

Abstract
Eukaryotic elongation factor 1A (eEF1A) is an essential, highly methylated protein that facilitates translational elongation by delivering aminoacyl-tRNAs to ribosomes. Here, we report a new eukaryotic protein N-terminal methyltransferase, Saccharomyces cerevisiae YLR285W, which methylates eEF1A at a previously undescribed high-stoichiometry N-terminal site and the adjacent lysine. Deletion of YLR285W resulted in the loss of N-terminal and lysine methylation in vivo, whereas overexpression of YLR285W resulted in an increase of methylation at these sites. This was confirmed by in vitro methylation of eEF1A by recombinant YLR285W. Accordingly, we name YLR285W as elongation factor methyltransferase 7 (Efm7). This enzyme is a new type of eukaryotic N-terminal methyltransferase as, unlike the three other known eukaryotic N-terminal methyltransferases, its substrate does not have an N-terminal [A/P/S]-P-K motif. We show that the N-terminal methylation of eEF1A is also present in human; this conservation over a large evolutionary distance suggests it to be of functional importance. This study also reports that the trimethylation of Lys79 in eEF1A is conserved from yeast to human. The methyltransferase responsible for Lys79 methylation of human eEF1A is shown to be N6AMT2, previously documented as a putative N(6)-adenine-specific DNA methyltransferase. It is the direct ortholog of the recently described yeast Efm5, and we show that Efm5 and N6AMT2 can methylate eEF1A from either species in vitro. We therefore rename N6AMT2 as eEF1A-KMT1. Including the present work, yeast eEF1A is now documented to be methylated by five different methyltransferases, making it one of the few eukaryotic proteins to be extensively methylated by independent enzymes. This implies more extensive regulation of eEF1A by this posttranslational modification than previously appreciated.
Protein methylation is emerging as one of the most prominent posttranslational modifications in the eukaryotic cell (1). Often showing high evolutionary conservation, it is increasingly recognized for its role in modulating protein–protein interactions (2). Indeed, it has been documented in protein interaction codes (3), such as those of the histones and p53 (4, 5), where it shows interplay with modifications such as acetylation and phosphorylation. Despite this, there remains a paucity of understanding of the enzymes that catalyze protein methylation. Many of the known methyltransferases target histones. However, many other methyltransferases have been discovered recently that act on nonhistone proteins (6).
While protein methylation predominantly occurs on lysine and arginine residues, it is also known to occur on glutamine, asparagine, glutamate, histidine, cysteine, and the N- and C termini of proteins. Although the presence of N-terminal methylation on numerous proteins has been known for decades (7), the first enzymes responsible for this methylation have only recently been discovered (8, 9). The Saccharomyces cerevisiae protein Tae1 and its human ortholog N-terminal methyltransferase 1 (NTMT1) catalyze N-terminal methylation of proteins with an N-terminal [A/P/S]-P-K motif (after methionine removal). Yet there is evidence that these enzymes may recognize a more general N-terminal motif (10). Human NTMT2 is a monomethyltransferase that methylates the same substrates as NTMT1 and may prime substrate proteins with monomethylation to assist subsequent trimethylation by NTMT1 (11).
The biological function of N-terminal methylation on some proteins has been recently revealed. For example, N-terminal methylation of regulator of chromatin condensation protein 1 (RCC1) is known to affect its binding to chromatin and thereby the correct chromosomal segregation during mitosis (12, 13), and N-terminal methylation of DNA damage-binding protein 2 (DDB2) is important for its role in UV-damaged DNA repair (14). Interestingly, there is evidence of interplay between N-terminal methylation and other posttranslational modifications (15), suggesting that, like lysine and arginine methylation, it may be incorporated into protein interaction codes (3). N-terminal methylation therefore appears to be a modification of functional importance in the cell.
Eukaryotic elongation factor 1A (eEF1A), and its bacterial ortholog EF-Tu, is an essential translation elongation factor that is found in all living organisms. Its canonical function is in facilitating delivery of aminoacyl-tRNAs to the ribosome; however, it is also known to have a role in many other cellular functions, such as actin bundling, nuclear export, and proteasomal degradation (16). A number of methyltransferases have been discovered in both S. cerevisiae and human that target translation elongation factors. In yeast, four of these elongation factor methyltransferases (EFMs) act on eEF1A, namely Efm1, Efm4, Efm5, and Efm6, generating monomethylated Lys30, dimethylated Lys316, trimethylated Lys79, and monomethylated Lys390, respectively (17–19). Human METTL10 is the ortholog of Efm4 in that it trimethylates eEF1A at Lys318, which is equivalent to Lys316 in yeast (20). Interestingly, eukaryotic elongation factor 2 (eEF2) is also methylated by a number of lysine methyltransferases. In yeast, Efm2 and Efm3 act on eEF2, generating dimethylated Lys613 and trimethylated Lys509, respectively (21–24). Human eEF2-KMT is the ortholog of Efm3 in that it trimethylates eEF2 at Lys525, which is equivalent to Lys509 in yeast eEF2 (23).
Here, we report the N-terminal methylation of eEF1A in S. cerevisiae and the identification of the methyltransferase that catalyzes this event. Using parallel reaction monitoring and MS/MS/MS (MS3), we unambiguously localize the modification to the N-terminal glycine and show it is conserved in the human cell. We also show that YLR285W, which we rename elongation factor methyltransferase 7 (Efm7), is responsible for this modification in yeast, as well as dimethylation at the adjacent lysine. We also characterize the methyltransferases responsible for methylation of lysine 79 in eEF1A. Human N6AMT2 is shown to be the ortholog of yeast Efm5 through its capacity to methylate yeast and human eEF1A at Lys79 in vitro. We therefore rename N6AMT2 as eEF1A-KMT1.
EXPERIMENTAL PROCEDURES
Yeast Strains, Culturing, and SDS-PAGE
The wild-type yeast strain used in this study was BY4741 (Open Biosystems, Huntsville, AL). Single gene deletion strains (ΔYLR285W, ΔYNL024C, ΔYGR001C, and ΔYNL092W) were obtained from (Euroscarf, Frankfurt, Germany). Strains were cultured and lysates obtained according to previous methods (22). The BG1805-YLR285W plasmid was modified for overexpression of YLR285W by site-direct, ligase-independent mutagenesis (25) to remove the HA-tag and the ZZ domain, leaving YLR285W with the C-terminal 6xHisTag and a short linker. Site-direct, ligase-independent mutagenesis was used again on this plasmid to remove the YLR285W gene, leaving only the 6xHisTag and the linker, to generate the empty vector control plasmid. Overexpression in BY4741 was done as per Mok et al. (26), except that the induction was done overnight. Lysates were separated by SDS-PAGE on 4–12% NuPAGE® Novex® gels (Life Technologies, Waltham, MA), fixed with 10% acetic acid/25% isopropanol (v/v) and stained with QC Colloidal Coomassie Stain (Bio-Rad, Hercules, CA).
Mass Spectrometry
Sample Preparation
Gel bands were excised, destained with 50% acetonitrile (ACN)/50% 20 mm ammonium bicarbonate (v/v) and dehydrated with 100% ACN. Gel bands were then rehydrated with 5 μl sequencing-grade trypsin (Promega, Fitchburg, WI 10 ng/μl), 10 μl endoproteinase AspN (Promega, 10 ng/μl) or LysargiNase (27) (40 ng/μl), and made up to 50 μl with 20 mm ammonium bicarbonate (for AspN and trypsin) or 20 mm ammonium bicarbonate/12.5 mm CaCl2 (for LysargiNase, to give a final concentration of 10 mm CaCl2). Digests were left at 37 °C overnight and peptides extracted from the gel bands with 50% ACN/50% 0.1% formic acid (v/v) for 30 min and then 100% ACN for 10 min. Peptides were dried in a SpeedVacTM (Thermo Fisher Scientific, Waltham, MA) for 2 h before being resuspended in 0.1% formic acid (v/v).
