

Abstract
Gambogic acid (GA) is an anticancer agent in phase IIb clinical trial in China. In HeLa cells, GA inhibited cell proliferation, induced cell cycle arrest at G2/M phase and apoptosis, as showed by results of MTT assay and flow cytometric analysis. Possible target-related proteins of GA were searched using comparative proteomic analysis (2-DE) and nine proteins at early (3 h) stage together with nine proteins at late (24 h) stage were found. Vimentin was the only target-related protein found at both early and late stage. Results of both 2-DE analysis and Western blotting assay suggested cleavage of vimentin induced by GA. MS/MS analysis of cleaved vimentin peptides indicated possible cleavage sites of vimentin at or near ser51 and glu425. Results of targeted proteomic analysis showed that GA induced change in phosphorylation state of the vimentin head domain (aa51–64). Caspase inhibitors could not abrogate GA-induced cleavage of vimentin. Over-expression of vimentin ameliorated cytotoxicity of GA in HeLa cells. The GA-activated signal transduction, from p38 MAPK, heat shock protein 27 (HSP27), vimentin, dysfunction of cytoskeleton, to cell death, was predicted and then confirmed. Results of animal study showed that GA treatment inhibited tumor growth in HeLa tumor-bearing mice and cleavage of vimentin could be observed in tumor xenografts of GA-treated animals. Results of immunohistochemical staining also showed down-regulated vimentin level in tumor xenografts of GA-treated animals. Furthermore, compared with cytotoxicity of GA in HeLa cells, cytotoxicity of GA in MCF-7 cells with low level of vimentin was weaker whereas cytotoxicity of GA in MG-63 cells with high level of vimentin was stronger. These results indicated the important role of vimentin in the cytotoxicity of GA. The effects of GA on vimentin and other epithelial-to-mesenchymal transition (EMT) markers provided suggestion for better usage of GA in clinic.
Presently, targeted anticancer therapies using monoclonal antibodies or synthetic protein kinase inhibitors are still deficient to meet the large and urgent need for novel cancer therapy agents, especially for solid tumors. Therefore, natural products continue to be attractive sources of new drug development. Gambogic acid (GA)1 is a natural product isolated from Garcinia hanburyi tree grown in Southeast Asia. The structure of GA (C38H44O8, molecular mass 628) (as shown in Fig. 1A) was elucidated by detailed NMR spectroscopic analysis and confirmed by x-ray crystallographic studies (1). Crude extract containing GA had been used in cancer therapy for a long time in China. In January 2004, GA was granted permission by State Food and Drug Administration of China for testing in clinical trial as a wide spectrum antitumor candidate. Up to now, the phase I and IIa trials of GA have finished and the IIb trial is undergoing to check the efficacy of GA in treatment of nonsmall cell lung, renal and colon cancers. However, the anticancer mechanism of GA had not been fully clarified. Lots of possible target-related proteins of GA such as p38 MAPK (2, 3), transferrin receptor (4), kir2.1 K+ channel (5), caspases (6), Bax and Bcl-2 (7), nucleophosmin and nucleoporins (8), NF-kappa B pathway (9) etc., had been reported. But, it was still not clear which target or signal pathway played critical role in the anticancer effects of GA.
Fig. 1.

GA exhibited cytotoxicity on HeLa cells. A, Chemical structure of gambogic acid (GA). B, Effects of GA on viability of HeLa cells. Cells were treated with 0.3125, 0.625, 1.25, 2.5, 5 and 10 μm GA for 24, 48, or 72 h and cell viability was determined by MTT assay. Data were statistical results of three independent experiments (n = 3, mean ± S.E.). C, GA induced G2/M cell cycle arrest in HeLa cells after 24 h treatment. Representative DNA histograms of HeLa cells obtained by flow cytometry analysis. D, Statistical analysis results of percentage of HeLa cells in different cell cycle phases after treated with GA at different doses for 24 h (n = 3, mean ± S.E.). E, GA induced apoptosis in HeLa cells after 24 h treatment. Apoptotic cells were quantified by double supravital staining with Annexin V-FITC and PI. Images were representative results of flow cytometry analysis. F, Statistical analysis results of percentage of apoptotic cells after treated with GA at different doses for 24 h (n = 3, mean ± S.E.).
Development of systems biology shed new light on the mechanism study of natural products. Systems pharmacology appeared as a new branch of pharmacology, which involved the application of systems biology approaches to the study of drugs, drug targets, and drug effects (10). In the present study, after checking the effects of GA on HeLa cells, proteomic methods including comparative proteomic method (2-DE analysis) and targeted proteomic method (nanoHPLC-ESI MS/MS analysis) were used to study important target-related proteins of GA. 2-DE analysis was used to unbiasedly search possible target-related proteins of GA at early stage (3 h) and late stage (24 h) of treatment. After finding vimentin as a possible important target-related protein of GA, cleavage of vimentin and phosphorylation of vimentin in GA-treated cells were further studied. The influence of increased expression of vimentin (by plasmid transfection) or decreased expression of vimentin (by siRNA transfection) on the cytotoxicity of GA was checked. Possible GA-activated p38 MAPK-HSP27-vimentin-cytoskeleton signal cascade pathway, which included published targets of GA such as p38 MAPK (2, 3) and cytoskeleton proteins (2) and new targets found in the present study such as HSP27 and vimentin, was predicted and then confirmed. In vivo effects of GA on tumor growth and vimentin expression in HeLa tumor-bearing mice were also observed. To confirm the role of vimentin in cytotoxicity of GA, cytotoxicity of GA in MCF-7 cells with low expression level of vimentin or in MG-63 cells with high expression level of vimentin was checked and compared with that in HeLa cells. Furthermore, because cellular vimentin was closely related to EMT, effects of GA on other EMT makers fibronectin, β-catenin, and E-cadherin were also checked.
EXPERIMENTAL PROCEDURES
Chemicals
GA with a purity of more than 97% was purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). GA was dissolved in dimethyl sulfoxide (DMSO) to the concentration of 0.1 m as stock solution and kept at −20 °C. It was then diluted in the culture medium to the final concentration indicated in every experiment. All reagents used in proteomic analysis were purchased from Bio-Rad Laboratories (Hercules, CA) and other chemical reagents, except where specially noted, were purchased from Sigma-Aldrich.
Cell Culture and MTT Assay
The human cervical carcinoma cell line HeLa (CCL-2), the human breast adenocarcinoma cell line MCF-7 (HTB-22) and the human osteosarcoma cell line MG-63 (CRL-1407) were obtained from the American Type Culture Collection (ATCC, Rockville, MD). HeLa cells were cultured in minimum essential medium supplemented with 10% fetal bovine serum, 100 U/ML penicillin, and 100 mg/L streptomycin (Invitrogen). MCF-7 cells and MG-63 cells were cultured in Eagle's minimum essential medium supplemented with 10% fetal bovine serum, 100 U/ML penicillin, and 100 mg/L streptomycin (Invitrogen).
Cell viability of cells with or without GA treatment was measured by MTT assay as described in our previous report (11). Briefly, cells were plated at a density of 9 × 102 cells/well in a 96-well plate and let to grow overnight. Then, the media were changed into fresh media containing various amount of GA for 24, 48, or 72 h. Control cells were treated with 0.1% DMSO (dose of GA at 0 μm). Cell viability of GA-treated cells was presented as % of control. Each experiment was performed in triplicate and results of three independent experiments were used for statistical analysis. Half-maximal inhibitory concentration (IC50 value) was calculated by the Logit method.
Flow Cytometric Analysis of Cell Cycle
Flow cytometric analysis of cell cycle was also carried out as reported before (11). Briefly, after treatment, both adherent and detached cells were collected, washed with PBS and then fixed in ice-cold 70% ethanol overnight at 4 °C. After centrifugation at 100 × g for 2 min, fixed cells were resuspended in hypotonic propidium iodide staining solution (0.1% Triton X-100, 10 μg/ml DNase-free RNase A, 50 μg/ml propidium iodide in PBS) for 30 min in dark. The stained cells were analyzed using a Becton Dickinson FACSCalibur Flow Cytometer and the percentage of cells in each phase of the cell cycle was quantitated using the ModFit software.
Flow Cytometric Analysis of Apoptosis
Apoptotic cells were quantified by using the apoptosis detection kit (Calbiochem, Gibbstown, NJ) as reported before (12). Briefly, after treatment, cells were collected, washed with PBS and then resuspended in binding buffer (2.5 mm CaCl2, 10 mm HEPES, and 140 mm NaCl, pH 7.5). Cells were let to incubate with Annexin V-FITC and propidium iodide for 10 min in the dark and then flow cytometric analysis was conducted. Data acquisition and analysis were performed in a FACSCalibur Flow Cytometer with CellQuest software.
2-DE Analysis and MS/MS Identification
For sample preparation, HeLa cells were cultured in 75 cm2 flasks at a density of 4 × 105 cells per flask. Cells were let to grow overnight and then treated with medium containing 2 μm GA (about the IC50 value) or 0.1% DMSO (solvent control). After incubation with GA for 3 h or 24 h, cells were washed three times with ice cold PBS and then scraped off with a cell scraper. After centrifugation for 5 min at 1500 × g, the supernatant was discarded and the cell pellet was resuspended in 200 μl lysis buffer containing 7 m urea, 2 m thiourea, 2% CHAPS, 1% DTT, 0.8% Pharmalyte, and a mixture of Protease inhibitors (all from Bio-Rad, Hercules, CA). Homogenization of the cells was achieved on ice by ultrasonication (10 strokes, low amplitude). The lysed cells were centrifuged at 15,000 × g for 30 min at 4 °C and the supernatant containing the solubilized proteins was used immediately or stored at −80 °C. Protein concentration of samples was determined using the Bio-Rad protein assay. Protein samples from three independent experiments were harvested for 2-DE assay. And, for each pair of protein samples (control and GA-treated), triplicate electrophoreses were performed to ensure reproducibility. 2-DE analysis and MS/MS identification were conducted similar to our previous reports (12–16).
2-DE was conducted with isoelectric focusing (IEF) and electrophoresis units (Bio-Rad). For the first dimensional separation, whole cell protein lysates (150 μg), mixed with rehydration buffer (8 m urea, 2% CHAPS, 65 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, and 0.2% pI 3–10 ampholytes) to a final volume of 300 μl, were applied on 17 cm immobilized pH 4 to 7 linear gradient strips (Bio-Rad) and focused. It was possible that proteins with pI>7 might be also involved in the effects of GA. Although, because most cellular proteins were acidic to natural proteins, pH 4 to 7 linear gradient strips were used in the present study. IEF protocol was performed using the following conditions: 250V, linear, 30 min; 1000V, rapid, 1 h; 10000V, linear, 5 h; 10,000V, rapid, 60,000 Vh. After IEF, pH gradient strips were subjected to a two-step equilibration in equilibration buffer (50 mm Tris-HCL, 30% glycerol; 7 m urea, 2% SDS, pH 8.8) with 1% dithiothreitol (the first step) and 4% iodoacetamide (the second step) for 15 min, respectively. For the second dimensional separation (SDS-PAGE), strips were transferred onto 1.0 mm-thick 12% polyacrylamide gels in a Bio-Rad PROTEAN II xi Cell system for electrophoresis at room temperature. All protein spots in the gels were visualized using Coomassie blue staining.
Stained gels were then scanned with a Densitometer GS-800 (Bio-Rad) and protein spots were quantified using the PDQuest 7.4.1 software (Bio-Rad). Paired (control and GA-treated) protein samples from three independent experiments were analyzed by 2-DE. And for each pair of protein samples, triplicate electrophoreses were performed to ensure reproducibility. Comparisons were made between gel images of protein profiles obtained from the GA-treated group and control group. Spot detection and matching between pairs of gels (control and GA-treated) were performed automatically, followed by manual matching. The individual protein spot quantity was normalized as follows: the raw quantity of each spot in a member gel was divided by the total quantity of the valid spots in the gel, and normalized spot intensities were expressed in ppm. A two-tailed Student's t test was performed to perform quantitative analysis between protein gels from control and GA-treated group. The significantly differentially expressed protein spots (p < 0.05) with 1.5 fold or more increased or decreased intensity between control and GA-treated group were punched out of the gels and used for further identification by MALDI-TOF MS/MS. Proteins of interest were excised from the gels with an EXQuest spot cutter (Bio-Rad) and placed into a 96-well microtiter plate.
MALDI-TOF MS/MS analysis was conducted with an Applied Biosystems 4700 Proteomics Analyzer (Framingham, MA) using the parameters as described in our previous report published in Molecular Cellular Proteomics (16). Briefly, the protein spots of interest were destained in a 1:1 solution of 100 mm sodium thiosulfate and 30 mm potassium ferricyanide for 2 min at room temperature and equilibrated in 50 mm ammonium bicarbonate to pH 8.0. After dehydrating with acetonitrile and drying in a SpeedVac, the spots were rehydrated in a minimal volume of trypsin solution (10 μg/ml in 25 mm ammonium bicarbonate) and incubated overnight at 37 °C. The supernatant containing extracted peptides was directly applied onto the sample plate with equal amounts of MALDI matrix (5 mg/ml CHCA diluted in 0.1% TFA/50% ACN) and spotted onto the 192-well stainless steel MALDI target plates after dried under the protection of N2.
