

Abstract
Cysteine occupies a unique place in protein chemistry. The nucleophilic thiol group allows cysteine to undergo a broad range of redox modifications beyond classical thiol-disulfide redox equilibria, including S-sulfenylation (-SOH), S-sulfinylation (-SO2H), S-sulfonylation (-SO3H), S-nitrosylation (-SNO), S-sulfhydration (-SSH), S-glutathionylation (-SSG), and others. Emerging evidence suggests that these post-translational modifications (PTM) are important in cellular redox regulation and protection against oxidative damage. Identification of protein targets of thiol redox modifications is crucial to understanding their roles in biology and disease. However, analysis of these highly labile and dynamic modifications poses challenges. Recent advances in the design of probes for thiol redox forms, together with innovative mass spectrometry based chemoproteomics methods make it possible to perform global, site-specific, and quantitative analyses of thiol redox modifications in complex proteomes. Here, we review chemical proteomic strategies used to expand the landscape of thiol redox modifications.
Among the protein-coding amino acids, cysteine is unique, owing both to its intrinsic nucleophilicity and redox sensitivity (1, 2). The nucleophilic thiol group allows cysteine to undergo a broad range of redox modifications, including S-sulfenylation (-SOH), S-sulfinylation (-SO2H), S-sulfonylation (-SO3H), S-nitrosylation (-SNO), S-sulfhydration (-SSH), and S-glutathionylation (-SSG) (Fig. 1) (3, 4). Most thiol redox modifications can be formed by the non-enzymatic reactions of reactive oxygen/nitrogen/sulfur species (ROS/RNS/RSS) with protein thiols, which has led to the widespread notion that these types of modifications are randomly distributed across the cysteine proteome. However, emerging evidence suggests that thiol redox modifications are well-controlled, site-specific cellular events, which play important roles in regulation of diverse protein functions, including catalytic or ligand binding activities (5–7), protein–protein interactions (8, 9), and protein stability (10, 11).
Fig. 1.
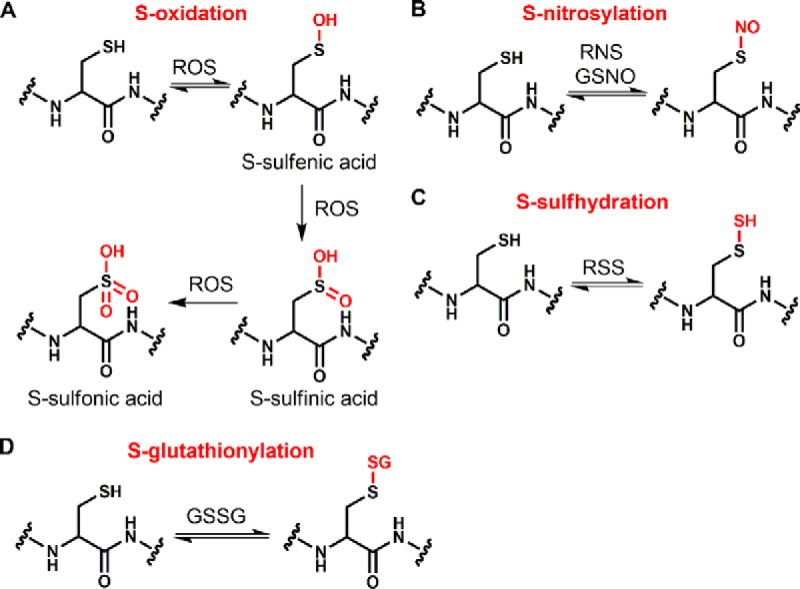
Thiol redox modifications. A, Protein thiol oxidation by ROS can lead to the formation of S-sulfenic acid, which can be subsequently oxidized to S-sulfenic acid or S-sulfonic acid. B, Protein thiols can be S-nitrosylated by reactive nitrogen species, such as nitric oxide (NO) or trans-nitrosylation by NO containing species such as S-nitrosoglutathione (GSNO). C, Protein thiols can be S-sulfhydrated by RSS such as the dissociated thiolate (HS−) or higher order polysulfides [H(S)nS−, n = 1–7]. D, Protein thiols can be S-glutathionylated by GSSG via a thiol/disulfide exchange. For a more comprehensive overview of formation and reaction of thiol redox modifications, the reader is referred to a recent review (4).
Identification of protein targets of thiol redox modifications is crucial to understanding their roles in biology and disease. However, the sites of specific protein thiol redox modifications have remained largely undefined. Technical advances in MS-based proteomics have greatly expand the inventories of several major post-translational modifications (PTM), including phosphorylation, ubiquitylation, and acetylation (12). Similar proteome-wide analysis of thiol redox modifications is complicated by the labile and highly dynamic nature of thiol redox forms. Nevertheless, the last decade has seen rapid growth of chemically-selective approaches to detect thiol redox modifications in concert with MS-based proteomics (13). In this review, we describe several major chemoproteomics strategies to globally profile thiol redox modifications.
Quantitative Thiol Reactivity Profiling
Historically, thiol oxidation in proteins has been monitored by loss of reactivity with electrophilic thiol-labeling agents containing reporter tags, such as radioisotopes or fluorescent groups (Fig. 2A). Once a cysteine residue is oxidized, it cannot react with these agents. Subsequently, a loss of signal for the modified cysteines in a protein can be detected by visualizing changes in the reporter group signal from these agents. For example, PTP1B was discovered as a oxidation-sensitive protein by determining the incorporation of radioactivity into the active cysteinyl thiol of this enzyme with a radiolabeled thiol-reactive agent (14).
Fig. 2.

Quantitative thiol-reactivity profiling for detection and identification of thiol oxidation. A, Thiol oxidation in proteins is monitored by loss of reactivity with thiol-reactive agents containing reporter tags, such as radiolabels or fluorescent groups. Thiol oxidation in the detected proteins results in a loss of signal in Western blot. B, ICAT-based thiol-reactivity profiling allows for identification of oxidant-sensitive thiols in complex proteomes. Thiol proteomes in samples treated with or without oxidant can be labeled with light and heavy ICAT, respectively. The light and heavy ICAT labeled samples are mixed, digested with trypsin, enriched by affinity purification, and analyzed by MS-based proteomics. C, Clickable electrophile probes in combination with a quantitative chemoproteomic platform enables global, in situ, site-specific profiling of redox sensitive thiols in complex proteomes. Thiol proteomes in two different samples are probed with alkyne-tagged electrophiles and then conjugated with an isotopically labeled, protease-cleavable azido-biotin via CuAAC reaction. The light and heavy protein samples are mixed, enriched by affinity purification, digested with two proteases. The resulting isotopically tagged peptides are detected and quantified by MS-based proteomics.