LC-MS/MS
Peptide samples were separated by nano-LC and eluting peptides ionized by nano-ESI following previously described methods (28) before analysis on either an LTQ Orbitrap Velos Pro or a Q Exactive Plus (Thermo Fisher Scientific) for all mass spectrometric analyses.
For analyses on the LTQ Orbitrap Velos Pro, survey scans m/z 350–1,750 were acquired in the Orbitrap (resolution = 30,000 at m/z 400) with an initial accumulation target value of 1 × 106 ions in the linear ion trap. The instrument was set to operate in data-dependent acquisition mode in combination with an inclusion list containing the m/z values of the relevant peptides for the methylation site(s) of interest. The five most abundant ions (>5,000 counts) within 10 ppm of any m/z value on the inclusion list were sequentially isolated and fragmented, followed by up to the five most abundant ions not within 10 ppm of any m/z value on the inclusion list. Precursor ions were fragmented via collision-induced dissociation with an activation time of 30 ms, normalized collision energy of 30% and at a target value of 10,000 ions, for analysis in the linear ion trap. Dynamic exclusion was enabled with an exclusion duration of 30 s.
For analyses on the Q Exactive Plus, survey scans m/z 350–1,750 (MS automated gain control target = 3 × 106) were acquired in the Orbitrap (resolution = 70,000 at m/z 200). The instrument was set to operate in data-dependent acquisition mode in combination with an inclusion list containing the m/z values of the relevant peptides for the methylation site(s) of interest. The 10 most abundant ions within 10 ppm of any m/z value on the inclusion list were sequentially isolated and fragmented via Higher-energy collisional dissociation (HCD) using the following parameters: normalized collision energy = 35%, maximum injection time = 125 ms, and automated gain control target = 1 × 105. Fragment ions were then analyzed in the Orbitrap (resolution = 17,500). “If idle” was set as “pick others” for most analyses, allowing selection and fragmentation of ions not within 10 ppm of any m/z value on the inclusion list, only after all inclusion list-selected ions have been selected if less than 10 consecutive MS2 scans have been performed. Dynamic exclusion was enabled for most analyses with an exclusion duration of 30 s.
All data were converted to Mascot Generic Format (.mgf) using either MassMatrix MS Data File Conversion (v. 3.9) or msConvert from the ProteoWizard Library and Tools collection (29). Data were then searched against the SwissProt database (2014_08, 546,238 sequences through to 2015_08, 549,008 sequences) using Mascot (v. 2.4, Matrix Sciences) hosted by the Walter and Eliza Hall Institute for Medical Research (Melbourne, Australia) with the following settings: enzyme: trypsin (trypsin-digested samples), AspN (AspN-digested samples), or none (LysargiNase-digested samples); two allowed missed cleavages; precursor tolerance: 4 ppm; fragment ion tolerance: 10 mmu (Q Exactive Plus) or 0.4 Da (LTQ Orbitrap Velos Pro); peptide charge: 2+, 3+, 4+ (except when analyzing the singly charged KFETS peptide); Instrument: Q-Exactive_Gen (Q Exactive Plus) or ESI-TRAP (LTQ Orbitrap Velos Pro); Variable modifications: oxidation (M), methyl (K), dimethyl (K), trimethyl (K), and methyl (DE). For samples pertaining to N-terminal methylation the following changes were made: Allowed missed cleavages was set to 0 and the following variable modifications were added: methyl (N-term), dimethyl (N-term), and propyl (N-term) (in lieu of any trimethyl N-term variable modification). Peptides were generally identified with Mascot scores >35. and then spectra and elution times were manually inspected and compared between different methylation states to confirm identification; the exception being the very short methylated KFETS peptides, that produced low Mascot scores due to their length (scores of 28–31), which were verified by manual inspection for additional, confirmatory peaks falling within 10 ppm of theoretical fragment ion m/z values. All Q Exactive Plus-derived MS2 spectra were manually annotated to only include fragment ions with 10 ppm error or less. Extracted ion chromatograms for peptides were obtained using Thermo Xcalibur Qual Browser 2.2 SP1.48 by setting mass windows of ±10 ppm of the relevant m/z value, and applying a scan filter to only analyze MS1 scans. Relevant data have been deposited to the ProteomeXchange Consortium (30) via the PRIDE partner repository with the dataset identifier PXD002941.
N-terminal Methylation Localization—Parallel Reaction Monitoring and MS/MS/MS (MS3)
For analysis of the abundance of y14 ions, samples were analyzed by parallel-reaction monitoring on a Q Exactive Plus using the Targeted MS2 method. An inclusion list containing the m/z values of all possible methylation states of the doubly charged N-terminal AspN peptide GKEKSHINVVVIGHV (unmethylated to pentamethylated) (List A) was used to generate full MS2 scans of precursors at these m/z values every ∼0.6 s across the entire LC run. The default MS2 scan settings were used except for the following changes: automated gain control target = 5 × 104, maximum injection time = 50 ms, isolation window = 1.6 m/z, normalized collision energy = 30%. Transitions from the methylated precursors to y14+1 ions were analyzed using Qual Browser. Mass windows of ±10 ppm for each y14 ion were analyzed with the relevant precursor ion set in the scan filter setting. Human eEF1A samples were analyzed in the same way except that the inclusion list contained only the m/z value of the doubly charged trimethylated N-terminal AspN peptide GKEKTHINIVVIGHV, and mass windows for y14 transitions were set at ±20 ppm.
For MS3 analysis of y14 ions, samples were analyzed on an LTQ Orbitrap Velos Pro using the Data Dependent Product MS3 method with the following settings: collision-induced dissociation activation (normalized collision energy = 30%, activation time = 10 ms, isolation width = 2.5 m/z, default charge state = 2+ (MS2) or 1+ (MS3)), minimum signal threshold = 5,000 (MS2) or 200 (MS3) ion counts. The MS2 event was set to use List A as its parent mass list. For the MS3 event, the product mass list was set to contain the m/z values of all possible methylation states of the y14+1 ion (unmethylated to pentamethylated). Data were analyzed by manual assignment of fragment peaks falling within 0.4 Da of theoretical fragment m/z values for MS2 precursors falling within 0.4 Da of theoretical precursor m/z values.
Heavy Methyl SILAC
Wild-type yeast cells were grown in synthetic complete media (2 g/l histidine and methionine drop-out mix (US Biological, Salem, MA), 1.7 g/l yeast nitrogen base without amino acids or ammonium sulfate (BD Biosciences, Franklin Lakes, NJ), 5 g/l ammonium sulfate, 20 g/l glucose, 82 mg/l histidine), with 82 mg/l unlabeled (light), or 13CD3-labeled (heavy) methionine (Sigma-Aldrich, St. Louis, MO). Cell lysates were prepared as above, protein concentration quantified, and lysates mixed 3:1 (light:heavy). Mixed lysates were separated by SDS-PAGE, and the band corresponding to eEF1A excised, digested by AspN, and analyzed by LC-MS/MS as above.
