

Protein crystals exhibit temperature-dependent autofluorescence when excited with visible light. The intensity of visible-light-excited autofluorescence is tenfold higher at cryogenic temperatures owing to temperature-dependent chromophore stabilization.
Keywords: protein crystallography, crystal screening, crystal visualization, fluorescence microscopy
Abstract
Fluorescence microscopy methods have seen an increase in popularity in recent years for detecting protein crystals in screening trays. The fluorescence-based crystal detection methods have thus far relied on intrinsic UV-inducible tryptophan fluorescence, nonlinear optics or fluorescence in the visible light range dependent on crystals soaked with fluorescent dyes. In this paper data are presented on a novel visible-light-inducible autofluorescence arising from protein crystals as a result of general stabilization of conjugated double-bond systems and increased charge delocalization due to crystal packing. The visible-light-inducible autofluorescence serves as a complementary method to bright-field microscopy in beamline applications where accurate crystal centering about the rotation axis is essential. Owing to temperature-dependent chromophore stabilization, protein crystals exhibit tenfold higher fluorescence intensity at cryogenic temperatures, making the method ideal for experiments where crystals are cooled to 100 K with a cryostream. In addition to the non-damaging excitation wavelength and low laser power required for imaging, the method can also serve a useful role for differentiating protein crystals from salt crystals in screening trays.
1. Introduction
In order to conduct a successful macromolecular crystallography experiment at an X-ray source, users need to be able to align the crystal in the X-ray beam while maintaining an accurate center of rotation about the φ axis. The accuracy becomes an increasingly important consideration when protein crystals are less than 10 µm in the longest dimension. Protein crystals at the beamline are traditionally imaged via bright-field microscopy, which is satisfactory with larger crystals. However, finding and centering crystals by traditional means in the mounting medium becomes increasingly difficult and time consuming with both ambient temperature and cryo-cooled crystals when the visibility is obstructed by excess cryoprotectant or heavy precipitate (skin), or when the vitrified medium is opaque in nature (e.g. lipidic cubic phase, LCP). Similarly, it is often difficult to gauge the precise outline of the crystal when mounted in a capillary. In addition to beamline use, a variety of methods have been developed over the past two decades for laboratory use to detect crystals in protein crystallization screening trays, including UV fluorescence (UVF) (Desbois et al., 2013 ▸), second-order nonlinear imaging of chiral crystals which utilizes second-harmonic generation (SHG) (Haupert & Simpson, 2011 ▸), two-photon-excited UV fluorescence (Madden et al., 2011 ▸), three-dimensional confocal Raman imaging (Nitahara et al., 2012 ▸), and methods that rely on visible light fluorescence due to covalently attached dyes or free dyes soaked into protein crystals (Watts et al., 2010 ▸; Khan et al., 2012 ▸; Meyer et al., 2015 ▸; Pusey et al., 2015 ▸). Although each of these methods can be beneficial both on the beamline and in laboratory settings, the methods are not without limitations, high cost or irregularities in fluorescence intensity.
Protein crystal detection methods relying on tryptophan UVF have been around for decades, and are limited to proteins that contain at least one or more Trp residues, with improved sensitivity and usefulness as the number of Trp residues increases. The method is pH insensitive, can enable differentiation of protein from salt crystals, and provides an increased contrast between background and crystal compared to bright-field microscopy (Desbois et al., 2013 ▸; Stewart & Mueller-Dieckmann, 2014 ▸; Meyer et al., 2015 ▸; Judge et al., 2005 ▸), the latter making it an ideal method for automated screening of large numbers of crystallization trays. It has also been shown that intrinsic UVF can be used as a potential scoring tool for protein crystal quality. The emission profile from UV-excited protein crystals can differ between poorly diffracting crystals and crystals that diffract well, seen as a difference in the fine structure of the emission spectrum from the more poorly diffracting crystals (Asanov et al., 2001 ▸). For these reasons, Trp-fluorescence-based methods are well established in screening applications and are also used on macromolecular crystallography beamlines. A second documented form of UV-inducible fluorescence from protein crystals that is not necessarily dependent on