that exhibited an increase in exon inclusion using siRNA targeting the alternative exon. However, modulation of the levels of these individual transcript variants did not significantly impact the invasion capacity of breast cancer cells (Supplemental Fig. S2C,D). This does not exclude the possibility that regulation of alternative splicing by MBNL1 contributes to invasion and metastasis, and modulation of multiple transcript variants simultaneously could have this effect. However, analysis of the transcripts identified here as having MBNL1-dependent changes in exon inclusion and being directly bound by MBNL1 revealed that modulation of each of these transcript isoform levels individually did not regulate the invasive capacity of breast cancer cells.
A key feature of RBP regulation is control of transcript stability. MBNL1 has been shown to modulate transcript stability in mouse myoblasts (Masuda et al. 2012). However, the effect of MBNL1 on transcript stability in cancer has not been reported. Therefore, to determine whether MBNL1 could mediate its effects on cancer progression by altering the stability of metastasis regulatory transcripts, we conducted transcriptome-wide analysis of RNA stability. In this experiment, MBNL1-depleted or control breast cancer cells were treated with α-amanitin, and RNA was isolated. Relative transcript levels were determined through transcriptomic profiling, revealing a set of transcripts whose stability was deregulated upon MBNL1 loss. Next, we asked whether transcripts directly bound by MBNL1 were significantly enriched among transcripts with MBNL1-dependent changes in stability. To this end, we analyzed the set of MBNL1-bound transcripts (from the HITS-CLIP data) along with transcript stability measurements obtained from the transcriptomic profiling of MBNL1 knockdown and control cells that had been treated with α-amanitin. We found that transcripts bound by MBNL1 in vivo were enriched in the group of transcripts destabilized by MBNL1 depletion compared with transcripts with no MBNL1-dependent changes in stability (Fig. 4B). These findings suggest that, in breast cancer, MBNL1 acts as an enhancer of transcript stability through direct binding of transcripts.
We next sought to identify MBNL1-bound transcripts that could mediate its effects on metastasis. As we had observed that MBNL1-bound transcripts were generally stabilized, we sought to identify transcripts bound by MBNL1, destabilized by MBNL1 depletion, and down-regulated at steady state in MBNL1-depleted cells. Through this analysis, we identified seven genes as potential regulators of breast cancer metastasis downstream from MBNL1. This gene set was comprised of COL6A1, DBNL (drebrin-like protein), IGFBP4, TACC1 (transforming acidic coiled-coil containing protein 1), TK1, TOX2, and VIPR1 (Fig. 4C). The steady-state levels of these transcripts were assessed in two MBNL1-depleted breast cancer lines. We observed that three of these transcripts (DBNL, TACC1, and TK1) exhibited reduced steady-state levels upon MBNL1 knockdown in both cell lines (Supplemental Fig. S3A,B). The protein levels of DBNL, TACC1, and TK1 were also decreased in MBNL1-depleted cells (Supplemental Fig S3C).
MBNL1-bound targets DBNL and TACC1 suppress metastatic invasion in breast cancer cells
Given that MBNL1 depletion reduced the stability and subsequent abundance of DBNL, TACC1, and TK1 transcripts, we sought to determine whether reconstituting the expression of these genes in cells depleted of MBNL1 could reverse the enhanced metastatic phenotype. We found that stable overexpression of DBNL and TACC1, but not TK1 or an mCherry control, significantly decreased the invasive capacity of MBNL1-depleted breast cancer cells (Fig. 5A; Supplemental Fig. S3D). Importantly, restoring the expression of DBNL and TACC1 in MBNL1-depleted cells significantly abrogated the enhanced metastatic lung colonization phenotype conferred by MBNL1 depletion alone, while overexpression of TK1 did not have a significant effect (Fig. 5B). These findings demonstrate that overexpression of either DBNL or TACC1 is sufficient to repress the metastatic lung colonization of MBNL1-depleted breast cancer cells in vivo as well as suppress transwell invasion in vitro. Consistent with these findings, overexpressing either DBNL or TACC1 in the CN34-Lm1a line, an independent highly metastatic breast cancer cell population, suppressed cell invasiveness as well (Fig. 5C). Interestingly, although simultaneous overexpression of DBNL and TACC1 in MBNL1-depleted breast cancer cells resulted in a significant decrease in invasion capacity, this effect was not significantly greater than the effect of overexpressing DBNL or TACC1 individually (Supplemental Fig. S3E). Notably, overexpression of either DBNL or TACC1 alone did not affect the level of the other protein (Supplemental Fig. S3F). Therefore, DBNL and TACC1 may function redundantly in reducing the invasive capacity of MBNL1-depleted cells.
Figure 5.