Cloning, Expression, and Purification of Methyltransferases and Elongation Factors
All relevant ORFs were cloned into pET15b for bacterial expression by Gibson assembly with the Gibson Assembly® Cloning Kit (New England BioLabs, Ipswich, MA). Primers were designed to insert a 6x HisTag at the N terminus for yeast EFM5 and human N6AMT2 and at the C terminus for all other ORFs. ORFs were amplified from wild-type yeast gDNA (YLR285W and SceEF1A1 (TEF1)), wild-type yeast cDNA (EFM5), or plasmids RC205604 (N6AMT2) and RC209697 (HseEF1A1) (Origene, Rockville, MD). SceEF1A truncations were cloned directly from gBlocksTM constructs (Integrated DNA Technologies, Coralville, IA) designed with C-terminal 6xHisTags and flanking regions for Gibson Assembly into pET15b. All plasmids were transformed into Escherichia coli Rosetta DE3 and recombinant proteins expressed and purified according to previous methods (22).
In Vitro Methylation
Purified SceEF1A (full length or truncated forms) or HseEF1A1 from E. coli (∼10 μm) were incubated with purified methyltransferase (Efm5, N6AMT2, or YLR285W/Efm7, all at ∼5 μm) in the presence of 500 μm S-adenosyl l-methionine (AdoMet) in in vitro methylation buffer (50 mm HEPES-KOH, 20 mm NaCl, 1 mm EDTA, pH 7.4) at 30 °C for 5 h (Efm5 and N6AMT2) or overnight (YLR285W/Efm7). GDP and a nonhydrolysable analog of GTP (guanosine 5′-[γ-thio] triphosphate) were added to a final concentration of 1 mm for the relevant assays. No enzyme was added for the negative controls. 6x SDS sample buffer (350 mm Tris-HCl, pH 6.8, 30% glycerol (v/v), 10% SDS (v/v), 0.6 m DTT, 0.012% bromphenol blue (w/v)) was added to stop reactions, which were then resolved by SDS-PAGE and prepared for mass spectrometry as above.
RESULTS
Yeast eEF1A Is Trimethylated at Its N Terminus and Dimethylated at Lysine Three
For over 20 years, yeast eEF1A has been known to have four different sites of methylation (31). It was therefore surprising to see two tryptic peptides of 682.39 and 691.73 m/z, from the N terminus of eEF1A but lacking the initiator methionine, carrying three or five methyl groups (Supplemental Fig. S1, Supplemental Table S1). To localize the methylation, we further analyzed eEF1A using AspN. This generated the N-terminal peptide GKEKSHINVVVIGHV, whereby the N-terminal glycine is numbered Gly2 and the adjacent lysine Lys3. Fragmentation revealed up to five methyl groups in this peptide (Fig. 1A; see Supplemental Table S1 for details of additional methylation states), whereby three of the five methyl groups were localized to Gly2 and the two others to Lys3. Heavy methyl SILAC confirmed that this methylation is enzyme-mediated (Supplemental Fig. S2). To further investigate the combinations of methylation present, we analyzed the y14 ion corresponding to the fragmentation of peptide GKEKSHINVVVIGHV after its N-terminal glycine. Parallel reaction monitoring and MS/MS/MS (MS3) revealed that the di- and trimethylated forms of the peptide only produced unmethylated y14 ions of 1,558.90 m/z (Figd. 1B and 1C, Supplemental Table S2). Furthermore, the tetra- and pentamethylated forms of the AspN peptide only produced mono- and dimethylated y14 ions, of 1,572.92 and 1,586.94 m/z respectively. Tri-, tetra-, and pentamethylated y14 ions were not observed in any case. Overall, this indicates that the N terminus of eEF1A is trimethylated at Gly2, following which—in some cases—it is further mono- and dimethylated on Lys3 to generate the tetra- and pentamethylated forms.
Fig. 1.

Yeast eEF1A is trimethylated at its N terminus before being mono- and dimethylated at lysine three. (A) Q Exactive Plus MS/MS spectrum of a doubly-charged AspN peptide of 843.51 m/z identifies trimethylation at the N terminus of eEF1A as well as dimethylation at lysine 3 (for mass-annotated spectrum see Supplemental Fig. S14). (B) Transitions from the di-, tri-, tetra- and pentamethylated AspN peptide (GKEKSHINVVVIGHV+2) to the un-, mono- and dimethylated forms of the y14 ion (KEKSHINVVVIGHV+1) indicate that y14 is unmethylated in the di- and trimethylated AspN peptide, monomethylated in the tetramethylated AspN peptide and dimethylated in the pentamethylated AspN peptide. This indicates that the N terminus is trimethylated before Lys3 is partially mono- and dimethylated. The monomethylated AspN peptide was not abundant enough for this analysis. (C) MS/MS/MS (MS3) spectra of MS2 precursors of 1,559.06 (i), 1,572.99 (ii) and 1,587.14 m/z (iii) confirm their identities as the y14 ion in its un-, mono- and dimethylated state, respectively (for mass-annotated spectra see Supplemental Figs. S15-S17).
YLR285W Is the Methyltransferase Responsible for N-terminal and Lys3 Methylation
Having established that the N-terminal Gly2 and Lys3 of eEF1A are methylated, we sought to identify the methyltransferase(s) responsible. Trypsinized eEF1A from knockouts of the putative protein methyltransferases YLR285W, YGR001C, YNL024C, and YNL092W (32) only revealed a loss of N-terminal Gly2 and Lys3 methylation in ΔYLR285W (Supplemental Fig. S3). AspN digests of eEF1A from wild-type and ΔYLR285W strains confirmed this, clearly showing a complete loss of both N-terminal Gly2 and Lys3 methylation (Fig. 2A). This also highlighted that the Gly2 N-terminal trimethylation, present in the tri-, tetra-, and pentamethylated forms of the peptide, was of very high stoichiometry (estimated to be >85%, Fig. 2A). We next investigated whether the overexpression of YLR285W in wild-type yeast leads to a gain of methylation on the N-terminal Gly2 and/or Lys3. Interestingly, YLR285W overexpression increased methylation at both sites. No dimethylated GKEKSHINVVVIGHV peptide was seen, as compared with the empty vector control (Fig. 2B), indicating increased N-terminal trimethylation (as per Fig. 1). An increase in the pentamethylated peptide was also observed, along with a dramatic degree of hexamethylation, indicating increased Lys3 di- and trimethylation (Fig. 2B). This hexamethylation is consistent with trimethylation of the N terminus and of Lys3 (Supplemental Table S1, see Supplemental Fig. S4 for MS/MS spectrum). Reanalysis of data obtained from wild-type yeast, without overexpressed YLR285W, also revealed the presence of small amounts of hexamethylation (<0.1%, data not shown). Together, these data strongly suggest that YLR285W is the sole enzyme responsible for N-terminal Gly2 and Lys3 modification. As a final confirmation, we investigated whether YLR285W could methylate eEF1A in vitro. Purified YLR285W (see Supplemental Fig. S5) was incubated with recombinant yeast eEF1A from E. coli, in the presence of S-adenosyl l-methionine (AdoMet). This resulted in the formation of mono- and dimethylation of eEF1A on the N-terminal Gly2 (Fig. 2C, Supplemental Fig. S6), which suggested that the conformation of eEF1A may be affecting its ability to be methylated by YLR285W in vitro. Based on knockout, overexpression, and in vitro methylation analyses, we conclude that YLR285W is an N-terminal and lysine methyltransferase that can methylate the N-terminal Gly2 and Lys3 of eEF1A. In line with the naming of methyltransferases that act on translation elongation factors, we propose YLR285W be named elongation factor methyltransferase 7 (Efm7).
Fig. 2.