the presence of tryptophan residues, but relies on more general electronic transitions of peptide-bond electrons delocalized through hydrogen bonding, has also been explored and has found use at a macromolecular crystallography beamline (Vernede et al., 2006 ▸; Shukla et al., 2004 ▸; Calero et al., 2014 ▸). However, regardless of the source of the UV-induced fluorescence, the exposure time to UV excitation must be minimized in order to limit structural damage due to the ionizing nature of UV light (Vernede et al., 2006 ▸). One way to reduce any deleterious effects of single-photon UV excitation is to use the nonlinear optical process of two-photon absorption and laser scanning microscopy which limits the ionizing radiation to a much thinner plane. This method has been applied using 560 nm fs pulses (two-photon equivalent of 280 nm) to excite aromatic residues in protein crystals (Madden et al., 2011 ▸). Owing to the elimination of out-of-plane excitation, the method provides an improved signal-to-noise ratio through improved background reduction. Another nonlinear optical imaging method growing in popularity is SHG, which has proved to be advantageous for imaging small crystals in highly scattering media (i.e. protein precipitate or vitrified LCP) (Haupert & Simpson, 2011 ▸; Kissick et al., 2011 ▸, 2013 ▸; Madden et al., 2013 ▸). However, one limitation of this method is that crystals belonging to higher-symmetry point groups do not produce detectable SHG signal (Padayatti et al., 2012 ▸; Kissick et al., 2011 ▸; Haupert & Simpson, 2011 ▸). For screening applications one must also consider that a number of salt crystals are also SHG active and thus produce false-positive results requiring further validation by a secondary method (e.g. UVF) (Haupert & Simpson, 2011 ▸; Closser et al., 2013 ▸). Additionally, two methods have been developed and are now routinely used for detecting microscopic membrane protein crystals in vitrified LCP that involve direct exposure of the protein crystal with an attenuated X-ray beam – X-ray rastering and radiography (Cherezov et al., 2009 ▸; Warren et al., 2013 ▸).
Here we describe a non-damaging imaging method that relies on native fluorescence of protein crystals generated using visible light excitation. The visible-light-induced native fluorescence was discovered somewhat serendipitously during a confocal imaging experiment with unstained vitrified protein crystals. The reason why fluorescence of this nature has remained previously unnoticed is probably related to its temperature dependence – although the initially detected fluorescence signal can be relatively weak at room temperature, there is an order of magnitude increase in intensity from vitrified crystals. The underlying source of the fluorescence signals is most likely identical to that in the previously published work utilizing UV-A for fluorescence excitation (Shukla et al., 2004 ▸). Our method uses simple optics and light sources (i.e. non-UV) and does not damage crystals.
2. Materials and methods
2.1. Crystallization of proteins
For protein and salt crystal mixtures, lysozyme crystals were grown in a microbatch crystallization tray under light mineral oil (Fisher), using a 1:1 ratio of protein to mother liquor. The first 72 conditions of the JCSG+ suite (Jena Bioscience) were explored for crystal growth. The lysozyme concentration for this screen was 40 mg ml−1. After the appearance of crystals, the tray was left to dry out to allow the observation of salt crystals where possible.
For the characterization of individual protein crystals the conditions utilized are listed below. All proteins were crystallized at ambient temperature. For lysozyme the protein concentration was 40 mg ml−1 and the mother liquor consisted of 0.6–1 M NaCl, 0.1 M NH4 acetate. Thermolysin crystals were grown via the hanging-drop vapor-diffusion method utilizing a 1:1 ratio of protein (25 mg ml−1) to mother liquor, consisting of 0.8–1 M (NH4)2SO4, 0.1 M Tris (pH 8.0), 12% glycerol. Thaumatin crystals were grown similarly, using 25 mg ml−1 of protein and a mother liquor consisting of 0.5–0.7 M KNa tartrate. The condition for trypsin crystals utilized 30 mg ml−1 of protein and mother liquor consisting of 12% PEG 4000, 0.1 M MES (pH 6.0), 0.1 M LiSO4, 15% ethylene glycol. Catalase crystals were grown with 20 mg ml−1 of protein and with a mother liquor consisting of 15–16% PEG 4000, 0.1 M HEPES (pH 7.5), 10% 2-propanol.
2.2. Crystallization of amino acids
The 20 standard naturally occurring amino acids were purchased from Sigma (St Louis, MO, USA). Saturated solutions of all amino acids were prepared and pH adjusted where necessary to increase or decrease solubility. Amino acids were recrystallized in a microbatch environment under light mineral oil.