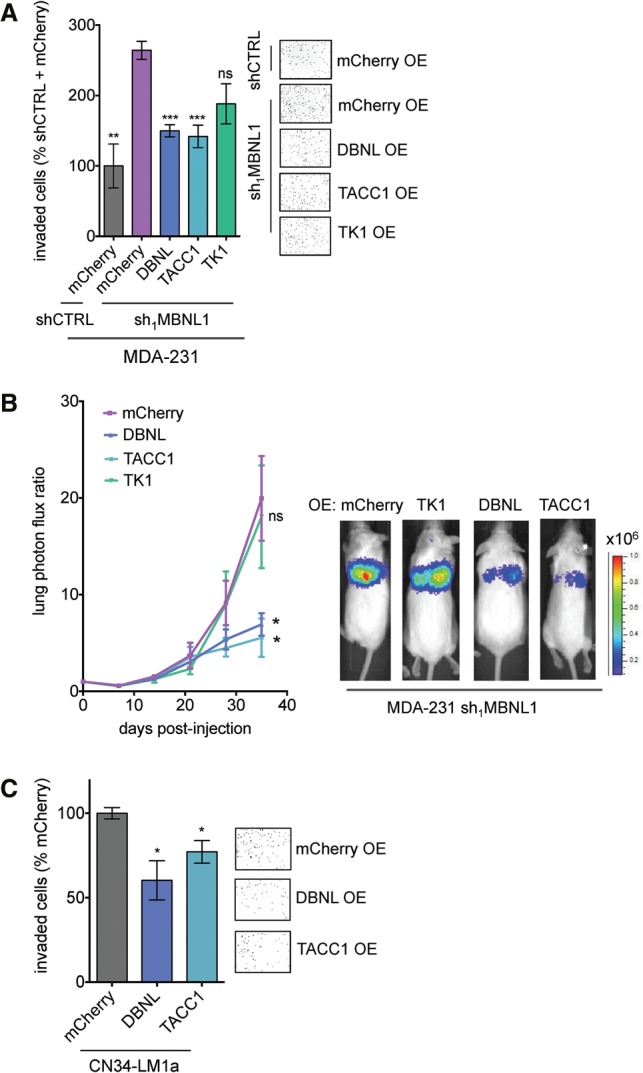
DBNL and TACC1 suppress metastatic invasion and colonization. (A) Candidate MBNL1-regulated genes were stably overexpressed in shMBNL1 MDA-231 cells. mCherry was overexpressed as a control. Transwell invasion assays were performed with these cells. n = 6. (B) MDA-231 cells (2 × 104) stably overexpressing DBNL, TACC1, TK1, or mCherry were injected into the venous circulation of mice. Lung colonization was monitored by bioluminescence imaging. n = 5. (C) Transwell invasion assays were performed with CN34-LM1a cells stably overexpressing DBNL, TACC1, or mCherry. n = 6. Data are shown as mean ± S.E.M.
The cell biological processes affected by DBNL and TACC1 could conceivably mediate the phenotypes that we observed upon MBNL1 depletion. DBNL is an F-actin-binding protein and has been reported to have roles in mammalian development, endocytosis, and the immune response (Larbolette et al. 1999; Kessels et al. 2000; Connert et al. 2006; Schymeinsky et al. 2009; Hepper et al. 2012). Although there is no reported role for DBNL in cancer progression, a recent study showed that DBNL depletion in Src transformed fibroblasts regulates podosome rosette formation and, interestingly, increases cell invasiveness (Boateng et al. 2012). TACC1 has been reported to associate with proteins involved in diverse cellular processes, including those involved in centrosome dynamics (chTOG, TRAP, and Aurora A, B, and C), transcription (Gas41, thyroid hormone receptor, and retinoid acid receptor α), and RNA processing (LSM7 and SmG) (Lauffart et al. 2002; Conte et al. 2003; Guyot et al. 2010; Gabillard et al. 2011). Intriguingly, TACC1 mRNA has been shown to be down-regulated in breast tumors compared with normal breast tissue, supporting the suppressive role that we identified for this gene in cancer progression (Conte et al. 2002).
DBNL and TACC1 are bound by MBNL1 and destabilized by MBNL1 depletion
Interestingly, the HITS-CLIP MBNL1-binding data revealed that MBNL1 binds the 3′ UTRs of DBNL and TACC1 (Fig. 6A), suggesting that MBNL1 may enhance the stability of these transcripts through direct interactions with their 3′ UTRs. To further validate this observation, the reduced stability of DBNL and TACC1 transcripts upon MBNL1 depletion was verified by quantitative RT–PCR (qRT–PCR), using α-amanitin to inhibit transcription (Fig. 6B). To validate the stabilization of these transcripts by MBNL1 using an independent method of transcription inhibition, we used dichlorobenzimidazole 1-β-D-ribofuranoside (DRB), an inhibitor of CDK9. Following DRB treatment, DBNL and TACC1 transcripts exhibited shorter half-lives in MBNL1-depleted cells relative to control cells (Fig. 6C). Importantly, mutating the major MBNL1-binding sites in the TACC1 3′ UTR, when cloned 3′ of an mCherry ORF, significantly decreased the levels of the mCherry reporter transcript compared with wild-type TACC1 3′ UTR-driven reporter transcript levels (Supplemental Fig. S4B).
Figure 6.