YLR285W is responsible for N-terminal and Lys3 methylation of yeast eEF1A in vivo and can N-terminally methylate eEF1A in vitro. (A) Deletion of YLR285W abolishes in vivo eEF1A methylation at the N terminus and Lys3 in yeast. Whole cell lysates from wild-type yeast (WT) and the single gene knockout of YLR285W were separated by SDS-PAGE and bands corresponding to eEF1A were analyzed by LC-MS/MS. The N terminus and Lys3 were both found to be unmethylated in the knockout of YLR285W, with the unmethylated form of the peptide (me0) being the only form of the peptide present. Trimethylation of the N terminus was estimated to be of high stoichiometry (>85%) by measuring the area under the curve for each methylation state. This does not, however, consider differences in ionization efficiency between the different methylation states. (B) Overexpression of YLR285W results in increased methylation at both the N-terminal Gly2 and Lys3 of eEF1A, as evidenced by the disappearance of the dimethylated peptide and the increase in abundance of the penta- and hexamethylated peptides. BG1805-YLR285W or the empty BG1805 vector were overexpressed in WT yeast and lysates were separated by SDS-PAGE and then bands corresponding to eEF1A were analyzed by LC-MS/MS. Un- and monomethylated eEF1A were below the limit of detection in both conditions. (C) YLR285W methylates eEF1A at its N terminus in vitro. Yeast eEF1A purified from E. coli was incubated with or without YLR285W in the presence of AdoMet. eEF1A was found to be mono- and dimethylated at the N terminus when incubated with YLR285W (see Supplemental Fig. S5 for MS/MS spectrum). For (A), (B), and (C), the methylation status of the N terminus/Lys3 was analyzed by taking mass windows (±10 ppm) corresponding to all relevant methylation states of the AspN peptide GKEKSHINVVVIGHV+2 (green). Peaks were normalized to the most abundant ion for each methylation state. The abundance of the eEF1A AspN peptide DAIEQPSRPT+2 is shown as an internal standard (black). Elution times of peptides are shaded; peaks outside shading are unrelated, near-isobaric ions.
Methylation by Efm7 Is Affected by the Conformation of eEF1A
The human N-terminal methyltransferase NTMT1 recognizes an N-terminal linear sequence motif and can methylate peptides (8). In contrast, we found that Efm7 was unable to methylate a synthetic peptide corresponding to the N-terminal 10 amino acids of eEF1A (GKEKSHINVV, data not shown). In consideration of this and the in vitro results above, we therefore investigated whether Efm7 recognizes conformational features of eEF1A during methylation. eEF1A binds GTP and hydrolyzes it to GDP as its source of energy, and the binding of these molecules causes conformational changes in the protein. We therefore tested whether the addition of GDP or a nonhydrolysable analog of GTP affects the ability of Efm7 to methylate recombinant eEF1A in vitro. Addition of either GDP or the GTP analog was found to approximately double the relative amount of dimethylation observed and, importantly, resulted in the formation of small amounts of trimethylation (Fig. 3A, Supplemental Fig. S7). These results indicate that the conformation of eEF1A is important for recognition and methylation by Efm7 and suggest that Efm7 may make critical contacts with eEF1A at sites apart from the site of methylation. We next sought to investigate which domains of eEF1A are important for this process. We analyzed the ability of Efm7 to methylate domain 1 of eEF1A in isolation (residues 1–238) or domains 1 + 2 together (residues 1–334) and compared this to the methylation of full length eEF1A (domains 1 + 2 + 3, residues 1–458). In vitro methylation of recombinant versions of these proteins revealed that Efm7 is capable of methylating both domain 1 and domains 1 + 2 (Fig. 3B, Supplemental Fig. S8). In fact, domain 1 and domains 1 + 2 were dimethylated more than full length eEF1A, with domain 1 also being trimethylated to a small degree (Fig. 3B, Supplemental Fig. S8). This indicates that while domain 1 alone is sufficient for methylation by Efm7, the presence of the other domains can affect the enzyme–substrate interaction. Overall, these results indicate that the conformation of eEF1A affects the capacity of Efm7 to methylate but that only domain 1 is required for methylation to occur.
Fig. 3.
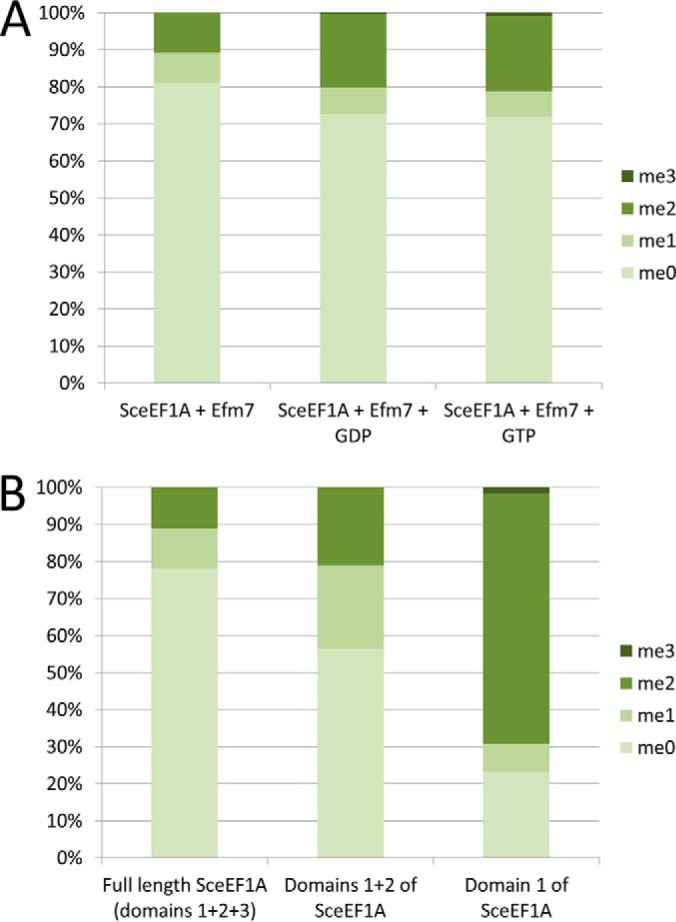
Methylation of eEF1A by Efm7 is affected by the structure and conformation of eEF1A. (A) In vitro methylation of eEF1A by Efm7 is enhanced by the addition of either GDP or GTP. Recombinant yeast eEF1A was in vitro methylated by Efm7 in the presence or absence of GDP or a GTP analog. eEF1A was found to be more dimethylated, and trimethylated to a small degree, in the presence of GDP or the GTP analog, indicating that the ability of Efm7 to methylate eEF1A is affected by its conformation. (B) In vitro methylation of recombinant full length and C-terminal truncations of eEF1A indicates that Efm7 does not require domains 2 or 3 in order to methylate eEF1A. For both (A) and (B), the graphs represents the relative amounts of each methylation state as determined by the area under the curve of the extracted ion chromatograms, which are shown as Supplemental Figs. S7 and S8 for (A) and (B), respectively.