2.3. Confocal fluorescence and reflection imaging of lysozyme crystals at room temperature
A Zeiss LSM510 laser scanning confocal microscope was used to image crystals in the dried-out tray (see supporting information). Imaging was carried out using a 20×/0.4 numerical aperture (NA) long-working-distance air-immersion objective lens (Olympus LMPLF20×) and 488 nm excitation (0.290 mW at the sample). The confocal pinhole was fully open (∼10 Airy units) to collect the maximum amount of fluorescence (500–590 nm) from the protein crystals. Identical settings were utilized for the imaging of salt crystals and the crystals of all 20 amino acids.
2.4. Fluorescence emission spectroscopy of protein crystals
Fluorescence emission spectra from protein crystals were recorded at room temperature on several different Zeiss confocal microscopes over the course of the research (an LSM 710 inverted microscope, an LSM 880 inverted microscope and a Zeiss LSM 880 upright system) using a 10× Zeiss objective lens (EC Plan-NEOFLUAR, working distance 5.2 mm, NA 0.3) and the 34-channel Quasar spectral detectors on each of the systems. Protein crystals were excited with a 405, 458 or a 488 nm laser and emission spectra were acquired from near the excitation wavelength out to 698 nm with 4 nm spectral resolution. The spectral collection efficiencies of each of three excitation systems were calibrated using a spectral calibration lamp (Ocean Optics LS-1-CAL lamp) and all spectra shown are corrected data. For this experiment, crystals in immersion oil were mounted on a glass cover slide and placed in the LSM 880 inverted microscope stage.
2.5. Confocal imaging of lysozyme crystals at cryogenic temperatures
For imaging protein crystals at cryogenic temperatures, an Oxford Cryosystems 700 Cryostream was mounted on the optical table next to the Zeiss 510 confocal microscope equipped with a long-working-distance air-immersion objective lens. For fluorescence temperature-dependence measurements, the flash-cooled crystals were warmed from 100 K at 3 K min−1 to reach 293 K. Fluorescence confocal images were collected at 1 K intervals with 2× averaging using 488 nm excitation and the same emission collection setting as described in §2.3.
2.6. Non-confocal fluorescence imaging of crystals at a beamline
To assess the feasibility of fluorescence imaging of protein crystals at a beamline, the off-axis bright-field microscope conventionally used for crystal centering was modified. An FGL435 (Thorlabs) long-pass Schott glass filter was mounted in front of the crystal centering telescope’s front aperture to block wavelengths shorter than 435 nm. Protein crystals were illuminated from the side with a 5 mW 405 nm laser (AixiZ) with a defocused (≤2 mm) or a focused (≤20 µm) laser beam (Fig. S4 in the supporting information). Prior to the experiment, the power output from the laser was calibrated with an optical power meter (Newport, 1918-C photodiode power meter). The crystal centering camera exposure and gain settings were adjusted accordingly to observe maximum fluorescence from protein crystals. Typically the crystals were imaged at a rate of 7 frames per second. To investigate thermal effects of laser illumination, lysozyme crystals in NVH immersion oil were illuminated both at room temperature and in the 100 K coldstream with either a defocused or a focused laser beam. Series of diffraction images of 1° oscillations were collected post laser illumination at the A1 station at CHESS (Cornell High Energy Synchrotron Source) utilizing a 12.7 keV monochromatic X-ray beam and a Q-210 detector (Area Detectors Systems Corporation). The overall R sym, I/σ and mosaicity were used as the estimate for the level of laser-induced damage to the crystal.
2.7. Fluorescence lifetime measurement in lysozyme crystals
For the measurement of fluorescence lifetime, tetragonal lysozyme crystals were grown in a 300 µl, 3 mm-path-length plastic cuvette (BioRad VersaFluor Microcuvettes), containing a 1:1 ratio of 50 mg ml−1 lysozyme (EMD Millipore) in 50 mM Na acetate buffer (pH 4.6) and a mother liquor consisting of 30%(w/v) PEG 5000 methyl ester, 100 mM Na acetate (pH 4.6), 1 M NaCl. Pulsed excitation at 440 nm [40 ps full width at half-maximum (FWHM) pulse width at 50 MHz] was used and the time-correlated single photon counting (TCSPC) data were collected using a Hamamatsu R3809U-50 microchannel plate detector (25 ps transit time spread) and a TCSPC card (Becker and Hickl, SPC-730) in a PC. Two different spectral regions were collected using 460–520 nm and 520–640 nm bandpass filters. Fluorescence lifetime measurements were carried out at room temperature.
