

Abstract
Modification of cytosine-guanine dinucleotides (CpGs) is a key part of mammalian epigenetic regulation and helps shape cellular identity. Tet enzymes catalyze stepwise oxidation of 5-methylcytosine (mC) in CpGs to 5-hydroxymethylcytosine (hmC), or onward to 5-formylcytosine (fC) or 5-carboxylcytosine (caC). The multiple mC oxidation products, while intricately linked, are postulated to play independent epigenetic roles, making it critical to understand how the products of stepwise oxidation are established and maintained. Using highly sensitive isotope-based studies, we newly show that Tet2 can yield fC and caC by iteratively acting in a single encounter with mC-containing DNA, without release of the hmC intermediate, and that the modification state of the complementary CpG has little impact on Tet2 activity. By revealing Tet2 as an iterative, de novo mC oxygenase, our study provides insight into how features intrinsic to Tet2 shape the epigenetic landscape.
In mammalian genomes, cytosine base modifications provide an epigenetic layer of information that can influence development, differentiation, and pluripotency. While 5-methylcytosine (mC) was long considered the predominant modification,1,2 the discovery of Tet family enzymes opened a new and expanded view of the epigenome.3 Tet enzymes are α-ketoglutarate-, Fe2+-dependent dioxygenases that can act on mC to generate 5-hydroxymethylcytosine (hmC) in genomic DNA (Figure 1), a modification readily detected in many cell types.4 Further, although hmC predominates, Tet enzymes can also catalyze stepwise oxidation of hmC to 5-formylcytosine (fC), and fC to 5-carboxylcytosine (caC), for a total of three oxidized mC (ox-mC) derivatives.5−7
Figure 1.
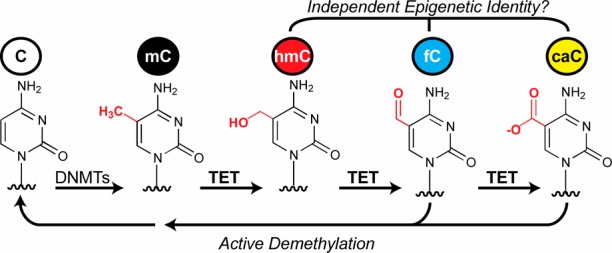
Tet enzymes act stepwise on mC to generate the extended epigenome. The oxidized mC bases—hmC, fC, and caC—could each play independent roles in epigenetic regulation. Only fC and caC are substrates for active DNA demethylation via base excision repair.
Now viewed as part of the extended epigenome, ox-mC bases appear to have distinct functions. Like mC, they could impact gene expression: ox-mCs interact with different sets of proteins, including transcription factors and RNA polymerase.8,9 They also have distinct genomic profiles, which can persist stably over time and differ by cell type.10−15 Additionally, ox-mCs can play different roles in the dynamic process of DNA demethylation. While all the ox-mCs may facilitate passive, replication-dependent demethylation, fC and caC, but not hmC, are specifically implicated in some proposed pathways for active demethylation, such as base excision repair mediated by thymine DNA glycosylase.4,6,16
In light of ox-mC modifications, CpGs can be considered complex units of epigenetic information, in which either DNA strand can contain unmodified cytosine or one of its four derivatives. A major question is how these marks are established and maintained. For the methylation code, this task is attributed to the coordinated action of DNA methyltransferases (DNMTs).17,18De novo DNMTs largely establish methylation patterns, showing similar preference for both unmodified (C/C) and hemi-methylated (mC/C) CpGs. In contrast, maintenance DNMTs show a strong preference for hemi-methylated CpGs and thereby function to maintain the CpG methylation code after genomic replication.