N-terminal Trimethylation of eEF1A Is Conserved in Human
Given the high degree of conservation of many other N-terminal methylation sites (8), we investigated whether the N-terminal methylation of eEF1A in yeast is conserved in human eEF1A. Utilizing an AspN digest of proteins from HEK293T cells, we identified an N-terminal peptide GKEKTHINIVVIGHV of 843.51 m/z from HseEF1A, carrying trimethylation on Gly2 (Fig. 4A, Supplemental Table S1). As with the yeast peptide, there was a consistently detectable singly charged y14 ion. Parallel reaction monitoring analysis of the trimethylated peptide confirmed that it only produces unmethylated y14 ions of 1,586.94 m/z, and that therefore the trimethylation of the peptide and HseEF1A is entirely N-terminal (Fig. 4B). MS3 analysis of an MS2 precursor of 1,586.99 m/z confirmed the identity of the unmethylated y14 ion (Fig. 4C, Supplemental Table S2). Unlike yeast, we did not observe the tetra- or pentamethylated forms of the AspN peptide, suggesting that Lys3 is unmethylated in HseEF1A. It was not possible to differentiate between the two human isoforms of eEF1A (eEF1A1 and eEF1A2), as they are identical in sequence for the first 81 amino acids. However, this conservation of the N-terminal sequence of eEF1A makes it likely that both human isoforms are N-terminally methylated. N-terminal trimethylation of eEF1A is therefore a highly conserved modification between yeast and human.
Fig. 4.

Human eEF1A is trimethylated at its N terminus. (A) Q Exactive Plus MS/MS spectrum of a doubly charged AspN peptide of 843.51 m/z identifies trimethylation at the N terminus of human eEF1A (for mass-annotated spectrum see Supplemental Fig. S18). (B) Transitions from the trimethylated AspN peptide (GKEKTHINIVVIGHV+2) to the un-, mono-, di- and trimethylated forms of the y14 ion (KEKTHINIVVIGHV+1) indicate that y14 is completely unmethylated in the trimethylated peptide. This indicates that the N terminus is trimethylated. The mono- and dimethylated AspN peptides were not abundant enough for this analysis. (C) MS/MS/MS (MS3) spectrum of an MS2 precursor of 1586.99 m/z confirms its identity as the unmethylated y14 ion (for mass-annotated spectrum see Supplemental Fig. S19).
Since we established Efm7 as the methyltransferase responsible for the N-terminal methylation of yeast eEF1A and that human eEF1A is also N-terminally methylated, we sought to investigate whether Efm7 could N-terminally methylate human eEF1A in vitro. Incubation of HseEF1A1 with Efm7 in the presence of AdoMet did not result in N-terminal methylation (Supplemental Fig. S9), although Efm7 was able to methylate SceEF1A in vitro under the same conditions (see above). This suggests that, although N-terminal methylation is conserved between yeast and human, the methyltransferases that catalyze the reaction have diverged and that the eEF1A from one species can only be N-terminally methylated by its own methyltransferase. It is not immediately obvious what protein is likely to be the methyltransferase responsible for N-terminal methylation of eEF1A in human. The closest aligning human protein to Efm7 is METTL21C (BLASTp (Basic Local Alignment Search Tool) expect value of 4 × 10−9). However, we note that the reciprocal is not true, as the closest aligning yeast protein to METTL21C is YNL024C/Efm6 (BLASTp expect value of 7 × 10−14). Efm7 is the second-closest aligning yeast protein to human METTL21C (BLASTp expect value of 9 × 10−11).
Methyltransferases That Act on Lysine 79 in eEF1A Are Conserved from Yeast to Human
In yeast, a recent report showed loss of eEF1A trimethylation at Lys79 upon knockout of YGR001C. This enzyme was then renamed Efm5 (18). We were able to confirm this result utilizing the recently described LysargiNase Archaea protease. This cleaves N-terminally to lysine and arginine and, importantly, allows the abundance of methylated and unmethylated peptides to be compared as it cuts effectively at methyl-lysines and -arginines (27). With LysargiNase, we observed a complete loss of mono-, di-, and trimethylation of Lys79 upon knockout of EFM5 (Supplemental Fig. S10).
Lysine 79 of human eEF1A has been reported as trimethylated in recent high-throughput studies (33–35). We confirmed this with LC-MS/MS analysis of HEK293T proteins (Supplemental Fig. S11, Supplemental Table S1). Since this methylation site is conserved between yeast and human, we searched for the human ortholog of Efm5. A BLASTp search of Efm5 against human proteins in SwissProt returned N6AMT2 (putative N(6)-adenine-specific DNA methyltransferase) as the best match with an expected value of 3 × 10−41; the reciprocal search returned Efm5 as the best match with an expected value of 7 × 10−42. The alignment of Efm5 and N6AMT2 showed very high sequence identity and the presence of the characteristic seven-beta-strand motifs I and Post-I (Fig. 5). N6AMT2 also showed a [D/N]XX[Y/F] motif, which is suggestive of nitrogen methylation activity (18). We therefore investigated whether N6AMT2 could methylate human and/or yeast eEF1A in vitro. Additionally, we sought to confirm Efm5 methyltransferase activity in vitro, as this has not yet been reported.
Fig. 5.

Sequence alignment of Efm5 and its human ortholog, N6AMT2. Efm5 and N6AMT2 were aligned using EMBOSS Stretcher (EMBL-EBI), indicating 36.1% identity and 53.4% similarity. Vertical lines (∣) indicate identical residues; double dots (:) indicate chemically similar residues; single dots (.) indicate dissimilar residues; dashes (-) indicate missing residues. The characteristic seven-beta-strand methyltransferase motifs I and Post-I are indicated according to (53), as well as the [D/N]XX[Y/F] motif proposed to be defining of nitrogen methylation (18). A BLASTp search of Efm5 against human proteins in SwissProt returns N6AMT2 as the best match with an expect value of 3 × 10−41; the reciprocal search returns Efm5 as the best match with an expect value of 7 × 10−42.
Incubation of purified Efm5 or N6AMT2 (see Supplemental Fig. S5) with recombinant unmethylated yeast eEF1A (Fig. 6A) or recombinant unmethylated human eEF1A1 (the predominant isoform of eEF1A in humans) (Fig. 6B) resulted in a gain of methylation at Lys79. MS/MS spectra of the trimethylated LysargiNase peptides from SceEF1A (KFETPKYQVTVIDAPGHof 658.02 m/z) and HseEF1A1 (KFETS of 653.35 m/z) are shown in Supplemental Figs. S12 and S13, respectively. Therefore, Efm5 and N6AMT2 can methylate both yeast and human eEF1A in vitro. This indicates that trimethylation of Lys79 in eEF1A and the methyltransferases that catalyze it are highly conserved in eukaryotes. Interestingly, Efm5 appeared to methylate SceEF1A more efficiently than HseEF1A1, and similarly, N6AMT2 appeared to methylate HseEF1A1 more efficiently than SceEF1A. This indicates that, despite strong conservation, there may be subtle differences between the enzymes and/or substrates that allow methylation to proceed more efficiently with enzymes and/or substrates from the same organism. This may be due to sequence variants in eEF1A, such as the 76A->76S substitution between yeast and human just upstream of the methylated Lys79 or structural features distal in sequence but proximal in three dimensions. In line with recent naming convention (23) and since there are likely to be numerous methyltransferases of eEF1A in human, we suggest N6AMT2 be renamed eEF1A-KMT1.
Fig. 6.

