

Abstract
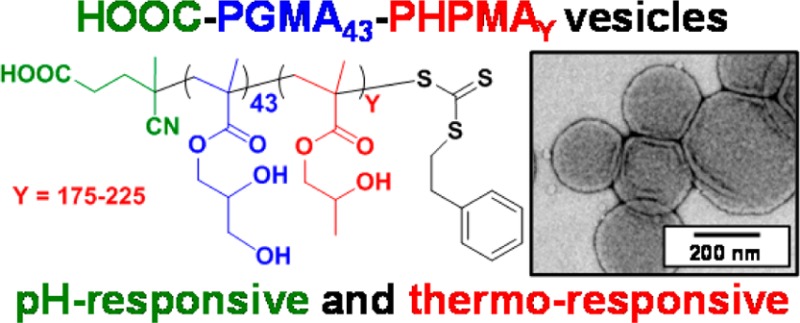
A series of non-ionic poly(glycerol monomethacrylate)–poly(2-hydroxypropyl methacrylate) (PGMA–PHPMA) diblock copolymer vesicles has been prepared by reversible addition–fragmentation chain transfer (RAFT) aqueous dispersion polymerization of HPMA at 70 °C at low pH using a carboxylic acid-based chain transfer agent. The degree of polymerization (DP) of the PGMA block was fixed at 43, and the DP of the PHPMA block was systematically varied from 175 to 250 in order to target vesicle phase space. Based on our recent work describing the analogous PGMA–PHPMA diblock copolymer worms [Lovett J. R.; et al. Angew. Chem. 2015, 54, 1279−1283], such diblock copolymer vesicles were expected to undergo an order–order morphological transition via ionization of the carboxylic acid end-group on switching the solution pH. Indeed, irreversible vesicle-to-sphere and vesicle-to-worm transitions were observed for PHPMA DPs of 175 and 200, respectively, as judged by turbidimetry, transmission electron microscopy (TEM), and dynamic light scattering (DLS) studies. However, such morphological transitions are surprisingly slow, with relatively long time scales (hours) being required at 20 °C. Moreover, no order–order morphological transitions were observed for vesicles comprising longer membrane-forming blocks (e.g., PGMA43–PHPMA225–250) on raising the pH from pH 3.5 to pH 6.0. However, in such cases the application of a dual stimulus comprising the same pH switch immediately followed by cooling from 20 to 5 °C, induces an irreversible vesicle-to-sphere transition. Finally, TEM and DLS studies conducted in the presence of 100 mM KCl demonstrated that the pH-responsive behavior arising from end-group ionization could be suppressed in the presence of added electrolyte. This is because charge screening suppresses the subtle change in the packing parameter required to drive the morphological transition.
Introduction
Over the past 50 years or so, there have been many reports of AB diblock copolymer self-assembly in solvents that are selective for only one block.1−12 In principle, varying the relative degrees of polymerization (DP) (and hence volume fractions) of each block enables a wide range of morphologies to be obtained in dilute solution, including spherical micelles, cylindrical micelles (e.g., rods or worms), or vesicles. In particular, stimulus-responsive nano-objects can be designed that undergo a morphological transition when exposed to an external stimulus such as temperature,13−18 light,19−23 salt,24−27 or pH.28−32 Rather less common are stimulus-responsive block copolymers that undergo two or more morphological transitions.33−37 Examples here include so-called “schizophrenic” diblock copolymers that can form two or more types of micelles in aqueous solution.34−37 Moreover, there are even fewer reports of morphological transitions driven by end-group effects. For example, O’Reilly and co-workers prepared a poly(N-isopropylacrylamide) (PNIPAM) based diblock copolymer using a quaternary amine-functionalized reversible addition–fragmentation transfer (RAFT) chain transfer agent (CTA) which self-assembled to form spherical micelles at 25 °C. However, heating above the copolymer lower critical solution temperature (LCST) induced a sphere-to-vesicle morphological transition, with the vesicles being stabilized by the cationic end-groups.38 The same team reported that hydrophilic polyamine homopolymers prepared by RAFT polymerization also underwent self-assembly provided that each end of the chain is capped with a hydrophobic end-group.39 Gibson and co-workers utilized pyridyl disulfide linkages in order to introduce hydrophilic end-groups so as to raise the LCST of PNIPAM. This strategy enabled a coil-to-globule transition to be achieved at constant temperature via selective cleavage of the hydrophilic end-group using glutathione.40 Recently, Du and co-workers found that PNIPAM and poly[oligo(ethylene glycol) methacrylate] homopolymers self-assembled to form various morphologies in aqueous solution by terminal alkynyl end-groups, apparently via a hydrogen bonding mechanism.41 Biocompatible nanoparticles that undergo either order–order or order–disorder morphology transitions upon exposure to a physiologically relevant stimulus are of particular interest for potential drug delivery applications.2,12,42−46 For example, the internal lumen of a vesicle may be used to encapsulate a payload, which can be released when subjected to a certain stimulus.47−51
Typically, diblock copolymer self-assembly requires postpolymerization processing via a pH or solvent switch, which is invariably conducted in dilute aqueous solution (<1 wt %).7,9,13,52 Recently, polymerization-induced self-assembly (PISA) has become widely recognized as a highly versatile route to diblock copolymer nano-objects.15,16,53−72 This approach is typically based on RAFT polymerization and enables nano-objects to be prepared at much higher concentrations (10–50% solids) in either aqueous solution,57−63 polar solvents,64−69 or non-polar solvents.15,16,70,71 For example, Armes and co-workers reported the chain extension of poly(glycerol monomethacrylate) (PGMA) macro-CTA using 2-hydroxypropyl methacrylate (HPMA) using a RAFT aqueous dispersion polymerization formulation.57 For a relatively narrow range of target diblock compositions and copolymer concentrations, a well-defined worm phase can be obtained.73 Such worms form soft, transparent, free-standing gels at 20 °C due to multiple inter-worm contacts and undergo a reversible worm-to-sphere transition on cooling to 4–5 °C.74,75 Recently, Lovett et al. reported that the preparation of such non-ionic PGMA–PHPMA diblock copolymers using a carboxylic acid-functionalized RAFT agent produced new worm gels that unexpectedly exhibited both thermo-responsive and pH-responsive behavior.75,76 More specifically, a reversible worm-to-sphere transition with concomitant degelation occurred on either cooling to 4 °C or on increasing the dispersion pH from around pH 3.5 to pH 6.0 at 20 °C. The former transition is the result of surface plasticization of the PHPMA core-forming block,62 whereas the latter transition is caused by ionization of a single terminal carboxylic acid group located at the end of the PGMA stabilizer block. In both cases there is a subtle shift in the geometric packing parameter, P, from the worm regime (0.33 < P < 0.50) to the sphere regime (P < 0.33).2,12,77
Herein we utilize the same RAFT aqueous dispersion polymerization formulation to prepare a series of four HOOC-PGMA43–PHPMAX vesicles with a fixed PGMA stabilizer DP and a variable PHPMA core-forming block DP (where X = 175, 200, 225, or 250). The pH- and thermo-responsive behavior of these non-ionic vesicles is examined in aqueous solution using TEM, DLS, turbidimetry, and rheology.