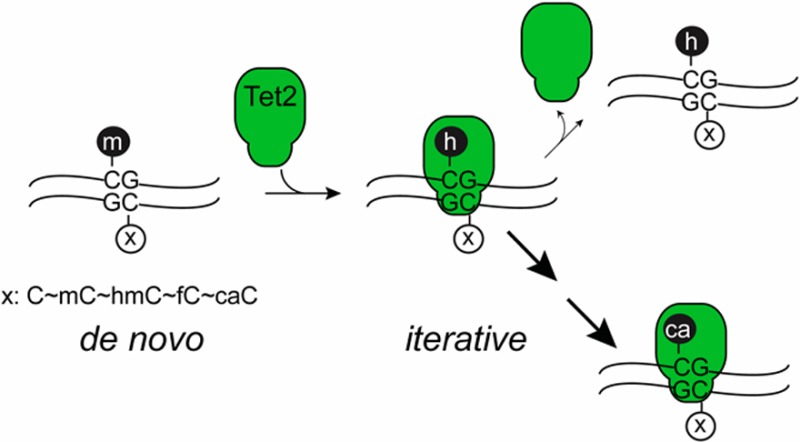
In contrast to methylation, the mechanisms involved in the generation and maintenance of specific ox-mCs remain unknown. These questions present a particular challenge since Tet enzymes catalyze not one but three reactions at CpGs. While it is now established that mC can be oxidized in a stepwise manner, it remains unknown if these events require multiple encounters between the enzyme and DNA (sequential model) or if caC can be generated in a single encounter with mC-containing DNA (iterative model). This issue is critical to resolve, as the prevalence of mC over hmC raises the question of how highly oxidized bases could be established at a given CpG.11,12,19 Similarly unexplored is the question of whether ox-mC marks, once established on one strand, can influence the activity of TET on the opposite CpG. This is important because propagation of epigenetic identity depends on maintenance, and it is unknown whether, akin to DNMTs, TET enzymes can maintain ox-mC marks across cellular generations.
Here, we focused on understanding how ox-mCs are established and maintained by Tet2. Sequential versus iterative and maintenance versus de novo models for Tet activity have not yet been resolved in part because prior assays have involved significant substrate depletion and were not designed to report on strand-specific modifications.3,5,6,20 To overcome these limitations, we devised highly sensitive, strand-specific, isotopologue-based assays.
Starting from a 27-nt oligonucleotide containing a central CpG moiety, we enzymatically introduced a single isotopically modified methyl group on the substrate (Supporting Information, Figures S1 and S2). Initially, a [14C]-mCpG-containing strand was hybridized to a complementary strand containing an unmodified CpG. After reacting this duplex with mouse Tet2 catalytic domain (mTet2-CD, hereafter referred to as Tet2; Figure S3A), the DNA was enzymatically digested to component nucleosides. The nucleoside mixture was subjected to HPLC fractionation and liquid scintillation counting (LSC) to track the kinetics and distribution of [14C]-mC oxidation with high sensitivity (Figure 2A). To describe the enzymatic total specific activity (TSA), we accounted for stepwise oxidation. As each detected fC product requires an undetected intermediate hmC, and caC requires intermediate hmC and fC, the observed specific activity (SA) values for fC and caC generation were multiplied to calculate TSA (eq 1).
| 1 |
Figure 2.
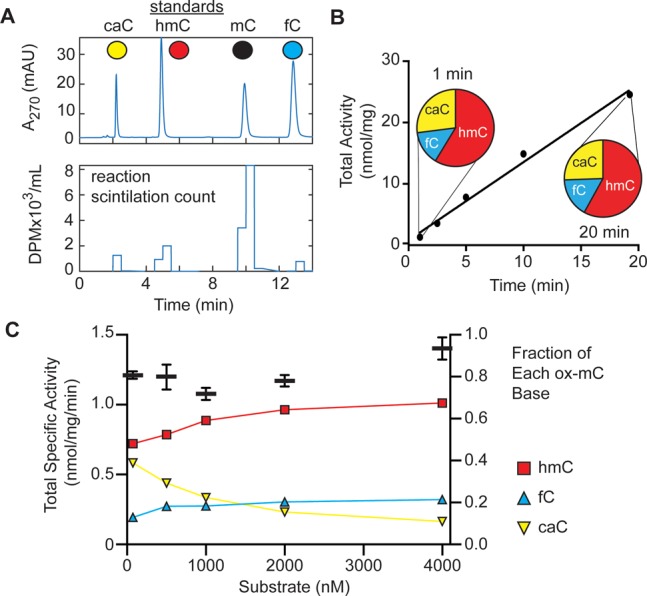
Tet2 generates fC and caC early and without a requirement for hmC accumulation. (A) Example traces for [14C]-mC Tet activity assay. DNA duplexes from Tet reactions were degraded and spiked with standards to delineate fractions containing each base. The fractions were then subjected to scintillation counting. Top: Chromatogram of nucleoside standards (10 μL of 10 μM each). Bottom: Corresponding LSC trace. (B) Time course of Tet2 (10 μg/mL) turnover of 500 nM [14C]-mCpG/CpG duplexed DNA showing total activity and fractions of each ox-mC at 1 and 20 min. (C) Titration of 75–4000 nM [14C]-mCpG/CpG with 5 μg/mL Tet2, reacted for 10 min. Total specific activity is plotted on the left y-axis (black bars) as mean and SD of duplicate experiments, along with fraction of each ox-mC base on the right y-axis.