Building upon this strategy, a number of quantitative thiol reactivity profiling techniques have been developed to globally identify protein targets sensitive to oxidative stress. Generally, thiol proteomes in samples under different oxidative states are directly labeled with the thiol reactivity profiling agents, which typically are biotinylated thiol-reactive chemicals. The biotinylated proteins then can be enriched by affinity purification and analyzed by quantitative proteomic techniques. Among the thiol reactivity profiling agents, isotope-coded affinity tag (ICAT)1 reagents were widely used, owing to their intrinsic thiol reactivity, quantitative capacity, and commercial availability (15). Typically, proteins from two different experimental samples are labeled with the light and heavy isotopically labeled ICAT reagents to compare reduced thiol content between cellular states (Fig. 2B). For example, this strategy has been successfully applied to mouse and rabbit heart tissues to identify protein thiols that are sensitive to hydrogen peroxide (H2O2) treatment (16–18). However, ICAT reagents have relatively bulky chemical structures, which incur steric hindrance that compromises the efficiency of thiol labeling.
To address this problem, several thiol-reactivity profiling probes with much simpler chemical structures have been developed (19). For example, a clickable iodoacetamide (IA)-alkyne probe was used to label reduced thiols in bacterial proteomes treated with and without H2O2, respectively (Fig. 2C) (20). An isotopically labeled, protease-cleavable azido-biotin tag was then conjugated to the alkyne-modified proteins using click chemistry (copper-catalyzed azide-alkyne cycloaddition reaction, CuAAC). The isotopically tagged light and heavy protein samples were mixed and subjected to successive tandem protease digestion and the resulting isotopically tagged, cysteine-containing peptides were analyzed by MS-based proteomics. This quantitative chemoproteomic workflow allowed for a proteome-wide identification of ∼200 proteins containing H2O2-sensitive thiols in bacteria (20). Interestingly, many oxidation-sensitive cysteines in bacterial proteomes were found in transcriptional regulators and metabolic enzymes (20).
However, the cytotoxicity and poor cell-membrane permeability of current IA-based thiol-labeling agents precludes in situ labeling, which is most desirable to avoid artifacts because of disruption of native cellular redox environment and protein structure. To circumvent these limitations of IA-based probes, a thiol-reactive probe with reduced cytotoxicity and spatial and temporal control of thiol labeling in living cells was developed recently (21). This novel chemical probe consists of a α-bromomethyl ketone (BK) electrophile caged with a photo-labile protecting group (o-nitrophenylethylene glycol) and an alkyne-tag as a bioorthogonal handle for incorporation of reporter tags using click chemistry (Fig. 2C). Using this probe in combination with the aforementioned quantitative chemoproteomic approach, the redox reactivity changes of ∼300 proteins were measured in living A431 cells in response to growth factor-dependent oxidative signaling (21).
The Biotin-switch Technique and Its Variations
Jaffrey et al. first introduced the biotin-switch technique (BST) to study protein S-nitrosylation (22). The original BST consists of three major steps to convert S-nitrosylthiols (SNO) into biotinylated thiols (Fig. 3A): First, free thiols are blocked with the thiol-blocking agent, methyl methanethiosulfonate (MMTS); second, S-nitrosylated thiols are reduced with ascorbate; third, the nascent thiols are labeled with a pyridyldithiol-activated, thiol-reactive biotin reagent, N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propionamide (biotin-HPDP). The biotin-switched, S-nitrosylated proteins or peptides then can be enriched by affinity purification and analyzed by MS-based proteomics. This strategy has been widely used in proteome-wide analyses of protein S-nitrosylation (23–27). The original BST also can be easily adapted to study other distinct reversible thiol modifications by changing the specific reducing agents. For example, arsenite and a recombinant glutaredoxin system can be used to reduce S-sulfenylated and S-glutathionylated proteins, respectively (28, 29). Furthermore, the original BST can be employed with a variety of quantitative proteomic techniques, including isobaric tags for relative and absolute quantification (28), stable isotope labeling of amino acids in cell culture (30), and label-free spectral counting (31), to compare thiol modification status in samples under different conditions. Alternatively, biotin-HPDP or biotin-maleimide commonly used in the original BST can be replaced with thiol-labeling agents with quantitative functions, such as ICAT and tandem mass tags (TMT).
Fig. 3.

Biotin-switch techniques (BST) and variations for the proteome-wide identification and quantification of thiol redox modifications. A, BST involve differential thiol labeling of reduced and oxidized thiols. Free thiols are first blocked with a small-molecule thiol-reactive agent such as iodoacetamide (IA), N-ethylmaleimide (NEM), or methyl methanethiosulfonate (MMTS). Reversibly oxidized thiols are reduced with a specific reducing agent, labeled with another thiol-reactive agent with a functionalized group, such as biotin, TMT, or resin, enriched by affinity purification or resin-assisted capture, and analyzed by MS-based proteomics. B, OxICAT is used to measure the relative content of oxidized thiols in thiol proteome. Free thiols are first labeled with the light (12C) ICAT (red), while oxidized proteins in the same sample are subsequently reduced and labeled with the heavy (13C) ICAT (blue). The ICAT labeled sample is then subjected to trypsin digestion and affinity purification. The resulting peptides are detected and quantified by MS-based proteomics. The extent of thiol oxidation in any given peptide is determined by the ratios of heavy to light precursor ion signal intensity.
For the ICAT-based BST, reversibly-modified thiols are reduced and labeled with light and heavy ICAT reagents (32). The two different samples can be compared after combination of the ICAT labeling, trypsin digestion, affinity purification, and MS-based proteomics. This strategy has been used to globally identify and quantify redox-modified thiols in a number of biological systems (33–36). For example, Winterbourn and colleagues employed the ICAT-based BST to quantify hundreds of redox-mediated changes in mouse heart thiol proteomes during ischemia/reperfusion (35). The ICAT-based BST can also be used to measure stoichiometry of reversible thiol oxidation in proteins, whereby the light and heavy ICAT reagents are used to differentially label oxidized versus reduced cysteine residues within a single sample (Fig. 3B) (37, 38). This method, termed as oxICAT, has been proven useful for determining changes in the extent of protein thiol oxidation in cells under stress conditions (37–39). oxICAT provides results that are unaffected by changes in the protein expression and stability. Thus, oxICAT is ideally suited to simultaneously monitor changes in the thiol oxidation status of proteins over time.