Experimental Section
Materials
Glycerol monomethacrylate (GMA; 99.8%) was donated by GEO Specialty Chemicals (Hythe, UK) and was used without further purification. 2-Hydroxypropyl methacrylate (HPMA) was purchased from Alfa Aesar (UK) and was used as received. 4,4′-Azobis(4-cyanopentanoic acid) (ACVA; V-501; 99%), ethanol (99%, anhydrous grade), methanol, and dichloromethane were purchased from Sigma-Aldrich UK and were used as received. All solvents were of HPLC quality and were purchased from Fisher Scientific (Loughborough, UK). 4-Cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) was prepared and purified as reported elsewhere.78
Synthesis of Poly(glycerol monomethacrylate) (HOOC-PGMA43) Macro-CTA
GMA (30.0 g, 187 mmol), PETTC (1.156 g, 3.4 mmol; target DP = 55), and ACVA (0.191 g, 0.68 mmol; PETTC/ACVA molar ratio = 5.0) were accurately weighed into a 250 mL round-bottomed flask. Anhydrous ethanol (previously purged with nitrogen for 1 h) was then added to produce a 50% w/w solution, which was placed in an ice bath and purged under nitrogen for 45 min at 0 °C. The sealed flask was immersed in an oil bath set at 70 °C to initiate the RAFT polymerization of GMA and stirred for 2 h at this temperature. The polymerization was then quenched at approximately 81% conversion by exposure to air, followed by cooling the reaction mixture to room temperature. Methanol (20 mL) was added to dilute the reaction solution, followed by precipitation into a 10-fold excess of dichloromethane in order to remove unreacted GMA monomer. The precipitate was isolated via filtration and washed with excess dichloromethane before being dissolved in methanol (50 mL). The crude polymer was precipitated for a second time by addition to excess dichloromethane and isolated by filtration. It was then dissolved in water and freeze-dried overnight to afford a yellow solid. 1H NMR studies indicated a mean degree of polymerization of 43 via end-group analysis (the integrated aromatic RAFT end-group signals at 7.1–7.4 ppm were compared to those assigned to the two oxymethylene protons at 3.5–4.4 ppm). DMF GPC studies (refractive index detector; calibrated against a series of 10 near-monodisperse poly(methyl methacrylate) standards) indicated an Mn of 15 400 g mol–1 and an Mw/Mn of 1.20.
Synthesis of HOOC-PGMA43–PHPMAX Diblock Copolymer Vesicles via RAFT Aqueous Dispersion Polymerization of HPMA
A typical protocol for the chain extension of HOOC-PGMA43 macro-CTA with 175 units of HPMA via RAFT aqueous dispersion polymerization of HPMA is as follows: PGMA43 macro-CTA (0.143 g, 0.020 mmol), HPMA monomer (0.50 g, 3.5 mmol), and ACVA (1.9 mg, 0.006 mmol; PGMA43 macro-CTA/ACVA molar ratio = 3.0) were added to a 25 mL round-bottomed flask, prior to addition of water to produce a 10% w/w solution. This reaction solution was purged with nitrogen gas for 30 min at 20 °C prior to immersion into an oil bath set at 70 °C. The reaction mixture was stirred for 4 h to ensure essentially complete conversion of the HPMA monomer (>99% by 1H NMR analysis) and was quenched by exposure to air, followed by cooling to ambient temperature. The resulting turbid free-flowing dispersion was characterized by DLS, TEM, and rheology without further purification.
Instrumentation
NMR Spectroscopy
1H NMR spectra were recorded using a 400 MHz Bruker Avance-500 spectrometer (64 scans averaged per spectrum).
Gel Permeation Chromatography (GPC)
Polymer molecular weights and polydispersities were determined using a DMF GPC setup operating at 60 °C and comprising two Polymer Laboratories PL gel 5 μm Mixed-C columns connected in series to a Varian 390-LC multidetector suite (only the refractive index detector was utilized) and a Varian 290-LC pump injection module. The GPC eluent was HPLC-grade DMF containing 10 mM LiBr at a flow rate of 1.0 mL min–1. DMSO was used as a flow-rate marker. Calibration was conducted using a series of 10 near-monodisperse poly(methyl methacrylate) standards (Mn = 625–2 480 000 g mol–1). Chromatograms were analyzed using Varian Cirrus GPC software (version 3.3).
DLS
DLS studies were conducted using a Malvern Zetasizer NanoZS instrument on 0.10% w/w aqueous dispersions at 25 °C in disposable cuvettes at a fixed scattering angle of 173°. The solution pH of the initially acidic copolymer dispersions was adjusted using 0.1 M KOH. Intensity-average hydrodynamic diameters were calculated via the Stokes–Einstein equation using a non-negative least-squares (NNLS) algorithm. All data were averaged over three consecutive runs.
Aqueous Electrophoresis
Measurements were performed on 0.10% w/w aqueous copolymer dispersions containing 10–3 mol dm–3 KCl as background electrolyte using a Malvern Zetasizer NanoZS at 25 °C. The solution pH of the initially acidic copolymer dispersions was adjusted using 0.1 M KOH. Zeta potentials were calculated from the Henry equation using the Smoluchowski approximation. All data were averaged over three consecutive runs.
Transmission Electron Microscopy (TEM)
Solutions were diluted 100-fold at either 20 or 5 °C to generate 0.10% w/w dispersions. Images obtained at lower pH were prepared by diluting dispersions using acidified water at the desired solution pH. Copper/palladium TEM grids (Agar Scientific, UK) were surface-coated in-house to yield a thin film of amorphous carbon. The grids were then plasma glow-discharged for 30 s to create a hydrophilic surface. Individual samples (0.10% w/w, 12 μL) were adsorbed onto the freshly glow-discharged grids for 60 s and then blotted with filter paper to remove excess solution. To stain the aggregates, a 9 μL drop of 0.75% w/w uranyl formate solution was soaked on the sample-loaded grid for 20 s and then carefully blotted to remove excess stain. The grids were then dried using a vacuum hose. Imaging was performed at 80 kV using a FEI Tecnai Spirit microscope equipped with a Gatan 1kMS600CW CCD camera.
Rheology Studies
Storage (G′) and loss (G″) moduli were determined between 25 and 4 °C for the PGMA43–HPMA200 diblock copolymer dispersion after a pH switch from 3.5 to 6.0 using a TA Instruments AR-G2 rheometer. A cone-and-plate geometry (40 mm 2° aluminum cone) was used for these measurements, which were conducted at a fixed strain of 1.0% and an angular frequency of 1.0 rad s–1.
Turbidimetry
Turbidimetry curves were recorded at 20 °C using a PerkinElmer Lambda 25 instrument operating in time drive mode at a fixed wavelength of 450 nm for 20 h. Prior to analysis, the HOOC-PGMA43–PHPMAX diblock copolymer vesicles were diluted to 0.10% w/w in aqueous solution at pH 3.5. Transmittance measurements were recorded every minute immediately after this solution pH was increased to pH 9.0 using KOH.
Results and Discussion
A low-polydispersity PGMA macro-CTA (Mw/Mn = 1.20) containing a terminal carboxylic acid was prepared in ethanol at 70 °C by RAFT solution polymerization of GMA using PETTC. The crude HOOC-PGMA macro-CTA was purified by precipitation into excess dichloromethane. 1H NMR spectroscopy indicated a mean degree of polymerization (DP) of 43 for this purified HOOC-PGMA macro-CTA by end-group analysis. This water-soluble macro-CTA was then chain-extended via RAFT aqueous dispersion polymerization of HPMA at 10% w/w solids and 70 °C. The target DP of the core-forming PHPMA block was systematically varied from 175 to 250 to produce a series of turbid, free-flowing vesicular dispersions (see Figure 1). According to 1H NMR analysis, all HPMA polymerizations reached high conversion (>99%). Furthermore, DMF GPC analysis (see Figure 2) indicated high blocking efficiencies and relatively narrow copolymer molecular weight distributions (Mw/Mn < 1.20), as expected based on previous reports.3,73,79
Figure 1.

(a) Synthesis of a HOOC-PGMA43 macro-CTA via RAFT solution polymerization of GMA using a 4-cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) RAFT agent and its subsequent chain extension with HPMA via RAFT aqueous dispersion polymerization to prepare a series of HOOC-PGMA43–PHPMAX diblock copolymer vesicles at pH 3.5 (where X = 175–250). (b) Illustration of the irreversible vesicle-to-sphere or vesicle-to-worm order–order transitions that occur when the terminal carboxylic acid on the PGMA stabilizer block becomes ionized as a result of a pH switch.
Figure 2.