MS measurements were carried out on an ABI 4700 Proteomics Analyzer with delayed ion extraction (Applied Biosystems, Fredrick, MD) in the Institutes of Biomedical Sciences, Fudan University, Shanghai, China. Instrument parameters were set using the 4000 Series Explorer software (Applied Biosystems). MS analysis was conducted using a Mass spectra and obtained in a mass range of 700–3200 Da by using a laser (355 nm, 200 Hz) as desorption ionization source. The accelerated voltage was operated at 20 kV, and the positive ion mass spectra were recorded. MS accuracy was internally calibrated with trypsin-digested peptides of horse myoglobin. The first five precursor ions with highest intensity were selected for fragmentation, excluding the trypsin autolysis peptides, human keratin peaks, and the matrix ion signals. For one main MS spectrum 25 subspectra with 125 shots per subspectrum were accumulated using a random search pattern. In MS/MS positive ion mode, for one main MS spectrum 50 subspectra with 50 shots per subspectrum were accumulated using a random search pattern. Collision energy was 1kV, collision gas was air and default calibration was set. Using the individual PMF spectra, peptides exceeding a signal-to-noise ratio of 20 that passed through a mass exclusion filter were submitted to fragmentation analysis. Total shots for each MS spectrum were 2000 whereas for MS/MS 3000 shots. MS/MS accuracy was calibrated against the MS/MS fragments of m/z 1606.85, which is one of the peaks generated in myoglobin PMF. Combined peptide mass fingerprinting PMF and MS/MS queries were performed by using the MASCOT search engine 2.2 (Matrix Science, London, United Kingdom) embedded into GPS-Explorer Software 3.6 (Applied Biosystems) on the NCBI database. Peptide differential modifications allowed during the search were oxidation of methionines and carbamidomethylation of cysteines. The maximum number of missed cleavages was set to 1 with trypsin as the protease. Peptide mass tolerance set was 100 ppm and fragment mass tolerance set was 0.4 Da. The database search was carried out by using the MASCOT search engine 2.2 (Matrix Science, London, United Kingdom) to screen the NCBI protein sequence database (updated on June 27, 2007, with 4182491 sequences and 1439956234 residues) restricted to human taxonomy. Protein homology identification of the top hit (first rank) with a relative score exceeding 95% probability and additional hits (second rank or more) with a relative score exceeding 98% probability threshold were retained. The probability-based protein score had to be more than 64 when submitting PMF data to the database and be more than 30 for individual peptide ions when submitting peptide sequence spectra, assuming that the observed match is significant (p < 0.05). Proteins that belonged to a protein family with multiple members were singled out based on the identification of unique and diagnostic peptides.
Western Blotting Analysis
Western blotting assay was conducted as described before (16). After treatment, cells were washed three times with cold TBS, harvested using a cell scraper, and lysed in 10 volume of cold lysis buffer (50 mm Tris-HCl, pH 7.2, 250 mm NaCl, 0.1% Nonidet P-40, 2 mm EDTA, 10% glycerol, 1 mm PMSF, 5 μg/ml Aprotinin, 5 μg/ml Leupeptin) on ice. The concentration of protein samples was quantified using the Bio-Rad protein assay. Lysates were centrifuged and then the supernatant protein was denatured by mixing with equal volume of 2× sample loading buffer and then boiling at 100 °C for 5 min. Equal amounts of protein samples (50 μg) were subjected to electrophoresis on 12% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane (Bio-Rad). After the nitrocellulose membrane was incubated with TBST buffer (TBS containing 0.1% Tween-20) and 10% nonfat milk for 1 h at 37 °C to block nonspecific protein binding, the membrane was incubated with primary antibodies overnight at 4 °C. The primary antibodies used were rabbit antiphospho(Thr180/Tyr182)-p38 MAPK monoclonal antibody (1:500), rabbit antiphospho(Thr334)-MAPKAPK 2 monoclonal antibody (1:500), rabbit antiphospho(Ser82)-HSP27 polyclonal antibody (1:500), mouse anti-HSP27 monoclonal antibody (1:500), mouse anti-vimentin (5G3F10) monoclonal antibody (1:500), rabbit anti-β-catenin polyclonal antibody (1:1000), rabbit anti-E-cadherin polyclonal antibody (1:600), all bought from Cell Signaling, Boston, MA. The primary antibody for fibronectin was rabbit anti-fibronectin polyclonal antibody (1:200), which was bought from Santa Cruz Biotechnology, Dallas, TX. The blots were washed three times with TBST and then incubated with HRP-conjugated goat anti-rabbit IgG (1:2000, Cell Signaling) or goat anti-mouse IgG (1:2000, Cell Signaling) for 1 h at 37 °C and then visualized using chemiluminescence (Pierce Biotechnology, Waltham, MA). All blots were stripped and reprobed with rabbit anti-GAPDH polyclonal antibody (1:1000, Cell Signaling, USA) to ascertain equal loading of protein.
MS/MS Analysis of the Protein Bands Detected in Western Blotting Assay as Vimentin
The protein bands identified as vimentin in Western blotting assay were cut and analyzed using MS/MS. Briefly, the 1-D gel protein bands of interest were cut into cubes (ca. 1 × 1 mm) and washed twice with 400 μl of 100 mm NH4HCO3/ACN (30%v/v). The Cys residues were firstly reduced by 100 μl of 10 mm DTT at 56 °C for 1 h and then alkylated by 100 μl of 60 mm iodoacetamide at room temperature for 20 min. After dehydration of the gel bands with ACN, the proteins were digested in gel overnight with 10 μl of 10 ng/μl trypsin (Promega) in 50 mm NH4HCO3 at room temperature. The generated peptides were extracted with 60% v/v ACN in 0.1% v/v formic acid. Afterward, the samples were completely dried in a vacuum centrifuge and then reconstituted in 0.1% formic acid. The nanoHPLC-ESI MS/MS analysis was carried out using a Tempo nanoMDLC System coupled to a 4000 Q TRAP system (AB Sciex Inc., Baltimore, MD). Each sample (5 μl) was loaded onto a trap column (PepMap™ 100, 300 μm x 5 mm), washed for 20 min, then eluted onto a C18 reversed phase column (PepMap100 C18, 75 μm x 5 μm × 15 cm) at a flow rate of 300 nL/min. Peptides were separated using a 120 min gradient ranging from 5% to 50% mobile phase B (mobile phase A: 2% acetonitrile, 0.1% formic acid; mobile phase B: 98% acetonitrile, 0.1% formic acid). The MS analysis was performed on the 4000 Q TRAP system in information dependent acquisition (IDA) mode with setting parameters: ion spray voltage: 2300 V; curtain gas: 20; ion source gas: 15; and heated interface: 150 °C. Tandem mass spectra were acquired over the mass range m/z 375–1500 using rolling collision energy for optimum peptide fragmentation. The mass spectrometry data files were then processed by ProteinPilot 3.0 (AB Sciex) using the Paragon algorithm. The mass spectrometry data were searched against all the protein sequence Homo sapiens of Uniprot_sprot_20100309 database. For ProteinPilot Paragon, Iodoacetamide was selected as the cysteine modification agent, trypsin as the digestion enzyme, “biological modifications” were selected as the “ID focus” and a “Thorough ID Search Effort” was selected. False discovery rate (FDR) analysis was performed using reversed protein sequences and was used to calculate the number of false positive proteins expected at a 95% confidence level. Peptides that passed a 1% FDR threshold were considered for protein identification.
MS/MS Analysis of the Phosphorylation State of Vimentin
For sample preparation, protein samples were extracted from control and GA-treated cells (2 μm GA for 3 h) using the same method as that of 2-DE analysis. The phosphorylation state of vimentin was checked using nanoHPLC-ESI MS/MS after enrichment of phosphopeptides. Phosphopeptides were enriched using TiO2 (17) and the nanoHPLC-ESI MS/MS analysis was performed with a TripleTOF 5600 system (AB Sciex) as reported before (18). Briefly, 1 mg protein sample was reduced with 100 mm ammonium bicarbonate and 100 mm dithiothreitol in a reaction volume of 250 μl by heating at 56 °C for 60 min. After cooling to room temperature, alkylation was induced by adding 200 mm iodoacetamide (250 μl) to each vial to get a final concentration of 100 mm and incubating for 20 min at room temperature. After alkylation, 1 μg/μl trypsin (10 μl) was added to each vial and digestion was allowed to occur at 37 °C overnight. Then, the sample was dried by evaporation at room temperature in a vacuum centrifuge and reconstituted to 30 μl with 0.1% formic acid in water and stored at 4 °C until analysis. β-casein was used as the internal standard for calibrating the phosphopeptide enrichment procedure. Briefly, 1 mg tryptic digested cell protein sample was mixed with 15 μg tryptic digested β-casein and then acidified by 0.1% trifluoroacetic acid followed by completely dried in a vacuum centrifuge. The samples were then reconstituted with loading buffer (35% glutamic acid saturated solution, 65% acetonitrile, 2% trifluoroacetic acid) to a total volume of 400 μl and incubated with 2 mg of TiO2 beads (5 μm) rotating (1000 rpm) at room temperature for 30 min. The peptide-loaded TiO2 beads were first washed in washing buffer containing 65% acetonitrile and 0.5% trifluoroacetic acid for three times and second washed in washing buffer containing 65% acetonitrile and 1% TFA for three times to remove unbound peptides. Then, the phosphopeptides were eluted from the TiO2 beads with ACN/water (1:1, v/v) solution containing 300 mm NH4OH. The eluent (200 μl) were completely dried in a vacuum centrifuge and then reconstituted in 30 μl 0.1% formic acid containing 50 fM digested β-galactosidase, the internal standard used for calibrating the MS analysis procedure. The nanoHPLC-ESI MS/MS analysis was carried out using an Eksigent cHiPLC-nanoflex system coupled to a quadrupole time-of-flight TripleTOF 5600 mass spectrometer (AB Sciex) equipped with an AB NanoSpray source and heated interface (AB Sciex). Each sample (5 μl) was loaded onto a trap column (Eksigent ChromXP C18-ClP, 200 μm × 0.5 mm, 3 μm, 120 Å) at a flow rate of 4 μl/min for 5 min. Peptide separation was carried out using a C18 column Eksigent ChromXP C18-Cl, 75 μm ×15 cm, 3 μm, 120 Å) at a flow rate of 300 nL/min. Phosphopeptides were separated with a 60 min gradient ranging from 5% to 50% B (mobile phase A/2% acetonitrile, 0.1% formic acid; mobile phase B/98% acetonitrile, 0.1% formic acid). The MS analysis was performed with the TripleTOF 5600 system. MS analysis was performed in positive ion mode over the mass range m/z 350–1250 with a 0.5 s accumulation time. The ionspray voltage was set to 2200 V, the curtain gas was set to 30, the nebulizer gas to 3 and the heated interface temperature was set to 150 °C. Tandem mass spectra were acquired over the mass range m/z 100–1500 using rolling collision energy for optimum peptide fragmentation.
An IDA mass spectrometry approach was utilized to identify vimentin phosphopeptides and β-casein phosphopeptides. In an IDA protocol, a full scan spectrum (TOF MS) was performed on the accurate mass, high-resolution 5600Triple TOF mass analyzer for precise mass measurement followed by collision-induced dissociation (CID) mass spectra. The IDA acquisition methods trigger MS/MS fragmentation enabled the identification of peptide species and modification such as phosphorylation. For localization of sites of phosphorylation, the mass spectrometry data files were processed by ProteinPilot 4.0 (AB/Sciex) using the Paragon algorithm. Ions in the parent list were only fragmented if they were detected above a specified minimum intensity (700 cps). From this initial analysis, a list of potential phosphorylated peptides was generated based on detected serine/threonine containing peptides of vimentin. The peptide identification was scored, formed into false and true positive distributions, and subsequently filtered to retain only the highest scoring identifications (Proteinpilot). The peptide sequences were compared with protein sequence databases (Swiss-Prot protein sequence databases) and the phosphorylation sites were suggested by the phosphate moiety in the peptides. Invalid and redundant peptides were removed and the PhosphoSTY probabilities above 75% were additionally set to further select for unique phosphopeptides, according to standard practices.