Using oxICAT, the global thiol redox status was determined at distinct time points in the life span of several model organisms (40–42). These studies established a clear connection between the thiol oxidation statues and aging in S. cerevisiae and C.elegans (40, 41). It was found that the yeast cells underwent a significant increase in global oxidation (i.e. ∼80% of the 300 proteins) several days before they died (40). A similar phenomenon was also observed during the lifespan of C. elegans, as the aged worms had significantly more oxidized protein thiols than young adults (41). However, it is particularly notable that high levels of thiol oxidation could also be observed in the early development of worms and that this elevated overall protein oxidation significantly decreased upon entering the reproductive period (41). Surprisingly, a recent oxICAT-based study revealed that thiol redox status did not become more oxidized with age in D .melanogaster (42). Despite these successful applications of oxICAT, the sensitivity of this method may not be sufficient for effective detection of low-abundance endogenous redox modifications in cells or tissues because of the highly abundant background thiol proteome. However, recent technological advances in MS-based proteomics have greatly improved the sensitivity of oxICAT. In a recent application of oxICAT in marine photosynthetic eukaryotes, the oxidative status of over 3800 protein thiols were quantified in parallel and approximately one tenth of these were identified to be highly redox-sensitive (43).
In addition to biotinylated thiol labeling agents (e.g. biotin-HPDP, ICAT), thiol-reactive TMT can be used for the thiol tag-switch labeling (44). More importantly, TMT enables multiplexed proteomic quantification of thiol redox modifications from different samples (45–47). For example, analysis of S-nitrosylation begins with free thiol alkylation, after which protein S-nitrosthiols are reduced with ascorbate and then labeled with TMT. After tryptic digestion, S-nitrosylated peptides from different samples can be mixed, enriched with antibody against TMT, and identified by MS-based proteomics. Van Eyk and colleagues first introduced this strategy to analyze protein S-nitrosylation in human pulmonary arterial endothelial cell lysates treated with reduced glutathione (GSH), glutathione disulfide (GSSG), and S-nitrosylglutathione (GSNO) in one single experiment (47). It is particularly important to note that, GSH and GSSG were considered as negative controls to discriminate potential artifacts, including instances of ascorbate-reduced protein disulfide bonds and S-glutathionylation. This analysis unambiguously identified a total of 220 S-nitrosylated sites on 179 proteins. Furthermore, the TMT-based tag-switch method enables simultaneous assessment of different types of redox modification (48, 49). In one such example, a total of 114 S-nitrosylation and S-sulfenylation sites were identified and quantified in E. coli under low and mild oxidative stress (48). Notably, most of these two modifications occupy the same sites, but their relative stoichiometries are quite different (48).
Redox modified thiols also can be tag-switched with a thiol-reactive resin and enriched directly without laborious affinity capture steps (e.g. avidin, anti-TMT antibody). This resin-assisted capture strategy was first introduced by Stamler and colleagues to globally profile protein S-nitrosylation dynamics in S-nitrosylcysteine-treated E. coli and HEK293 cells, in combination with isobaric tags for relative and absolute quantification as the quantification approach (50). This strategy was further improved by Qian and colleagues and used to identify hundreds of S-nitrosylated or S-glutathionylated thiols in cells and tissues with 95% of the enriched peptides containing cysteine residues (51, 52). More recently, the strategy was applied to measure total reversible thiol oxidation in an oxygenic photosynthetic prokaryote. The redox changes of ∼2100 thiols in 1060 proteins under light/dark conditions were quantified (53). In this study, most protein thiols were observed to be less oxidized under continuous light, but became significantly more oxidized in the dark or simulating dark phase (53).
Despite of the great success of the BST in proteome-wide analyses of thiol redox modifications, it is important to note its limitations. The principal issue is that the thiol-blocking agents commonly used in BST methods display reactivity with cysteine S-sulfenic acid, which is a key intermediate in thiol oxidation (54, 55). Uncertainty regarding the species labeled by thiol-blocking agents introduces ambiguity in interpretation. Moreover, the BSTs for analyzing distinct thiol redox modifications rely highly on the specificity of the reducing reagents, which is difficult to establish unambiguously under relevant experimental conditions (56).
Chemoselective Probe-based Approaches
Advances in understanding the unique chemistries of different thiol redox forms have led to the development of chemoselective probes, which have enabled new, direct labeling approaches to the analysis of thiol redox chemistry. Chemoproteomic strategies based on these selective probes are emerging as alternatives to the indirect analysis approaches described above. In these chemoproteomic approaches, functionalized groups (e.g. biotin or clickable tags) used for the subsequent detection and capture can either be metabolically incorporated into these modifications or synthetically incorporated into the chemospecific probe molecules (Fig. 4). Coupled with MS-based proteomics, chemoselective probe-based approaches have significantly broadened the study of S-glutathionylation, S-nitrosylation, S-sulfenylation, S-sulfinylation, and S-sulfhydration.
Fig. 4.

Chemoselective probe-based approaches enable the selective incorporation of affinity tags for distinct types of thiol redox modifications. A, BioGSH or BioGSSG is used to metabolically label S-glutathionylated proteins via disulfide-exchange. B, Azido-Ala is transferred by an engineered glutathione synthetase to label S-glutathionylated proteins. The azido-tagged proteins then can be conjugated with alkyne-biotin via CuAAC reaction. C, The phenylmercury-biotin or agarose resins are used to directly label and capture S-nitrosylated proteins via stable S-Hg bond formation. D, The one-step reductive ligation allows for the coupling of S-nitrosylated proteins with a biotin-linked phosphine probe. E, Bioorthogonal reactions in combination with azide and alkyne analogs of dimedone enable in situ labeling and detection of protein S-sulfenylation. F, S-sulfinylated proteins can be detected or enriched by chemoselective ligation with a biotinylated aryl-nitroso compound. G, S-sulfhydrated proteins are first tagged with methylsulfonyl benzathiazole and the persulfide adducts can then be selectively reacted with a biotin-linked cyanoacetate probe.
Protein S-glutathionylation
Direct approaches (e.g. radiolabeling and antibody-based methods) to detect protein S-glutathionylation were described two decades ago (56). However, these approaches lacked sensitivity and were not suitable for proteomic studies. To overcome these limitations, several biotinyl analogs of GSH or GSSG have been developed (57, 58). Adding biotinylated glutathione to biological systems mimics an increase in GSSG in the cell, tissue, or organ under oxidative stress, thereby leading to thiol-disulfide exchange. Thus, the cellular targets of protein S-glutathionylation can be metabolically labeled by biotin, which allows for their detection and identification (Fig. 4A). For protein-level identification, biotinylated proteins can be enriched by affinity purification and eluted by treatment with reducing agents to release the bound proteins, followed by subsequent MS-based proteomics. For site-level identification, biotinylated proteins can be first digested with trypsin and S-glutathionylated peptides are enriched with affinity purification, eluted with reducing agents, and analyzed by MS-based proteomics. These approaches have been successfully applied to several biological systems to identify S-glutathionylated proteins or sites (59, 60). For example, biotinylated glutathione disulfide (BioGSSG) was applied to analyze the S-glutathionylome in Synechocystis (59). About 350 proteins were identified as S-glutathionylation targets and the modified sites were determined for 125 targets (59). Nevertheless, these approaches have two major limitations. First, biotinylated glutathione cannot be used to study native protein S-glutathionylation in cells, in which the intrinsic oxidation status may be perturbed by the addition of this reagent. Second, the bulky structure of biotin group limits access to some targets of S-glutathionylation.