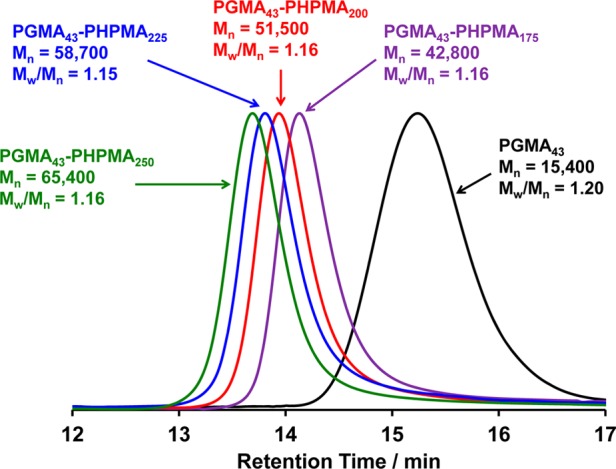
DMF GPC curves obtained for a HOOC-PGMA43 macro-CTA (black curve) and the corresponding HOOC-PGMA43–PHPMAX diblock copolymer vesicles (where X = 175–250). In all cases high blocking efficiencies (>95%) and low final copolymer polydispersities (Mw/Mn < 1.20) were obtained.
TEM studies of the turbid diblock copolymer dispersions prepared at 10% w/w solids at pH 3.5 (after dilution to 0.1% w/w using an acidified aqueous solution at the same pH) confirmed the presence of polydisperse vesicles of 150–500 nm diameter in each case (see Figure 3a). According to studies on a series of closely related PGMA54–PHPMAX vesicles by Warren and co-workers, the range of PHPMA DPs (175–250) targeted in the present work should produce mean vesicle membrane thicknesses of around 10–15 nm.79 TEM studies are consistent with this estimated range (see Figure 3a). On increasing the solution pH of these vesicular dispersions from pH 3.5 to pH 6.0 using 0.5 M KOH, a physical change from an initially turbid free-flowing dispersion to either a transparent free-flowing dispersion or a free-standing translucent gel was observed after approximately 12 h for HOOC-PGMA43–PHPMA175 and HOOC-PGMA43–PHPMA200, respectively. In contrast, no physical change was observed for the HOOC-PGMA43–PHPMA225 or HOOC-PGMA43–PHPMA250 diblock copolymer vesicle dispersions when subjected to the same pH switch. Subsequent TEM studies indicated a vesicle-to-sphere and a vesicle-to-worm transition for HOOC-PGMA43–PHPMA175 and HOOC-PGMA43–PHPMA200, respectively (see Figure 3b). Like the previously reported worm-to-sphere transition,76 these two order−order morphological transitions are the result of ionization of a single terminal carboxylic acid group, which increases the effective volume fraction of the hydrophilic PGMA stabilizer block and hence lowers the packing parameter, P, for the copolymer chains.12 Conversely, TEM images obtained for the HOOC-PGMA43–PHPMA225 and HOOC-PGMA43–PHPMA250 diblock copolymer nano-objects at pH 6.0 indicated no pH-responsive behavior; the original vesicles are retained more or less intact. However, close inspection reveals some evidence for the presence of hemi-vesicles and possibly some degree of aggregation. Thus these preliminary studies suggest that there is a critical DP for the hydrophobic PHPMA block above which the vesicles no longer exhibit pH-responsive behavior. This is reasonable, because increasing the PHPMA block DP is expected to increase the packing parameter such that P significantly exceeds 0.50, which leads to the formation of vesicles that are further removed from the vesicle/worm phase boundary. Thus the enhanced hydrophilic character gained by the PGMA stabilizer block as a result of ionization of its terminal carboxylic acid group is no longer sufficient to induce an order–order transition.
Figure 3.

TEM images (recorded after dilution to 0.10% w/w solids using an aqueous solution of either pH 3.5 or pH 6.0) and corresponding digital photographs obtained for HOOC-PGMA43–PHPMAX diblock copolymer nano-objects: (a) at pH 3.5 and (b) at pH 6.0.
In contrast, targeting shorter PHPMA DPs of either 175 or 200 for PGMA–PHPMA diblock copolymer vesicles enables access to either worm (0.33 < P < 0.50) or sphere (P < 0.33) phase space after a pH switch. It is perhaps worth emphasizing the subtle nature of the observations summarized in Figure 3: deprotonation of a single terminal carboxylic acid group on a diblock copolymer chain with a mean molecular weight of more than 35 000 g mol–1 is sufficient enough to induce a morphological transition. Moreover, it is noteworthy that this pH-response is irreversible in all cases; adding acid to return the solution pH from pH 6.0 to pH 3.5 merely produces an insoluble white paste, rather than a free-flowing turbid dispersion. This is believed to be because the worm phase constitutes a significant kinetic barrier to vesicle reformation. A worm-to-vesicle transition is well-documented for PGMA–PHPMA chains during PISA syntheses, but in this latter case there is excess unreacted HPMA monomer present at intermediate monomer conversions to plasticize the hydrophobic PHPMA chains and hence ensure their high mobility.
To further examine these order–order morphological transitions, dynamic light scattering (DLS) and aqueous electrophoresis studies were conducted on 0.10% w/w HOOC-PGMA43–PHPMA175–250 vesicles as a function of dispersion pH (see Figure 4). In the case of the HOOC-PGMA43–PHPMA175 vesicles, a significant reduction in the intensity-average mean particle diameter from 150 to 35 nm is observed on increasing the dispersion pH from 3.5 to 5.0, which provides good evidence for a vesicle-to-sphere transition (see Figure 4a). Moreover, this morphological transition occurs over a similar pH range to that previously reported for a worm-to-sphere transition. In both cases the pKa of the terminal carboxylic acid is around 4.7.76 A similar trend was observed for the HOOC-PGMA43–PHPMA200 diblock copolymer, which undergoes a vesicle-to-worm transition with a corresponding reduction in apparent particle diameter from 240 to 130 nm after the same pH switch (Figure 4b). In this latter case, it is noteworthy that DLS reports a “sphere-equivalent” diameter for the final worm phase that corresponds to neither their mean length nor width. Conversely, the HOOC-PGMA43–PHPMA225 and HOOC-PGMA43–PHPMA250 diblock copolymer vesicles exhibit an increase in particle diameter over the same pH range, although the latter is less pronounced than the former (see Figure 4c,d). This is attributed to a more extended PGMA stabilizer layer when the terminal carboxylic acid groups become ionized. This suggests that these two types of vesicles do not undergo any morphological transition during a pH switch, which is corroborated by the TEM studies shown in Figure 3. Moreover, in the case of the HOOC-PGMA43–PHPMA225 vesicles, DLS studies provide some evidence for vesicle aggregation. In all cases, ionization of the terminal carboxylic acid group above its pKa results in greater anionic character for the nano-objects. Thus aqueous electrophoresis studies indicate that the zeta potential increases in each case from around −10 mV for the original vesicles at pH 3.5 to approximately −25 mV at pH 8.0 for the final diblock copolymer nano-objects.
Figure 4.

Variation of intensity-average hydrodynamic particle diameter (measured by dynamic light scattering) and zeta potential with dispersion pH (starting at pH 3.5) recorded at 25 °C for 0.1% w/w aqueous dispersions of (a) HOOC-PGMA43–PHPMA175 vesicles, (b) HOOC-PGMA43–PHPMA200 vesicles, (c) HOOC-PGMA43–PHPMA225 vesicles, and (d) HOOC-PGMA43–PHPMA250 vesicles.