We determined optimized enzyme conditions (Figure S3) and in an initial assay observed a TSA of 1.3 nmol·min–1·mg–1 (Figure 2B), which puts an approximate lower limit for turnover at 0.13 min–1. Notably, we detected minimal loss in activity over 20 min, in contrast to prior reports of time-dependent loss of activity.5,20 The sensitivity achieved by our assays also revealed two additional relevant features. First, even at the earliest time points with <1% product formation, we can readily detect the formation of caC. Second, the distribution of the products between hmC, fC, and caC were virtually unchanged at early versus late time points. These factors suggest a proficiency for Tet2-catalyzed stepwise oxidation under our reaction conditions.
A closer examination revealed interesting features at both lower and higher substrate concentrations (Figure 2C). On the low end, when reacting 5 μg/mL (maximally 50 nM) Tet2 with as low as 75 nM substrate, activity was near maximal levels. The result suggests a KM,DNA which is in the low nanomolar range; otherwise, a greater substrate dependence would be expected. Consistent with this observation, the TSA plateaus as the substrate concentration increases further. Notably, the increase in hmC at higher substrate concentrations appears limited relative to fC and caC. Thus, large amounts of fC and caC are formed even when mC is in vast and increasing excess of hmC and fC, respectively. For example, under these reaction conditions with 2000 nM substrate, Tet2 generates approximately 25 nM hmC, 8 nM fC, and 6 nM caC. These observations suggest one of two (non-exclusive) possibilities: a sequential oxidation model where mC, hmC, and fC substrates have substantially different kcat and KM values, or an iterative model where Tet2 remains bound to DNA in proximity to the reactive site to establish more highly oxidized bases.
To differentiate between models for how Tet2 establishes ox-mCs, we drew on techniques used previously to examine substrate channeling of metabolites between enzymes.21 In several metabolic pathways, the product of one reaction is directly fed to the next enzyme without diffusion into bulk solution. Distinct isotopic labels on substrates and products can be used to confirm this molecular hand-off. Given the analogy to the possible models for Tet activity, we set up an isotope-based competition assay that relies upon measuring the isotopic composition of fC and caC produced from [13C2H3]-mC (heavy; *mC) substrates mixed with natural-isotope hmC-containing substrates (light; Figure 3A). In the sequential oxidation model, if Tet2 releases the heavy hmC-containing duplex (*hmC) formed from *mC, the isotope will be diluted by the light hmC-containing substrate. Thus, the fraction of downstream heavy fC and caC (*fC and *caC, respectively) products can be no greater than the simultaneous fraction of *hmC/hmC as measured by LC-MS/MS. However, with the iterative oxidation model, if the *hmC product frequently remains bound to Tet2, then the downstream heavy products could be at a higher ratio. At an extreme, if iterative oxidation is highly efficient, we would expect the ratio of *fC/fC and *caC/caC to reflect the initial *mC/hmC ratios.
Figure 3.
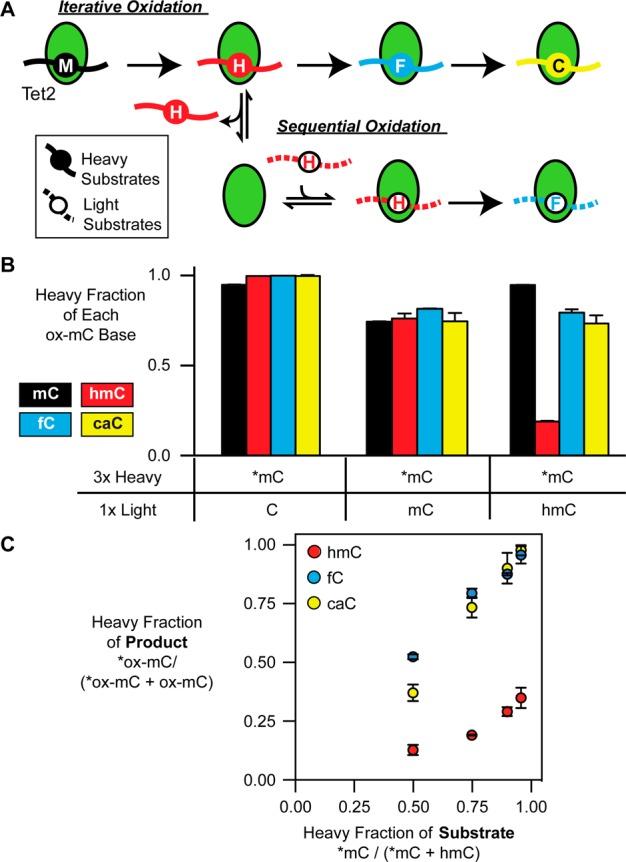
fC and caC are formed from iterative oxidation of mC without release of hmC. (A) Iterative versus sequential oxidation models. Tet2 is shown in green, heavy substrates as filled circles, and light as open circles with dotted lines. Tet2 complexed with heavy *mC-containing DNA can directly proceed to heavy fC and caC by iterative oxidation. In sequential oxidation, release of heavy hmC would result in mixing in solution with light hmC, generating predominantly light fC and caC. (B) 5 μg/mL Tet2 was incubated for 10 min with heavy *mCpG-containing duplexes mixed in a 3:1 ratio with light CpG-, mCpG-, or hmCpG-containing duplexes (100 nM total). Shown is the fraction of heavy isotope for each modification, as analyzed by LC-MS/MS. (C) Under similar conditions, Tet2 was incubated with varying ratios of *mC-containing DNA to light hmC-containing DNA. Shown is the heavy fraction of products, compared to the heavy fraction of substrate. The mean and SD are shown from triplicate experiments.