MBNL1 depletion destabilizes DBNL and TACC1, and DBNL and TACC1 expression levels are associated with clinical outcome in breast cancer. (A) MBNL1 HITS-CLIP read density mapped onto the 3′ UTRs of DBNL and TACC1. The two colors indicate the two biological MBNL1 HITS-CLIP replicates. (B) qRT–PCR of DBNL and TACC1 in MDA-231 and CN34 shMBNL1 and shCTRL cells at 0 and 9 h after α-amanitin treatment. 18S was used as an endogenous control. n = 3. (C) qRT–PCR of DBNL and TACC1 in MDA-231 shMBNL1 and shCTRL cells at the times indicated after treatment of cells with DRB. n = 3. (D) Kaplan-Meier curve showing relapse-free survival of breast cancer patients with tumors expressing high (red) or low (black) levels of the TACC1 transcript. n = 3455. (E) Levels of DBNL were assessed by qRT–PCR from RNA obtained from a panel of staged breast cancer tumors. β-Actin was used as an endogenous control. n = 48.
MBNL1 has a known role in modulating poly(A) site selection (Batra et al. 2014), and the location of the MBNL1-binding sites, immediately 3′ of the coding sequence stop codons in the DBNL and TACC1 transcripts, suggested that MBNL1 might regulate the stability of these transcripts through modulation of poly(A) site choice. Therefore, we tested whether the 3′ UTR lengths of the DBNL and TACC1 transcripts were altered in MBNL1 knockdown cells. We did not find a significant difference in the 3′ UTR lengths of these transcripts upon MBNL1 depletion (Supplemental Fig. S4A). These findings reveal that, in breast cancer cells, MBNL1 binds to the 3′ UTRs of DBNL and TACC1 transcripts and promotes their stability.
DBNL and TACC1 expression levels are associated with breast cancer clinical outcome
The expression levels of TACC1 in human cancer samples can be inferred from microarray data sets, since it is represented by an informative probe. Analysis of a data set comprising 3455 tumor samples representing multiple cohorts revealed a significant positive correlation between TACC1 expression and relapse-free survival in breast cancer patients (Fig. 6D), consistent with TACC1 acting as a suppressor of breast cancer metastasis. DBNL, however, was not represented by a probe on the microarray platform used to generate the large breast cancer data sets analyzed. Therefore, to determine whether an association exists between DBNL and breast cancer progression, we performed qRT–PCR on a panel of commercially available (Origene) cDNAs generated from breast tumor-derived RNA. Consistent with its uncovered metastasis suppressor role, metastatic stage IV tumors expressed significantly lower levels of DBNL relative to early stage, localized tumors (stages I and II) (Fig. 6E; Supplemental Fig. S4C).
Discussion
Using a combination of biochemical, functional, and computational approaches, we identified the RBP MBNL1 as a suppressor of breast cancer metastasis in models of human breast cancer progression studied here. We also elucidated a mechanism through which MBNL1 mediates, in part, its suppressive effect. We found that, in breast cancer cells, MBNL1 globally enhances the stability of the transcripts it binds. Among these transcripts are DBNL and TACC1, targets that encode suppressors of invasion and metastatic colonization. DBNL and TACC1 are examples of MBNL1-bound transcripts that are directly regulated by MBNL1 and act as metastasis suppressors. We speculate that DBNL may exert its invasion-suppressing effects through its modulation of podosome rosette formation. Podosomes, cell protrusions enriched in F-actin, have been shown to regulate cell invasion by localized secretion of extracellular matrix-degrading factors, and this action is usually associated with increased cell invasiveness (Linder 2007). Alternatively, DBNL may modulate cell invasiveness through a mechanism independent of podosome formation. TACC1 is a scaffolding protein that has been shown to interact with a variety of proteins, including centrosome-associated factors, nuclear hormone receptors, and RNA processing proteins (Lauffart et al. 2002; Conte et al. 2003; Guyot et al. 2010; Gabillard et al. 2011). This protein could potentially localize these interacting factors in order to mediate the observed metastatic phenotype.
RNA-interacting proteins are abundant, with >800 mRNA-interacting proteins reported in human cells (Castello et al. 2012). Given their ability to coordinately regulate sets of transcripts, proteins belonging to this group have the potential to confer attributes that affect the metastatic potential of cancer cells. Indeed, recent studies have noted just such effects on breast cancer progression for a variety of RBPs. The metastasis-modulating effects of these RBPs were shown to arise through diverse mechanisms, including direct modulation of transcript stability (Goodarzi et al. 2014; Vanharanta et al. 2014) as well as regulation of alternative splicing patterns (Xu et al. 2014). MBNL1 affects many steps of RNA maturation and expression. While MBNL1 could impact gene expression and phenotypic output of cancer cells through multiple mechanisms, our findings reveal that its effects are mediated in part through its regulation of TACC1 and DBNL. Here, we found that MBNL1 enhances the stability of the DBNL and TACC1 transcripts and interacts with their 3′ UTRs. Although it has been reported previously that MBNL1 globally destabilizes transcripts (Masuda et al. 2012), this previous study both used a different method to assess global transcript stability in response to MBNL1 depletion and found this effect in mouse myoblasts. In the current study, we present evidence that MBNL1 directly interacts with a large number of transcripts in human breast cancer cells and can stabilize a subset of these transcripts. However, as MBNL1 binds numerous transcripts and as MBNL1 binding to a transcript does not result in a binary output, MBNL1 binding would not be expected to impart the same effect on the entire set of RNAs with which it interacts. This is consistent with general RBP regulatory paradigms, where the combined influences of the subcellular compartment and the local RNA sequence/structure determine the expression output of an RBP interaction with a transcript. We found that, in breast cancer cells, MBNL1 interacts with a subset of transcripts to promote their stability, which may occur through inhibiting their interactions with destabilizing factors such as components of the mRNA decay machinery or miRNAs.