The worm-to-sphere transition reported by Lovett and co-workers was relatively rapid, occurring over a time scale of minutes.76 In contrast, the vesicle-to-sphere and vesicle-to-worm transitions observed herein occur over much longer time scales (hours). We believe that this difference is related to the shorter (and hence less hydrophobic) PHPMA DP required for worms compared to that for vesicles. In the present study, the change from vesicles to worms (or spheres) is accompanied by a significant change in the visual appearance of the dispersions. The initial vesicles are relatively large and hence scatter light strongly, resulting in turbid dispersions. On the other hand, the resulting worms or spheres are smaller and so scatter light much more weakly, leading to semi-transparent dispersions. In principle, this physical change can be utilized to probe the time scales of these morphological transitions by turbidimetry. However, such experiments must be conducted on relatively dilute dispersions (0.10% w/w at pH 3.5) because 10% w/w dispersions are too turbid to be analyzed. The transmittance at a fixed wavelength of 450 nm was monitored for dilute copolymer dispersions over a 20 h period after a pH switch from 3.5 to 9.0 (see Figure 5). As expected, no discernible change in transmittance is observed if the PHPMA DP is either 225 or 250. This is consistent with our TEM and DLS observations discussed earlier: such vesicles cannot undergo an order–order morphological transition on ionization of the terminal carboxylic acid on the PGMA stabilizer chains. However, if the PHPMA DP is either 175 or 200, then a pH switch from pH 3.5 to pH 9.0 leads to a significant increase in transmittance being observed over time. These turbidimetry studies indicate that the vesicle-to-worm transition for the HOOC-PGMA43–PHPMA200 diblock copolymer is remarkably slow, with approximately 15 h being required to reach completion. In contrast, the vesicle-to-sphere transformation observed for the HOOC-PGMA43–PHPMA175 diblock copolymer is complete within just 2 h under the same conditions. We do not fully understand the differing time scales required for these two order–order morphology transitions. However, we hypothesize that the likely explanation is related to the differing DP of the membrane-forming PHPMA block. This parameter dictates how far the vesicles lie from the respective vesicle/worm and vesicle/sphere phase boundaries. Furthermore, longer PHPMA blocks should have more inter-chain entanglements, thus presenting a higher kinetic barrier to a stimulus-induced morphology transition. Therefore vesicles comprising longer PHPMA blocks respond more slowly to a pH switch.
Figure 5.
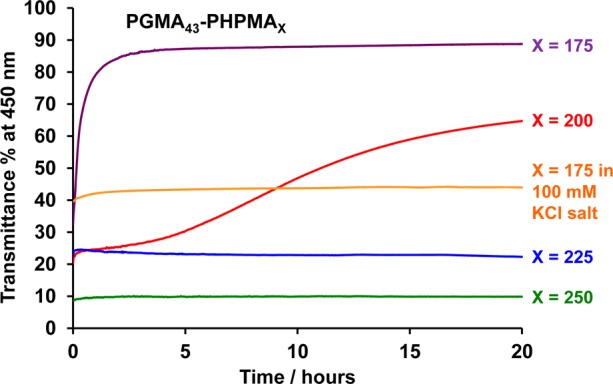
Change in transmittance % at a fixed wavelength of 450 nm for 0.10% w/w aqueous dispersions of HOOC-PGMA43–PHPMA175–250 nano-objects over 20 h at 20 °C after a pH switch from pH 3.5 to pH 9.0 using KOH.
It is noteworthy that such order–order morphological transitions are much slower compared to the characteristic time scale 4 ms to 6 s required for the acid-induced swelling of microgel particles reported in the literature.80,81 However, this pronounced difference is really not surprising: the copolymer chains in a conventional pH-responsive microgel (or soluble polymer) typically undergo extensive protonation (or ionization) during a pH switch to produce a highly hydrophilic polyelectrolyte. In contrast, the PHPMA block remains weakly hydrophobic both before and after the pH switch.
If the same turbidimetry experiment is conducted on a 0.10% w/w aqueous dispersion of HOOC-PGMA43–PHPMA175 vesicles prepared in the presence of 100 mM KCl, no increase in transmittance is observed over the same time period (see Figure 5). This suggests that added salt leads to pH-insensitive vesicles. It is well documented that the behavior of many pH-responsive polymers can be suppressed (or altered) by addition of salt.27,59,82−85 Of particular relevance to the current study, the worm-to-sphere transition previously reported for HOOC-PGMA56–PHPMA155 does not occur in the presence of 100 mM KCl.76 Thus HOOC-PGMA43–PHPMA175 vesicles were prepared via PISA in the absence and presence of 100 mM KCl. DLS studies indicated a constant particle diameter of approximately 150 nm between pH 3.5 and 8.5 in the presence of this electrolyte (see red data set in Figure 6). TEM studies confirmed that the original vesicle morphology observed at pH 3.5 was retained at pH 8.5 (compare Figures 6b and 6c; N.B.: the small dark crystals observed in these images are KCl nanocrystals). The corresponding data obtained for the same copolymer obtained under the same conditions in the absence of salt is included in Figures 6d and 6e as a reference. In summary, the addition of salt screens the additional solvation associated with the ionization of the terminal carboxylic acid and hence suppresses the vesicle-to-sphere transition.
Figure 6.
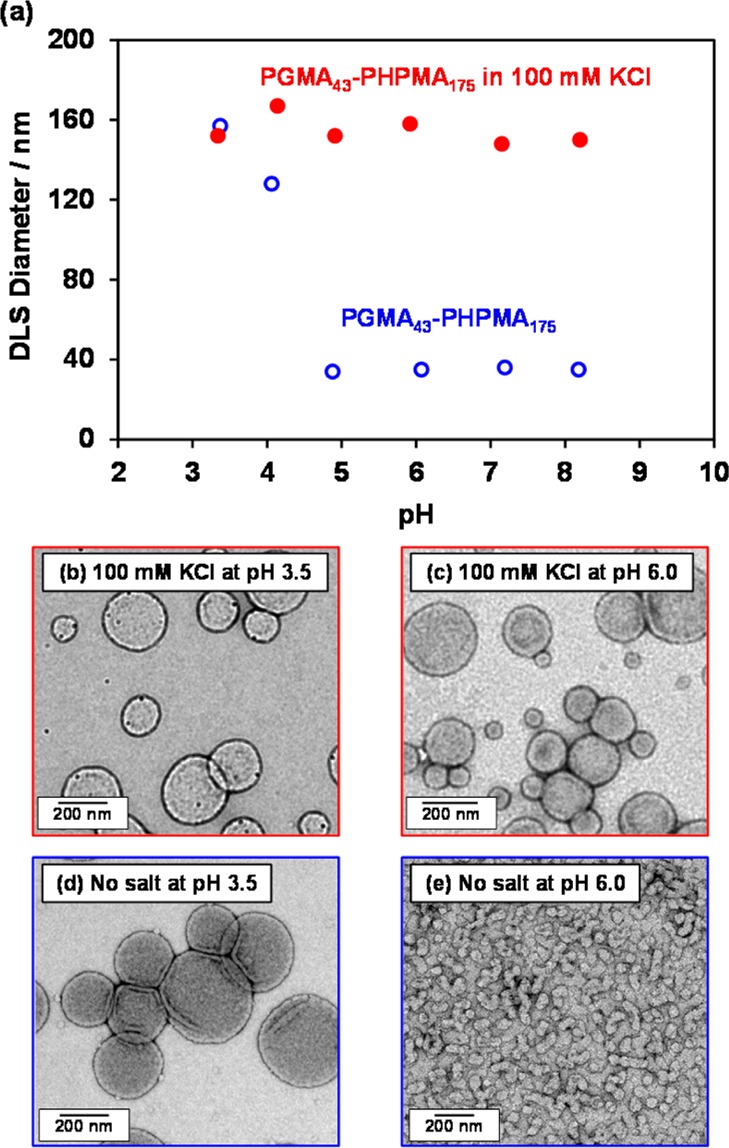
(a) Variation of the hydrodynamic particle diameter measured by dynamic light scattering with dispersion pH recorded for 0.1% w/w aqueous dispersions of HOOC-PGMA43–PHPMA175 diblock copolymer vesicles starting at pH 3.5 in the absence of salt (open blue circles) and in the presence of 100 mM KCl (closed red circles). TEM images obtained for HOOC-PGMA43–PHPMA175 diblock copolymer nano-objects in the presence of 100 mM KCl salt at (b) pH 3.5 and (c) pH 6.0 and in the absence of salt at (d) pH 3.5 and (e) pH 6.0.