Normalization and quantification of phosphopeptides were conducted using method similar to previous study (19). Quantification of phosphopeptides was conducted using label-free quantification method with a high resolution TOF-MS survey scan followed by product Ion scans. Protein samples from three independent experiments were used for quantification of vimentin phosphopeptides in control and GA-treated group. And, for each pair of protein samples (control and GA-treated), triplicate technological repeats were performed to ensure reproducibility. Briefly, targeted ions were fragmented in each product ion segment with a maximum accumulation time of 0.1 ms. The parent ion list and the charge states of peptides were selected from the prominent ions observed in the IDA tandem mass spectrometry analysis. The relative amounts of vimentin phosphopeptides in control and GA-treated samples were normalized. The raw peaks were processed, and extracted ion chromatograms (XIC) were generated from the full-scan MS using MultiQuant™ software (AB SCIEX). Peak areas of vimentin phosphopeptides were then normalized by peak areas of tryptic digested peptides of internal standards, β-casein and β-galactosidase. The three β-casein phosphopeptides used as internal standard were FQS[Pho]EEQQQTEDELQDK (687.9/3+), FQS[Pho]EEQQQTEDELQDKIHPF (852.7/3+) and IEKFQS[Pho]EEQQQTEDELQDK (811.4/3+). The three β-galactosidase peptides used as internal standard were YSQQQLMETSHR (503.2/3+), VDEDQPFPAVPK (671.3/2+) and APLDNDIGVSEATR (729.4/2+). The normalized phosphorylation rate was calculated as (area of vimentin phosphopeptides/area of the average of three β-casein phosphopeptides) or (area of vimentin phosphopeptides/area of the average of three β-galactosidase peptides).
Caspase-3 Activity in GA-treated Cells and Influence of Caspases Inhibitors on Vimentin Cleavage
After treated with 2 μm GA or 0.1% DMSO (solvent control) for 24 h, cells were collected and the cellular caspase-3 activities were assayed using the colorimetric caspase-3 assay kit (Sigma) according to the manufacturer's protocol. Briefly, after treated with 2 μm GA or 0.1% DMSO (solvent control) for 24 h, cells were collected, washed with PBS and then lysed in 200 μl of the lysis buffer containing 50 mm HEPES (pH 7.5), 5 mm EDTA, 10 mm dithiothreitol, 10 μg/ml proteinase K, 100 μg/ml phenylmethysulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml pepstatin. Cells in lysis solution were kept in −80 °C and 4 °C in turn for four times until cells were lysed. After centrifugation at 12,000 × g for 30 min at 4 °C, the supernatant was collected and used for caspase-3 activity assay. The protein concentration of each sample was measured using Bradford method. Two hundred micro grams cellular protein sample was incubated with 2 X reaction buffer containing 100 μm Ac-DEVD- pNA (substrate of caspase-3) for 2 h at 37 °C. The OD of the reaction mixture was observed at an absorbance wavelength of 405 nm. To confirm the credibility of caspase-3 activity assay, a group of cells was pretreated with 100 μm caspase-3 inhibitor Ac-DEVD-CHO for 4 h before GA treatment and caspase-3 activity in these cells was checked. Furthermore, to study whether vimentin cleavage was mediated by caspases or not, vimentin cleavage in GA-treated cells (2 μm for 24 h) with or without pretreatment of pan-caspases inhibitor z-DEVD-FMK (100 μm for 4 h) or caspase-3 inhibitor Ac-DEVD-CHO (100 μm for 4 h) was checked using Western blotting assay as described above.
Influence of Overexpression of Vimentin on Cytotoxicity of GA
Cells with over-expression of vimentin were obtained by transfection with a pcDNA3.1 (+) plasmid encoding full-length human vimentin cDNA as described before (11). Plasmid encoding full-length human vimentin cDNA (pCMV-SPORT6 plasmid, Catalog No. MHS4768–99609365, Clone ID. 2985712) was purchased from Thermo Fisher Scientific, Inc. After cut down by restriction enzyme, the KpnI-NotI fragment of the vimentin was subcloned into pcDNA3.1 (+) plasmid. Negative control cells were transfected with empty pcDNA3.1 (+) plasmid. For plasmid transfection, 5 × 104 cells were seeded into each well of a six-well plate until the cells reached ∼80% confluence after cultured for 24 h. Eight micrograms plasmid DNA was diluted in 400 μl serum-free medium and then incubated for 20 min at room temperature with a mixture of 10 μl Lipofectamine 2000 (Invitrogen) and 400 μl serum-free medium. The resultant complex mixture was then added to each well of the cell culture plate. After 6 h incubation, the complex medium was replaced with fresh minimum essential medium supplemented with 10% (v/v) FBS and let to grow overnight. Then, G418 (Merck) was added to the medium to reach a final concentration equal to the minimum fatal dose (200 μg/ml). The cells were allowed to grow and passage in the presence of G418 for over 2 weeks. Expression level of vimentin in the transfected cells was checked using Western blotting assay and compared with that of wild-type cells. The cytotoxicity of GA on cells with overexpression of vimentin was then checked and compared with that of negative control cells using MTT assay and flow cytometric analysis as described above.
Immunofluorescence Staining of Cytoskeleton Proteins
HeLa cells were inoculated onto glass cover slips coated with poly-l-lysine in six-well plates and cultured overnight. Cells were exposed to 2 μm GA for 3 h or 24 h. Then, cells were stained with fluorescence labeled antibody against vimentin, F-actin or β-tubulin and then observed using immunofluorescence laser scanning confocal microscopy (Olympus, Japan). Briefly, after treatment, cells were gently rinsed with PBS and fixed with 4% paraformaldehyde for 15 min at 4 °C. After washing with PBS, cells were permeabilized by incubation with 0.1% Triton X-100 in PBS at room temperature for 15 min. Nonspecific antibody binding was inhibited by incubation of cells in blocking buffer containing 3% BSA at room temperature for 30 min. Vimentin in cell cytoskeleton was stained by incubating cells with primary rabbit antivimentin antibody in blocking buffer overnight at 4 °C and then followed by incubation with Alexa Flour 488 labeled anti-rabbit IgG (1:500, Cell Signaling) at room temperature for 2 h. F-actin in cytoskeleton was stained by incubating cells with Alexa Flour 546 labeled phalloidin (1:200, Cell Signaling) in blocking buffer at room temperature for 2 h. β-tubulin in cytoskeleton was stained by incubating cells with Alexa Flour 488 labeled anti-β-tubulin (1:100, Cell Signaling) in blocking buffer at room temperature for 2 h. DAPI fluorescent dye (final concentration: 1 μg/ml, Sigma), used for staining of nucleic DNA, was added to the cover slips at 5 min before the end of incubation. Cover slips were then washed twice with PBS and mounted onto microscope slides with glycerol gelatin (Sigma) containing 0.1 m N-propyl gallate (Sigma) to prevent bleaching. Observation was made under the immunofluorescence laser scanning confocal microscopy (Olympus, Japan). Fluorescence images were collected using excitation wavelengths of 488 nm (green), 555 nm (red) and 405 nm (blue) and emission wavelengths at 520 nm (green), 618 nm (red), and 461 nm (blue), respectively.
Influence of siRNA Knock-down of p38, HSP27, or Vimentin on Cytotoxicity of GA
To confirm the role of p38, HSP27 and vimentin in the effects of GA, cytotoxicity of GA was checked in cells treated with siRNAs against p38, HSP27 or vimentin. Cells treated with scrambled siRNA were used as negative control. The siRNAs against p38 (Cat. #6564) and HSP27 (Cat. #6356) were bought from Cell Signaling Technology Company. The vimentin siRNA (SMARTpool: ON-TARGETplus VIM siRNA, Cat. l-003551–00-0005) and scramble siRNA (ON-TARGETplus Nontargeting Control siRNAs, Cat. d-001810–0X) were bought from Thermo Fisher Scientific, Inc. For siRNA treatment, 2 × 105/well HeLa cells were seeded into each well of a six-well plate in complete MEM medium without antibiotics. After cultured overnight, cells were transfected in MEM with 40 pm siRNA for 24 h using Lipofectamine RNAiMAX according to the manufacturer's instructions. After siRNA treatment, the expression level of p38, HSP27 or vimentin in siRNA-treated cells was checked using Western blotting assay to ensure the efficiency of siRNA treatment. The cytotoxicity of GA on cells treated with siRNA against p38, HSP27 or vimentin was then checked using flow cytometric analysis as described above.
In Vivo Tumor Model Using Nude Mice
Male athymic nude mice (BALB/cA nu/nu) aged between 4 and 6 weeks were obtained from the Shanghai Institute of Materia Medica (Shanghai, China) and all experiments were performed according to institutional ethical guidelines on animal care. HeLa cells (8 × 106 cells in 0.2 ml of sterile saline solution) were subcutaneously inoculated into the right flank of each mouse. As the tumor volume reached ∼300 mm3, the mice were randomly assigned into control and GA-treated groups (five mice/group). The mice were injected intraperitoneal with 2 mg/kg/day GA (GA-treated group) or vehicle control (control group). The tumor size was measured in three dimensions with calipers every 3–4 days, and tumor volume was calculated using the following formula: tumor volume (mm3) = length × (width)2/2. 36 days after inoculation, animals were euthanized and the weight of tumor xenografts was measured. Then, for each tumor xenograft, half of the tissue was used for Western blotting assay and the other half of the tissue was used for immunohistochemical staining. Western blotting assay of vimentin in the tumor xenografts was conducted as described above. For immunohistochemical staining, tumor tissues were fixed with 10% neutral formalin, desiccated and embedded in paraffin. After stained with hematoxylin and eosin or anti-vimentin antibody (1:80, rabbit anti-vimentin(R28) polyclonal antibody, Cell signaling, CST#3932), tissue sections (4 μm) were examined under a light microscope. The anti-vimentin antibody could only detect the head domain of vimentin, which exists in un-cleaved vimentin protein. The images were acquired under a Leica DM 4000B fluorescence microscope equipped with a digital camera. For the quantitative analysis, a Histo score (H score) was calculated based on the staining intensity and percentage of stained cells. The intensity score was defined as follows: 0, no appreciable staining in cells; 1, weak staining in cells comparable with stromal cells; 2, intermediate staining; 3, strong staining. The fraction of positive cells was scored as 0–100%. The H score was calculated by multiplying the intensity score and the fraction score, producing a total range of 0–300. A cutoff of 30 was used for vimentin positivity. Tissue sections were examined and scored separately by two independent investigators blinded to the clinicopathologic data.
Cytotoxicity of GA in MCF-7 Cells with Low Level of Vimentin Expression
The expression level of vimentin in MCF-7 was checked using Western blotting assay and compared with that of HeLa cells. Cytotoxicity of GA in MCF-7 cells was checked using MTT assay as described above and compared with cytotoxicity of GA in HeLa cells.
Effects of GA on EMT Markers and Cytotoxicity of GA in MG-63 Cells With High Level of Vimentin Expression
The effects of GA on EMT markers including fibronectin, β-catenin and E-cadherin in HeLa cells and MG-63 cells, human osteoblast-like cells of tumor origin, were checked using Western blotting assay as described above. And, the expression level of vimentin in MG-63 was also checked using Western blotting assay. Cytotoxicity of GA in MG-63 cells was checked using MTT assay as described above and compared with cytotoxicity of GA in HeLa cells.
Statistical Analysis
Values were expressed as mean ± S.E., except where specially noted. The statistical significance of differences between control and treated groups was evaluated by a nonpaired two-tailed Student's t test (GraphPadPrism). All comparisons are made relative to untreated controls and p < 0.05 was considered statistically significant. For each variable three independent experiments were carried out unless otherwise indicated.
RESULTS
GA Exhibited Cytotoxicity on HeLa Cells
As shown in Fig. 1B, MTT assay results suggested that proliferation of HeLa cells was inhibited in a time- and dose-dependent manner by treatment with different concentrations (0.3125, 0.625, 1.25, 2.5, 5, 10 μm) of GA for 24, 48, or 72 h. The IC50 value of GA was 4.17 ± 0.30, 2.19 ± 0.11, and 1.59 ± 0.05 μm for 24, 48, and 72 h treatment, respectively. Results of flow cytometric analysis of cell cycle were shown in Fig. 1C (representative results) and Fig. 1D (statistical results). As shown in Fig. 1C and Fig. 1D, GA treatment dose-dependently induced an increase in the number of cells at G2/M phase. Representative result of AnnexinV-FITC/PI double-labeled flow cytometry analysis of apoptosis was shown in Fig. 1E (representative results) and Fig. 1F (statistical results). Statistical analysis results indicated that, in cells treated with GA at 2 μm or 4 μm for 24 h, the early apoptosis rate was 6.37 ± 1.05% and 11.47 ± 1.09%, respectively, which were statistically different from the control (2.64 ± 0.61%) with p value of 0.037 and 0.006, respectively. The late apoptosis rate was 5.36 ± 0.84% and 26.25 ± 4.89%, respectively, which were statistically different from the control (2.32 ± 0.25%) with p value of 0.026 and 0.013, respectively. These results demonstrated that GA induced apoptosis of HeLa cells in a dose-dependent manner.