It is interesting to note that while MBNL1 has not been implicated as a regulator of cancer progression, it has been well established that MBNL1 is clinically important in the pathogenesis of myotonic dystrophy, a genetic disease that has been extensively studied. In myotonic dystrophy, a disease-specific CTG repeat expansion in the 3′ UTR of DMPK in type 1 or a CCTG repeat expansion in the first intron of CNBP in type 2 results in the accumulation of transcripts with these repeat expansions (Brook et al. 1992; Liquori et al. 2001). These aberrant RNAs have been shown to bind and sequester MBNL1, leading to the subsequent misregulation of MBNL1 RNA targets. There is considerable evidence that this loss-of-function-like mechanism is a key contributor to the pathogenesis of myotonic dystrophy (Miller et al. 2000). Although myotonic dystrophy is primarily characterized by muscle weakness along with cardiac and neurological abnormalities (Udd and Krahe 2012), a higher incidence of primary tumor development in individuals affected with myotonic dystrophy has been observed (Gadalla et al. 2011; Win et al. 2012). In light of our findings concerning a suppressive role for MBNL1 in cancer progression, future studies into metastatic relapse outcomes in these myotonic dystrophy patients are warranted.
Materials and methods
Animal experiments
All mouse studies were conducted according to a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Rockefeller University. Intravenous injections of cancer cells were performed on age-matched (4- to 6-wk-old) female NOD-Scid mice. Metastatic lung colonization was monitored by in vivo bioluminescence imaging and measuring photon flux in the region of interest. In vivo tumor growth assays were performed by injecting 5 × 105 cancer cells with 100 μL of Matrigel into bilateral mammary fat pads of NOD-Scid mice. Tumor volume was assessed by caliper measurements. Two-way ANOVA was used to assess significance of the metastatic colonization assays, and a Student's two-tailed t-test was used to assess significance of the tumor volume measurements.
Cell culture
All cell lines were maintained at 37°C and 5% CO2. The human embryonic kidney 293T, MDA-MB-231, and CN34 cell lines were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 μg/mL streptomycin, and amphotericin B.
For cell counting, cell viability was assessed by Trypan blue, and only cells that excluded the dye were counted as viable. A Student's two-tailed t-test was used to determine significance.
Gene knockdown
Lentivirus was produced by transfecting 293T cells that had been seeded in six-well plates and allowed to become 60% confluent with 2 μg of pLKO.1 shRNA-containing vector, 2 μg of vector K, and 1 μg of vector A. TransIT-293T (Mirus Bio) was used for transfection according to the manufacturer's protocol. Virus was harvested at 48 h after transfection and passed through a 0.45-μm syringe filter to remove 293T cells.
Target cancer cells were transduced by incubation with the lentivirus for 6 h in the presence of 8 μg/mL polybrene. After transduction, the medium was changed to normal growth medium. Selection was started 48 h after transduction by adding puromycin to a final concentration of 1.5 μg/mL. Cells were kept under selection for 48 h.
For siRNA-mediated gene knockdown studies, cells were seeded at 5 × 104 per well in 12-well plates. Twenty hours after seeding, cells were transfected with 30 pmol of siRNA using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions.
shRNA target sequences were as follows: sh1MBNL1, 5′-GCCAACCAGATACCCATAATA-3′; sh2MBNL1, 5′-GCCTGCTTTGATTCATTGAAA-3′; and shCTRL, 5′-CAACAAGATGAAGAGCACCAA-3′. siRNA target sequences were as follows: siFGFR1-SE, 5′-TGACACCACCTACTTCTCCGTCAATGT-3′; siMYL6-SE, 5′-AAGCGTTTGTGAGGCATATCCTGTCGG-3′; siPICALM-SE, 5′-AACAATGAATGGCATGCATTTTCCACA-3′; siSEC31A-SE, 5′-CTCCTCCTACTTCAAACGCTTACCCTA-3′; and siCTRL, 5′-CGTTAATCGCGTATAATACGCGTAT-3′.
Retroviral- and lentiviral-mediated protein overexpression
Retrovirus was produced by transfecting 293T cells seeded in six-well plates with 2 μg of pBabe containing the ORF of interest, 2 μg of Gag/Pol, and 1 μg of VSV-G. Transfection was carried out using TransIT-293T (Mirus Bio) according to the manufacturer's protocol. Virus was harvested 48 h after transfection and passed through a 0.45-μm syringe filter to remove cells. For C-terminal Flag-tagged MBNL1 overexpression, target cells were transduced by incubation with retrovirus for 6 h in the presence of 8 μg/mL polybrene. At this time, the medium was changed to normal growth medium. Selection was started 48 h after transduction by adding puromycin to a final concentration of 1.5 μg/mL or hygromycin to a final concentration of 350 μg/mL. Puromycin selection was maintained for 48 h, while hygromycin selection was carried out for 11–14 d.