To overcome these limitations, Ahn and colleagues recently developed a novel approach to detect S-glutathionylation by metabolically tagging intracellular glutathione with small clickable functionality (Fig. 4B) (61). To this end, glutathione synthetase, a key enzyme that catalyzes coupling of γGlu-Cys to Gly to form glutathione, was genetically mutated to produce an enzyme that catalyzed addition of azido-Ala in place of Gly. Cells were transfected with the engineered glutathione synthetase and incubated with azido-Ala, thereby generating the azide-containing glutathione derivative, γGlu-Cys-azido-Ala. Thereafter, this clickable glutathione was conjugated to substrates of protein S-glutathionylation in cells. The S-glutathionylated product could be further conjugated to different functionalized groups, such as biotin and fluorescein, using click chemistry (CuAAC), which allowed for selective and sensitive detection of this modification (Fig. 4B). In the future, this approach could be combined with MS-based proteomics to globally identify S-glutathionylated proteins and modification sites in situ.
Protein S-nitrosylation
It is known that phenylmercury compounds can react with SNO groups to form a relatively stable thiol-mercury bond (S-Hg) (62). Building upon this chemistry, two novel phenylmercury-based SNO-capture reagents have been developed to directly capture S-nitrosylated proteins (63). As shown in Fig. 4C, these reagents mainly consist of a phenylmercury moiety and agarose resins or a biotin group. Free thiols within the sample first are blocked, and the S-nitrosylated proteins and peptides then can be selectively reacted with these reagents to form S-Hg bonds and be enriched by either resin-assisted capture or avidin-based affinity purification. For protein-level capture, the bound S-nitrosylated-proteins are eluted by reduction of the S-Hg bond with 2-mercaptoethanol. For peptide-level capture, captured proteins are subjected to on-resin tryptic digestion and S-nitrosylated peptides then are eluted by oxidation of cysteine with performic acid to generate sulfonic acids at the S-Hg Cys residues and the released peptides are analyzed by MS-based proteomics. Using these approaches, over 300 endogenous S-nitrosylation sites on ∼200 proteins were identified from mouse liver (63). Ischiropoulos and colleagues further utilized this phenylmercury-based SNO-capture approach to identify up to 1,000 S-nitrosylation sites on hundreds of proteins in various mouse tissues (64, 65). The number of S-nitrosylation sites in vivo to nitric oxide were found to be substantially dependent on endothelial nitric oxide synthase (eNOS) or neuronal nitric oxide synthase (nNOS) (64, 65).
In addition to phenylmercury, a variety of phosphine-based chemistries have been developed for direct labeling of SNO groups, including reductive ligation, bis-ligation, one-step disulfide formation, reductive elimination, and S-alkylphosphonium formation. More comprehensive discussion of these reactions has been presented in several recently published reviews (66–68). These reactions have been successfully explored to detect small molecular S-nitrosothiols (69, 70) or single S-nitrosylated protein (71). However, development of phosphine-based probes for the enrichment and identification of S-nitrosylated targets in complex proteomes remains a significant challenge, since selectivity and bio-compatibility of the phosphine-based chemistry for labeling SNO still needs improvement. Until recently, only a few probes have been developed to detect S-nitrosylated targets in complex proteomes. In one such example, based on the one-step reductive ligation of S-nitrosothiols, Xian and colleagues developed a biotin-linked phosphine probe to globally detect protein S-nitrosylation in CysNO treated COS-7 cells using Western blot (Fig. 4B) (72). In principle, this probe could be coupled with MS-based proteomics to capture and identify S-nitrosylated proteins.
Protein S-sulfenylation
The distinct chemical reactivity of cysteine sulfenic acid makes it possible to react with a variety of nucleophiles and electrophiles (4, 73). In 1974, Benitez and Allison first reported that a cyclic 1,3-diketone carbon nucleophile, 5,5-dimethyl-1,3-cyclohexadione (dimedone), reacts with sulfenic acid (74). Since then, many dimedone-based probes for detecting S-sulfenylated proteins have been developed (75–79). Among these probes, cell-permeable alkyne and azide analogs of dimedone are most often used, as they enable in situ labeling of protein S-sulfenylation. These probes have been successfully applied to identify epidermal growth factor receptor, threonyl-tRNA synthetase, and many ovarian tumor deubiquitinases as targets of S-sulfenylation in mammalian cells (7, 10, 80). Furthermore, these clickable probes were utilized for proteomic analysis of protein S-sulfenylation in intact cells. In the first step, S-sulfenylated proteins are selectively tagged and, in the second step, probe-labeled proteins are captured by bioorthogonal chemistry (e.g. Staudinger ligation or CuAAC) and identified by MS-based proteomics (Fig. 4C). This strategy identified ∼200 potential targets of protein S-sulfenylation in the unstimulated HeLa cells or H2O2-treated Arabidopsis cells, respectively (81, 82). However, these protein-level analyses did not identify specific sites of this modification, which increases the possibility of false-positive identifications. Moreover, analyses based on protein-level capture do not provide information about multiple redox changes on a single protein. To address these issues, we recently developed a novel site-specific S-sufenylcysteine mapping strategy based on an alkynyl-functionalized dimedone probe, DYn-2, and a fully optimized chemoproteomic workflow (83, 84). Specifically, S-sulfenylated proteins were labeled in intact cells by DYn-2. Cell proteins were then digested with trypsin and labeled peptides were conjugated to azide biotin via CuAAC, affinity purified on streptavidin resin, and released by photocleavage of the biotin linker. The released, DYn-2 modified peptides were subsequently analyzed by MS-based proteomics. Using this chemoproteomic strategy, a total of 1105 S-sulfenylated sites from 778 proteins were identified from human cells (83). Furthermore, the difference of S-sulfenylation levels in cells with or without stimuli was compared with the use of unlabeled (light) and deuterated (heavy) DYn-2 (83). This quantitative, site-specific S-sulfenylome analysis thus provides a means to simultaneously measure different susceptibility of S-sulfenylation events in intact cells and to detect differential redox reactivity within proteins under oxidative stress.