Comparative Proteomic Analysis Results of Control and GA-treated HeLa Cells at Different Time Points
Representative 2-DE images for control and GA-treated cells were shown in Fig. 2. The gel pair showed in Fig. 2A was representative gel pair of nine replicate gel pairs collected from three independent experiments. Differentially expressed spots were shown by the arrows. Enlarged 2-DE images in Fig. 2A were also provided as supplemental Fig. S1–S4. Fig. 2B showed the expanded region of differentially expressed protein spots. The proteins within the circles were the differentially expressed proteins. Eighteen significantly differently expressed protein spots (the MS and MS/MS spectra are listed in supplemental Table S1) were found including 9 protein spots (spots 1–9) at 3 h treatment and another nine protein spots (spots 10–18) at 24 h treatment. The average intensity values and standard errors of the spots, the statistical assay results, and the fold differences between control and GA-treated group were shown in Table I. The fold difference was represented by the ratio of the mean intensity value of GA-treated group to the mean value of the control group. Results of MS/MS identification of the spots were summarized in Table II. The protein name, protein score, coverage, number of identified peptides and best ion score of each spot were all shown in Table II. The result of MALDI-TOF MS/MS analysis of spot 1 was shown in supplemental Fig. S5 as an example. To be noted, spot 3 and spot 11 were both identified as the same protein, vimentin. The molecular weight value of the two spots (spot 3 at 50–60 kDa and spot 11 at 30–40 kDa) suggested that spot 3 might be the intact form of vimentin (57 kDa) whereas spot 11 might be a cleaved form of vimentin. The result that GA treatment caused decrease in the level of spot 3 (intact vimentin) and increase in the level of spot 11 (cleaved vimentin) suggested that GA might induce cleavage of vimentin.
Fig. 2.

Comparative proteomic analysis results of control and GA-treated HeLa cells at different time points. A, The representative 2-DE images of control HeLa cells and cells treated with 2 μm GA for 3 or 24 h. The gel pair showed here was representative gel pair of nine replicate gel pairs collected from three independent experiments. Differentially expressed spots were shown by the arrows. B, The expanded region of differentially expressed protein spots. The proteins within the circles were the differentially expressed proteins.
Table I. Summary of differentially expressed proteins in GA-treated HeLa cells compared with control for 3 hour treatment (from Spot 1 to Spot 9) or 24 hour treatment (from Spot 10 to Spot 18). *p < 0.05 vs the control group. The fold difference was represented by the ratio of the intensity value of GA-treated group to the value of the control group.
| Spot number | Protein name | Spot volume 3 h (ppm) (Mean ± S.E.) |
Fold difference (3 h) | Spot volume 24 h (ppm) (Mean ± S.E.) |
Fold difference (24 h) | Pairs of gels (n) | ||
|---|---|---|---|---|---|---|---|---|
| Control 3 h | GA-treated 3 h | Control 24 h | GA-treated 24 h | |||||
| 1 | Heat shock 27kDa protein 1 | 1398.48 ± 183.52 | 825.11 ± 167.50* | 0.59 | 595.39 ± 121.67 | 470.27 ± 177.51 | 0.79 | 9 |
| 2 | Calumenin | 79.14 ± 9.8000 | 8.57 ± 3.55*00 | 0.11 | 159.34 ± 19.30 | 91.61 ± 19.25 | 0.57 | 9 |
| 3 | Vimentin | 1427.08 ± 232.59 | 763.68 ± 201.54* | 0.54 | 1534.01 ± 253.54 | 948.69 ± 106.89 | 0.62 | 9 |
| 4 | Eukaryotic translation elongation factor 1 beta 2 | 3696.80 ± 489.88 | 2017.80 ± 355.32* | 0.55 | 3437.39 ± 594.30 | 3143.14 ± 816.55 | 0.91 | 9 |
| 5 | DJ-1 protein | 5835.58 ± 908.47 | 3125.98 ± 579.06* | 0.54 | 3061.27 ± 506.97 | 2509.36 ± 356.85 | 0.82 | 9 |
| 6 | BAG 2 | 378.24 ± 102.91 | 140.18 ± 38.37*0 | 0.37 | 334.34 ± 58.37 | 164.57 ± 53.19 | 0.49 | 9 |
| 7 | Chaperonin containing TCP1, subunit 3 (gamma) | 330.93 ± 43.130 | 169.54 ± 30.70*0 | 0.51 | 470.71 ± 55.28 | 295.93 ± 52.77 | 0.63 | 9 |
| 8 | Nucleosome assembly protein 1-like 4 | 479.41 ± 70.010 | 279.63 ± 58.41*0 | 0.58 | 404.47 ± 93.57 | 432.61 ± 108.69 | 1.07 | 9 |
| 9 | Transaldolase 1 | 139.42 ± 30.680 | 331.68 ± 66.95*0 | 2.38 | 245.68 ± 25.44 | 381.61 ± 51.06 | 1.55 | 9 |
| 10 | Ribosomal protein, large, P0 | 372.08 ± 117.75 | 489.48 ± 156.17 | 1.32 | 375.91 ± 94.040 | 901.86 ± 200.18* | 2.40 | 9 |
| 11 | Vimentin | 1120.24 ± 154.02 | 2101.91 ± 436.00 | 1.88 | 1020.91 ± 535.06 | 2658.72 ± 412.91* | 2.60 | 9 |
| 12 | Nascent-polypeptide-associated complex alpha polypeptide | 1647.08 ± 209.19 | 1199.29 ± 294.10 | 0.73 | 1641.53 ± 330.63 | 685.21 ± 144.82* | 0.42 | 9 |
| 13 | Gelsolin-like capping protein | 594.03 ± 100.26 | 519.42 ± 61.65 | 0.87 | 929.04 ± 151.96 | 418.91 ± 106.75* | 0.45 | 9 |
| 14 | Keratin 18 | 375.44 ± 47.48 | 367.77 ± 49.97 | 0.98 | 518.90 ± 112.78 | 236.01 ± 63.60*0 | 0.45 | 9 |
| 15 | Eukaryotic translation elongation factor 1 delta isoform 2 | 659.33 ± 110.97 | 860.26 ± 98.14 | 1.30 | 368.70 ± 78.100 | 803.61 ± 112.32* | 2.18 | 9 |
| 16 | Beta actin | 292.80 ± 27.70 | 269.6 ± 81.65 | 0.92 | 267.26 ± 62.810 | 101.89 ± 21.65*0 | 0.38 | 9 |
| 17 | Tubulin alpha 6 | 236.27 ± 20.92 | 337.42 ± 85.02 | 1.43 | 243.86 ± 65.040 | 990.48 ± 175.03* | 4.06 | 9 |
| 18 | Keratin 8 | 759.39 ± 112.72 | 1032.57 ± 140.88 | 1.36 | 190.11 ± 151.600 | 1184.11 ± 299.06* | 6.23 | 9 |
Table II. The results of protein identifications of differentially expressed proteins using MALDI-TOF MS/MS.
| Spot | Target protein | NCBI accession number | Theoretical molecular mass (kDa)/pI | Protein score | Sequence coverage (%) | Number of peptides matched/unmatched | Best ion score/unique peptides |
|---|---|---|---|---|---|---|---|
| 1 | Heat shock 27kDa protein 1 | 4504517 | 22.8/5.98 | 269 | 41 | 9/17 | 104/2 |
| 2 | Calumenin | 49456627 | 37.1/4.48 | 193 | 24 | 6/13 | 65/3 |
| 3 | Vimentin | 37852 | 53.7/5.06 | 518 | 58 | 23/15 | 80/4 |
| 4 | Eukaryotic translation elongation factor 1 beta 2 | 4503477 | 24.7/4.50 | 112 | 24 | 13/47 | 30/2 |
| 5 | DJ-1 protein | 42543006 | 19.8/6.33 | 197 | 68 | 18/50 | 80/2 |
| 6 | BAG 2 | 49065418 | 23.8/6.02 | 140 | 32 | 16/68 | 47/2 |
| 7 | Chaperonin containing TCP1, subunit 3 (gamma) | 14124984 | 60.4/6.10 | 153 | 31 | 18/22 | 41/2 |
| 8 | Nucleosome assembly protein 1-like 4 | 5174613 | 42.8/4.60 | 113 | 29 | 12/84 | 38/2 |
| 9 | Transaldolase 1 | 48257056 | 37.4/6.35 | 136 | 45 | 18/47 | 65/2 |
| 10 | Ribosomal protein, large, P0 | 12654583 | 34.3/5.42 | 301 | 47 | 16/16 | 54/2 |
| 11 | Vimentin | 340234 | 35.0/4.70 | 106 | 26 | 21/18 | 38/2 |
| 12 | Nascent-polypeptide-associated complex alpha polypeptide | 5031931 | 23.3/4.52 | 287 | 30 | 8/10 | 70/4 |
| 13 | Gelsolin-like capping protein | 63252913 | 38.5/5.82 | 154 | 29 | 7/10 | 78/2 |
| 14 | Keratin 18 | 30311 | 47.3/5.27 | 206 | 25 | 12/29 | 48/2 |
| 15 | Eukaryotic translation elongation factor 1 delta isoform 2 | 25453472 | 31.1/4.90 | 208 | 37 | 10/17 | 48/2 |
| 16 | Beta actin | 15277503 | 40.2/5.55 | 334 | 49 | 19/69 | 69/3 |
| 17 | Tubulin alpha 6 | 14389309 | 49.9/4.96 | 156 | 39 | 14/67 | 47/2 |
| 18 | Keratin 8 | 4504919 | 53.7/5.52 | 170 | 36 | 13/3 | 50/2 |
GA Induced Vimentin Cleavage and Caspase-3 Activation
Results of Western blotting assay (Fig. 3A) showed that GA induced considerable vimentin cleavage in cultured HeLa cells at both 3 h treatment and 24 h treatment. The protein bands exhibited molecular weights of about 57 kDa, 50 kDa, 45 kDa, and 38 kDa. The result was consistent with the 2-DE analysis result in which a cleaved vimentin protein (spot 11) with molecular weight of 30–40 kDa was found to be increased by GA treatment. Furthermore, as shown in Fig. 3B, caspase-3 activity was significantly increased in GA-treated cells. Caspase-3 inhibitor Ac-DEVD-CHO could inhibit both the caspase-3 activity in control cells and in GA-treated cells. The results suggested the involvement of caspase activation in apoptosis induced by GA. Although the cytotoxicity of GA could be ameliorated in cells with pre-treatment of caspases inhibitor z-DEVD-FMK or caspase-3 inhibitor Ac-DEVD-CHO before GA treatment (as shown in supplemental Fig. S6), interestingly, GA-induced cleavage of vimentin could not be significantly inhibited by caspase-3 inhibitor Ac-DEVD-CHO or pan-caspases inhibitor z-DEVD-FMK (Fig. 3C). The results indicated that caspases might not be the critical protease in the cleavage of vimentin.
Fig. 3.

GA induced vimentin cleavage and caspase-3 activation. A, Western blotting assay of vimentin in HeLa cells treated with 2 μm GA for 3 h or 24 h. B, Caspase-3 activity in cells treated for 24 h with solvent control, 2 μm GA, 100 μm caspase-3 inhibitor CHO, or 2 μm GA with pretreatment of CHO (100 μm for 4 h). C, Western blotting assay of vimentin in HeLa cells treated for 24 h with solvent control, 2 μm GA, 2 μm GA with pretreatment of caspases inhibitor FMK (100 μm for 4 h) or 2 μm GA with pretreatment of CHO (100 μm for 4 h). D, Illustration of sequence coverage in MS/MS identification of the protein bands detected as vimentin in Western blotting assay. Identified amino acids in MS/MS analysis were highlighted in red. E, A map of vimentin protein showing possible cleavage sites and calculated molecular weight values of the possible cleaved vimentin fragments.
MS/MS analysis results of protein bands that were detected in Western blotting assay were showed in supplemental Table S2 and the coverage of the identified peptides in the whole sequence of vimentin were showed in Fig. 3D. Protein bands with molecular weight of about 57 kDa, 50 kDa, and 45 kDa were successfully identified. Although, maybe because of low level of protein amount, MS analysis results of protein band with molecular weight of 38 kDa exhibited low confidence rate and were not included in the report. As showed in Fig. 3D, in MS analysis of the protein band with molecular weight of about 57 kDa, identified peptides were almost covered full sequence of vimentin. The results suggested that the protein might be the intact form of vimentin. On the contrary, in proteins with molecular weight of about 50 kDa and 45 kDa, sequences before ser51 and/or after arg424 were not identified. The results suggested possible cleavage sites of vimentin at or near ser51 and glu425. As shown in Fig. 3E, calculated molecular weight values of vimentin peptides cleaved at or near ser51 and glu425 were consistent with the molecular weight values of protein bands detected in Western blotting assay.