Lentivirus was produced by transfecting 293T cells seeded in six-well plates with 2 μg of pLX302 or pLX304 containing the ORF of interest with a C-terminal V5 tag, 2 μg of vector K, and 1 μg of vector A. TransIT-293T (Mirus Bio) was used to transfect the DNA per the manufacturer's protocol. Virus was harvested at 48 h after transfection and passed through a 0.45-μm syringe filter to remove 293T cells. For DBNL, TACC1, TK1, and mCherry overexpression, target cancer cells were transduced by incubation with the lentivirus for 6 h in the presence of 8 μg/mL polybrene. After transduction, the medium was changed to normal growth medium. Selection was started 48 h after transduction by adding puromycin to a final concentration of 1.5 μg/ml or blasticidin to a final concentration of 10 μg/mL. Puromycin selection was maintained for 48 h, while blasticidin selection was carried out for 5–6 d.
Transcriptome sequencing and alternative splicing analysis
Whole-transcriptome sequencing libraries were constructed using the ScriptSeq version 2 kit (Epicentre) per the manufacturer's instructions. The input RNA was isolated with a spin column-based kit, including an on-column DNase treatment per the manufacturer's instructions (Norgen), and then depleted of rRNA using the Ribo-Zero kit (Epicentre). The libraries were sequenced at the Rockefeller University Genomics Resource Center on a HiSeq 2000 (Illumina). Reads were first trimmed to remove linker sequences and low-quality bases using Cutadapt (version 1.2.1). TopHat2 (version 2.0.8) was then used to map the reads to the human transcriptome (RefSeq transcriptome index hg19). Cufflinks (version 2.0.2) was then used to estimate RPKM (reads per kilobase per million mapped reads) values and compare shControl and shMBNL1 samples.
For alternative splicing analysis, mapped reads from each of the two MBNL1 targeting shRNA-expressing MDA-231 cell lines were compared with the shCTRL-expressing cell line using MISO (version 0.4.9) (Katz et al. 2010), in conjunction with the provided annotations for skipped exons, to quantitate modulations in alternative splicing events.
qRT–PCR
RNA was isolated using a total RNA isolation kit, including an on-column DNase treatment (Norgen). cDNA synthesis was carried out using the SuperScript III reverse transcriptase kit using a mixture of oligodT and random hexamers for priming (Life Technologies). qRT–PCR was carried out using Fast SYBR Green master mix (Applied Biosystems), and fluorescence was monitored using a 7900HT Fast real-time instrument (Applied Biosystems). Data were analyzed using the ΔΔCt method. The endogenous control transcripts that were used for normalization are indicated in each figure. Statistical significance was determined using a one-tailed Student's t-test. The sequences of the primers used for all qRT–PCR assays are in Supplemental Table S2.
Semiquantitative RT–PCR
RNA was isolated using a total RNA isolation kit, including an on-column DNase treatment (Norgen). cDNA synthesis was carried out using the SuperScript III reverse transcriptase kit using oligodT for priming (Life Technologies). PCR amplification was carried out using high-fidelity platinum Taq polymerase according to the manufacturer's protocol (Life Technologies). The reactions were separated on 1.5% or 2% agarose gels, depending on the expected amplicon sizes, and stained with ethidium bromide. Band intensity was quantified using ImageJ (National Institutes of Health). The sequences of the primers used for all semiquantitative RT–PCR experiments are in Supplemental Table S2.
Matrigel transwell invasion assays
Cancer cells were serum-starved in medium containing 0.2% FBS for 20 h. The starved cancer cells were seeded at 5 × 104 cells per well in Matrigel-coated invasion chambers with 8.0-μm pore size (BD Biosciences) that were pre-equilibrated in 0.2% FBS-containing medium. After the cells had been allowed to invade for 20 h at 37°C, the Matrigel-coated inserts were washed with PBS, and the cells on the top side of each insert were scraped off. The cells were fixed in 4% paraformaldehyde for 15 min at 37°C, and the inserts were cut out and mounted onto slides using VectaShield with DAPI (Vector Laboratories). The number of cells invaded through the Matrigel was quantified by imaging each insert using an inverted fluorescence microscope (Zeiss Axiovert 40 CFL). Five images were taken per insert at 10× magnification. ImageJ (National Institutes of Health) was used to quantify the number of invaded cells. Statistical significance was determined using a one-tailed Student's t-test.
Transendothelial and migration assays
Cancer cells were serum-starved in medium containing 0.2% FBS for 12 h. For transwell migration assays, 5 × 104 serum-starved cancer cells were seeded into 3.0-μm porous HTS Fluoroblock transwell migration inserts (BD Biosciences) and allowed to migrate for 12 h at 37°C, after which the inserts were processed, imaged, and quantified as described above for the Matrigel invasion assays. For transendothelial migration assays, 5 × 104 human umbilical vein endothelial cells (HUVECs) were seeded into collagen-precoated 3.0-μm porous HTS Fluoroblock transwell migration inserts. After the HUVECs reached a monolayer state (typically after 2 d), the endothelial cells were serum-starved overnight in EGM-2 medium containing 0.2% FBS. The following day, 5 × 104 serum-starved cancer cells were seeded into 3.0-μm porous inserts containing HUVEC monolayers. The cancer cells were allowed to migrate through the HUVEC monolayer for 20 h at 37°C. Following completion of the assay, the inserts were processed, imaged, and quantified as described above for the Matrigel invasion assays. Statistical significance was determined using a one-tailed Student's t-test.