2.8. Crystal structure of l-glutamic acid
A series of 80 5° oscillations were collected from a single crystal of glutamic acid at the F1 beamline utilizing a 13.5 keV monochromatic X-ray beam. Images were recorded using a Q-270 detector (Area Detector Systems Corporation). The limiting resolution was 1.11 Å. Images were indexed, refined and integrated with XDS (Kabsch, 2010 ▸), then scaled and merged with POINTLESS and AIMLESS (Evans, 2006 ▸). The space group was P212121, with unit-cell dimensions 5.15 × 6.94 × 17.27 Å. Approximately 10% of the spots on the images included some saturated pixels, with the result that the intensities for the strongest reflections were poorly determined, but inclusion of these reflections was essential to structure solution. Processing statistics are given in Table 1 ▸.
Table 1. Data collection and refinement statistics for L-Glu.
| Observations | 3449 (330 with saturated pixels) |
| Unique reflections | 315 (including 19 overloads) |
| Space group | P212121 |
| Unit-cell dimensions (Å) | 5.15 × 6.94 × 17.27 |
| Resolution range (Å) | 17.27–1.11 |
| R merge | 0.071 (0.058)† |
| R p.i.m. | 0.023 (0.021)† |
| Completeness (%) | 98.0 (93.4)† |
| Multiplicity | 10.4 (7.9)† |
| Reflections for SnB | 299 accepted, 100 used (min. E 1.085) |
| Initial R factor | 0.342 |
| Refined R factor (REFMAC5) | 0.118 |
| RMS error in bond lengths (Å) | 0.023 |
| RMS error in bond angles (°) | 4.24 |
| RMS error in chiral volumes (Å3) | 0.153 |
Values in parentheses correspond to the highest-resolution shell.
The program SnB (Miller et al., 1994 ▸) was used to determine the structure from the merged F 2. Nine of the ten non-hydrogen atoms in glutamic acid were correctly placed. After adding the missing atom, the structure was refined with SHELXL (Sheldrick, 2015 ▸) and REFMAC5 (Murshudov et al., 2011 ▸), applying suitable geometric restraints for Glu. Statistics from REFMAC5 are given in Table 1 ▸. The Glu molecules are tightly packed, with multiple salt bridges holding them together; no solvent atoms were found.
3. Results
In this paper we characterize the visible-light-excited intrinsic fluorescence native to protein crystals without added dyes. To gain a better understanding of the nature of this fluorescence, emission scans of lysozyme crystals at multiple excitation wavelengths were carried out. These scans showed a broad emission peak with an FWHM of 80–120 nm for all excitation wavelengths used. Lysozyme crystals exhibit a decrease in the Stokes shift at lower excitation energies. In order to further elucidate this phenomenon, emission scans for crystals of three additional enzymes were acquired (Fig. 1 ▸) with the excitation wavelength of either 405, 458 or 488 nm. In all cases, the strongest fluorescence signal was observed under 405 nm excitation, with a peak emission at around 485 nm.
Figure 1.

Fluorescence emission spectra and normalized total fluorescence profiles of typical protein crystal standards at a macromolecular crystallography beamline. (a), (b) and (c) correspond to 405, 458 and 488 nm excitation wavelengths, respectively. (d) Fluorescence confocal images of (left to right) hen egg white lysozyme, thaumatin, thermolysin and trypsin crystals in hanging-drop crystallization trays. Crystals were imaged through cover slips utilizing the Zeiss LSM 880 upright confocal microscope using 405 nm as the excitation wavelength.
Over all of the protein crystals we measured from a variety of enzymes, the fluorescence intensities varied by close to an order of magnitude. The lack of any obvious single type of chromophore in all of the explored cases suggests that the source of the intrinsic fluorescence is the general stabilization of conjugated double bonds and increased electron delocalization arising from crystal packing. The broad emission spectrum and broad excitation band found support this model (Lakowicz, 2006 ▸). As a control to test this hypothesis, concentrated lysozyme solution was also excited with a 405 nm laser under the same conditions, but no detectable fluorescence was observed from the protein solution alone.