N6AMT2 is the human ortholog of Efm5. N6AMT2 or Efm5 was incubated with either SceEF1A (A) or HseEF1A1 (B), both purified from E. coli, in the presence of AdoMet. The methylation status of Lys79 was analyzed by taking mass windows (±10 ppm) corresponding to all relevant methylation states of the LysargiNase peptides KFETPKYQVTVIDAPGH+3 (SceEF1A) or KFETS+1 (HseEF1A1) (red). Peaks were normalized to the most abundant ion for each methylation state. The abundance of the eEF1A LysargiNase peptide KIGGIGTVPVG+2 is shown as an internal standard (black). Elution times of peptides are shaded; peaks outside shading are unrelated, near-isobaric ions. Lys79 from both SceEF1A (A) and HseEF1A1 (B) was found to be mono-, di- and trimethylated only when incubated with Efm5 or N6AMT2, with the exception that trimethylation of Lys79 was not detected on HseEF1A1 when incubated with Efm5. Therefore, Efm5 and N6AMT2 were able to methylate both yeast eEF1A and human eEF1A1 at lysine 79 in vitro. Trimethylation of lysine 79 on eEF1A is therefore a highly conserved modification catalyzed by a methyltransferase that is conserved from yeast to human. We propose N6AMT2 be renamed eEF1A-KMT1.
DISCUSSION
Here, we have shown that the new yeast enzyme Efm7, which was previously predicted to be a nicotinamide N-methyltransferase (36), is in fact a protein N-terminal and lysine methyltransferase of eEF1A. It represents a new type of N-terminal methyltransferase as it belongs to the Family 16 group of methyltransferases (Pfam family PF10294), unlike the three other known eukaryotic N-terminal methyltransferases that belong to a family of proteins all predicted to be N-terminal methyltransferases (Pfam family PF05891). So far, all Family 16 methyltransferases have proved to be lysine specific (21, 23, 37–40). Efm7 therefore also represents the first Family 16 methyltransferase that is not lysine specific. It is not surprising that Efm7 is the methyltransferase responsible for both N-terminal and Lys3 methylation of yeast eEF1A. This is because there is very high local chemical similarity between the N-terminal glycine and the lysine sidechain (Fig. 7). Hence, although the α-amine of the N-terminal glycine is the favored substrate, the ε-amine of the lysine sidechain is a secondary substrate that is only methylated when the α-amine is saturated. Nonetheless, Efm7 represents the first methyltransferase that can methylate at both the N terminus of a protein as well as another, adjacent residue.
Fig. 7.

Methylation of yeast eEF1A by five methyltransferases. The structure of yeast eEF1A (PDB ID: 1F60) showing the five methyltransferases that act on it and their substrate residues. eEF1A is shown as a yellow ribbon structure; methylated residues are shown as stick structures (N-terminal Gly2 and Lys3 (K3): green; Lys30 (K30), Lys316 (K316), and Lys390 (K390): black; Lys79 (K79): red), with added methyl groups shown in orange. Inset: the chemical structure of the trimethylated N terminus and dimethylated Lys3 is shown, showing the local chemical similarity between the N-terminal α-amine of the glycine and the sidechain ε-amine of the lysine. eEF1A was visualized in PyMOL (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC.).
It is possible that eEF1A is the only substrate of Efm7. Previous in vitro methylation of yeast lysate with YLR285W/Efm7 showed methylation of a single ∼50kDa band, which corresponds to the mass of eEF1A (41). However, given that eEF1A is one of the most abundant proteins in the cell, it is possible that other, much less abundant substrates may have gone undetected. Additionally, in vitro conditions may not have been conducive to methylation of all substrates. It therefore remains possible that Efm7 has other substrates; indeed, there are 35 other yeast proteins with an N-terminal glycine and then a lysine, assuming removal of the initiating methionine. However, as Efm7 requires structural features for recognition and methylation of its substrate, it would be difficult to predict what these would be. If Efm7 has only one substrate, this would be consistent with that observed for a number of other Family 16 methyltransferases (23, 37, 39). While it is costly for the cell to produce a methyltransferase just to modify a single protein, a marginal increase in, for example, protein translation efficiency by the modification of eEF1A could outweigh this and provide an evolutionary advantage.
Functionally, ΔYLR285W/EFM7 has been reported to have decreased silencing at the rDNA locus compared with wild type (36). This was thought to be related to its putative function as a nicotinamide N-methyltransferase. However, in light of its protein methyltransferase activity, it could that this phenotype is related to the loss of methylation on eEF1A or it could be due to loss of methylation on another, as yet unknown substrate of Efm7. Structurally, the N-terminal methylation catalyzed by Efm7 is located in domain 1 of eEF1A, which is its GTPase domain. However, the site is not located near the GTP binding pocket and, hence, is unlikely to affect the GTPase activity of eEF1A. Rather, it is located next to the tRNA binding cleft of eEF1A. As trimethylation introduces a permanent positive charge on the α-amino group of the N terminus, it has been suggested this may facilitate binding to the negatively charged phosphate backbone of polynucleotides (7). Indeed, it has been shown that N-terminal methylation can affect the interaction of proteins with DNA (42). The trimethylation may therefore enhance the tRNA-binding capacity of eEF1A. Alternatively, it may affect the protein–protein interactions of eEF1A in a manner similar to that of lysine and arginine methylation of proteins (2, 43, 44). Given the conservation of this modification between yeast and human, it will be of interest to investigate the functional role of eEF1A N-terminal methylation in more detail.
The closest aligning human protein to Efm7 is METTL21C. However, the reciprocal BLAST returns the recently described Efm6, which methylates eEF1A at Lys390 (19). It is therefore unclear if METTL21C is indeed the ortholog of Efm7 and therefore the human eEF1A N-terminal methyltransferase. Rather, it may be that another closely aligning human methyltransferase of no known substrate is the ortholog of Efm7, such as METTL21B or METTL23. These are also members of the Family 16 group of methyltransferases.
This study also reports N6AMT2 as the human ortholog of Efm5. Both were shown to act as lysine methyltransferases specific to eEF1A Lys79 in vitro, extending previous observations of the loss of methylation on knockout of EFM5 (18). In yeast, Efm5 is cytosolic and exists in medium abundance (45). In human, N6AMT2/eEF1A-KMT1 is expressed in most tissues at medium to high levels and is also predominantly cytosolic (46, 47). This localization matches that of eEF1A. Interestingly, N6AMT2/eEF1A-KMT1 is overexpressed in many cancers (48), which may reflect a translational activating function of the methylation of Lys79 in eEF1A due to the increased protein synthesis needs of fast-growing cells.
Until recently, N6AMT2/eEF1A-KMT1 and Efm5 were annotated to be N(6)-adenine-specific DNA methyltransferases. There is another example of misclassification of N(6)-adenine-specific DNA methyltransferases; the human protein N6AMT1 was found to be a protein glutamine methyltransferase specific to Q185 of peptide chain release factor subunit 1 (49). The above errors in classification, along with our disproving the notion that Family 16 methyltransferases are lysine specific, demonstrate the difficulty in assigning methyltransferase substrate specificity based on sequence alone. Nevertheless, it was recently proposed that a (D/N)XX(Y/F) motif, which is present in Efm5, eEF1A-KMT1, and Efm7, may be a general motif for the recognition and methylation of nitrogen atoms (18). This is similar to the (D/E)XX(Y/F) motif characteristic of Family 16 methyltransferases (37) and suggests that a general (D/E/N)XX(Y/F) motif may be associated with nitrogen methylation.