Immunoblotting
Whole-cell lysate was prepared by washing trypsinized cells twice with cold PBS, resuspending the cell pellet in ice-cold RIPA buffer with 1× Complete protease inhibitor cocktail (Roche), and incubating for 20 min on ice. The lysate was sonicated and then cleared by spinning in a microcentrifuge at maximum speed for 15 min at 4°C. The total protein concentration of the resulting cleared lysate was determined using the BCA assay method (Thermo Scientific). Equal amounts of total protein, typically 20 μg per lane, were run on 4%–12% Bis-Tris NuPAGE gels in MOPS buffer (Life Technologies) under reducing conditions. Proteins were transferred to 0.2 μm of PVDF (Millipore) and blocked with 5% nonfat milk in PBS. Antibodies used in this study include custom-made polyclonal rabbit anti-MBNL1 (Yenzyme, raised to amino acids 363–378 of NP_066368), anti-Flag (Sigma, F1804), anti-DBNL (Abcam, ab86708), anti-TACC1 (Abcam, ab187358), anti-TK1 (Cell Signaling Technology, 8960), and anti-β-tubulin (Cell Signaling Technology, 2128). The blots were incubated with horseradish peroxidase-conjugated secondary antibodies and developed using the ECL method (Thermo Scientific).
HITS-CLIP
HITS-CLIP for endogenous MBNL1 was carried out as described previously (Jensen and Darnell 2008) with the following modifications. MDA-231 cells were cross-linked by irradiation with 400 mJ/cm2 at 254 nm. A custom antibody raised against MBNL1 was conjugated to protein A Dynabeads (Life Technologies) and used to immunoprecipitate endogenous MBNL1–RNA complexes. Biological duplicate libraries were prepared, and the samples were sequenced on an Illumina HiSeq 2000 instrument at the Rockefeller University Genomics Resource Center. The sequencing reads were first trimmed to remove linker sequences and low-quality bases using Cutadapt (version 1.2.1) with parameters -q 15 and -m 25. The resulting reads were then aligned to the human genome (build hg19) using Bowtie2 (version 2.1.0). The analytical package CIMS (Zhang and Darnell 2011) was then used to identify CLIP peaks in each of the biological replicates (false discovery rate < 10%). The peaks from the two replicates were then overlapped (intersectBed) to generate a high-confidence list of MBNL1-binding sites. Sequences were extracted and analyzed for motif discovery using ChIPseeqer (Giannopoulou and Elemento 2011). Briefly, a randomized background set was generated using a first-order Markov model to control for length and dinucleotide frequency in the real binding sites. The resulting “real” and “scrambled” sequences were then analyzed using FIRE (finding informative regulatory elements) (Elemento et al. 2007) to identify the best representation of the MBNL1-binding site.
Transcript stability assays
For the α-amanitin microarray data, MDA-231 MBNL1 knockdown or control cells were treated with 10 μg/mL α-amanitin (Sigma). Nine hours after α-amanitin addition, RNA was isolated from the cells using a total RNA isolation kit with on-column DNase treatment (Norgen). RNA was labeled using the TargetAmp nano-labeling kit according to the manufacturer's protocol (Epicentre) and hybridized to Illumina beadchip arrays. Lumi package (R) was used to normalize and compare Illumina HT12 beadchip raw signals for shControl and sh1MBNL1 samples at 0 and 9 h after α-amanitin treatment. The differences between the sh1MBNBL1/shControl log fold changes were used as a measure of stability. To discretize the input values, transcripts with log fold change difference of less than −0.1 were labeled as “destabilized.” The computational tool TEISER, in nondiscovery mode, was then used to assess the stability of MBNL1-bound transcripts in this data set (as described in Goodarzi et al. 2014). For validation of the beadchip array results, relative transcript levels were assessed by qRT–PCR, and 18S was used as an endogenous normalization control. Statistical significance was determined using a one-tailed Student's t-test.
Cells were seeded at 2 × 105 per well in six-well plates. Eighteen hours after seeding, DRB (Sigma) was added to the cells to a final concentration of 100 μM. RNA was isolated at 0, 2, 4, 6, and 8 h after DRB addition using a total RNA isolation kit with on-column DNase treatment (Norgen). Relative levels of the transcripts of interest were assessed by qRT–PCR, using 18S as the endogenous control. Half-life calculations were done using the formula t1/2 = ln 2/kdecay, where the decay constant was determined by plotting the data on a semilog scale and using nonlinear regression to find the best fit line (Graphpad Prism version 6).
For the reporter assay, a truncated mCherry ORF with all YGCY motifs mutated was cloned into the pCMV6-AC vector (Origene) and used as an exogenous reporter gene. The 3′ UTR of TACC1 was cloned downstream from the mCherry ORF stop codon. The MBNL1-binding sites, as determined by CIMS analysis of the MBNL1 HITS-CLIP data, were mutated (scrambled) while keeping the dinucleotide frequency constant. For each of these constructs, 100 ng was transiently transfected into 7.5 × 104 MDA-231 cells in 12-well plates, and total RNA was isolated 48 h after transfection. The relative level of the mCherry reporter was assessed by qRT–PCR, and the neomycin resistance transcript (transcribed from the same vector) was used as the normalization control. Statistical significance was determined using a one-tailed Student's t-test.