Fluorescence lifetime measurements of tetragonal lysozyme crystals further confirmed that the observed emission is most likely due to a collection of heterogeneously structured chromophores. Using 440 nm pulsed excitation and time-correlated single photon counting, we measured the fluorescence lifetime of lysozyme crystals in the blue and green–yellow spectral region (Fig. 2 ▸). In both cases we found that a minimum of two lifetime components in the nanosecond range were required for a satisfactory fit (Table 2 ▸), indicating a complex collection of chromophores and that the emission is fluorescence and not phosphorescence. Although we could achieve statistically acceptable χ2 values with a two-decay-component fit, the fractional amplitudes and decay times only represent weighted averages of a complex set of decays. Similar to spectroscopy carried out on complex heterogeneous fluorophores such as lipofuscin (a cross-linked aggregate of protein and oxidized lipids prevalent in aged tissues) (Marmorstein et al., 2002 ▸), protein crystal fluorescence decay kinetics are not single exponential. Additional evidence, albeit unfortunate, for the lipofuscin-like fluorescence arising from protein crystals is the fact that aggregated material within the crystallization drop (i.e. precipitate, skin) produces high background fluorescence (data not shown).
Figure 2.

Typical fluorescence lifetime measurement of lysozyme crystals excited at 440 nm. The best fit (χ2 = 1.03) yielded approximately one half of the emission (480 nm) decaying rapidly with a sub-nanosecond lifetime and one half exhibiting a slower 3 ns decay. [Data were fitted to 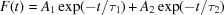 , A
1 = 0.55, τ1 = 0.35 ns, A
2 = 0.45, τ2 = 3.16 ns.]
, A
1 = 0.55, τ1 = 0.35 ns, A
2 = 0.45, τ2 = 3.16 ns.]
Table 2. Fluorescence lifetime measurements of lysozyme crystals (data are the average ± standard deviation of two measurements at each emission band).
| Emission wavelength (nm) | A 1 | τ1 (ns) | A 2 | τ2 (ns) | χ2 |
|---|---|---|---|---|---|
| 460–520 | 0.53 ± 0.04 | 0.37 ± 0.03 | 0.48 ± 0.04 | 3.06 ± 0.09 | 1.04 ± 0.01 |
| 520–640 | 0.43 ± 0.09 | 0.46 ± 0.05 | 0.57 ± 0.09 | 2.47 ± 0.12 | 1.07 ± 0.02 |
We also tested whether this method is a viable means to detect protein crystals in screening trays. Second-order nonlinear imaging techniques such as SHG or two-photon UVF (TP-UVF) can be used to visualize protein crystals but those techniques can also ‘light up’ certain salt crystals (Closser et al., 2013 ▸). Thus, we set forth to test salts that had previously been reported as SHG/TP-UVF active; no fluorescence was observed (Fig. S1), which we consider an advantage for protein crystal screening. To investigate this potential discriminatory ability further, we tested an aged tray of lysozyme crystals. Although the tray contained salt crystals in the majority of the wells, only protein crystals exhibited fluorescence (Fig. S2).
We also tested all 20 naturally occurring amino acids for visible-light-excited fluorescence. As expected, some amino acids exhibited fluorescence both at 405 nm and at 488 nm excitation wavelengths. Not surprisingly, the largest amount of fluorescence was observed in the case of l-Trp and l-His, but it was also present in the case of l-Glu (Fig. S3). In order to better understand the source of fluorescence in biological crystals l-Glu was examined further as it does not contain an obvious chromophore. The X-ray crystal structure of l-Glu, originally determined in 1955 (Hirokawa, 1955 ▸), was re-determined by modern methods in order to verify details of crystal packing and salt bridges within the crystal lattice. The amino group of l-Glu makes three salt bridges to two symmetry-related γ-carboxylate groups and one salt bridge to the α-carboxylate group of the glutamate molecule. The γ-carboxylate group also makes a salt bridge to one of the O atoms of a symmetry-related α-carboxylate group. Inferred from the crystal structure, the largest charge stabilization, though, comes from the α-carboxylate group which hydrogen bonds to three α-amino groups and one O atom from the symmetry-related γ-carboxylate group. We therefore hypothesize that the major contributing factor to the fluorescence from the l-Glu crystals comes from the stabilization of the delocalized charge at the α-carboxylate group (Fig. 3 ▸).
Figure 3.