Protein S-sulfinylation
Since the discovery of sulfiredoxin, which reduces S-sulfinic acid to the thiol in an ATP-dependent reaction (85, 86), protein S-sulfinylation has emerged as another important, reversible PTM event in cells. Although S-sulfinic acid on a particular protein can be directly detected by MS or antibodies raised against specific sulfinic acid-modified peptides, global analysis of protein S-sulfinylation remains a major challenge (3). Inspired by reactions with aromatic sulfinic acids and aryl-nitroso compounds, we developed a ligation reaction for selectively converting S-sulfinic acid moieties into stable conjugates under physiological conditions (87). Building upon this ligation reaction, we further developed the first probe for detection of protein S-sulfinylation. This probe, termed as NO-Bio, combines an aryl-nitroso moiety for labeling of the sulfinic acid with a biotin tag (Fig. 4D) (88). Following thiol blockage, S-sulfinylated protein can be selectively labeled by NO-Bio and detected by Western blot. This novel approach has been applied to evaluate global protein S-sulfinylation levels in HeLa cells treated with H2O2 as well as to compare S-sulfinylation among human NSCLC tumors and matched normal lung tissue (88). More importantly, this probe should make it possible to perform a proteomic survey of protein S-sulfinylation in biological systems in the future, as S-sulfinylated proteins labeled by NO-Bio can be enriched by affinity purification and identified by MS-based proteomics.
Protein S-sulfhydration
Direct and selective detection of protein S-sulfhydration has long been a significant challenge, since the persulfide group shows reactivity similar to that of other sulfur species, particularly free thiols. Snyder and colleagues developed the first method for this purpose, which employs an thiol-reactive agent MMTS to differentially label thiols and persulfides (89). Following the labeling reaction, the unreacted S-sulfhydrated proteins are subsequently conjugated to biotin-HPDP and identified by MS-based proteomics. Using this method, a number of proteins were identified as potential targets for S-sulfhydration and the basal S-sulfhydration level of some proteins was estimated to be as high as 25% (89). However, a key concern about this approach is the selectivity of MMTS for thiols versus persulfides (90). To overcome this limitation, Xian and colleagues recently developed a selective two-step approach to detect protein S-sulfhydration (Fig. 4E) (91, 92). In the first step, nucleophilic protein thiol species (-SH and -SSH) are tagged with the electrophile, methylsulfonyl benzathiazole. Thereafter the sample is treated with a biotin-linked cyanoacetate probe to selectively label the persulfide adducts. This novel technique has been successfully applied to analyze protein S-sulfhydration in a model protein (e.g. glyceraldehyde-3-phosphate dehydrogenase) in cell lysates and cells exposed to Na2S (91). In this proof-of-concept study, the authors also identified heat shock protein 70 as a novel target of protein S-sulfhydration (91). The probe-labeled proteins also can be captured and subject to MS-based proteomic analysis. This two-step labeling approach was recently utilized to globally identify persulfide and polysulfide-modified proteins in A549 cells overexpressing cystathionine γ-lyase, an enzyme used for converting L-cysteine to hydrogen sulfide (93). Of interest, most of targets of protein S-sulfhydration identified in this study could also be S-sulfenylated (83), such as protein disulfide isomerase, heat shock proteins, aldo-keto reductase, glyceraldehyde-3-phosphate dehydrogenase, enolase, and phosphoglycerate kinase.
Site-specific Mapping of Thiol Redox Modifications
As discussed above, the vast majority of thiol redox proteomic data were acquired via protein-level capture and indirect or unselective methods. Therefore, these data should be treated with caution. Fortunately, technological advances in chemoproteomics, such as novel probes with MS-compatibility, efficient enrichment methods, sensitive MS detection, and reliable database search algorithms, make it possible to precisely pinpoint sites of thiol redox modifications in proteins. Moreover, like small molecule cysteine conjugates, the chemically modified Cys-containing peptides often produce distinct reporter ions in MS/MS spectra through the characteristic C-S fragmentations, which increase the reliability of site-level identification of thiol redox modifications. With current reagents and instrumentation, a redox proteomic experiment has the potential to identify thousands of modified Cys-containing peptides and their corresponding sites. Nevertheless, automated site identifications should be viewed with caution. It is challenging to automatically estimate the site FDR of these identifications using the common target-decoy database search strategy, because of limitations of the sample size. Thus, automated site localization results should be critically inspected. Moreover, a small proportion of peptides identified in a redox proteomic study contain more than one cysteine. To ensure unambiguous site localization, manual evaluation of these identifications is typically required with at least three modification-specific fragment ions annotated in the corresponding MS/MS spectra.
CONCLUSIONS
The landscape of thiol redox modifications is rapidly expanding, due both to recent advances in MS-based proteomics and thiol chemistry. Dramatic improvements in MS instrumentation significantly improves the sensitivity, analytical throughput, and reliability of chemical approaches for global and site-specific mapping of thiol redox modifications. Advances in our understanding of thiol chemistry have led to the development of chemoselective probes, which enable direct detection, efficient enrichment, identification, and quantification of distinct types of thiol redox modifications. These chemical proteomic strategies have not only expanded the inventory of thiol redox modifications, but also afforded novel insights into how oxidative stress and redox-mediated signaling regulate cell homeostasis during physiological and/or pathological processes.
Future work in this field should improve the sensitivity and quantitative accuracy of MS-based proteomics and expand the repertoire of selective chemical probes for in situ or in vivo labeling of distinct thiol redox modifications. Recently described analysis platforms and expected improvements will provide the means to address important unanswered questions in the field of redox biology:
What is the stoichiometry of distinct types of thiol redox modification in physiological or pathological states?
How does the cross-talk between redox modifications on thiols contribute to redox regulation networks in cells?
How does the thiol proteome sense organelle-specific redox changes in cells induced by endogenous stimuli?
As the methodologies for thiol redox proteomics evolve, these and other questions can likely be addressed in the near future. Meanwhile, it is important to realize that the data generated from a large-scale redox proteomic surveys will require further adaptation of informatics infrastructure to integrate proteome-scale data with biochemistry and cell biology experiments. A variety of computational structural biology tools (94–96), bioinformatics (97, 98), and databases (99, 100) can be used for this purpose, thereby providing a greater understanding of redox regulation of protein functions.
Footnotes
Author contributions: J.Y., K.S.C., and D.C.L. analyzed data; J.Y., K.S.C., and D.C.L. wrote the paper; J.Y., K.S.C., and D.C.L. reviewed literature.
* This work was supported by grants from the National Natural Science Foundation of China (No. 31500666 and No. 81573395 to J.Y.) and the National Institutes of Health (U24CA159988 to D.C.L. and R01GM102187 and R01CA174864 to K.S.C.).
1 The abbreviations used are:
- ICAT
- isotope-coded affinity tag
- TMT
- tandem mass tags
- BST
- biotin-switch technique
- GSH
- reduced glutathione
- GSSG
- glutathione disulfide.