For our isotope dilution experiment, we enzymatically generated heavy S-adenosyl-l-methionine (SAM).22 Following our protocol for generating radiolabeled substrate, we analogously prepared a 27-nt duplex containing a single heavy *mC opposite an unreactive CpG (Figure S1A). We also prepared duplexes containing light CpG, mCpG, or hmCpG opposite an unreactive CpG. We reacted 5 μg/mL Tet2 with 100 nM of total duplex DNA containing various mixtures of the *mC duplex with light C, mC, or hmC duplex for 10 min. Reaction products were purified and degraded to nucleosides, and the heavy/light nucleoside ratios were determined with high-precision nano-LC-MS/MS (Figure S4).
As a control, reacting *mC-containing substrate in a 3:1 ratio with non-reactive, unmodified CpG duplex gave >95% yield of heavy *hmC, *fC, and *caC, consistent with the isotopic labeling ratio of the *mC (Figure 3B), and, as expected, no light or heavy ox-mC products were detected in the absence of Tet2. When Tet2 was reacted with a 3:1 mixture of the heavy *mC- and light mC-containing substrates, the heavy:light ratios of hmC, fC, and caC were all approximately 3:1, suggesting the absence of any dominant isotope effects. Strikingly, when the *mC-containing substrate was mixed 3:1 with light hmC-containing duplex, the ratios of heavy:light fC and caC were both ∼3:1. The accumulation of heavy *fC and *caC is most consistent with the iterative oxidation model, where *mC can be converted to higher ox-mCs without obligate release and dilution of the *hmC intermediate.
To determine the extent to which iterative oxidation was occurring, we varied the ratio of heavy *mC to light hmC substrates and quantified the isotopic composition of the resulting products (Figure 3C). Across the 1:1, 3:1, 9:1, and 23:1 ratios evaluated, heavy *hmC is generated. However, while *hmC never exceeded light hmC, *fC/fC and *caC/caC ratios were always in great excess of *hmC/hmC. Indeed, these ratios increasingly approach the initial *mC/hmC mixture, consistent with a dominant role for iterative oxidation in establishing the higher ox-mC products. When viewed alongside the [14C] experiments, these results also suggest that fC and caC generation under low-turnover conditions was a consequence of iterative activity, as opposed to significantly increased catalytic activity of Tet2 on hmC- or fC-containing duplexes.
In all of the above experiments, we examined duplexes with a single reactive substrate and a non-reactive opposite strand (unmodified CpG). Given the implications for maintenance of the extended epigenome, we next exploited our assays to examine the effects of opposite strand modifications on Tet2 reactivity. Utilizing our [14C]-mCpG assay, which cleanly reports on oxidation of the labeled strand only, we hybridized our original 27-mer [14C]-mCpG strand with complementary strands containing non-radioactive CpG, mCpG, hmCpG, fCpG, or caCpG. We then reacted 75 nM of each duplex with 5 μg/mL Tet2 for 10 min, purified, digested, and analyzed using HPLC and LSC as before. Across the substrates, we found that the TSAs for each reaction were very similar, with the largest difference occurring between the opposite strands fC and C, which differ only by a factor of 2 (Figure 4). Given the analogy to de novo DNMTs, which show minimal differences based on the methylation status of the opposite strand CpG, we suggest that Tet2 is therefore best classified as a de novo mC dioxygenase. Moreover, we note that the relative amounts of hmC, fC, and caC formed were very similar regardless of the identity of the opposite strand CpG (Figure 4). Thus, not only is overall activity largely unaffected, but stalling or iterative oxidation to generate the various ox-mCs is not dictated by the opposite strand CpG.