Left: stereo representation of l-Glu crystal packing; the 2F o − F c map for l-Glu is contoured at the 1σ level. Right: stabilization of the delocalized charge of the α-carboxylate group is facilitated by three α-ammonium and one γ-carboxylate oxygen group from the symmetry-related l-Glu molecules. Hydrogen bonds are shown for interactions less than 3 Å.
It has been estimated that 98% of all protein structures in the Protein Data Bank (PDB) have been determined at 100 K (Garman, 2003 ▸); therefore we investigated the intensity of protein crystal autofluorescence as a function of temperature. Fluorescence images of lysozyme crystals excited at 488 nm were acquired as crystals warmed to room temperature from 100 K. As expected, the results indicate a strong temperature dependence of crystal autofluorescence, with decreasing fluorescence with increasing temperature (Fig. 4 ▸) and a phase transition at around 200 K (Fig. 4 ▸). The more than tenfold increase in fluorescence intensity at 100 K indicates that this method is ideal for imaging cryo-cooled crystals at macromolecular crystallography beamlines, where this technique could serve as a useful tool in crystal positioning.
Figure 4.

Tetragonal lysozyme crystal autofluorescence as a function of temperature. Averaged fluorescence (488 nm excitation) versus temperature curves from experiments in which lysozyme crystals were allowed to warm to 293 K (room temperature) and then cooled back down to 100 K. Plot shown is the mean ± standard error of the mean from data from three measurements (two from 100 K to RT, one from RT to 100 K; no significant differences were seen in curves taken in alternate cooling directions).
To test the usefulness of this simple-to-implement imaging method at the beamline, the crystal centering microscopes at the A1 and F1 beamlines of CHESS were modified as described in §2. Examples of the achievable precision of sample location can be seen in Fig. 5 ▸(b). Comparing regular bright-field microscopy images with those that were obtained in fluorescence mode for our tested cases revealed much clearer crystal boundaries within the drop or the mounting material.
Figure 5.

(a) Thermolysin crystal mounted in a nylon loop using NVH immersion oil as the cryoprotectant, imaged at 100 K; (b) lysozyme crystal mounted in a glass capillary during a room-temperature data collection. BFM – bright-field microscopy, FM – fluorescence microscopy. Thermolysin crystals (top) were illuminated with a 405 nm narrow bandwidth LED; lysozyme crystals in a capillary (bottom) were illuminated with a 5 mW CW 405 nm laser. White scale bar is 100 µm.
We also investigated the thermal effects of laser illumination on diffraction quality. Four modes of illumination were tested including a focused 20 µm laser beam and a defocused 2 mm laser beam at either ambient or cryogenic temperatures. The optical power density of the focused laser beam had negligible effects on diffraction quality when crystals were imaged at 100 K, but destroyed the crystal almost instantaneously when illuminated at ambient temperature. The 5 mW defocused laser beam had no effects on the data collection statistics at either temperature (Table 3 ▸).
Table 3. Data collection statistics for tetragonal lysozyme crystals that were exposed to 405 nm laser light at 5 mW of total optical power either focused or diffuse on the sample (exp = laser exposure, RT = room temperature).
| Setup | Crystal | R merge | I/σ | Mosaicity range (°) | Resolution range (Å) |
|---|---|---|---|---|---|
| RT 2 mm laser | Lysozyme 2 no exp | 0.083 (0.2) | 10.2 (2.8) | 0.23–0.23 | 40–2.5 |
| Lysozyme 2 10 min exp | 0.098 (0.361) | 10.0 (2.1) | 0.28–0.28 | 40–2.5 | |
| Lysozyme 3 no exp | 0.053 (0.112) | 16.8 (9.8) | 0.19–0.19 | 40–2.5 | |
| Lysozyme 3 10 min exp | 0.040 (0.045) | 17.8 (9.6) | 0.29–0.29 | 40–2.5 | |
| Lysozyme 4 no exp | 0.051 (0.114) | 12.0 (4.2) | 0.18–0.18 | 40–2.5 | |
| Lysozyme 4 10 min exp | 0.059 (0.302) | 9.6 (2.4) | 0.32–0.32 | 40–2.5 | |
| Lysozyme 5 no exp | 0.068 (0.116) | 9.5 (3.2) | 0.12–0.12 | 40–2.5 | |
| Lysozyme 5 10 min exp | 0.101 (0.375) | 6.7 (2.0) | 0.14–0.14 | 40–2.5 | |
| 100 K 20 µm laser | Lysozyme no exp | 0.031 (0.043) | 50.2 (32.5) | 0.32–0.44 | 50–2.0 |
| Lysozyme 45 min exp | 0.032 (0.047) | 49.5 (31.7) | 0.33–0.44 | 50–2.0 |
Values in parentheses correspond to the highest-resolution shell.