REFERENCES
- 1.Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B., Bachovchin D. A., Mowen K., Baker D., and Cravatt B. F. (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poole L. B. (2015) The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 80, 148–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baez N. O., Reisz J. A., and Furdui C. M. (2015) Mass spectrometry in studies of protein thiol chemistry and signaling: opportunities and caveats. Free Radic. Biol. Med. 80, 191–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paulsen C. E., and Carroll K. S. (2013) Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 113, 4633–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dotsey E. Y., Jung K. M., Basit A., Wei D., Daglian J., Vacondio F., Armirotti A., Mor M., and Piomelli D. (2015) Peroxide-Dependent MGL Sulfenylation Regulates 2-AG-Mediated Endocannabinoid Signaling in Brain Neurons. Chem. Biol. 22, 619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anastasiou D., Poulogiannis G., Asara J. M., Boxer M. B., Jiang J. K., Shen M., Bellinger G., Sasaki A. T., Locasale J. W., Auld D. S., Thomas C. J., Vander Heiden M. G., and Cantley L. C. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulsen C. E., Truong T. H., Garcia F. J., Homann A., Gupta V., Leonard S. E., and Carroll K. S. (2012) Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Z., Kozlov S., Lavin M. F., Person M. D., and Paull T. T. (2010) ATM activation by oxidative stress. Science 330, 517–521 [DOI] [PubMed] [Google Scholar]
- 9.Sobotta M. C., Liou W., Stocker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 [DOI] [PubMed] [Google Scholar]
- 10.Kulathu Y., Garcia F. J., Mevissen T. E., Busch M., Arnaudo N., Carroll K. S., Barford D., and Komander D. (2013) Regulation of A20 and other OTU deubiquitinases by reversible oxidation. Nat. Commun. 4, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J. G., Baek K., Soetandyo N., and Ye Y. (2013) Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat. Commun. 4, 1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsen J. V., and Mann M. (2013) Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 12, 3444–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y., Fonslow B. R., Shan B., Baek M. C., and Yates J. R. 3rd. (2013) Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 113, 2343–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S. R., Kwon K. S., Kim S. R., and Rhee S. G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372 [DOI] [PubMed] [Google Scholar]
- 15.Gygi S. P., Rist B., Gerber S. A., Turecek F., Gelb M. H., and Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 16.Sethuraman M., McComb M. E., Huang H., Huang S., Heibeck T., Costello C. E., and Cohen R. A. (2004) Isotope-coded affinity tag (ICAT) approach to redox proteomics: identification and quantitation of oxidant-sensitive cysteine thiols in complex protein mixtures. J Proteome Res. 3, 1228–1233 [DOI] [PubMed] [Google Scholar]
- 17.Sethuraman M., McComb M. E., Heibeck T., Costello C. E., and Cohen R. A. (2004) Isotope-coded affinity tag approach to identify and quantify oxidant-sensitive protein thiols. Mol. Cell. Proteomics 3, 273–278 [DOI] [PubMed] [Google Scholar]
- 18.Fu C., Hu J., Liu T., Ago T., Sadoshima J., and Li H. (2008) Quantitative analysis of redox-sensitive proteome with DIGE and ICAT. J. Proteome Res. 7, 3789–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weerapana E., Simon G. M., and Cravatt B. F. (2008) Disparate proteome reactivity profiles of carbon electrophiles. Nat. Chem. Biol. 4, 405–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng X., Weerapana E., Ulanovskaya O., Sun F., Liang H., Ji Q., Ye Y., Fu Y., Zhou L., Li J., Zhang H., Wang C., Alvarez S., Hicks L. M., Lan L., Wu M., Cravatt B. F., and He C. (2013) Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe. 13, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abo M., and Weerapana E. (2015) A Caged Electrophilic Probe for Global Analysis of Cysteine Reactivity in Living Cells. J. Am. Chem. Soc. 137, 7087–7090 [DOI] [PubMed] [Google Scholar]
- 22.Jaffrey S. R., Erdjument-Bromage H., Ferris C. D., Tempst P., and Snyder S. H. (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193–197 [DOI] [PubMed] [Google Scholar]
- 23.Murray C. I., Kane L. A., Uhrigshardt H., Wang S. B., and Van Eyk J. E. (2011) Site-mapping of in vitro S-nitrosation in cardiac mitochondria: implications for cardioprotection. Mol. Cell. Proteomics 10, M110 004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sangwung P., Greco T. M., Wang Y., Ischiropoulos H., Sessa W. C., and Iwakiri Y. (2012) Proteomic identification of S-nitrosylated Golgi proteins: new insights into endothelial cell regulation by eNOS-derived NO. PLoS ONE 7, e31564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaccarin M., Falda M., Roveri A., Bosello-Travain V., Bordin L., Maiorino M., Ursini F., and Toppo S. (2014) Quantitative label-free redox proteomics of reversible cysteine oxidation in red blood cell membranes. Free Radic. Biol. Med. 71, 90–98 [DOI] [PubMed] [Google Scholar]
- 26.Lam Y. W., Yuan Y., Isaac J., Babu C. V., Meller J., and Ho S. M. (2010) Comprehensive identification and modified-site mapping of S-nitrosylated targets in prostate epithelial cells. PLoS ONE 5, e9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato H., Takemoto D., and Kawakita K. (2013) Proteomic analysis of S-nitrosylated proteins in potato plant. Physiol. Plant 148, 371–386 [DOI] [PubMed] [Google Scholar]
- 28.McDonagh B., Martinez-Acedo P., Vazquez J., Padilla C. A., Sheehan D., and Barcena J. A. (2012) Application of iTRAQ Reagents to Relatively Quantify the Reversible Redox State of Cysteine Residues. Int. J. Proteomics 2012, 514847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kehr S., Jortzik E., Delahunty C., Yates J. R. 3rd, Rahlfs S., and Becker K. (2011) Protein S-glutathionylation in malaria parasites. Antioxid. Redox. Signal. 15, 2855–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X., Han P., Li J., Zhang X., Huang B., Ruan H. Q., and Chen C. (2010) ESNOQ, proteomic quantification of endogenous S-nitrosation. PLoS ONE 5, e10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X., Huang B., and Chen C. (2012) SNO spectral counting (SNOSC), a label-free proteomic method for quantification of changes in levels of protein S-nitrosation. Free Radic. Res. 46, 1044–1050 [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Santamarina S., Boronat S., Domenech A., Ayte J., Molina H., and Hidalgo E. (2014) Monitoring in vivo reversible cysteine oxidation in proteins using ICAT and mass spectrometry. Nat. Protoc. 9, 1131–1145 [DOI] [PubMed] [Google Scholar]
- 33.Fares A., Rossignol M., and Peltier J. B. (2011) Proteomics investigation of endogenous S-nitrosylation in Arabidopsis. Biochem. Biophys. Res. Commun. 416, 331–336 [DOI] [PubMed] [Google Scholar]
- 34.Puyaubert J., Fares A., Reze N., Peltier J. B., and Baudouin E. (2014) Identification of endogenously S-nitrosylated proteins in Arabidopsis plantlets: effect of cold stress on cysteine nitrosylation level. Plant Sci. 215–216, 150–156 [DOI] [PubMed] [Google Scholar]