Of particular interest is the vesicle-to-worm transition observed for the HOOC-PGMA43–PHPMA200 diblock copolymer after a pH switch from 3.5 to 6.0. Unlike the relatively large phase space occupied by vesicles (and spheres), the worm phase space is typically very narrow.73 Thus it is perhaps not surprising that a pure worm phase can only be obtained from a pure vesicle phase for a rather narrow range of PHPMA DP. After end-group ionization at pH 6.0, HOOC-PGMA43–PHPMA200 worms are believed to form a soft free-standing gel due to multiple inter-worm contacts, rather than the inter-worm entanglements suggested for surfactant worm gels.86 Rheological studies conducted on a 10% w/w HOOC-PGMA43–PHPMA200 worm gel at pH 6.0 indicate a storage modulus (G′) of approximately 60 Pa at 25 °C (see Figure 7). This is slightly lower than the moduli reported by Blanazs et al. for a 10% w/w non-ionic PGMA54–PHPMA150 diblock copolymer worm gel.74 We hypothesize that this is the result of electrostatic repulsion between the former anionic worms, resulting in weaker/fewer interworm contacts. Temperature-dependent rheological studies indicate that the HOOC-PGMA43–PHPMA200 worm gel undergoes degelation on cooling to approximately 4 °C. The critical gelation temperature (CGT) is defined as the point where the loss modulus (G″) exceeds the storage modulus (G′), indicating the formation of a viscoelastic fluid. For this HOOC-PGMA43–PHPMA200 worm gel, the CGT was found to be approximately 5 °C. Verber and co-workers reported that the CGT values of their non-ionic PGMA54–PHPMAX diblock copolymer worm gels decreased monotonically from 20 to 7 °C as the PHPMA DP (X) was increased from 135 to 170.74 This is due to longer PHPMA DPs requiring a greater degree of hydration to induce a worm-to-sphere transition. Thus it might be expected that the HOOC-PGMA43–PHPMA200 worm gel possesses a lower CGT due to its higher PHPMA DP.
Figure 7.
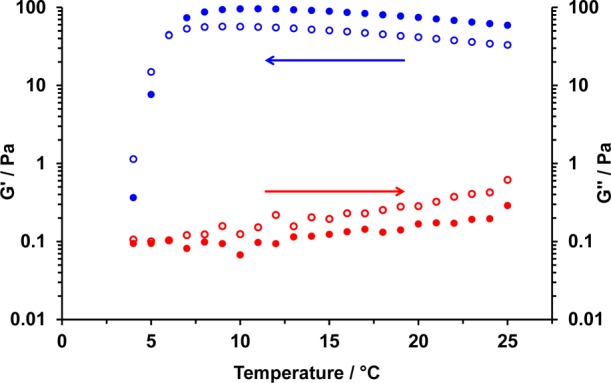
Variation of the storage modulus (G′, denoted by full circles) and loss modulus (G″, denoted by open circles) for a 10% w/w aqueous dispersion of HOOC-PGMA43–PHPMA200 diblock copolymer nano-objects as a function of temperature, after a pH switch from 3.5 to 6.0 to induce a vesicle-to-worm transition. In each case, the blue data represent decreasing temperature and the red data represent increasing temperature. Conditions: 1.0 rad s–1 angular frequency at an applied strain of 1.0%.
Blanazs and co-workers used variable temperature 1H NMR spectroscopy to show that the PHPMA core-forming block becomes significantly more hydrated on cooling;75 this is consistent with surface plasticization of the PGMA–PHPMA worm cores, which leads to a reduction in the packing parameter and hence accounts for the observed worm-to-sphere transition. Blanazs and co-workers also demonstrated that this sol–gel thermal transition was fully reversible as judged by small-angle X-ray scattering (SAXS) and rheology (although the latter technique suggested some degree of hysteresis).74,75 In contrast, rheological studies of the HOOC-PGMA43–PHPMA200 worm gel formed from vesicles after a pH switch suggests irreversible thermo-responsive behavior for this system. Thus after cooling to 5 °C and returning to 25 °C, regelation does not occur: the loss modulus remains larger than the storage modulus (see red data on Figure 7), which is characteristic of free-flowing spherical micelles. It is hypothesized that these anionic micelles are mutually repulsive (zeta potential ∼ −25 mV) and hence are unable to undergo the multiple 1D fusion events required for worm reconstitution. Moreover, if the HOOC-PGMA43–PHPMA200 nano-objects are returned to pH 3.5 after a 25 °C–5 °C–25 °C thermal cycle, then a white insoluble paste is produced, rather than the original colloidally stable vesicles. Again, it appears that the worm phase provides an effective kinetic barrier to vesicle reformation.
The thermo-responsive behavior of PGMA–PHPMA diblock copolymer worms has been studied in some detail.74,75 However, to date there have been no analogous studies of PGMA–PHPMA diblock copolymer vesicles. Thus the four HOOC-PGMA43–PHPMA175–250 diblock copolymer vesicles were studied to examine the effect of varying the PHPMA DP on their thermo-responsive behavior. Verber and co-workers reported that PGMA54–PHPMAX worms exhibited lower CGTs when targeting higher X values.74 This was attributed to the longer (and hence more hydrophobic) PHPMA blocks requiring a higher degree of hydration to induce a worm-to-sphere transition, which can only be attained at lower temperatures. By analogy, PGMA–PHPMA diblock copolymer vesicles prepared using a sufficiently high PHPMA DP might be expected to possess no thermo-responsive behavior. Moreover, Kocik et al. used SAXS to show that PGMA57–PHPMA140 worms underwent a worm-to-sphere transition at around 5 °C, but further cooling to −2 °C resulted in near-molecular dissolution of the spheres.87 In view of these observations, the lower limit temperature in the present study was restricted to 5 °C. Perhaps surprisingly, only the shortest HOOC-PGMA43–PHPMA175 diblock copolymer switched from a turbid, free-flowing dispersion (at pH 3.5) to a translucent, free-standing gel on cooling to 5 °C (see Figure 8). Moreover, this thermal transition was irreversible: an insoluble white paste was obtained on returning to 25 °C. TEM studies on grids prepared at 5 °C using 0.10% w/w copolymer dispersions are consistent with a vesicle-to-worm transition (see Figure 8). In contrast, representative TEM images obtained at 5 °C for the other three diblock copolymers suggest that their original vesicular morphologies remain unchanged. At first sight it is perhaps surprising that ionization of a single terminal carboxylic acid group leads to pH-responsive behavior for HOOC-PGMA43–PHPMA200, yet the same copolymer exhibits no thermo-responsive behavior (at pH 3.5). On reflection, this discrepancy is not unreasonable: carboxylic acid group ionization makes the relatively short PGMA stabilizer significantly more hydrophilic, whereas lowering the temperature leads to greater hydration (i.e., reduced hydrophobic character) for the relatively long PHPMA core-forming block. In the latter case, this effect is negated if the PHPMA DP is too high.
Figure 8.

TEM images (for grids prepared at 5 °C after dilution to 0.10% w/w copolymer at pH 3.5) and corresponding digital photographs obtained for HOOC-PGMA43–PHPMAX diblock copolymer nano-objects for X = 175, 200, 225, or 250.
Subjecting the series of four HOOC-PGMA43–PHPMAX vesicles to a pH switch from 3.5 to 6.0 followed by immediate cooling to 5 °C leads to an order–order transition in each case. The original dispersions become significantly less turbid, while remaining free-flowing dispersions (see Figure 9). TEM studies conducted on the HOOC-PGMA43–PHPMAX nano-objects after this dual stimulus confirmed that the vesicles are transformed into a mixture of spheres and spherical dimers,75 with mean particle width dimensions estimated to be 21–30 nm (based on analysis of at least 100 particles in each case). Similarly, DLS studies conducted at 5 °C indicate that the final copolymer dispersions have a mean hydrodynamic diameter of approximately 40 nm at pH 6.0, which is substantially lower than that of the original vesicles.
Figure 9.