Change in Phosphorylation State of Vimentin in GA-treated HeLa Cells
The MS data files were processed by ProteinPilot 4.0 (AB/Sciex) using the Paragon algorithm. The MS data were searched against all the Homo sapiens protein sequence of Uniprot_sprot_201105 database. For ProteinPilot Paragon, iodoacetamide was selected as the cysteine modification agent, trypsin as the digestion enzyme, “biological modifications” were selected as the “ID focus” and a “Thorough ID Search Effort” was selected. FDR analysis was performed using reversed protein sequences and was used to calculate the number of false positive proteins expected at a 95% confidence level. Peptides that passed a 1% FDR threshold were considered for protein identification. Vimentin phosphopeptides with phosphorylation at ser55, ser56, ser412, ser419, ser420 were identified and verified by MS/MS. The vimentin phosphopeptides were listed in supplemental Table S3 and the CID-MS/MS product ion spectrums of the vimentin phosphopeptides, which indicated the phosphorylation sites, were shown in supplemental Fig. S7. The parent ion list and the charge states of the internal standards, three phosphorylated β-casein peptides and the three β-galactosidase peptides, were selected from the prominent ions observed in the IDA tandem mass spectrometry analysis as listed in supplemental Table S4. The peak area transitions for each phosphopeptides were obtained by integration of the appropriate reconstructed ion chromatograms. Peak areas for three phosphorylated β-casein peptides and three β-galactosidase peptides were also determined. The average of peak areas of three phosphorylated β-casein peptides and three β-galactosidase peptides was used for normalization of peak areas of vimentin phosphopeptides. The normalized peak areas of vimentin phosphopeptides were shown in Table III. No matter normalized with phosphorylated β-casein peptides or normalized withβ-galactosidase peptides (as shown in Table III), the normalized peak areas of vimentin peptide SLYASSPGGVYATR (aa51–64) with phosphorylation at ser55 or ser56 in GA-treated cells were significantly lower than that of control cells. On the contrary, no significant difference was observed between the peak areas of vimentin peptide ISLPLPNFSSLNLR (aa411–424) with phosphorylation at ser412, ser419 or ser420 in GA-treated cells and that in control cells.
Table III. Normalized peak areas of vimentin phosphopeptides SLYASSPGGVYATR and ISLPLPNFSSLNLR.
| Vimentin phosphoSTY probabilities (%) | Normalized to β-casein phosphopeptides (Mean ± S.E.) |
Normalized to β-galactosidase non-phosphopeptides (Mean ± S.E.) |
||||
|---|---|---|---|---|---|---|
| Control | GA-treated | P value | Control | GA-treated | P value | |
| SLYAS[4.7%]S[95.3%]PGGVYATR | 0.265 ± 0.076 | 0.197 ± 0.024 | 0.009* | 1.314 ± 0.328 | 0.963 ± 0.086 | 0.016* |
| IS[90.5%]LPLPNFS[2.1%]S[7.4%]LNLR | 0.010 ± 0.005 | 0.007 ± 0.004 | 0.218 | 0.049 ± 0.028 | 0.034 ± 0.018 | 0.117 |
Cytotoxicity of GA on HeLa Cells With Overexpression of Vimentin
Cytotoxicity of GA was checked in HeLa cells transfected with plasmid encoding vimentin. The levels of vimentin expression in wild-type cells, negative control cells (cells transfected with pcDNA3.1(+) plasmid without vimentin cDNA) and cells with over-expression of vimentin (cells transfected with pcDNA3.1(+) plasmid encoding vimentin cDNA) were checked using Western blotting (Fig. 4A). The cytotoxicity of GA in HeLa cells with over-expression of vimentin was shown in Fig. 4B and Fig. 4C. As shown in Fig. 4B, overexpression of vimentin ameliorated the inhibitive effects of GA on viability of HeLa cells. And, overexpression of vimentin also decreased the apoptosis rate of HeLa cells treated by GA (Fig. 4C).
Fig. 4.

Cytotoxicity of GA on HeLa cells with over-expression of vimentin. A, Western blotting assay results of expression levels of vimentin in wild type cells, negative control cells (cells transfected with empty plasmid) and cells transfected with plasmid encoding vimentin. B, Effects of GA on viability of negative control cells or cells transfected with plasmid encoding vimentin. Cells were treated with 0.25, 0.5, 1, 2, or 4 μm GA for 72 h and cell viability was determined by MTT assay. Data were statistical results of three independent experiments (n = 3, mean ± S.E.). C, Induction of apoptosis by 24 h treatment of GA in negative control cells or cells transfected with plasmid encoding vimentin. Apoptotic cells were quantified by supravital double staining with Annexin V-FITC and PI. Showed were representative results of three independent experiments.
Prediction and Confirmation of Possible Signal Pathway Activated by GA
Based on the results of 2-DE analysis and previous reports of GA, the possible signal pathway activated by GA in cancer cells was predicted (as shown in Fig. 5A). In our proteomic study, vimentin was the only protein whose expression was found to be significantly regulated both at early stage (3 h) and late stage (24 h) of GA treatment. These results suggested that vimentin might play an important role in the effects of GA. Furthermore, HSPB1 (heat shock 27 kDa protein 1), also called as HSP27, was the only target-related protein activated by GA at early stage (3 h treatment), which had been reported to have direct connections with vimentin (detailed information was provided in Discussion section). And, activation of HSP27 could be regulated by MAPKAPK2 (mitogen-activated protein kinase-activated protein kinase 2), which belonged to p38 MAPK pathway. Therefore, the signal pathway from p38-MAPK, HSP27 to vimentin was predicted to be the pathway through which GA exhibited regulative effects on vimentin. Then, change in vimentin might cause cytoskeleton dysfunction, which finally induced cell death.
Fig. 5.
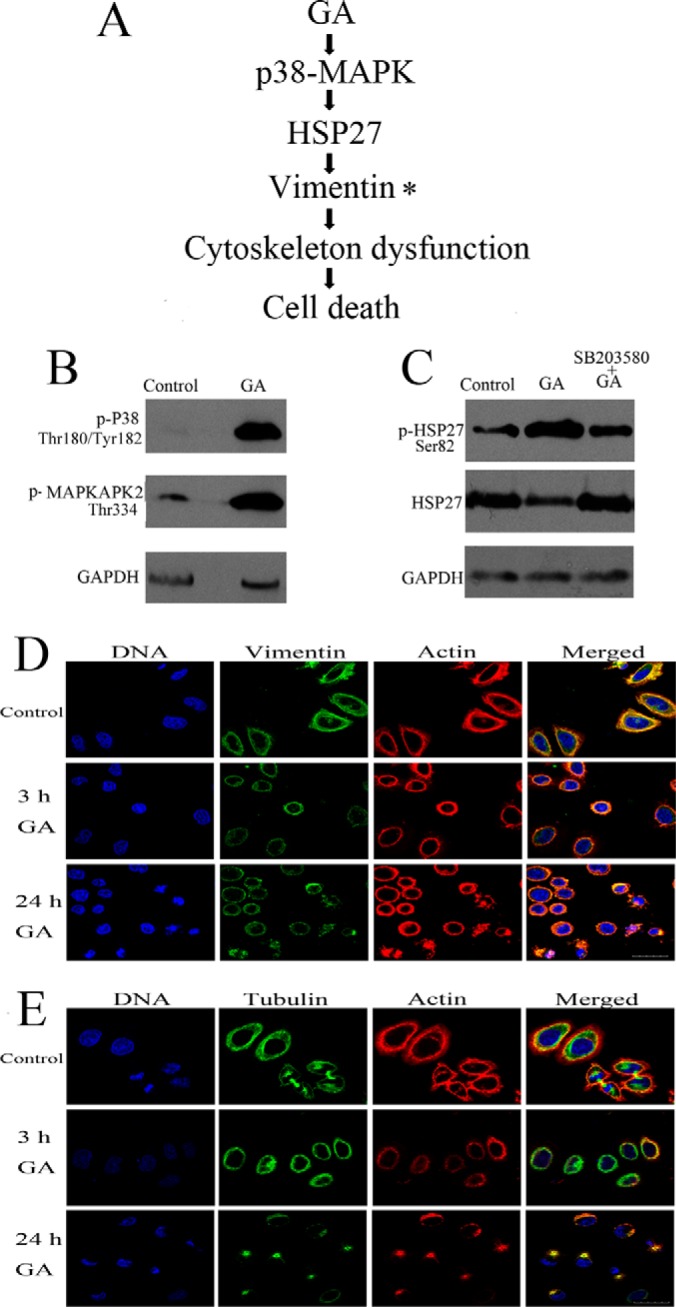
Prediction and confirmation of possible signal pathway activated by GA. A, Predicted signal pathway activated by GA. B, Western blotting assay of levels of phosphorylated p38 (Thr334), phosphorylated MAPKAPK2 (Thr180/Tyr182) in HeLa cells treated with 2 μm GA for 3 h. Each blot was the representative result of three independent experiments (n = 3, mean ± S.E.). C, Western blotting assay of levels of phosphorylated HSP27 (Ser82) and HSP27 in HeLa cells treated with 2 μm GA for 3 h. The influence of p38 MAPK inhibitor SB203580 on the effects of GA was also checked. Each blot was the representative result of three independent experiments. D, Representative images of observation of intermediate filament (vimentin, stained with Alexa Fluor 488 conjugated antibody, green), microfilaments (actin, stained with Alexa Fluor 546 phalloidin, red) and nuclear chromatin (stained with DAPI, blue) in HeLa cells treated with 2 μm GA for 3 or 24 h. The intermiediate filaments network was faint and reorganized in GA-treated HeLa cells. E, Representative images of observation of microtubules (tubulin, stained with Alexa Fluor 488 conjugated antibody, green), microfilaments (actin, stained with Alexa Fluor 546 phalloidin, red) and nuclear chromatin (stained with DAPI, blue) in HeLa cells treated with 2 μm GA for 3 or 24 h. The microtubules and microfilaments were re-arranged in GA-treated HeLa cells. Scale bar = 30 μm. Showed were representative results of three independent experiments.
Western blotting assay was employed to assess the activation of p38-MAPK pathway and regulation of HSP27 and vimentin level by GA. Primary antibodies used were antibodies against phospho(Thr180/Tyr182)-p38, phospho(Thr334)-MAPKAPK2, phospho(Ser82)-HSP27, HSP27, vimentin, and GAPDH. As shown in Fig. 5B, after incubation with 2 μm GA for 3 h, the phosphorylation level of p38 MAPK and MAPKAPK2 was markedly elevated. As shown in Fig. 5C, 3 h GA treatment also induced increase in phosphorylation of HSP27 and decrease in the total protein level of HSP27. Furthermore, the effects of GA on HSP27 could be ameliorated by SB203580, an inhibitor of p38 MAPK. The results confirmed the activation of signal cascade from p38 MAPK to HSP27 in GA-treated HeLa cells. The full length images of Western blotting assay of phospho (Thr180/Tyr182)-p38, phospho(Thr334)-MAPKAPK2, and phospho (Ser82)-HSP27 were provided as supplemental Fig. S8.
The effects of GA (3 h and 24 h treatment) on cytoskeleton configuration of HeLa cells were shown in Fig. 5D (vimentin and F-actin) and Fig. 5E (β-tubulin and F-actin). Results of immunofluorescence staining of vimentin (intermediate filaments), actin (microfilaments) and tubulin (microtubules) showed that, after GA treatment, cells rounded up and exhibited change in distribution of cytoskeleton proteins. GA treatment induced faint and reorganized intermediate filaments (vimentin) network. And, GA treatment also induced re-arrangement of microtubules and microfilaments.
Influence of siRNA Knock-down of p38, HSP27 or Vimentin on Cytotoxicity of GA
As shown in Fig. 6A, siRNAs against p38, HSP27, or vimentin could effectively decrease the expression levels of p38, HSP27, or vimentin. Results of FCM analysis of apoptosis were shown in Fig. 6B. As shown in Fig. 6B, siRNA treatments exhibited cytotoxicity on cells and the increase in cell apoptosis, including both early apoptosis and late apoptosis. The increase in apoptosis rate was significant for knock-down of p38 and vimentin. For cells treated with 2 μm GA, pretreatment of siRNAs for p38, HSP27, or vimentin increased the cytotoxicity of GA and the increase was significant for knock-down of p38. These results suggested the important roles of p38, HSP27, and vimentin in cell growth and death. Interestingly, in cells treated with 4 μm GA, pretreatment of siRNA for vimentin significantly decreased the apoptosis rate of GA-treated cells. The results suggested the critical role of vimentin in the cytotoxicity of GA.
Fig. 6.