Figure 4.
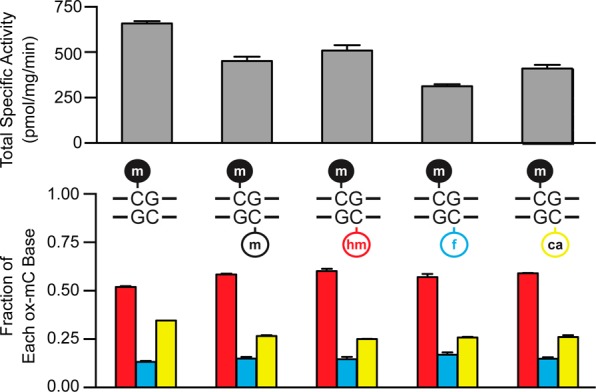
Tet2 is a de novo methylcytosine oxygenase: 75 nM [14C]-mCpG was duplexed to an unlabeled strand containing either CpG, mCpG, hmCpG, fCpG, or caCpG and then reacted with 5 μg/mL Tet2 for 10 min. TSA and the fraction of each ox-mC were measured by HPLC-LSC. Shown are the mean and SD from duplicate experiments.
Our results provide insight into how Tet2 helps establish the extended epigenome. We show that Tet2 can catalyze multiple ox-mC modifications in a single enzyme-substrate encounter, following the iterative oxidation model. Iterative oxidation helps to explain how fC and caC can be established in genomic DNA despite the relative abundance of mC over hmC or fC. Further, it has implications for the role of ox-mC’s either as independent marks or in active DNA demethylation. The fact that Tet2 can iteratively convert a single CpG to fC or caC means that these independent roles can be accessed without an obligatory stable, functional existence as hmC. Additionally, mC bases can be primed for demethylation by iterative oxidation to fC/caC, and this need not occur only at sites that first stably exist as hmC.
As noted earlier, in genomic DNA, hmC is far more prevalent than fC and caC.4−7 Our results indicate that fC and caC, when generated, can derive from a single encounter between Tet2 and mC; however, the result does not resolve the questions of how or why stalling at hmC is frequently seen in vivo, rather than progression to fC and caC. It is feasible that altering the Tet2-DNA encounter lifetime or chromatin accessibility mediates the accumulation of hmC in cells. The extent of iterative oxidation could also be influenced by levels of metabolites such as α-ketoglutarate, interactions with partner proteins, or the non-catalytic domains of Tet2, which could modulate or inhibit activity.
Our results also shed light on possible mechanisms by which ox-mCs are maintained in the extended epigenome. We show that CpG modifications on one strand neither impact overall Tet2 activity on the opposite strand nor skew the progression through stepwise oxidation. Thus, Tet2 appears capable of establishing oxidative marks wherever substrates are available, implying that all the permutations of CpG states are biochemically feasible members of the epigenetic repertoire. Notably, while the stable mapping of ox-mCs in various cell types implies maintenance,10−14 our data suggest that substrate preferences intrinsic to Tet2 do not offer a mechanism for such maintenance. Our results imply that alternative cellular factors or the coordinated activity of different Tet isoforms is more likely to be involved in restoring specific ox-mC marks at a given CpG after cellular division. Indeed, we anticipate that further studies on the mechanisms by which Tet enzymes target specific CpGs, regulate iterative oxidation, and coordinate with each other will shed additional light into the generation, maintenance, and functional roles of the extended epigenome.
Acknowledgments
This work was supported by the Rita Allen Foundation Scholar Award (R.M.K.) and NIH grants (R01GM110174, B.A.G.; F30CA196097, M.Y.L.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b10554.