Representative TEM images obtained for HOOC-PGMA43-PHPMAX dispersions after dilution at 5 °C to dilution to 0.10% w/w copolymer at pH 6.5 and (inset) the corresponding digital photographs of their visual appearance at 10% w/w copolymer.
It is worth emphasizing that the HOOC-PGMA43–PHPMA250 diblock copolymer vesicles only undergo a morphological transition when subjected to both a pH switch and a temperature switch; exposure to either stimulus alone results in no morphological transition. However, regardless of the route taken to return to the original conditions (i.e., heating followed by a pH switch, or vice versa), these order–order morphological transitions always proved to be irreversible. TEM images obtained after dilution of the insoluble white paste revealed a mixed phase of vesicles and worms (see Figure S1 in the Supporting Information).
Given the rather modest variation in the PHPMA DP, the stimulus-responsive nature of this series of four HOOC-PGMA43–PHPMAX vesicles is unexpectedly complex, and their overall behavior is summarized in Table 1. For X = 225 or 250, no pH-responsive behavior is observed on raising the pH from pH 3.5 to pH 6.0. On the other hand, a vesicle-to-sphere (plus spherical dimers) transition is observed after a pH switch for X = 175, while a vesicle-to-worm transition is found for X = 200. Only the former vesicles exhibit a thermally triggered transition, which produces a free-standing worm gel at 5 °C. All four HOOC-PGMA43–PHPMA175–250 vesicles undergo morphological transitions to give a mixture of spheres and spherical dimers when subjected to a dual stimulus (i.e., a pH switch followed by immediate cooling to 5 °C). In all cases, these morphological transitions proved to be irreversible. This is believed to be because the worm phase acts as an effective kinetic barrier that prevents the original vesicle morphology from being reformed.
Table 1. Summary of Data Obtained for HOOC-PGMA43–HPMAX Diblock Copolymer Vesicles Illustrating Their pH- and Thermo-responsive Behavior.
| PHPMA DP | Mna (g mol–1) | Mn/Mwa | particle diameter at pH 3.5 and 25 °Cb (nm) | PDIb | pH responsive? | morphology after 3.5 to 6.0 pH switchc | temperature responsive? | morphology after 20 to 5 °C temperature switchc | dual responsive? | morphology after temperature and pH switchc |
|---|---|---|---|---|---|---|---|---|---|---|
| 175 | 42800 | 1.16 | 157 | 0.218 | yes | spheres and spherical dimers | yes | worms | yes | spheres and spherical dimers |
| 200 | 51500 | 1.16 | 237 | 0.264 | yes | worms | no | vesicles | yes | spheres and spherical dimers |
| 225 | 58700 | 1.15 | 232 | 0.108 | no | vesicles | no | vesicles | yes | spheres and spherical dimers |
| 250 | 65400 | 1.16 | 211 | 0.058 | no | vesicles | no | vesicles | yes | spheres and spherical dimers |
Measured by DMF GPC using a series of near-monodisperse PMMA calibration standards.
Measured using dynamic light scattering (DLS).
Determined by TEM.
There are many literature examples of the use of pH- or thermo-responsive vesicles for potential biomedical applications such as drug delivery.2,8,42−46 In principle, the vesicles can be loaded in situ during their preparation via PISA, with exposure to an external stimulus resulting in an order–order morphological transition, loss of the membrane structure, and hence subsequent release of the payload. Furthermore, vesicles that only undergo a morphological transition when exposed to two or more stimuli may offer greater control in terms of specificity compared to vesicles that can respond to just one stimulus. These possibilities will be examined in future studies. However, in this context it is noteworthy that the weakly hydrated nature of the PHPMA membrane-forming block suggests that PGMA–PHPMA vesicles are unlikely to retain water-soluble small molecules over long time periods.79 Given this limitation, it may be more fruitful to focus on the encapsulation of organic nanoparticles such as globular proteins (e.g., enzymes, antibodies, etc.).
Conclusions
In summary, we demonstrate that PGMA–PHPMA diblock copolymer vesicles prepared using a carboxylic acid-functionalized RAFT agent exhibit complex stimulus-responsive behavior in aqueous solution. By fixing the DP of the PGMA stabilizer block at 43, vesicles can be prepared by targeting PHPMA block DPs of 175, 200, 225, or 250. Switching the solution pH from 3.5 to 6.0 induces ionization of the terminal carboxylic acid on the PGMA stabilizer block, which increases its hydrophilic character. This results in a vesicle-to-sphere transition for HOOC-PGMA43–PHPMA175 and a vesicle-to-worm transition for HOOC-PGMA43–PHPMA200, respectively. However, if the DP of the PHPMA block is longer (either 225 or 250), no morphological transformation occurs, as judged by TEM and DLS. In this case, the vesicles lie further from the vesicle/worm phase boundary, which makes vesicle dissociation more difficult. Turbidimetry studies conducted on dilute vesicle dispersions indicate that these vesicle-to-sphere and vesicle-to-worm transitions are relatively slow, typically requiring time scales of hours at 20 °C. However, if the original vesicles are subjected to the same pH switch in the presence of added salt, charge screening results in no order–order transition being observed. Only the HOOC-PGMA43–PHPMA175 vesicles undergo an order–order transition to form worms simply on cooling to 5 °C. However, subjecting the HOOC-PGMA43–PHPMAX vesicles to both a pH switch and a temperature switch causes a vesicle-to-sphere transition in each case, as judged by TEM studies. In summary, the stimulus-responsive behavior of HOOC-PGMA43–PHPMAX vesicles is unexpectedly complex and critically depends on the DP of the core-forming PHPMA block.
Acknowledgments
J.R.L. thanks the University of Sheffield for a PhD studentship. GEO Specialty Chemicals is thanked for providing the GMA monomer and for partial financial support. S.P.A. thanks the EPSRC (Platform grant EP/J007846/1 and HIPS EP/L024160/1) for postdoctoral support of NJW and also the European Research Council for a five-year ERC Advanced Investigator grant (PISA 320372).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.5b02470.