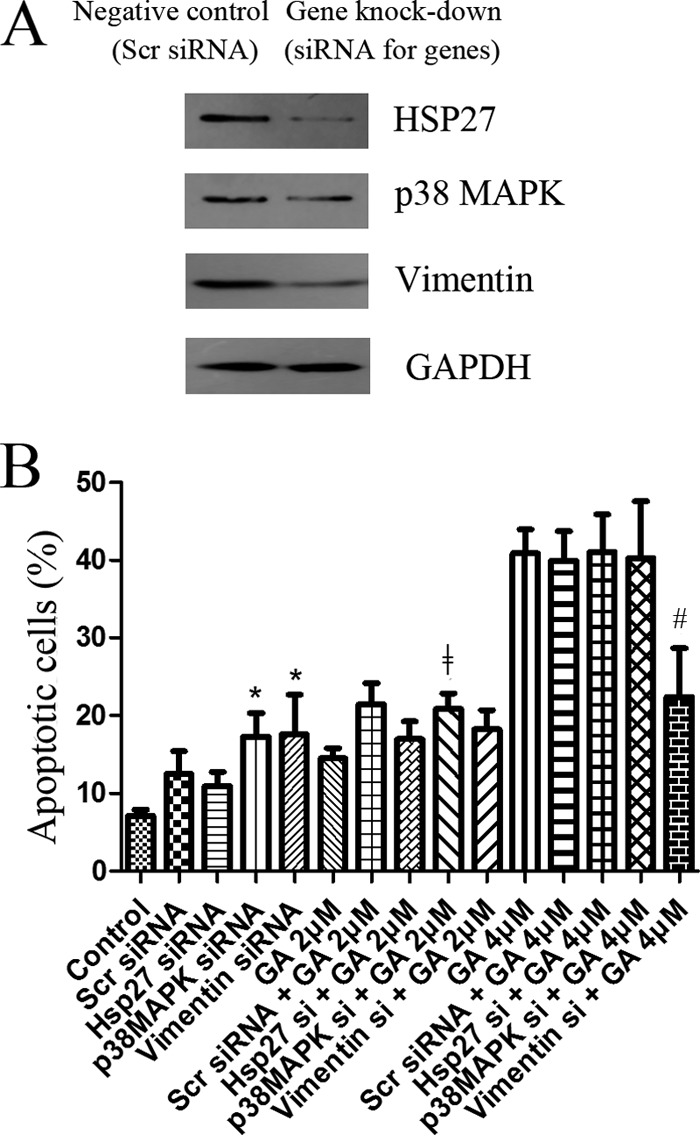
Influence of siRNA knock-down of p38, HSP27 or vimentin on cytotoxicity of GA. A, Western blotting assay of levels of p38, HSP27 or vimentin in cells treated with siRNA against p38, HSP27 or vimentin. Cells treated with scrambled siRNA were used as negative control. B, Apoptosis of cells treated for 24 h by GA (at 2 or 4 μm) with or without pretreatment of siRNAs. siRNAs used were scrambled siRNA or siRNA against p38, HSP27 or vimentin. Apoptotic cells were quantified by supravital double staining with Annexin V-FITC and PI. Showed were representative results of three independent experiments. *p < 0.05 versus control cells. ‡p < 0.05 versus cells treated with 2 μm GA. #p < 0.05 versus cells treated with 4 μm GA.
Effects of GA on In Vivo Tumor Growth and Induction of Vimentin Cleavage
As shown in Fig. 7A and Fig. 7B, GA treatment inhibited tumor growth (both tumor volume and tumor weight) in nude mice inoculated with HeLa cells. Importantly, GA treatment also induced vimentin cleavage in vivo. As shown in Fig. 7C, though some extent of vimentin cleavage could be observed in the tumor tissues of control group, the cleavage of vimentin was more obvious in GA-treated group. Moreover, H&E staining of tumor xenografts (Fig. 7D) also indicated that, in control group, the tumor cells were irregular and had large and deformity nuclei, abundant cytoplasm and high nucleocytoplasmic ratio. Mitotic and amphinucleolus could be observed in control group. However, in GA-treated group, mitotic and amphinucleolus was rarely and the nucleolus was smaller and more regular (Fig. 7D). In addition, the immunohistochemical staining of vimentin (Fig. 7E) showed that, the levels of vimentin (uncleaved form) in GA-treated group were lower than that of control group. As shown in Fig. 7F, the Histo score (H score) of vimentin expression from the GA-treated group was 94.50 ± 11.77, which were statistically different from the control group (199.50 ± 10.82) with p value < 0.05.
Fig. 7.

Effects of GA on in vivo tumor growth and induction of vimentin cleavage. A, Tumor growth in nude mice treated with vehicle control or 2 mg/kg GA. B, Weight of tumor xenograts in nude mice treated with vehicle control or 2 mg/kg GA. n = 5, mean ± S.E., *p < 0.05 compared with that of control. C, Western blotting assay results of vimentin in tumor grafts of nude mice treated with vehicle control or 2 mg/kg GA. D, H&E staining analysis results of tumor xenografts of nude mice treated with vehicle control or 2 mg/kg GA. E, Immunohistochemical analysis results of vimentin expression in tumor xenografts of nude mice treated with vehicle control or 2 mg/kg GA. Showed were representative results. F, Statistical analysis results of vimentin expression in immunohistochemical staining of tumor xenografts of nude mice treated with vehicle control or 2 mg/kg GA (n = 5, mean ± S.E.).
Cytotoxicity of GA in MCF-7 Cells with Low Vimentin Expression
Results of Western blotting assay of vimentin levels in MCF-7 cells and HeLa cells with or without GA treatment (2 μm GA for 3 h or 24 h) were shown in Fig. 8. As shown in Fig. 8A, if we used the same Western blotting assay parameters as that for detection of vimentin in HeLa cells, vimentin expression in MCF-7 cells was almost undetectable. The results indicated that vimentin level in MCF-7 cells was much lower than that of HeLa cells. Although, if we increased the time of exposure, weak expression of vimentin could be detected in MCF-7 cells (Fig. 8B). Comparison of the cytotoxicity of GA in the two kinds of cells (Fig. 8C) suggested that GA exhibited weaker cytotoxicity in MCF-7 cells compared with in HeLa cells.
Fig. 8.
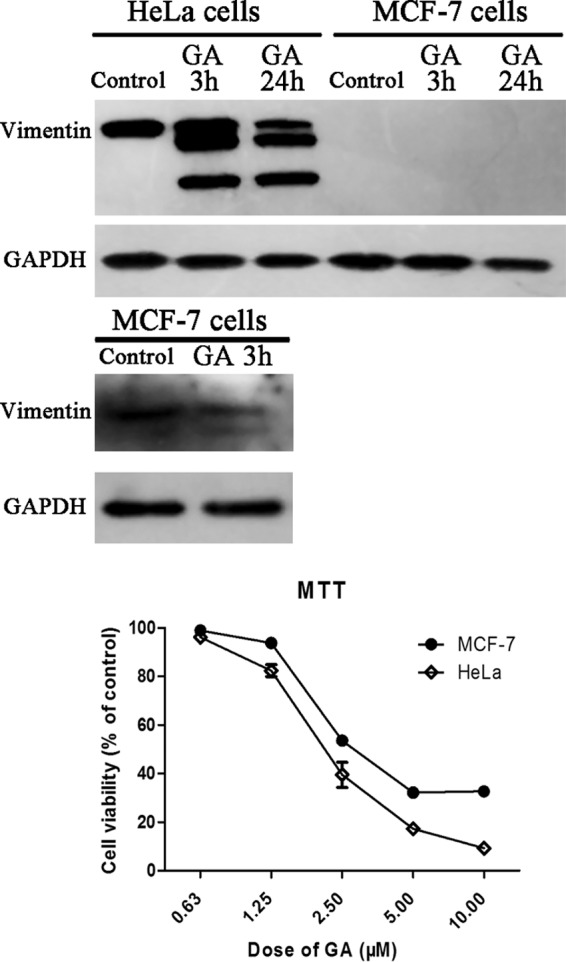
Cytotoxicity of GA in MCF-7 cells with low vimentin expression. A, Western blotting assay of levels of vimentin in HeLa cells and MCF-7 cells with or without GA treatment (2 μm GA for 3 h or 24 h). B, Western blotting assay with increased exposure time to detect vimentin in MCF-7 cells with or without GA treatment (2 μm GA for 3 h). C, Effects of GA on viability of HeLa cells or MCF-7 cells. Cells were treated with different doses of GA for 72 h and cell viability was determined by MTT assay. Data were statistical results of three independent experiments (n = 3, mean ± S.E.).
Effects of GA on EMT Markers and Cytotoxicity of GA in MG-63 Cells
Results of Western blotting assay of levels of EMT makers such as fibronectin, β-catenin and E-cadherin in HeLa cells with or without GA treatment (2 μm GA for 24 h) were shown in Fig. 9A. As shown in Fig. 9A, GA treatment decreased the level of fibronectin and β-catenin and increased the level of E-cadherin in HeLa cells. The results indicated that GA exhibited inhibitive effects on EMT. Similar results (Fig. 9B) could be observed in MG-63 cells, human osteoblast-like cells of tumor origin. Notably, as shown in Fig. 9B, MG-63 cells exhibited high level of vimentin expression whereas GA treatment could induce cleavage of vimentin. MTT assay results of comparing cytotoxicity of GA in HeLa cells and in MG-63 cells (Fig. 9C) showed that GA exhibited stronger cytotoxicity on MG-63 cells than on HeLa cells, especially at doses of 1.25 and 2.5 μm.
Fig. 9.
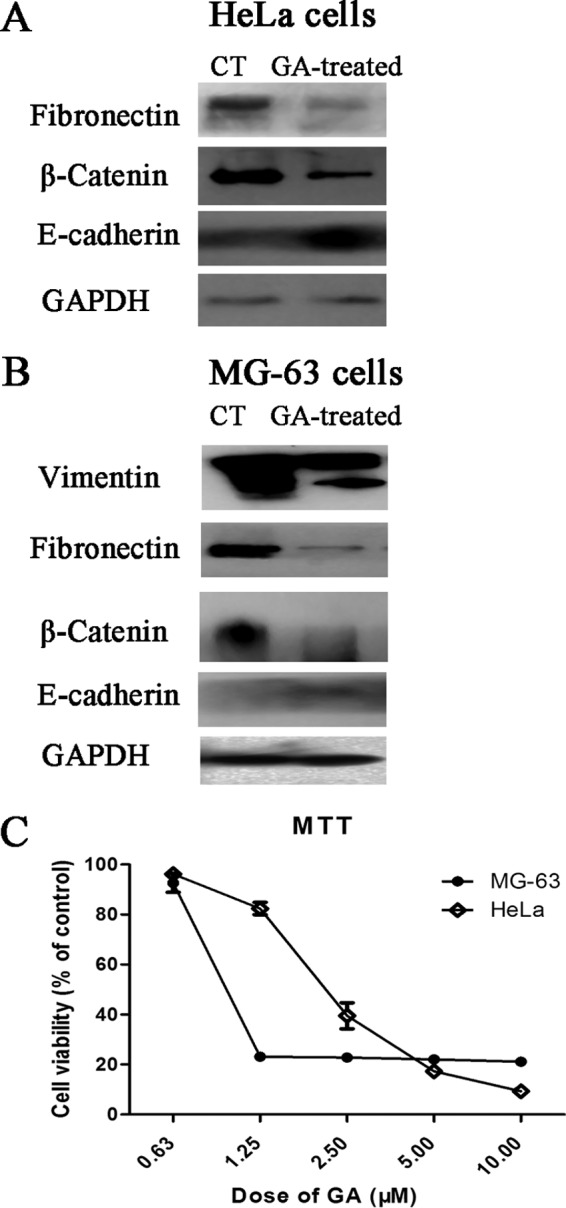
Effects of GA on EMT markers and cytotoxicity of GA in MG-63 cells. A, Western blotting assay of levels of fibronectin, β-catenin and E-cadherin in HeLa cells with or without GA treatment (2 μm GA for 24 h). B, Western blotting assay of levels of vimentin, fibronectin, β-catenin and E-cadherin in MG-63 cells with or without GA treatment (2 μm GA for 24 h). C, Effects of GA on viability of HeLa cells or MG-63 cells. Cells were treated with different doses of GA for 72 h and cell viability was determined by MTT assay. Data were statistical results of three independent experiments (n = 3, mean ± S.E.).
DISCUSSION
In the present study, anticancer effects of GA, a natural product in phase IIb trial in China, were checked in HeLa cells. Our results indicated that GA inhibited proliferation, induced G2/M cell cycle arrest and apoptosis in HeLa cells. These results were consistent with previous reports of GA (2, 20–22). By comparing the protein expression profile of GA-treated cells with that of control cells, 18 significantly differentially expressed proteins (possible target-related proteins of GA) were found including 9 proteins found at early stage of GA treatment (3 h) and nine proteins found at late stage of GA treatment (24 h). To be noted, the most interesting finding in the present study was the observation of the role of vimentin in the effects of GA. Among these possible target-related proteins of GA, vimentin was the only protein that was differentially expressed both at early stage and later stage of GA treatment. Vimentin, a 57-kDa protein, is a member of the type III intermediate filament protein family. As one of the key components of the cellular cytoskeleton, vimentin played an important role in maintaining cell shape and integrity and was also involved in cytoskeletal interactions such as adhesion and migration (23). Importantly, vimentin intermediate filament network is able to serve as platforms/scaffolds for signaling molecules therefore vimentin could participate in various cellular functions by interacting with a large number of signaling proteins (24). Lots of reports had shown that vimentin was overexpressed in different cancer cell lines/tissues (25). Accordingly, vimentin was considered as a possible biomarker of cancer (26, 27). In the present study, cleavage of vimentin induced by GA was suggested by 2-DE analysis results and confirmed by Western blotting assay results. In 2-DE analysis, two spots (spot 3 and spot 11) were both identified as vimentin whereas GA induced decrease in vimentin with high molecular weight (spot 3) and increase in vimentin with low molecular weight (spot 11). Results of Western blotting assay clearly showed that there was only one protein band of vimentin (at about 57 kDa) in control cells whereas four protein bands (at about 57, 50, 45, 38 kDa) were detected as protein bands of vimentin in GA-treated cells. To be noted, because of low level of protein at 38 kDa, sometimes only three protein bands of vimentin (at about 57, 50, 45 kDa) could be observed in Western blotting assay. MS/MS analysis results of vimentin protein bands further confirmed the cleavage of vimentin induced by GA and suggested possible cleavage sites at or near ser51 and glu425. Our results were consistent with previous report that found cleavage sites of human recombinant vimentin at or near ser51 and asn422 (28).