- 35.Kumar V., Kleffmann T., Hampton M. B., Cannell M. B., and Winterbourn C. C. (2013) Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free Radic. Biol. Med. 58, 109–117 [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Santamarina S., Boronat S., Espadas G., Ayte J., Molina H., and Hidalgo E. (2011) The oxidized thiol proteome in fission yeast–optimization of an ICAT-based method to identify H2O2-oxidized proteins. J. Proteomics 74, 2476–2486 [DOI] [PubMed] [Google Scholar]
- 37.Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., and Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Go Y. M., Roede J. R., Walker D. I., Duong D. M., Seyfried N. T., Orr M., Liang Y., Pennell K. D., and Jones D. P. (2013) Selective targeting of the cysteine proteome by thioredoxin and glutathione redox systems. Mol. Cell. Proteomics 12, 3285–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brandes N., Reichmann D., Tienson H., Leichert L. I., and Jakob U. (2011) Using quantitative redox proteomics to dissect the yeast redoxome. J. Biol. Chem. 286, 41893–41903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brandes N., Tienson H., Lindemann A., Vitvitsky V., Reichmann D., Banerjee R., and Jakob U. (2013) Time line of redox events in aging postmitotic cells. Elife 2, e00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knoefler D., Thamsen M., Koniczek M., Niemuth N. J., Diederich A. K., and Jakob U. (2012) Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol. Cell 47, 767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menger K. E., James A. M., Cocheme H. M., Harbour M. E., Chouchani E. T., Ding S., Fearnley I. M., Partridge L., and Murphy M. P. (2015) Fasting, but Not Aging, Dramatically Alters the Redox Status of Cysteine Residues on Proteins in Drosophila melanogaster. Cell. Rep. 11, 1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenwasser S., Graff van Creveld S., Schatz D., Malitsky S., Tzfadia O., Aharoni A., Levin Y., Gabashvili A., Feldmesser E., and Vardi A. (2014) Mapping the diatom redox-sensitive proteome provides insight into response to nitrogen stress in the marine environment. Proc. Natl. Acad. Sci. U.S.A. 111, 2740–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson A., Schafer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A. K., and Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 45.McGarry D. J., Chen W., Chakravarty P., Lamont D. J., Wolf C. R., and Henderson C. J. (2015) Proteome-wide identification and quantification of S-glutathionylation targets in mouse liver. Biochem. J. 469, 25–32 [DOI] [PubMed] [Google Scholar]
- 46.Qu Z., Meng F., Bomgarden R. D., Viner R. I., Li J., Rogers J. C., Cheng J., Greenlief C. M., Cui J., Lubahn D. B., Sun G. Y., and Gu Z. (2014) Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res. 13, 3200–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murray C. I., Uhrigshardt H., O'Meally R. N., Cole R. N., and Van Eyk J. E. (2012) Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol. Cell. Proteomics 11, M111 013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wojdyla K., Williamson J., Roepstorff P., and Rogowska-Wrzesinska A. (2015) The SNO/SOH TMT strategy for combinatorial analysis of reversible cysteine oxidations. J. Proteomics 113, 415–434 [DOI] [PubMed] [Google Scholar]
- 49.Pan K. T., Chen Y. Y., Pu T. H., Chao Y. S., Yang C. Y., Bomgarden R. D., Rogers J. C., Meng T. C., and Khoo K. H. (2014) Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxid. Redox. Signal. 20, 1365–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forrester M. T., Thompson J. W., Foster M. W., Nogueira L., Moseley M. A., and Stamler J. S. (2009) Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 27, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su D., Shukla A. K., Chen B., Kim J. S., Nakayasu E., Qu Y., Aryal U., Weitz K., Clauss T. R., Monroe M. E., Camp D. G. 2nd, Bigelow D. J., Smith R. D., Kulkarni R. N., and Qian W. J. (2013) Quantitative site-specific reactivity profiling of S-nitrosylation in mouse skeletal muscle using cysteinyl peptide enrichment coupled with mass spectrometry. Free Radic. Biol. Med. 57, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su D., Gaffrey M. J., Guo J., Hatchell K. E., Chu R. K., Clauss T. R., Aldrich J. T., Wu S., Purvine S., Camp D. G., Smith R. D., Thrall B. D., and Qian W. J. (2014) Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic. Biol. Med. 67, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo J., Nguyen A. Y., Dai Z., Su D., Gaffrey M. J., Moore R. J., Jacobs J. M., Monroe M. E., Smith R. D., Koppenaal D. W., Pakrasi H. B., and Qian W. J. (2014) Proteome-wide light/dark modulation of thiol oxidation in cyanobacteria revealed by quantitative site-specific redox proteomics. Mol. Cell. Proteomics 13, 3270–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reisz J. A., Bechtold E., King S. B., Poole L. B., and Furdui C. M. (2013) Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 280, 6150–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paulech J., Solis N., and Cordwell S. J. (2013) Characterization of reaction conditions providing rapid and specific cysteine alkylation for peptide-based mass spectrometry. Biochim. Biophys. Acta 1834, 372–379 [DOI] [PubMed] [Google Scholar]
- 56.Couvertier S. M., Zhou Y., and Weerapana E. (2014) Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim. Biophys. Acta 1844, 2315–2330 [DOI] [PubMed] [Google Scholar]
- 57.Brennan J. P., Miller J. I., Fuller W., Wait R., Begum S., Dunn M. J., and Eaton P. (2006) The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol. Cell. Proteomics 5, 215–225 [DOI] [PubMed] [Google Scholar]
- 58.Sullivan D. M., Wehr N. B., Fergusson M. M., Levine R. L., and Finkel T. (2000) Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry 39, 11121–11128 [DOI] [PubMed] [Google Scholar]
- 59.Chardonnet S., Sakr S., Cassier-Chauvat C., Le Marechal P., Chauvat F., Lemaire S. D., and Decottignies P. (2015) First proteomic study of S-glutathionylation in cyanobacteria. J. Proteome Res. 14, 59–71 [DOI] [PubMed] [Google Scholar]
- 60.Zaffagnini M., Bedhomme M., Groni H., Marchand C. H., Puppo C., Gontero B., Cassier-Chauvat C., Decottignies P., and Lemaire S. D. (2012) Glutathionylation in the photosynthetic model organism Chlamydomonas reinhardtii: a proteomic survey. Mol. Cell. Proteomics 11, M111 014142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samarasinghe K. T., Munkanatta Godage D. N., VanHecke G. C., and Ahn Y. H. (2014) Metabolic synthesis of clickable glutathione for chemoselective detection of glutathionylation. J. Am. Chem. Soc. 136, 11566–11569 [DOI] [PubMed] [Google Scholar]
- 62.Saville B. (1958) A scheme for the colorimetric determination of microgram amounts of thiols. Analyst 83, 670–672 [Google Scholar]