4. Discussion
The described method can serve as a complementary tool to regular bright-field, UVF or nonlinear-optics-based techniques to image protein crystals. Traditionally, when crystals appear in screening trays one can make an educated guess, based on the mother liquor and protein storage buffer composition, as to whether the crystal is salt or protein. At other times, one can add a dye to the drop to see whether the crystal will absorb the dye and change color, an indication of a protein crystal owing to its large water channels. An advantage of the described method over UVF and nonlinear-optics-based methods is the fact that it uses non-ionizing light at much lower illumination power. The method is simple to implement and relatively cost effective and does not require complicated optics or light sources. Unfortunately some crystals do not fluoresce at all or do so at very low intensity in our visible-light-excited fluorescence scheme. A larger pool of crystals of different proteins differing in primary sequence, bound prosthetic groups, space group and resolution should be explored in order to gain a better understanding of the source of visible-light-excited fluorescence in biological crystals.
5. Conclusions
Visible-light-excited fluorescence is a novel method for imaging protein crystals. The detection of crystals is achieved without the use of fluorescent dyes. Although the fluorescence intensity varied by close to an order of magnitude among the tested protein crystals, it proved to be a useful tool for differentiating salt from protein crystals since it did not produce false-positive signal from salt crystals as methods relying on nonlinear optics often do.
We also show that this method can be used at macromolecular crystallography beamlines for visualizing crystals that are otherwise difficult to see via regular bright-field microscopy. Unlike SHG, UV-fluorescence-based methods, X-ray rastering or X-ray radiography, the method described here does not cause damage to protein crystals at the low laser powers required for imaging (diffuse 5 mW laser beam). At cryogenic temperatures, the fluorescence intensity arising from protein crystals is about tenfold higher than at ambient temperature owing to temperature-dependent chromophore stabilization.
Supplementary Material
Supplementary Fig. 1. DOI: 10.1107/S160057671502419X/te5008sup1.pdf
Supplementary Fig. 2. DOI: 10.1107/S160057671502419X/te5008sup2.pdf
Supplementary Fig. 3. DOI: 10.1107/S160057671502419X/te5008sup3.pdf
Supplementary Fig. 4. DOI: 10.1107/S160057671502419X/te5008sup4.pdf
Supporting information file. DOI: 10.1107/S160057671502419X/te5008sup5.mcf
Supporting information file. DOI: 10.1107/S160057671502419X/te5008sup6.pdf
Acknowledgments
The authors would like to thank Irina Kriksunov from MacCHESS for providing protein crystals and Rebecca Williams from Cornell BRC imaging facility for helpful advice. CHESS is supported by the NSF and NIH/NIGMS via NSF award DMR-1332208, and the MacCHESS resource is supported by NIH/NIGMS award GM-103485. WRZ acknowledges support from Shared Instrumentation Grant S10-OD018516 from the NIH for use of the Zeiss LSM 880.
References
- Asanov, A. N., McDonald, H. M., Oldham, P. B., Jedrzejas, M. J. & Wilson, W. W. (2001). J. Cryst. Growth, 232, 603–609.
- Calero, G., Cohen, A. E., Luft, J. R., Newman, J. & Snell, E. H. (2014). Acta Cryst. F70, 993–1008. [DOI] [PMC free article] [PubMed]
- Cherezov, V., Hanson, M. A., Griffith, M. T., Hilgart, M. C., Sanishvili, R., Nagarajan, V., Stepanov, S., Fischetti, R. F., Kuhn, P. & Stevens, R. C. (2009). J. R. Soc. Interface, 6, S587–S597. [DOI] [PMC free article] [PubMed]
- Closser, R. G., Gualtieri, E. J., Newman, J. A. & Simpson, G. J. (2013). J. Appl. Cryst. 46, 1903–1906. [DOI] [PMC free article] [PubMed]
- Desbois, S., Seabrook, S. A. & Newman, J. (2013). Acta Cryst. F69, 201–208. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Garman, E. (2003). Curr. Opin. Struct. Biol. 13, 545–551. [DOI] [PubMed]
- Haupert, L. M. & Simpson, G. J. (2011). Methods, 55, 379–386. [DOI] [PMC free article] [PubMed]
- Hirokawa, S. (1955). Acta Cryst. 8, 637–641.