Additional TEM images of vesicle/worm mixtures obtained after pH cycling (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang L.; Eisenberg A. Science 1995, 268, 1728–1731. 10.1126/science.268.5218.1728. [DOI] [PubMed] [Google Scholar]
- Antonietti M.; Förster S. Adv. Mater. 2003, 15, 1323–1333. 10.1002/adma.200300010. [DOI] [Google Scholar]
- Blanazs A.; Madsen J.; Battaglia G.; Ryan A. J.; Armes S. P. J. Am. Chem. Soc. 2011, 133, 16581–16587. 10.1021/ja206301a. [DOI] [PubMed] [Google Scholar]
- Jain S.; Bates F. S. Science 2003, 300, 460–464. 10.1126/science.1082193. [DOI] [PubMed] [Google Scholar]
- Cui H.; Chen Z.; Zhong S.; Wooley K. L.; Pochan D. J. Science 2007, 317, 647–650. 10.1126/science.1141768. [DOI] [PubMed] [Google Scholar]
- Charleux B.; Delaittre G.; Rieger J.; D’Agosto F. Macromolecules 2012, 45, 6753–6765. 10.1021/ma300713f. [DOI] [Google Scholar]
- Zhang L.; Eisenberg A. Polym. Adv. Technol. 1998, 9, 677–699. . [DOI] [Google Scholar]
- Tuzar Z.; Kratochvíl P. Adv. Colloid Interface Sci. 1976, 6, 201–232. 10.1016/0001-8686(76)80009-7. [DOI] [Google Scholar]
- Lim Soo P.; Eisenberg A. J. Polym. Sci., Part B: Polym. Phys. 2004, 42, 923–938. 10.1002/polb.10739. [DOI] [Google Scholar]
- Hayward R. C.; Pochan D. J. Macromolecules 2010, 43, 3577–3584. 10.1021/ma9026806. [DOI] [Google Scholar]
- Chu B. Langmuir 1995, 11, 414–421. 10.1021/la00002a009. [DOI] [Google Scholar]
- Blanazs A.; Armes S. P.; Ryan A. J. Macromol. Rapid Commun. 2009, 30, 267–277. 10.1002/marc.200800713. [DOI] [PubMed] [Google Scholar]
- Cammas S.; Suzuki K.; Sone C.; Sakurai Y.; Kataoka K.; Okano T. J. Controlled Release 1997, 48, 157–164. 10.1016/S0168-3659(97)00040-0. [DOI] [Google Scholar]
- Fielding L. A.; Lane J. A.; Derry M. J.; Mykhaylyk O. O.; Armes S. P. J. Am. Chem. Soc. 2014, 136, 5790–5798. 10.1021/ja501756h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y.; Thurairajah L.; Sugita O. R.; Lowe A. B. Macromolecules 2015, 48, 236–244. 10.1021/ma502230h. [DOI] [Google Scholar]
- Pei Y.; Sugita O. R.; Thurairajah L.; Lowe A. B. RSC Adv. 2015, 5, 17636–17646. 10.1039/C5RA00274E. [DOI] [Google Scholar]
- Sundararaman A.; Stephan T.; Grubbs R. B. J. Am. Chem. Soc. 2008, 130, 12264–12265. 10.1021/ja8052688. [DOI] [PubMed] [Google Scholar]
- Bauri K.; Narayanan A.; Haldar U.; De P. Polym. Chem. 2015, 6, 6152–6162. 10.1039/C5PY00919G. [DOI] [Google Scholar]
- Zhao Y. Macromolecules 2012, 45, 3647–3657. 10.1021/ma300094t. [DOI] [Google Scholar]
- Lee H.-i.; Wu W.; Oh J. K.; Mueller L.; Sherwood G.; Peteanu L.; Kowalewski T.; Matyjaszewski K. Angew. Chem., Int. Ed. 2007, 46, 2453–2457. 10.1002/anie.200604278. [DOI] [PubMed] [Google Scholar]
- Gohy J.-F.; Zhao Y. Chem. Soc. Rev. 2013, 42, 7117–7129. 10.1039/c3cs35469e. [DOI] [PubMed] [Google Scholar]
- Schumers J.-M.; Fustin C.-A.; Gohy J.-F. Macromol. Rapid Commun. 2010, 31, 1588–1607. 10.1002/marc.201000108. [DOI] [PubMed] [Google Scholar]
- Pearson S.; Vitucci D.; Khine Y. Y.; Dag A.; Lu H.; Save M.; Billon L.; Stenzel M. H. Eur. Polym. J. 2015, 69, 616–627. 10.1016/j.eurpolymj.2015.04.001. [DOI] [Google Scholar]
- Magnusson J. P.; Khan A.; Pasparakis G.; Saeed A. O.; Wang W.; Alexander C. J. Am. Chem. Soc. 2008, 130, 10852–10853. 10.1021/ja802609r. [DOI] [PubMed] [Google Scholar]
- He W. N.; Xu J. T.; Du B. Y.; Fan Z. Q.; Wang X. S. Macromol. Chem. Phys. 2010, 211, 1909–1916. 10.1002/macp.201000184. [DOI] [Google Scholar]
- Yu K.; Eisenberg A. Macromolecules 1998, 31, 3509–3518. 10.1021/ma971419l. [DOI] [Google Scholar]
- Zhang L.; Eisenberg A. Macromolecules 1996, 29, 8805–8815. 10.1021/ma961376t. [DOI] [Google Scholar]
- Maiti C.; Banerjee R.; Maiti S.; Dhara D. Langmuir 2015, 31, 32–41. 10.1021/la504165e. [DOI] [PubMed] [Google Scholar]
- Sumerlin B. S.; Lowe A. B.; Thomas D. B.; McCormick C. L. Macromolecules 2003, 36, 5982–5987. 10.1021/ma034088n. [DOI] [Google Scholar]
- Ma Y.; Tang Y.; Billingham N. C.; Armes S. P.; Lewis A. L.; Lloyd A. W.; Salvage J. P. Macromolecules 2003, 36, 3475–3484. 10.1021/ma021762c. [DOI] [Google Scholar]
- Du J.; Tang Y.; Lewis A. L.; Armes S. P. J. Am. Chem. Soc. 2005, 127, 17982–17983. 10.1021/ja056514l. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Hernández J.; Lecommandoux S. J. Am. Chem. Soc. 2005, 127, 2026–2027. 10.1021/ja043920g. [DOI] [PubMed] [Google Scholar]
- Jochum F. D.; Theato P. Chem. Soc. Rev. 2013, 42, 7468–7483. 10.1039/C2CS35191A. [DOI] [PubMed] [Google Scholar]
- Bütün V.; Liu S.; Weaver J. V. M; Bories-Azeau X.; Cai Y.; Armes S. P. React. Funct. Polym. 2006, 66, 157–165. 10.1016/j.reactfunctpolym.2005.07.021. [DOI] [Google Scholar]
- Roy D.; Cambre J. N.; Sumerlin B. S. Chem. Commun. 2009, 2106–2108. 10.1039/b900374f. [DOI] [PubMed] [Google Scholar]
- Quek J. Y.; Zhu Y.; Roth P. J.; Davis T. P.; Lowe A. B. Macromolecules 2013, 46, 7290–7302. 10.1021/ma4013187. [DOI] [Google Scholar]
- Zhou Y. N.; Zhang Q.; Luo Z. H. Langmuir 2014, 30, 1489–1499. 10.1021/la402948s. [DOI] [PubMed] [Google Scholar]
- Moughton A. O.; O’Reilly R. K. Chem. Commun. 2010, 46, 1091–1093. 10.1039/B922289H. [DOI] [PubMed] [Google Scholar]
- Du J.; Willcock H.; Patterson J. P.; Portman I.; O’Reilly R. K. Small 2011, 7, 2070–2080. 10.1002/smll.201100382. [DOI] [PubMed] [Google Scholar]
- Summers M. J.; Phillips D. J.; Gibson M. I. Chem. Commun. 2013, 49, 4223–4225. 10.1039/C2CC34236G. [DOI] [PubMed] [Google Scholar]
- Liu T.; Tian W.; Zhu Y.; Bai Y.; Yan H.; Du J. Polym. Chem. 2014, 5, 5077–5088. 10.1039/C4PY00501E. [DOI] [Google Scholar]
- Blum A. P.; Kammeyer J. K.; Rush A. M.; Callmann C. E.; Hahn M. E.; Gianneschi N. C. J. Am. Chem. Soc. 2015, 137, 2140–2154. 10.1021/ja510147n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C. L.; Zhuang X. L.; Tang Z. H.; Tian H. Y.; Chen X. S. Adv. Healthcare Mater. 2012, 1, 48–78. 10.1002/adhm.201100008. [DOI] [PubMed] [Google Scholar]
- Alarcon C. D. H.; Pennadam S.; Alexander C. Chem. Soc. Rev. 2005, 34, 276–285. 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- Bajpai A. K.; Shukla S. K.; Bhanu S.; Kankane S. Prog. Polym. Sci. 2008, 33, 1088–1118. 10.1016/j.progpolymsci.2008.07.005. [DOI] [Google Scholar]