- 63.Doulias P. T., Greene J. L., Greco T. M., Tenopoulou M., Seeholzer S. H., Dunbrack R. L., and Ischiropoulos H. (2010) Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 107, 16958–16963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doulias P. T., Tenopoulou M., Greene J. L., Raju K., and Ischiropoulos H. (2013) Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 6, rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raju K., Doulias P. T., Evans P., Krizman E. N., Jackson J. G., Horyn O., Daikhin Y., Nissim I., Yudkoff M., Nissim I., Sharp K. A., Robinson M. B., and Ischiropoulos H. (2015) Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci. Signal. 8, ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang H., and Xian M. (2011) Chemical methods to detect S-nitrosation. Curr. Opin. Chem. Biol. 15, 32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Y. J., Ching W. C., Lin Y. P., and Chen Y. J. (2013) Methods for detection and characterization of protein S-nitrosylation. Methods 62, 138–150 [DOI] [PubMed] [Google Scholar]
- 68.Bechtold E., and King S. B. (2012) Chemical methods for the direct detection and labeling of S-nitrosothiols. Antioxid. Redox. Signal. 17, 981–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seneviratne U., Godoy L. C., Wishnok J. S., Wogan G. N., and Tannenbaum S. R. (2013) Mechanism-based triarylphosphine-ester probes for capture of endogenous RSNOs. J. Am. Chem. Soc. 135, 7693–7704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang D., Chen W., Miao Z., Ye Y., Zhao Y., King S. B., and Xian M. (2014) A reductive ligation based fluorescent probe for S-nitrosothiols. Chem. Commun. 50, 4806–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bechtold E., Reisz J. A., Klomsiri C., Tsang A. W., Wright M. W., Poole L. B., Furdui C. M., and King S. B. (2010) Water-soluble triarylphosphines as biomarkers for protein S-nitrosation. ACS Chem. Biol. 5, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J., Li S., Zhang D., Wang H., Whorton A. R., and Xian M. (2010) Reductive ligation mediated one-step disulfide formation of S-nitrosothiols. Org. Lett. 12, 4208–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta V., and Carroll K. S. (2014) Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 1840, 847–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benitez L. V., and Allison W. S. (1974) The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J. Biol. Chem. 249, 6234–6243 [PubMed] [Google Scholar]
- 75.Pan J., and Carroll K. S. (2014) Chemical biology approaches to study protein cysteine sulfenylation. Biopolymers 101, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Charles R. L., Schroder E., May G., Free P., Gaffney P. R., Wait R., Begum S., Heads R. J., and Eaton P. (2007) Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol. Cell. Proteomics 6, 1473–1484 [DOI] [PubMed] [Google Scholar]
- 77.Poole L. B., Klomsiri C., Knaggs S. A., Furdui C. M., Nelson K. J., Thomas M. J., Fetrow J. S., Daniel L. W., and King S. B. (2007) Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug. Chem. 18, 2004–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Paulsen C. E., and Carroll K. S. (2009) Chemical dissection of an essential redox switch in yeast. Chem. Biol. 16, 217–225 [DOI] [PubMed] [Google Scholar]
- 79.Reddie K. G., Seo Y. H., Muse Iii W. B., Leonard S. E., and Carroll K. S. (2008) A chemical approach for detecting sulfenic acid-modified proteins in living cells. Mol. Biosyst. 4, 521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu J., Fan Y., and Ling J. (2014) Mechanism of oxidant-induced mistranslation by threonyl-tRNA synthetase. Nucleic Acids Res. 42, 6523–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leonard S. E., Reddie K. G., and Carroll K. S. (2009) Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem. Biol. 4, 783–799 [DOI] [PubMed] [Google Scholar]
- 82.Akter S., Huang J., Bodra N., De Smet B., Wahni K., Rombaut D., Pauwels J., Gevaert K., Carroll K., Van Breusegem F., and Messens J. (2015) DYn-2 Based Identification of Arabidopsis Sulfenomes. Mol. Cell. Proteomics 14, 1183–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang J., Gupta V., Carroll K. S., and Liebler D. C. (2014) Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 5, 4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang J., Gupta V., Tallman K. A., Porter N. A., Carroll K. S., and Liebler D. C. (2015) Global, in situ, site-specific analysis of protein S-sulfenylation. Nat. Protoc. 10, 1022–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Biteau B., Labarre J., and Toledano M. B. (2003) ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425, 980–984 [DOI] [PubMed] [Google Scholar]
- 86.Woo H. A., Chae H. Z., Hwang S. C., Yang K. S., Kang S. W., Kim K., and Rhee S. G. (2003) Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300, 653–656 [DOI] [PubMed] [Google Scholar]
- 87.Lo Conte M., and Carroll K. S. (2012) Chemoselective ligation of sulfinic acids with aryl-nitroso compounds. Angew. Chem. Int. Ed. Engl. 51, 6502–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lo Conte M., Lin J., Wilson M. A., and Carroll K. S. (2015) A Chemical Approach for the Detection of Protein Sulfinylation. ACS Chem. Biol. 10, 1825–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., and Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pan J., and Carroll K. S. (2013) Persulfide reactivity in the detection of protein s-sulfhydration. ACS Chem. Biol. 8, 1110–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang D., Macinkovic I., Devarie-Baez N. O., Pan J., Park C. M., Carroll K. S., Filipovic M. R., and Xian M. (2014) Detection of protein S-sulfhydration by a tag-switch technique. Angew Chem. Int. Ed. Engl. 53, 575–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park C. M., Macinkovic I., Filipovic M. R., and Xian M. (2015) Use of the “tag-switch” method for the detection of protein S-sulfhydration. Methods Enzymol. 555, 39–56 [DOI] [PubMed] [Google Scholar]
- 93.Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., Yamamoto M., Ono K., Devarie-Baez N. O., Xian M., Fukuto J. M., and Akaike T. (2014) Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7606–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rostkowski M., Olsson M. H., Sondergaard C. R., and Jensen J. H. (2011) Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 11, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Petersen B., Petersen T. N., Andersen P., Nielsen M., and Lundegaard C. (2009) A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marino S. M., and Gladyshev V. N. (2012) Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 287, 4419–4425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 98.Shi Z., Wang J., and Zhang B. (2013) NetGestalt: integrating multidimensional omics data over biological networks. Nat. Methods 10, 597–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen Y. J., Lu C. T., Su M. G., Huang K. Y., Ching W. C., Yang H. H., Liao Y. C., Chen Y. J., and Lee T. Y. (2015) dbSNO 2.0: a resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation. Nucleic Acids Res. 43, D503–D511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun M. A., Wang Y., Cheng H., Zhang Q., Ge W., and Guo D. (2012) RedoxDB–a curated database for experimentally verified protein oxidative modification. Bioinformatics 28, 2551–2552 [DOI] [PubMed] [Google Scholar]