There are now a total of five known methyltransferases that target eEF1A in yeast (Fig. 7). While the exact function of their resulting methylation sites is unknown, the fact that they act upon one protein suggests they may modulate various functions of eEF1A (16). The differences in stoichiometry of the methylation sites, which ranges from ∼85% for the N-terminal methylation to between 2.5 and 13% for other sites (50), is also suggestive of this. However, the stoichiometry observed here for the N-terminal methylation should be confirmed by a more accurate measurement, such as absolute quantification, to allow a direct comparison with previous studies (50). It is likely that all methylation sites can co-occur, as previous whole protein mass spectrometric analysis found that eEF1A exists at an average mass roughly equal to what is expected for all lysine methylation sites plus N-terminal acetylation (17), now known to be N-terminal trimethylation. To investigate the function of each methylation site, the knockout of each corresponding methyltransferase could be compared with wild-type yeast for a number of different cellular functions that eEF1A is known to be involved in, such as protein translation, actin bundling, and proteasomal degradation (16). It is interesting to note that there are few other proteins known to be methylated by so many methyltransferases. One such example is the tumor suppressor protein p53, which is methylated at four distinct sites by five different methyltransferases (51). Each of these sites is known to have different roles in modulating the activity of p53 (51). Another example is histone H3, which is methylated at nine sites by many different methyltransferases, all of which have distinct roles in modulating transcription (52). It is therefore plausible that the eEF1A methyltransferases have distinct roles in modulating the activity of eEF1A. This may be by activating or inactivating eEF1A or by switching which cellular process it partakes in by modulating its interactions, given that lysine methylation is known to modulate protein–protein interactions (44).
Supplementary Material
Acknowledgments
Mass spectrometric results were obtained at the Bioanalytical Mass Spectrometry Facility within the Analytical Centre of the University of New South Wales. HEK293T cell lysate was a kind gift from Dr. Winnie Luu and Prof. Andrew Brown.
Footnotes
Author contributions: J.J.H., D.L.W., G.H., and M.R.W. designed the research; J.J.H., D.L.W., and D.Y. performed the research; C.M.O. contributed new reagents or analytic tools; J.J.H. analyzed data; and J.J.H., D.L.W., D.Y., C.M.O., G.H., and M.R.W. wrote the paper.
* This work was supported by various grants. J.J.H., D.L.W. and D.Y. acknowledge the support of Australian Postgraduate Awards. J.J.H. and D.L.W. acknowledge additional support from the University of New South Wales. M.R.W. and G.H.S. acknowledge support from the Australian Research Council. G.H.S. acknowledges additional support from the University of New South Wales. C.M.O. is supported as a Canada Research Chair in Protease Proteomics and Systems Biology.
 This article contains supplemental material Supplemental Tables S1 and S2 and Supplemental Figs. S1-S19.
This article contains supplemental material Supplemental Tables S1 and S2 and Supplemental Figs. S1-S19.
1 The abbreviations used are:
- SceEF1A
- Saccharomyces cerevisiae eukaryotic elongation factor 1A
- HseEF1A
- Homo sapiens eukaryotic elongation factor 1A
- KMT
- lysine methyltransferase
- SILAC
- stable isotope labeling by amino acids in cell culture.
REFERENCES
- 1.Khoury G. A., Baliban R. C., and Floudas C. A. (2011) Proteome-wide post-translational modification statistics: Frequency analysis and curation of the Swiss-prot database. Sci. Rep. 1, Article number: 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erce M. A., Pang C. N., Hart-Smith G., and Wilkins M. R. (2012) The methylproteome and the intracellular methylation network. Proteomics 12, 564–586 [DOI] [PubMed] [Google Scholar]
- 3.Winter D. L., Erce M. A., and Wilkins M. R. (2014) A web of possibilities: Network-based discovery of protein interaction codes. J. Proteome Res. 13, 5333–5338 [DOI] [PubMed] [Google Scholar]
- 4.Jenuwein T., and Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 5.Gu B., and Zhu W. G. (2012) Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 8, 672–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarke S. G. (2013) Protein methylation at the surface and buried deep: Thinking outside the histone box. Trends Biochem. Sci. 38, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stock A., Clarke S., Clarke C., and Stock J. (1987) N-terminal methylation of proteins: Structure, function and specificity. FEBS Lett. 220, 8–14 [DOI] [PubMed] [Google Scholar]
- 8.Tooley C. E., Petkowski J. J., Muratore-Schroeder T. L., Balsbaugh J. L., Shabanowitz J., Sabat M., Minor W., Hunt D. F., and Macara I. G. (2010) NRMT is an alpha-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature 466, 1125–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webb K. J., Lipson R. S., Al-Hadid Q., Whitelegge J. P., and Clarke S. G. (2010) Identification of protein N-terminal methyltransferases in yeast and humans. Biochemistry 49, 5225–5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petkowski J. J., Schaner Tooley C. E., Anderson L. C., Shumilin I. A., Balsbaugh J. L., Shabanowitz J., Hunt D. F., Minor W., and Macara I. G. (2012) Substrate specificity of mammalian N-terminal alpha-amino methyltransferase NRMT. Biochemistry 51, 5942–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petkowski J. J., Bonsignore L. A., Tooley J. G., Wilkey D. W., Merchant M. L., Macara I. G., and Schaner Tooley C. E. (2013) NRMT2 is an N-terminal monomethylase that primes for its homologue NRMT1. Biochem. J. 456, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen T., Muratore T. L., Schaner-Tooley C. E., Shabanowitz J., Hunt D. F., and Macara I. G. (2007) N-terminal alpha-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat. Cell Biol. 9, 596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hao Y., and Macara I. G. (2008) Regulation of chromatin binding by a conformational switch in the tail of the Ran exchange factor RCC1. J. Cell Biol. 182, 827–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai Q., Fu L., Wang Z., Gan N., Dai X., and Wang Y. (2014) alpha-N-methylation of damaged DNA-binding protein 2 (DDB2) and its function in nucleotide excision repair. J. Biol. Chem. 289, 16046–16056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey A. O., Panchenko T., Sathyan K. M., Petkowski J. J., Pai P. J., Bai D. L., Russell D. H., Macara I. G., Shabanowitz J., Hunt D. F., Black B. E., and Foltz D. R. (2013) Posttranslational modification of CENP-A influences the conformation of centromeric chromatin. Proc. Natl. Acad. Sci. U.S.A. 110, 11827–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mateyak M. K., and Kinzy T. G. (2010) eEF1A: Thinking outside the ribosome. J. Biol. Chem. 285, 21209–21213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipson R. S., Webb K. J., and Clarke S. G. (2010) Two novel methyltransferases acting upon eukaryotic elongation factor 1A in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 500, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dzialo M. C., Travaglini K. J., Shen S., Loo J. A., and Clarke S. G. (2014) A new type of protein lysine methyltransferase trimethylates Lys-79 of elongation factor 1A. Biochem. Biophys. Res. Commun. 455, 382–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakobsson M. E., Davydova E., Malecki J., Moen A., and Falnes P. Ø. (2015) Saccharomyces cerevisiae eukaryotic elongation factor 1A (eEF1A) is methylated at Lys-390 by a METTL21-like methyltransferase. PLoS ONE 10, e0131426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimazu T., Barjau J., Sohtome Y., Sodeoka M., and Shinkai Y. (2014) Selenium-based S-adenosylmethionine analog reveals the mammalian seven-beta-strand methyltransferase METTL10 to be an EF1A1 lysine methyltransferase. PloS One 9, e105394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couttas T. A., Raftery M. J., Padula M. P., Herbert B. R., and Wilkins M. R. (2012) Methylation of translation-associated proteins in Saccharomyces cerevisiae: Identification of methylated lysines and their methyltransferases. Proteomics 12, 960–972 [DOI] [PubMed] [Google Scholar]