- Murthy N.; Campbell J.; Fausto N.; Hoffman A. S.; Stayton P. S. Bioconjugate Chem. 2003, 14, 412–419. 10.1021/bc020056d. [DOI] [PubMed] [Google Scholar]
- Sanson C.; Schatz C.; Le Meins J.-F.; Soum A.; Thévenot J.; Garanger E.; Lecommandoux S. J. Controlled Release 2010, 147, 428–435. 10.1016/j.jconrel.2010.07.123. [DOI] [PubMed] [Google Scholar]
- Ahmed F.; Pakunlu R. I.; Srinivas G.; Brannan A.; Bates F.; Klein M. L.; Minko T.; Discher D. E. Mol. Pharmaceutics 2006, 3, 340–350. 10.1021/mp050103u. [DOI] [PubMed] [Google Scholar]
- Pegoraro C.; Cecchin D.; Gracia L. S.; Warren N.; Madsen J.; Armes S. P.; Lewis A.; MacNeil S.; Battaglia G. Cancer Lett. 2013, 334, 328–337. 10.1016/j.canlet.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Lomas H.; Canton I.; MacNeil S.; Du J.; Armes S. P.; Ryan A. J.; Lewis A. L.; Battaglia G. Adv. Mater. 2007, 19, 4238–4243. 10.1002/adma.200700941. [DOI] [Google Scholar]
- Li M.-H.; Keller P. Soft Matter 2009, 5, 927–937. 10.1039/b815725a. [DOI] [Google Scholar]
- Kita-Tokarczyk K.; Grumelard J.; Haefele T.; Meier W. Polymer 2005, 46, 3540–3563. 10.1016/j.polymer.2005.02.083. [DOI] [Google Scholar]
- Charleux B.; Delaittre G.; Rieger J.; D’Agosto F. Macromolecules 2012, 45, 6753–6765. 10.1021/ma300713f. [DOI] [Google Scholar]
- Sun J.-T.; Hong C.-Y.; Pan C.-Y. Polym. Chem. 2013, 4, 873–881. 10.1039/C2PY20612A. [DOI] [Google Scholar]
- Zetterlund P. B.; Thickett S. C.; Perrier S.; Bourgeat-Lami E.; Lansalot M. Chem. Rev. 2015, 115, 9745–9800. 10.1021/cr500625k. [DOI] [PubMed] [Google Scholar]
- Rieger J. Macromol. Rapid Commun. 2015, 36, 1458–1471. 10.1002/marc.201500028. [DOI] [PubMed] [Google Scholar]
- Li Y.; Armes S. P. Angew. Chem., Int. Ed. 2010, 49, 4042–4046. 10.1002/anie.201001461. [DOI] [PubMed] [Google Scholar]
- Liu G.; Qiu Q.; Shen W.; An Z. Macromolecules 2011, 44, 5237–5245. 10.1021/ma200984h. [DOI] [Google Scholar]
- Boissé S.; Rieger J.; Pembouong G.; Beaunier P.; Charleux B. J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 3346–3354. 10.1002/pola.24771. [DOI] [Google Scholar]
- Semsarilar M.; Ladmiral V.; Blanazs A.; Armes S. P. Langmuir 2012, 28, 914–922. 10.1021/la203991y. [DOI] [PubMed] [Google Scholar]
- Chaduc I.; Girod M.; Antoine R.; Charleux B.; D’Agosto F.; Lansalot M. Macromolecules 2012, 45, 5881–5893. 10.1021/ma300875y. [DOI] [Google Scholar]
- Warren N. J.; Armes S. P. J. Am. Chem. Soc. 2014, 136, 10174–10185. 10.1021/ja502843f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Xiao X.; Li S.; Dan M.; Wang X.; Zhang W. Polym. Chem. 2014, 5, 578–587. 10.1039/C3PY00995E. [DOI] [Google Scholar]
- Wan W.-M.; Hong C.-Y.; Pan C.-Y. Chem. Commun. 2009, 5883–5885. 10.1039/b912804b. [DOI] [PubMed] [Google Scholar]
- Semsarilar M.; Jones E. R.; Blanazs A.; Armes S. P. Adv. Mater. 2012, 24, 3378–3382. 10.1002/adma.201200925. [DOI] [PubMed] [Google Scholar]
- Pei Y. W.; Lowe A. B. Polym. Chem. 2014, 5, 2342–2351. 10.1039/C3PY01719B. [DOI] [Google Scholar]
- Karagoz B.; Boyer C.; Davis T. P. Macromol. Rapid Commun. 2014, 35, 417–421. 10.1002/marc.201300730. [DOI] [PubMed] [Google Scholar]
- Kang Y.; Pitto-Barry A.; Willcock H.; Quan W.-D.; Kirby N.; Sanchez A. M.; O’Reilly R. K. Polym. Chem. 2015, 6, 106–117. 10.1039/C4PY01074D. [DOI] [Google Scholar]
- Yeow J.; Xu J.; Boyer C. ACS Macro Lett. 2015, 4, 984–990. 10.1021/acsmacrolett.5b00523. [DOI] [PubMed] [Google Scholar]
- Fielding L. A.; Derry M. J.; Ladmiral V.; Rosselgong J.; Rodrigues A. M.; Ratcliffe L. P. D.; Sugihara S.; Armes S. P. Chem. Sci. 2013, 4, 2081–2087. 10.1039/c3sc50305d. [DOI] [Google Scholar]
- Derry M. J.; Fielding L. A.; Armes S. P. Polym. Chem. 2015, 6, 3054–3062. 10.1039/C5PY00157A. [DOI] [Google Scholar]
- Zhang Q.; Zhu S. ACS Macro Lett. 2015, 4, 755–758. 10.1021/acsmacrolett.5b00360. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Ryan A. J.; Armes S. P. Macromolecules 2012, 45, 5099–5107. 10.1021/ma301059r. [DOI] [Google Scholar]
- Verber R.; Blanazs A.; Armes S. P. Soft Matter 2012, 8, 9915–9922. 10.1039/c2sm26156a. [DOI] [Google Scholar]
- Blanazs A.; Verber R.; Mykhaylyk O. O.; Ryan A. J.; Heath J. Z.; Douglas C. W. I.; Armes S. P. J. Am. Chem. Soc. 2012, 134, 9741–9748. 10.1021/ja3024059. [DOI] [PubMed] [Google Scholar]
- Lovett J. R.; Warren N. J.; Ratcliffe L. P.; Kocik M. K.; Armes S. P. Angew. Chem., Int. Ed. 2015, 54, 1279–1283. 10.1002/anie.201409799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelachvili J. N.; Mitchell D. J.; Ninham B. W. J. Chem. Soc., Faraday Trans. 2 1976, 72, 1525–1568. 10.1039/f29767201525. [DOI] [Google Scholar]
- Jones E. R.; Semsarilar M.; Blanazs A.; Armes S. P. Macromolecules 2012, 45, 5091–5098. 10.1021/ma300898e. [DOI] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Ryan A. J.; Williams M.; Doussineau T.; Dugourd P.; Antoine R.; Portale G.; Armes S. P. J. Am. Chem. Soc. 2015, 137, 1929–1937. 10.1021/ja511423m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin D.; Rosselgong J.; Armes S. P.; Routh A. F. Langmuir 2007, 23, 4035–4041. 10.1021/la063278z. [DOI] [PubMed] [Google Scholar]
- Morse A. J.; Armes S. P.; Mills P.; Swart R. Langmuir 2013, 29, 15209–15216. 10.1021/la403626s. [DOI] [PubMed] [Google Scholar]
- Lee A. S.; Buetuen V.; Vamvakaki M.; Armes S. P.; Pople J. A.; Gast A. P. Macromolecules 2002, 35, 8540–8551. 10.1021/ma0114842. [DOI] [Google Scholar]
- Geng Y.; Ahmed F.; Bhasin N.; Discher D. E. J. Phys. Chem. B 2005, 109, 3772–3779. 10.1021/jp0459559. [DOI] [PubMed] [Google Scholar]
- An S. W.; Thirtle P. N.; Thomas R. K.; Baines F. L.; Billingham N. C.; Armes S. P.; Penfold J. Macromolecules 1999, 32, 2731–2738. 10.1021/ma981146+. [DOI] [Google Scholar]
- Fujii S.; Dupin D.; Araki T.; Armes S. P.; Ade H. Langmuir 2009, 25, 2588–2592. 10.1021/la804212y. [DOI] [PubMed] [Google Scholar]
- Dreiss C. A. Soft Matter 2007, 3, 956–970. 10.1039/b705775j. [DOI] [PubMed] [Google Scholar]
- Kocik M. K.; Mykhaylyk O. O.; Armes S. P. Soft Matter 2014, 10, 3984–3992. 10.1039/c4sm00415a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.