- Judge, R. A., Swift, K. & González, C. (2005). Acta Cryst. D61, 60–66. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Khan, I., Gillilan, R., Kriksunov, I., Williams, R., Zipfel, W. R. & Englich, U. (2012). J. Appl. Cryst. 45, 936–943. [DOI] [PMC free article] [PubMed]
- Kissick, D. J., Dettmar, C. M., Becker, M., Mulichak, A. M., Cherezov, V., Ginell, S. L., Battaile, K. P., Keefe, L. J., Fischetti, R. F. & Simpson, G. J. (2013). Acta Cryst. D69, 843–851. [DOI] [PMC free article] [PubMed]
- Kissick, D. J., Wanapun, D. & Simpson, G. J. (2011). Annu. Rev. Anal. Chem. 4, 419–437. [DOI] [PMC free article] [PubMed]
- Lakowicz, J. R. (2006). Principles of Fluorescence Spectroscopy, 3rd ed. New York: Springer.
- Madden, J. T., DeWalt, E. L. & Simpson, G. J. (2011). Acta Cryst. D67, 839–846. [DOI] [PMC free article] [PubMed]
- Madden, J. T. et al. (2013). J. Synchrotron Rad. 20, 531–540. [DOI] [PMC free article] [PubMed]
- Marmorstein, A. D., Marmorstein, L. Y., Sakaguchi, H. & Hollyfield, J. G. (2002). Invest. Ophthalmol. Vis. Sci. 43, 2435–2441. [PubMed]
- Meyer, A., Betzel, C. & Pusey, M. (2015). Acta Cryst. F71, 121–131. [DOI] [PMC free article] [PubMed]
- Miller, R., Gallo, S. M., Khalak, H. G. & Weeks, C. M. (1994). J. Appl. Cryst. 27, 613–621.
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nitahara, S., Maeki, M., Yamaguchi, H., Yamashita, K., Miyazaki, M. & Maeda, H. (2012). Analyst, 137, 5730–5735. [DOI] [PubMed]
- Padayatti, P., Palczewska, G., Sun, W. Y., Palczewski, K. & Salom, D. (2012). Biochemistry, 51, 1625–1637. [DOI] [PMC free article] [PubMed]
- Pusey, M., Barcena, J., Morris, M., Singhal, A., Yuan, Q. & Ng, J. (2015). Acta Cryst. F71, 806–814. [DOI] [PMC free article] [PubMed]
- Shaw Stewart, P. & Mueller-Dieckmann, J. (2014). Acta Cryst. F70, 686–696. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Shukla, A., Mukherjee, S., Sharma, S., Agrawal, V., Radha Kishan, K. V. & Guptasarma, P. (2004). Arch. Biochem. Biophys. 428, 144–153. [DOI] [PubMed]
- Vernede, X., Lavault, B., Ohana, J., Nurizzo, D., Joly, J., Jacquamet, L., Felisaz, F., Cipriani, F. & Bourgeois, D. (2006). Acta Cryst. D62, 253–261. [DOI] [PubMed]
- Warren, A. J., Armour, W., Axford, D., Basham, M., Connolley, T., Hall, D. R., Horrell, S., McAuley, K. E., Mykhaylyk, V., Wagner, A. & Evans, G. (2013). Acta Cryst. D69, 1252–1259. [DOI] [PMC free article] [PubMed]
- Watts, D., Müller-Dieckmann, J., Tsakanova, G., Lamzin, V. S. & Groves, M. R. (2010). Acta Cryst. D66, 901–908. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. DOI: 10.1107/S160057671502419X/te5008sup1.pdf
Supplementary Fig. 2. DOI: 10.1107/S160057671502419X/te5008sup2.pdf
Supplementary Fig. 3. DOI: 10.1107/S160057671502419X/te5008sup3.pdf
Supplementary Fig. 4. DOI: 10.1107/S160057671502419X/te5008sup4.pdf
Supporting information file. DOI: 10.1107/S160057671502419X/te5008sup5.mcf
Supporting information file. DOI: 10.1107/S160057671502419X/te5008sup6.pdf