MBNL1, DBNL, and TACC1 expression correlation with clinical outcomes
Relative MBNL1 transcript levels in a set of 117 primary breast cancers and 36 distal metastases were taken from previously published data sets in which cancer samples were transcriptomically profiled using a common platform (as describe in Png et al. 2011). The KMplotter tool was used to assess the correlation between MBNL1 and TACC1 expression and distant metastasis-free survival or relapse-free survival in breast cancer patients (Gyorffy et al. 2013). For both, high and low gene expression levels were determined by dividing at the median, and a log rank P-value was used to assess significance. DBNL correlation with clinical outcome in breast cancer was assessed by qRT–PCR in a commercially available set of cDNA from 48 staged breast tumor samples (Origene), using β-actin as the endogenous control.
Data analysis
Statistical comparisons were carried out using the test indicated for each experiment type. In all figures, P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***). Statistical significance was concluded at P < 0.05.
Accession number
All high-throughput sequencing and Illumina beadchip array data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE76488.
Supplementary Material
Acknowledgments
We thank the members of our laboratory who contributed insightful comments to past versions of this manuscript. We thank Tina Marney and the Darnell laboratory for technical advice on the MBNL1 HITS-CLIP protocol. S.F.T. conceived the project and supervised all research. L.F. and S.F.T. wrote the manuscript. L.F., N.P., H.G., H.T., M.Y., and S.F.T. designed, performed, and analyzed the experiments.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.270645.115.
References
- Artero R, Prokop A, Paricio N, Begemann G, Pueyo I, Mlodzik M, Perez-Alonso M, Baylies MK. 1998. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev Biol 195: 131–143. [DOI] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, et al. 2014. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell 56: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann G, Paricio N, Artero R, Kiss I, Perez-Alonso M, Mlodzik M. 1997. muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development 124: 4321–4331. [DOI] [PubMed] [Google Scholar]
- Boateng JR, Coresio CL, Huttenlocher A. 2012. Src-mediated phosphorylation of mammalian Abp1 (DBNL) regulates podosome rosette formation in transformed fibroblasts. J Cell Sci 125: 1329–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68: 799–808. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149: 1393–1406. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. 2011. A perspective on cancer cell metastasis. Science 331: 1559–1564. [DOI] [PubMed] [Google Scholar]
- Cheng AW, Shi J, Wong P, Luo KL, Trepman P, Wang ET, Choi H, Burge CB, Lodish HF. 2014. Muscleblind-like 1 (Mbnl1) regulates pre-mRNA alternative splicing during terminal erythropoiesis. Blood 124: 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang AC, Massague J. 2008. Molecular basis of metastasis. N Engl J Med 359: 2814–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connert S, Wienand S, Thiel C, Krikunova M, Glyvuk N, Tsytsyura Y, Hilfiker-Kleiner D, Bartsch JW, Klingauf J, Wienands J. 2006. SH3P7/mAbp1 deficiency leads to tissue and behavioral abnormalities and impaired vesicle transport. EMBO J 25: 1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte N, Charafe-Jauffret E, Delaval B, Adelaide J, Ginestier C, Geneix J, Isnardon D, Jacquemier J, Birnbaum D. 2002. Carcinogenesis and translational controls: TACC1 is down-regulated in human cancers and associates with mRNA regulators. Oncogene 21: 5619–5630. [DOI] [PubMed] [Google Scholar]
- Conte N, Delaval B, Ginestier C, Ferrand A, Isnardon D, Larroque C, Prigent C, Seraphin B, Jacquemier J, Birnbaum D. 2003. TACC1–chTOG–Aurora A protein complex in breast cancer. Oncogene 22: 8102–8116. [DOI] [PubMed] [Google Scholar]
- Elemento O, Slonim N, Tavazoie S. 2007. A universal framework for regulatory element discovery across all genomes and data types. Mol Cell 28: 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabillard JC, Ulisse S, Baldini E, Sorrenti S, Cremet JY, Coccaro C, Prigent C, D'Armiento M, Arlot-Bonnemains Y. 2011. Aurora-C interacts with and phosphorylates the transforming acidic coiled-coil 1 protein. Biochem Biophys Res Commun 408: 647–653. [DOI] [PubMed] [Google Scholar]
- Gadalla SM, Lund M, Pfeiffer RM, Gortz S, Mueller CM, Moxley RT, Kristinsson SY, Bjorkhom M, Shebl FM, Hilbert JE, et al. 2011. Cancer risk among patients with myotonic muscular dystrophy. JAMA 306: 2480–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannopoulou EG, Elemento O. 2011. An integrated ChIP-seq analysis platform with customizable workflows. BMC Bioinformatics 12: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goers ES, Purcell J, Voelker RB, Gates DP, Berglund JA. 2010. MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res 38: 2467–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Zhang S, Buss CG, Fish L, Tavazoie S, Tavazoie SF. 2014. Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins. Nature 513: 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massague J. 2007. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 446: 765–770. [DOI] [PubMed] [Google Scholar]