Vimentin cleavage could be conducted by caspases (29) or by other proteases such as calpain (30) or cathepsin (31). Activation of caspase-3 was observed in GA-treated HeLa cells in the present study. Although, the results that both pan-caspases inhibitor and caspase-3 inhibitor could not abolish GA-induced vimentin cleavage suggested that caspases might not be the critical protease in vimentin cleavage. Our efforts of trying to find proteases responsible for GA-induced cleavage of vimentin using several inhibitors of calpain or cathepsin exhibited ambiguous results (data not shown). Further study is necessary to clarify the cleavage process of vimentin in GA-treated cells. Although, the very early appearance of vimentin cleavage after only 3 h of GA treatment suggested the important role of vimentin cleavage in the effects of GA.
Importantly, site-specific phosphorylation of intermediate filament proteins including vimentin could affect assembly and disposition of the intermediate filament structure and also played major roles in regulating the connection between intermediate filament and its associated proteins (32). The phosphorylation state of vimentin was an important factor in the functions of vimentin (32–34). Our results in the present study showed that, GA treatment induced significant decrease in phosphorylation of vimentin peptide SLYASSPGGVYATR (aa51–64) but exhibited no influence on phosphorylation of vimentin peptide ISLPLPNFSSLNLR (aa411–424). The structure of intermediate filament proteins included a conserved tripartite domain structure consisting of a large α-helical central rod domain flanked by head and tail domains (35). Peptide of aa51–64 was at the head domain (1–94) of vimentin whereas aa411–412 was at the tail domain (407–466) of vimentin. Our results showed GA induced change in phosphorylation of head domain (aa51–64) but not tail domain (aa411–424) were consistent with previous finding that vimentin head domain was the major site of phosphorylation involved in important functions such as disassembly and re-organization of cytoskeleton in mitosis (36). Interestingly, aa51 (ser51) was also suggested in the present study to be a possible site of vimentin cleavage induced by GA. Previous report showed that phosphorylation of vimentin peptide prevented its hydrolysis (28). It might be possible that GA-induced decrease in phosphorylation of vimentin peptide aa51–64 contributed to the cleavage of vimentin at amino acid 51.
To confirm the role of vimentin in the effects of GA, HeLa cells with overexpression of vimentin (by plasmid transfection) were used in the present study. Effects of GA were checked in these cells and were compared with that of control. The fact that overexpression of vimentin by plasmid transfection could ameliorate the cytotoxicity of GA confirmed the role of vimentin in the effects of GA. Based on the results of present study and previous studies, which reported GA-induced p38-MAPK activation and cytoskeleton change (2, 3), signal pathway from p38-MAPK, HSP27, vimentin, other cytoskeleton proteins and then to induction of cell death (as shown in Fig. 5A) was predicted to be the possible signal pathway activated by GA. HSP27, a small stress protein, was an ATP-independent chaperone crucial for the maintenance of cellular integrity and played diverse roles as regulators of protein folding, cell motility, cytoskeletal stability, cellular stress, and cell death (37, 38). In the present study, HSP27 was found to be a target-related protein of GA at early stage (3 h) of treatment. HSP27 could be regulated by p38 MAPK pathway (39) and could interact directly with vimentin (40, 41). Results of Western blotting assay confirmed that GA treatment induced increase in phosphorylation of p38 and MAPKAPK2, the kinase that could phosphorylate HSP27 after activation of p38 MAPK pathway (42). And, GA treatment also induced increase in phosphorylation of HSP27 and decrease in the total protein level of HSP27. Furthermore, SB203580, an inhibitor of p38 MAPK, ameliorated the regulative effects of GA on HSP27. The results confirmed the activation of signal cascade from p38 MAPK to HSP27 by GA. In the present study, efforts were also devoted to check the roles of p38, HSP27, and vimentin in effects of GA using cells with siRNA knock-down of these genes. Unfortunately, great influence of knock-down of these genes to cell viability made it difficult to detect possible difference between the effects of GA in siRNA knock-down cells and the effects of GA in control cells. Only siRNA knock-down of vimentin showed ameliorating effect on cytotoxicity of 4 μm GA. The result confirmed the involvement of vimentin in effects of GA.
Notably, among the target-related proteins of GA found in late stage (24 h) of treatment in our proteomic analysis, seven proteins including vimentin were cytoskeleton-related proteins. The other six cytoskeleton-related proteins, besides vimentin, were β-actin, tubulin α6, keratin 8, keratin 18, gelsolin-like capping protein, and calumenin. β-actin and gelsolin-like capping protein were related to microfilaments. Gelsolin-like capping protein was a member of the actin-binding protein, which was crucial for the organization of the actin cytoskeleton. Evidence indicated that gelsolin-like capping protein was involved in the control of cell migration or invasion (43). Tubulin α6 was microtubule-related protein whereas vimentin, keratin 8, and keratin 18 were intermediate filament-related proteins. Calumenin was involved in regulation of intracellular calcium level and thus organization of cytoskeleton and carcinoma metastasis (44). Finding these cytoskeleton-related proteins as target-related proteins of GA was consistent with the previous reports that showed the regulative effects of GA on cytoskeleton structure (2). In our study, influence of GA on cytoskeleton structure of HeLa cells was confirmed.
Our in vivo study results showed that GA treatment inhibited tumor growth in nude mice inoculated with HeLa cells and also induced cleavage of vimentin in tumor tissues by Western blotting assay and immunohistochemical staining analysis. Furthermore, effects of GA on MCF-7 cells with low expression level of vimentin and MG-63 cells with high expression level of vimentin were also checked in the present study. The fact that GA exhibited weaker cytotoxicity in MCF-7 cells than in HeLa cells and stronger cytotoxicity in MG-63 cells than in HeLa cells further confirmed the critical role of vimentin in effects of GA. And, Western blotting assay results of EMT markers in GA-treated cells showed the inhibitive effect of GA on EMT.
In all, in the present study, vimentin was found to play an important role in the anticancer effects of GA. The pathway including p38 MAPK, HSP27, vimentin, and dysfunction of cytoskeleton was suggested to be activated by GA. Clarifying the important role of vimentin in the anticancer effects of GA might contribute to better use of GA in clinic. Up to now, results of phase I clinical study of GA had displayed that GA showed a good safety profile and tolerance in cancer patients. The maximum tolerance dose of intravenous GA was estimated to be 55 mg/m2 and the dose-restrictive toxicities included pain and liver damage occurred at 70 mg/m2 (45). In phase IIa clinical study, intravenous GA at 45 mg/m2 showed anticancer activities against advanced malignant cancers such as nonsmall cell lung, renal and colon cancers (46). Results of the present study showing the important role of vimentin in effects of GA suggested possible promising usage of GA in treatment of tumors with high level of vimentin such as osteosarcoma. Although, it has to be mentioned that other targets of GA also had to be considered in the use of GA. For example, MT1, a metabolite of GA, could bind proteasomal β5 subunit and exhibit proteasome-inhibiting effects (47). Further study clarifying the anticancer mechanisms of GA and more results of clinical use of GA might be necessary for successful development of GA as a new anticancer agent.
Supplementary Material
Acknowledgments
We thank Dr. Jiefei Tong (Program in Molecular Structure and Function, Hospital for Sick Children, Princess Margaret Cancer Centre, and Department of Molecular Genetics, Toronto, University of Toronto, Canada) and Dr. Yang Zhang (Institutes of Biomedical Sciences, Fudan University) for their help in revising the manuscript.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD002975 and PXD002985.
Footnotes
Author contributions: X.L. and D.G. designed research; Q.Y., L.F., B.C., M.L., and D.Z. performed research; W.W., B.J., and M.Y. contributed new reagents or analytic tools; Q.Y., L.F., B.C., M.L., and D.Z. analyzed data; Q.Y., X.L., and D.G. wrote the paper.
* This work was supported in part by grants from the Twelfth Five-Year National Science & Technology Support Program (2012BAI29B06), Shanghai Science & Technology Support Program (13431900401), China Postdoctoral Science Foundation funded project (2012M510907), Shanghai Postdoctoral Scientific Program (13R21417800), the Postdoctor Research Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (2012KIP516), the Sanofi-Aventis-Shanghai Institutes for Biological Sciences Scholarship Program, the National Nature Science Foundation (81302809, 81373964), the fund (syzrc2014-001, syz2014-005) and the grant constructed multi-disciplinary team for oncology in No. 3 People's Hospital, School of Medicine, Shanghai Jiaotong University.
 This article contains supplemental Figs. S1 to S8 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S8 and Tables S1 to S4.
1 The abbreviations used are:
- GA
- gambogic acid
- HSP27
- heat shock protein 27
- EMT
- epithelial-to-mesenchymal transition
- DMSO
- dimethyl sulfoxide
- IEF
- isoelectric focusing
- CHCA
- α-cyano-4-hydroxycinnamic acid
- TFA
- trifluoroacetic acid
- ACN
- acetonitrile
- CID
- collision-induced dissociation
- FDR
- false discovery rate; information dependent acquisition
- IDA
- information dependent acquisition
- HSPB1
- heat shock 27kDa protein 1
- MAPKAPK2
- mitogen-activated protein kinase-activated protein kinase 2.