- 22.Zhang L., Hamey J. J., Hart-Smith G., Erce M. A., and Wilkins M. R. (2014) Elongation factor methyltransferase 3—A novel eukaryotic lysine methyltransferase. Biochem. Biophys. Res. Commun. 451, 229–234 [DOI] [PubMed] [Google Scholar]
- 23.Davydova E., Ho A. Y., Malecki J., Moen A., Enserink J. M., Jakobsson M. E., Loenarz C., and Falnes P. Ø. (2014) Identification and characterization of a novel evolutionarily conserved lysine-specific methyltransferase targeting eukaryotic translation elongation factor 2 (eEF2). J. Biol. Chem. 289, 30499–30510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dzialo M. C., Travaglini K. J., Shen S., Roy K., Chanfreau G. F., Loo J. A., and Clarke S. G. (2014) Translational roles of elongation factor 2 protein lysine methylation. J. Biol. Chem. 289, 30511–30524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu J., Tillett D., Dawes I. W., and March P. E. (2008) Site-directed, ligase-independent mutagenesis (SLIM) for highly efficient mutagenesis of plasmids greater than 8kb. J. Microbiol. Methods 73, 195–198 [DOI] [PubMed] [Google Scholar]
- 26.Mok J., Im H., and Snyder M. (2009) Global identification of protein kinase substrates by protein microarray analysis. Nat. Protoc. 4, 1820–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huesgen P. F., Lange P. F., Rogers L. D., Solis N., Eckhard U., Kleifeld O., Goulas T., Gomis-Rüth F. X., and Overall C. M. (2015) LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification. Nat. Methods 12, 55–58 [DOI] [PubMed] [Google Scholar]
- 28.Hart-Smith G., and Raftery M. J. (2012) Detection and characterization of low abundance glycopeptides via higher-energy C-trap dissociation and Orbitrap mass analysis. J. Am. Soc. Mass. Spectrom. 23, 124–140 [DOI] [PubMed] [Google Scholar]
- 29.Kessner D., Chambers M., Burke R., Agus D., and Mallick P. (2008) ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavallius J., Zoll W., Chakraburtty K., and Merrick W. C. (1993) Characterization of yeast EF-1 alpha: Non-conservation of post-translational modifications. Biochim. Biophys. Acta 1163, 75–80 [DOI] [PubMed] [Google Scholar]
- 32.Szczepińska T., Kutner J., Kopczynski M., Pawlowski K., Dziembowski A., Kudlicki A., Ginalski K., and Rowicka M. (2014) Probabilistic approach to predicting substrate specificity of methyltransferases. PLoS Comput. Biol. 10, e1003514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K. A., Yang V., Aguiar M., Kornhauser J., Jia X., Ren J., Beausoleil S. A., Silva J. C., Vemulapalli V., Bedford M. T., and Comb M. J. (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 13, 372–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bremang M., Cuomo A., Agresta A. M., Stugiewicz M., Spadotto V., and Bonaldi T. (2013) Mass spectrometry-based identification and characterisation of lysine and arginine methylation in the human proteome. Mol. Biosyst. 9, 2231–2247 [DOI] [PubMed] [Google Scholar]
- 35.Cao X. J., Arnaudo A. M., and Garcia B. A. (2013) Large-scale global identification of protein lysine methylation in vivo. Epigenetics 8, 477–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., and Sinclair D. A. (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kernstock S., Davydova E., Jakobsson M., Moen A., Pettersen S., Maelandsmo G. M., Egge-Jacobsen W., and Falnes P. Ø. (2012) Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nat. Commun. 3, 1038. [DOI] [PubMed] [Google Scholar]
- 38.Cloutier P., Lavallee-Adam M., Faubert D., Blanchette M., and Coulombe B. (2013) A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS Genet. 9, e1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jakobsson M. E., Moen A., Bousset L., Egge-Jacobsen W., Kernstock S., Melki R., and Falnes P. Ø. (2013) Identification and characterization of a novel human methyltransferase modulating Hsp70 protein function through lysine methylation. J. Biol. Chem. 288, 27752–27763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magnani R., Dirk L. M., Trievel R. C., and Houtz R. L. (2010) Calmodulin methyltransferase is an evolutionarily conserved enzyme that trimethylates Lys-115 in calmodulin. Nat. Commun. 1, 43. [DOI] [PubMed] [Google Scholar]
- 41.Wlodarski T., Kutner J., Towpik J., Knizewski L., Rychlewski L., Kudlicki A., Rowicka M., Dziembowski A., and Ginalski K. (2011) Comprehensive structural and substrate specificity classification of the Saccharomyces cerevisiae methyltransferome. PloS One 6, e23168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai X., Otake K., You C., Cai Q., Wang Z., Masumoto H., and Wang Y. (2013) Identification of novel alpha-n-methylation of CENP-B that regulates its binding to the centromeric DNA. J. Proteome Res. 12, 4167–4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erce M. A., Abeygunawardena D., Low J. K., Hart-Smith G., and Wilkins M. R. (2013) Interactions affected by arginine methylation in the yeast protein-protein interaction network. Mol. Cell. Proteomics 12, 3184–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winter D. L., Abeygunawardena D., Hart-Smith G., Erce M. A., and Wilkins M. R. (2015) Lysine methylation modulates the protein-protein interactions of yeast cytochrome C Cyc1p. Proteomics 15, 2166–2176 [DOI] [PubMed] [Google Scholar]
- 45.Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., and Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 46.Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., and Pontén F. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 47.Fagerberg L., Stadler C., Skogs M., Hjelmare M., Jonasson K., Wiking M., Abergh A., Uhlén M., and Lundberg E. (2011) Mapping the subcellular protein distribution in three human cell lines. J. Proteome Res. 10, 3766–3777 [DOI] [PubMed] [Google Scholar]
- 48.Uhlen M., Björling E., Agaton C., Szigyarto C. A., Amini B., Andersen E., Andersson A. C., Angelidou P., Asplund A., Asplund C., Berglund L., Bergstrom K., Brumer H., Cerjan D., Ekström M., Elobeid A., Eriksson C., Fagerberg L., Falk R., Fall J., Forsberg M., Björklund M. G., Gumbel K., Halimi A., Hallin I., Hamsten C., Hansson M., Hedhammar M., Hercules G., Kampf C., Larsson K., Lindskog M., Lodewyckx W., Lund J., Lundeberg J., Magnusson K., Malm E., Nilsson P., Odling J., Oksvold P., Olsson I., Oster E., Ottosson J., Paavilainen L., Persson A., Rimini R., Rockberg J., Runeson M., Sivertsson A., Sköllermo A., Steen J., Stenvall M., Sterky F., Stromberg S., Sundberg M., Tegel H., Tourle S., Wahlund E., Waldén A., Wan J., Wernérus H., Westberg J., Wester K., Wrethagen U., Xu L. L., Hober S., and Pontén F. (2005) A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 4, 1920–1932 [DOI] [PubMed] [Google Scholar]
- 49.Figaro S., Scrima N., Buckingham R. H., and Heurgué-Hamard V. (2008) HemK2 protein, encoded on human chromosome 21, methylates translation termination factor eRF1. FEBS Lett. 582, 2352–2356 [DOI] [PubMed] [Google Scholar]
- 50.Hart-Smith G., Chia S. Z., Low J. K., McKay M. J., Molloy M. P., and Wilkins M. R. (2014) Stoichiometry of Saccharomyces cerevisiae lysine methylation: Insights into non-histone protein lysine methyltransferase activity. J Proteome Res 13, 1744–1756 [DOI] [PubMed] [Google Scholar]
- 51.West L. E., and Gozani O. (2011) Regulation of p53 function by lysine methylation. Epigenomics 3, 361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greer E. L., and Shi Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrossian T. C., and Clarke S. G. (2009) Multiple motif scanning to identify methyltransferases from the yeast proteome. Mol. Cell. Proteomics 8, 1516–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.