- Gurtan AM, Sharp PA. 2013. The role of miRNAs in regulating gene expression networks. J Mol Biol 425: 3582–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyot R, Vincent S, Bertin J, Samarut J, Ravel-Chapuis P. 2010. The transforming acidic coiled coil (TACC1) protein modulates the transcriptional activity of the nuclear receptors TR and RAR. BMC Mol Biol 11: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, Lanczky A. 2013. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8: e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, David L, Golipour A, Gabut M, Michael IP, Nachman EN, et al. 2013. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498: 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepper I, Schymeinsky J, Weckbach LT, Jakob SM, Frommhold D, Sixt M, Laschinger M, Sperandio M, Walzog B. 2012. The mammalian actin-binding protein 1 is critical for spreading and intraluminal crawling of neutrophils under flow conditions. J Immunol 188: 4590–4601. [DOI] [PubMed] [Google Scholar]
- Ho TH, Charlet-B N, Poulos MG, Singh G, Swanson MS, Cooper TA. 2004. Muscleblind proteins regulate alternative splicing. EMBO J 23: 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KB, Darnell RB. 2008. CLIP: crosslinking and immunoprecipitation of in vivo RNA targets of RNA-binding proteins. Methods Mol Biol 488: 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. 2003a. A muscleblind knockout model for myotonic dystrophy. Science 302: 1978–1980. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Urbinati CR, Crusselle VJ, Luo D, Lee YJ, Harrison JK, Oh SP, Swanson MS. 2003b. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr Patterns 3: 459–462. [DOI] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD. 2007. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8: 533–543. [DOI] [PubMed] [Google Scholar]
- Kessels MM, Engqvist-Goldstein AE, Drubin DG. 2000. Association of mouse actin-binding protein 1 (mAbp1/SH3P7), an Src kinase target, with dynamic regions of the cortical actin cytoskeleton in response to Rac1 activation. Mol Biol Cell 11: 393–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larbolette O, Wollscheid B, Schweikert J, Nielsen PJ, Wienands J. 1999. SH3P7 is a cytoskeleton adapter protein and is coupled to signal transduction from lymphocyte antigen receptors. Mol Cell Biol 18: 1539–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffart B, Howell SJ, Tasch JE, Cowell JK, Still IH. 2002. Interaction of the transforming acidic coiled-coil 1 (TACC1) protein with ch-TOG and GAS41/NuBI1 suggests multiple TACC1-containing protein complexes in human cells. Biochem J 363: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. 2010. Resolving RNA complexity to decipher regulatory rules governing biological networks. Nat Rev Genet 11: 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. 2006. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15: 2087–2097. [DOI] [PubMed] [Google Scholar]
- Linder S. 2007. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol 17: 107–117. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293: 865–867. [DOI] [PubMed] [Google Scholar]
- Llobet-Navas D, Rodriguez-Barrueco R, Castro V, Ugalde AP, Sumazin P, Jacob-Sendler D, Demircan B, Castillo-Martin M, Putcha P, Marshall N, et al. 2014. The miR-424(322)/503 cluster orchestrates remodeling of the epithelium in the involuting mammary gland. Genes Dev 28: 765–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K. 2012. CUGBP1 and MBNL1 preferentially bind to 3′UTRs and facilitate mRNA decay. Sci Rep 2: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. 2000. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19: 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. 2005. Genes that mediate breast cancer metastasis to lung. Nature 436: 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SF, Parker R. 2014. Principles and properties of eukaryotic mRNPs. Mol Cell 54: 547–558. [DOI] [PubMed] [Google Scholar]
- Pascual M, Vicente M, Monferrer L, Artero R. 2006. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation 74: 65–80. [DOI] [PubMed] [Google Scholar]
- Pencheva N, Tavazoie SF. 2013. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol 15: 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Png KJ, Halberg N, Yoshida M, Tavazoie SF. 2011. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 481: 190–194. [DOI] [PubMed] [Google Scholar]
- Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et al. 2011. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 18: 840–845. [DOI] [PubMed] [Google Scholar]
- Schymeinsky J, Gerstl R, Mannigel I, Niedung K, Frommhold D, Panthel K, Heesemann J, Sixt M, Quast T, Kolanus W, et al. 2009. A fundamental role of mAbp1 in neutrophils: impact on β(2) integrin-mediated phagocytosis and adhesion in vivo. Blood 114: 4209–4220. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J. 2008. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udd B, Krahe R. 2012. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 10: 891–905. [DOI] [PubMed] [Google Scholar]
- Vajda NA, Brimacombe KR, LeMasters KE, Ladd AN. 2009. Muscleblind-like 1 is a negative regulator of TGF-β-dependent epithelial-mesenchymal transition of atrioventricular canal endocardial cells. Dev Dyn 238: 3266–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, Marney CB, Shu W, Valiente M, Zou Y, Mele A, Darnell RB, Massague J. 2014. Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. Elife 3: e02734–e02734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NAL, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. 2012. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150: 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win AK, Perattur PG, Pulido JS, Pulido CM, Lindor NM. 2012. Increased cancer risks in myotonic dystrophy. Mayo Clin Proc 87: 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Gao XD, Lee JH, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, et al. 2014. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev 28: 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Darnell RB. 2011. Mapping in vivo protein–RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29: 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ma L. 2012. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev 31: 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.