REFERENCES
- 1.Ren Y., Yuan C., Chai H. B., Ding Y., Li X. C., Ferreira D., and Kinghorn A. D. (2011) Absolute configuration of (-)-gambogic acid, an antitumor agent. J. Nat. Products 74, 460–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J., Gu H. Y., Lu N., Yang Y., Liu W., Qi Q., Rong J. J., Wang X. T., You Q. D., and Guo Q. L. (2008) Microtubule depolymerization and phosphorylation of c-Jun N-terminal kinase-1 and p38 were involved in gambogic acid induced cell cycle arrest and apoptosis in human breast carcinoma MCF-7 cells. Life Sci. 83, 103–109 [DOI] [PubMed] [Google Scholar]
- 3.Xu Y., Ouyang J., Zhang Q. G., Zhou M., Li J., Chen M. M., and Xu Y. Y. (2012) [Gambogic acid induces apoptosis of Jurkat cell through the MAPK signal pathway]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 20, 587–591 [PubMed] [Google Scholar]
- 4.Zhu X., Zhang H., Lin Y., Chen P., Min J., Wang Z., Xiao W., and Chen B. (2009) Mechanisms of gambogic acid-induced apoptosis in non-small cell lung cancer cells in relation to transferrin receptors. J. Chemother. 21, 666–672 [DOI] [PubMed] [Google Scholar]
- 5.Zaks-Makhina E., Li H., Grishin A., Salvador-Recatala V., and Levitan E. S. (2009) Specific and slow inhibition of the kir2.1 K+ channel by gambogic acid. J. Biol. Chem. 284, 15432–15438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rahman M. A., Kim N. H., and Huh S. O. (2013) Cytotoxic effect of gambogic acid on SH-SY5Y neuroblastoma cells is mediated by intrinsic caspase-dependent signaling pathway. Mol. Cell. Biochem. 377, 187–196 [DOI] [PubMed] [Google Scholar]
- 7.Xu X., Liu Y., Wang L., He J., Zhang H., Chen X., Li Y., Yang J., and Tao J. (2009) Gambogic acid induces apoptosis by regulating the expression of Bax and Bcl-2 and enhancing caspase-3 activity in human malignant melanoma A375 cells. Int. J. Dermatol. 48, 186–192 [DOI] [PubMed] [Google Scholar]
- 8.Shu W., Chen Y., Li R., Wu Q., Cui G., Ke W., and Chen Z. (2008) Involvement of regulations of nucleophosmin and nucleoporins in gambogic acid-induced apoptosis in Jurkat cells. Basic Clin. Pharmacol. Toxicol. 103, 530–537 [DOI] [PubMed] [Google Scholar]
- 9.Pandey M. K., Sung B., Ahn K. S., Kunnumakkara A. B., Chaturvedi M. M., and Aggarwal B. B. (2007) Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood 110, 3517–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boran A. D., and Iyengar R. (2010) Systems pharmacology. Mount Sinai J. Med. 77, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yue Q. X., Xie F. B., Guan S. H., Ma C., Yang M., Jiang B. H., Liu X., and Guo D. A. (2008) Interaction of Ganoderma triterpenes with doxorubicin and proteomic characterization of the possible molecular targets of Ganoderma triterpenes. Cancer Sci. 99, 1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng L., Zhang D., Fan C., Ma C., Yang W., Meng Y., Wu W., Guan S., Jiang B., Yang M., Liu X., and Guo D. (2013) ER stress-mediated apoptosis induced by celastrol in cancer cells and important role of glycogen synthase kinase-3beta in the signal network. Cell Death Dis. 4, e715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshikawa T., Ogata N., Izuta H., Shimazawa M., Hara H., and Takahashi K. (2011) Increased expression of tight junctions in ARPE-19 cells under endoplasmic reticulum stress. Curr. Eye Res. 36, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 14.Yue Q. X., Song X. Y., Ma C., Feng L. X., Guan S. H., Wu W. Y., Yang M., Jiang B. H., Liu X., Cui Y. J., and Guo D. A. (2010) Effects of triterpenes from Ganoderma lucidum on protein expression profile of HeLa cells. Phytomedicine 17, 606–613 [DOI] [PubMed] [Google Scholar]
- 15.Ma C., Yue Q. X., Guan S. H., Wu W. Y., Yang M., Jiang B. H., Liu X., and Guo D. A. (2009) Proteomic analysis of possible target-related proteins of cyclophosphamide in mice thymus. Food Chem. Toxicol. 47, 1841–1847 [DOI] [PubMed] [Google Scholar]
- 16.Yue Q. X., Cao Z. W., Guan S. H., Liu X. H., Tao L., Wu W. Y., Li Y. X., Yang P. Y., Liu X., and Guo D. A. (2008) Proteomics characterization of the cytotoxicity mechanism of ganoderic acid D and computer-automated estimation of the possible drug target network. Mol. Cell. Proteomics 7, 949–961 [DOI] [PubMed] [Google Scholar]
- 17.Zeng Y. Y., Chen H. J., Shiau K. J., Hung S. U., Wang Y. S., and Wu C. C. (2012) Efficient enrichment of phosphopeptides by magnetic TiO(2)-coated carbon-encapsulated iron nanoparticles. Proteomics 12, 380–390 [DOI] [PubMed] [Google Scholar]
- 18.Nagaraj S. H., Harsha H. C., Reverter A., Colgrave M. L., Sharma R., Andronicos N., Hunt P., Menzies M., Lees M. S., Sekhar N. R., Pandey A., and Ingham A. (2012) Proteomic analysis of the abomasal mucosal response following infection by the nematode, Haemonchus contortus, in genetically resistant and susceptible sheep. J. Proteomics 75, 2141–2152 [DOI] [PubMed] [Google Scholar]
- 19.Lam M. P., Scruggs S. B., Kim T. Y., Zong C., Lau E., Wang D., Ryan C. M., Faull K. F., and Ping P. (2012) An MRM-based workflow for quantifying cardiac mitochondrial protein phosphorylation in murine and human tissue. J. Proteomics 75, 4602–4609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu P., Li J., Shi L., Selke M., Chen B., and Wang X. (2013) Synergetic effect of functional cadmium-tellurium quantum dots conjugated with gambogic acid for HepG2 cell-labeling and proliferation inhibition. Int. J. Nanomed. 8, 3729–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao W., Zhou S. F., Zhang Z. P., Xu G. P., Li X. B., and Yan J. L. (2011) Gambogic acid inhibits the growth of osteosarcoma cells in vitro by inducing apoptosis and cell cycle arrest. Oncology Reports 25, 1289–1295 [DOI] [PubMed] [Google Scholar]
- 22.Yu J., Guo Q. L., You Q. D., Zhao L., Gu H. Y., Yang Y., Zhang H. W., Tan Z., and Wang X. (2007) Gambogic acid-induced G2/M phase cell-cycle arrest via disturbing CDK7-mediated phosphorylation of CDC2/p34 in human gastric carcinoma BGC-823 cells. Carcinogenesis 28, 632–638 [DOI] [PubMed] [Google Scholar]
- 23.Dave J. M., and Bayless K. J. (2014) Vimentin as an integral regulator of cell adhesion and endothelial sprouting. Microcirculation 21, 333–344 [DOI] [PubMed] [Google Scholar]
- 24.Pallari H. M., and Eriksson J. E. (2006) Intermediate filaments as signaling platforms. Science's STKE 366, pe53. [DOI] [PubMed] [Google Scholar]
- 25.Satelli A., and Li S. (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 68, 3033–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shirahata A., Sakuraba K., Kitamura Y., Yokomizo K., Gotou T., Saitou M., Kigawa G., Nemoto H., Sanada Y., and Hibi K. (2012) Detection of vimentin methylation in the serum of patients with gastric cancer. Anticancer Res. 32, 791–794 [PubMed] [Google Scholar]
- 27.Song B. P., Jain S., Lin S. Y., Chen Q., Block T. M., Song W., Brenner D. E., and Su Y. H. (2012) Detection of hypermethylated vimentin in urine of patients with colorectal cancer. J. Mol. Diagn. 14, 112–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tozser J., Bagossi P., Boross P., Louis J. M., Majerova E., Oroszlan S., and Copeland T. D. (1999) Effect of serine and tyrosine phosphorylation on retroviral proteinase substrates. Eur. J. Biochem. 265, 423–429 [DOI] [PubMed] [Google Scholar]
- 29.Byun Y., Chen F., Chang R., Trivedi M., Green K. J., and Cryns V. L. (2001) Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 8, 443–450 [DOI] [PubMed] [Google Scholar]
- 30.Nakajima E., Hammond K. B., Rosales J. L., Shearer T. R., and Azuma M. (2011) Calpain, not caspase, is the causative protease for hypoxic damage in cultured monkey retinal cells. Invest. Ophthalmol. Visual Sci. 52, 7059–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura N., Tsuru A., Hirayoshi K., and Nagata K. (1992) Purification and characterization of a vimentin-specific protease in mouse myeloid leukemia cells. Regulation during differentiation and identity with cathepsin G. Eur. J. Biochem. 205, 947–954 [DOI] [PubMed] [Google Scholar]
- 32.Sihag R. K., Inagaki M., Yamaguchi T., Shea T. B., and Pant H. C. (2007) Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp. Cell Res. 313, 2098–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ando S., Tanabe K., Gonda Y., Sato C., and Inagaki M. (1989) Domain- and sequence-specific phosphorylation of vimentin induces disassembly of the filament structure. Biochemistry 28, 2974–2979 [DOI] [PubMed] [Google Scholar]
- 34.Eriksson J. E., He T., Trejo-Skalli A. V., Harmala-Brasken A. S., Hellman J., Chou Y. H., and Goldman R. D. (2004) Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117, 919–932 [DOI] [PubMed] [Google Scholar]
- 35.Aziz A., Hess J. F., Budamagunta M. S., Voss J. C., and Fitzgerald P. G. (2010) Site-directed spin labeling and electron paramagnetic resonance determination of vimentin head domain structure. J. Biol. Chem. 285, 15278–15285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lahat G., Zhu Q. S., Huang K. L., Wang S., Bolshakov S., Liu J., Torres K., Langley R. R., Lazar A. J., Hung M. C., and Lev D. (2010) Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PloS One 5, e10105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Guo K., Kang N. X., Li Y., Sun L., Gan L., Cui F. J., Gao M. D., and Liu K. Y. (2009) Regulation of HSP27 on NF-kappaB pathway activation may be involved in metastatic hepatocellular carcinoma cells apoptosis. BMC Cancer 9, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acunzo J., Katsogiannou M., and Rocchi P. (2012) Small heat shock proteins HSP27 (HspB1), alphaB-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death. Int. J. Biochem. Cell Biol. 44, 1622–1631 [DOI] [PubMed] [Google Scholar]
- 39.Clements R. T., Feng J., Cordeiro B., Bianchi C., and Sellke F. W. (2011) p38 MAPK-dependent small HSP27 and alphaB-crystallin phosphorylation in regulation of myocardial function following cardioplegic arrest. Am. J. Physiol. 300, H1669–H1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia Y., Wu S. L., Isenberg J. S., Dai S., Sipes J. M., Field L., Zeng B., Bandle R. W., Ridnour L. A., Wink D. A., Ramchandran R., Karger B. L., and Roberts D. D. (2010) Thiolutin inhibits endothelial cell adhesion by perturbing Hsp27 interactions with components of the actin and intermediate filament cytoskeleton. Cell Stress Chaperones 15, 165–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J. S., Zhang M. H., Yun E. K., Geum D., Kim K., Kim T. H., Lim Y. S., and Seo J. S. (2005) Heat shock protein 27 interacts with vimentin and prevents insolubilization of vimentin subunits induced by cadmium. Exp. Mol. Med. 37, 427–435 [DOI] [PubMed] [Google Scholar]
- 42.Zheng C., Lin Z., Zhao Z. J., Yang Y., Niu H., and Shen X. (2006) MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J. Biol. Chem. 281, 37215–37226 [DOI] [PubMed] [Google Scholar]
- 43.Nomura H., Uzawa K., Ishigami T., Kouzu Y., Koike H., Ogawara K., Siiba M., Bukawa H., Yokoe H., Kubosawa H., and Tanzawa H. (2008) Clinical significance of gelsolin-like actin-capping protein expression in oral carcinogenesis: an immunohistochemical study of premalignant and malignant lesions of the oral cavity. BMC Cancer 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Honore B., and Vorum H. (2000) The CREC family, a novel family of multiple EF-hand, low-affinity Ca(2+)-binding proteins localised to the secretory pathway of mammalian cells. FEBS Letts. 466, 11–18 [DOI] [PubMed] [Google Scholar]
- 45.Zhou Z. T., and Wang J. W. (2007) Phase I human tolerability trail of Gambogic acid. Chin. New Drugs 16, 435–437 [Google Scholar]
- 46.Chi Y., Zhan X. K., Yu H., Xie G. R., Wang Z. Z., Xiao W., Wang Y. G., Xiong F. X., Hu J. F., Yang L., Cui C. X., and Wang J. W. (2013) An open-labeled, randomized, multicenter phase IIa study of gambogic acid injection for advanced malignant tumors. Chin. Med. J. 126, 1642–1646 [PubMed] [Google Scholar]
- 47.Li X., Liu S., Huang H., Liu N., Zhao C., Liao S., Yang C., Liu Y., Zhao C., Li S., Lu X., Liu C., Guan L., Zhao K., Shi X., Song W., Zhou P., Dong X., Guo H., Wen G., Zhang C., Jiang L., Ma N., Li B., Wang S., Tan H., Wang X., Dou Q. P., and Liu J. (2013) Gambogic acid is a tissue-specific proteasome inhibitor in vitro and in vivo. Cell Reports 3, 211–222 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
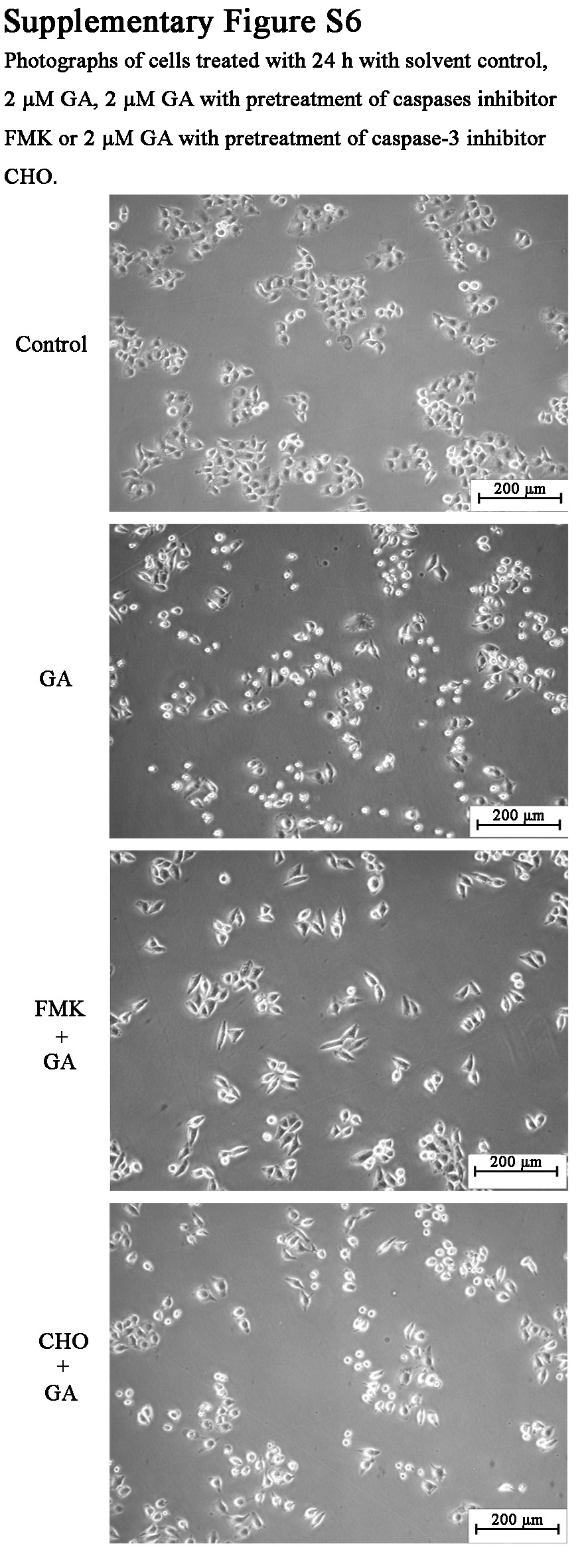{kind=link}
{kind=link}
{kind=link}