Experimental protocols, optimized reaction conditions, experimental schemes, and representative data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Klose R. J.; Bird A. P. Trends Biochem. Sci. 2006, 31, 89–97. 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Schubeler D. Nature 2015, 517, 321–326. 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- Tahiliani M.; Koh K. P.; Shen Y.; Pastor W. A.; Bandukwala H.; Brudno Y.; Agarwal S.; Iyer L. M.; Liu D. R.; Aravind L.; Rao A. Science 2009, 324, 930–935. 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R. M.; Zhang Y. Nature 2013, 502, 472–479. 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; Shen L.; Dai Q.; Wu S. C.; Collins L. B.; Swenberg J. A.; He C.; Zhang Y. Science 2011, 333, 1300–1303. 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. F.; Li B. Z.; Li Z.; Liu P.; Wang Y.; Tang Q.; Ding J.; Jia Y.; Chen Z.; Li L.; Sun Y.; Li X.; Dai Q.; Song C. X.; Zhang K.; He C.; Xu G. L. Science 2011, 333, 1303–1307. 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffeneder T.; Hackner B.; Truss M.; Munzel M.; Muller M.; Deiml C. A.; Hagemeier C.; Carell T. Angew. Chem., Int. Ed. 2011, 50, 7008–7012. 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- Spruijt C. G.; Gnerlich F.; Smits A. H.; Pfaffeneder T.; Jansen P. W.; Bauer C.; Munzel M.; Wagner M.; Muller M.; Khan F.; Eberl H. C.; Mensinga A.; Brinkman A. B.; Lephikov K.; Muller U.; Walter J.; Boelens R.; van Ingen H.; Leonhardt H.; Carell T.; Vermeulen M. Cell 2013, 152, 1146–1159. 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhou Y.; Xu L.; Xiao R.; Lu X.; Chen L.; Chong J.; Li H.; He C.; Fu X. D.; Wang D. Nature 2015, 523, 621–625. 10.1038/nature14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Hon G. C.; Szulwach K. E.; Song C. X.; Zhang L.; Kim A.; Li X.; Dai Q.; Shen Y.; Park B.; Min J. H.; Jin P.; Ren B.; He C. Cell 2012, 149, 1368–1380. 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth M. J.; Marsico G.; Bachman M.; Beraldi D.; Balasubramanian S. Nat. Chem. 2014, 6, 435–440. 10.1038/nchem.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Wu X.; Shen L.; Zhang Y. Nat. Biotechnol. 2014, 32, 1231–1240. 10.1038/nbt.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman M.; Uribe-Lewis S.; Yang X.; Williams M.; Murrell A.; Balasubramanian S. Nat. Chem. 2014, 6, 1049–1055. 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman M.; Uribe-Lewis S.; Yang X.; Burgess H. E.; Iurlaro M.; Reik W.; Murrell A.; Balasubramanian S. Nat. Chem. Biol. 2015, 11, 555–557. 10.1038/nchembio.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffeneder T.; Spada F.; Wagner M.; Brandmayr C.; Laube S. K.; Eisen D.; Truss M.; Steinbacher J.; Hackner B.; Kotljarova O.; Schuermann D.; Michalakis S.; Kosmatchev O.; Schiesser S.; Steigenberger B.; Raddaoui N.; Kashiwazaki G.; Muller U.; Spruijt C. G.; Vermeulen M.; Leonhardt H.; Schar P.; Muller M.; Carell T. Nat. Chem. Biol. 2014, 10, 574–581. 10.1038/nchembio.1532. [DOI] [PubMed] [Google Scholar]
- Maiti A.; Drohat A. C. J. Biol. Chem. 2011, 286, 35334–35338. 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll M. G.; Bestor T. H. Annu. Rev. Biochem. 2005, 74, 481–514. 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Liu Y.; Upadhyay A. K.; Chang Y.; Howerton S. B.; Vertino P. M.; Zhang X.; Cheng X. Nucleic Acids Res. 2012, 40, 4841–4849. 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F.; Incarnato D.; Krepelova A.; Rapelli S.; Anselmi F.; Parlato C.; Medana C.; Dal Bello F.; Oliviero S. Cell Rep. 2015, 10, 674–683. 10.1016/j.celrep.2015.01.008. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Chen W.; Iyer L. M.; Hu J.; Wang G.; Fu Y.; Yu M.; Dai Q.; Aravind L.; He C. J. Am. Chem. Soc. 2014, 136, 4801–4804. 10.1021/ja500979k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivey H. O.; Ovadi J. Methods 1999, 19, 306–321. 10.1006/meth.1999.0858. [DOI] [PubMed] [Google Scholar]
- Ottink O. M.; Nelissen F. H.; Derks Y.; Wijmenga S. S.; Heus H. A. Anal. Biochem. 2010, 396, 280–283. 10.1016/j.ab.2009.09.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.