

Abstract
The inter-α-trypsin inhibitor complex is a macromolecular arrangement of structurally related heavy chain proteins covalently cross-linked to the chondroitin sulfate (CS) chain of the proteoglycan bikunin. The inter-α-trypsin inhibitor complex is abundant in plasma and associated with inflammation, kidney diseases, cancer and diabetes. Bikunin is modified at Ser-10 by a single low-sulfated CS chain of 23–55 monosaccharides with 4–9 sulfate groups. The innermost four monosaccharides (GlcAβ3Galβ3Galβ4Xylβ-O-) compose the linkage region, believed to be uniform with a 4-O-sulfation to the outer Gal. The cross-linkage region of the bikunin CS chain is located in the nonsulfated nonreducing end, (GalNAcβ4GlcAβ3)n, to which heavy chains (H1-H3) may be bound in GalNAc to Asp ester linkages. In this study we employed a glycoproteomics protocol to enrich and analyze light and heavy chain linkage and cross-linkage region CS glycopeptides derived from the IαI complex of human plasma, urine and cerebrospinal fluid samples. The samples were trypsinized, enriched by strong anion exchange chromatography, partially depolymerized with chondroitinase ABC and analyzed by LC-MS/MS using higher-energy collisional dissociation. The analyses demonstrated that the CS linkage region of bikunin is highly heterogeneous. In addition to sulfation of the Gal residue, Xyl phosphorylation was observed although exclusively in urinary samples. We also identified novel Neu5Ac and Fuc modifications of the linkage region as well as the presence of mono- and disialylated core 1 O-linked glycans on Thr-17. Heavy chains H1 and H2 were identified cross-linked to GalNAc residues one or two GlcA residues apart and H1 was found linked to either the terminal or subterminal GalNAc residues. The fragmentation behavior of CS glycopeptides under variable higher-energy collisional dissociation conditions displays an energy dependence that may be used to obtain complementary structural details. Finally, we show that the analysis of sodium adducts provides confirmatory information about the positions of glycan substituents.
Glycosylation is the most complex protein post-translational modification known today but unfortunately glycomics characterization is often kept apart from the proteomic characterization of proteins of biological tissues and samples. Such dual approaches may be complementary but also limiting because they will give neither the whole picture of all protein iso/glycoforms in a tissue nor the detailed structure of any of the single proteins included. Glycoproteomics approaches are now becoming available bridging these two fields by keeping the glycan and the peptide parts together in giving simultaneous and specific information on the glycan structures, their attachment sites and the identities of the core proteins (1). Here we report on new liquid chromatography-tandem MS (LC-MS/MS) protocols that can be used to decipher the structural heterogeneity of proteoglycans and even proteoglycan complexes, i.e. glycoproteins known to be inherently demanding to structurally characterize either alone or in mixtures. Because of the theoretically immense but biologically limited number of glycan structures appearing in nature, and given the limitations of MS analysis of glycopeptides (defining types, numbers and sequences of monosaccharides rather than monosaccharide identities, linkage positions and configurations), it is important to build on earlier experience from analyses of single isolated proteins and to use nomenclature, abbreviations and symbols that are well accepted and understood. Throughout this report we have, used the CFG nomenclature of glycan structures (http://www.functionalglycomics.org/static/consortium/Nomenclature.shtml) to increase the intelligibility of our findings (Fig. 1).
Fig. 1.

Schematic depiction of the inter-α-trypsin inhibitor complex and its degradation by chondroitinase ABC. Both the linkage and cross-linkage regions are highlighted as well as the molecular details of the GalNAc to aspartic acid ester bond that cross-links the heavy chains to the CS chain of bikunin. In addition to CS-glycopeptides the endolytic activity of the chondroitinase ABC digestion is also expected to generate free disaccharides of the CS chain (faded region). The formation of unsaturated GlcA by chondrotinase ABC is shown below the complex and the monosaccharide symbols are explained in the upper left quadrant.
Proteoglycans constitute a group of O-glycosylated proteins all carrying one or more complex glycan (glycosaminoglycan or GAG)1 chains attached to Ser residues of the core proteins through a common linkage tetrasaccharide, called the GAG linkage region, which is composed of the innermost four monosaccharides (GlcAβ3Galβ3Galβ4Xylβ-O-) at the reducing end of the GAG chain (GlcA is glucuronic acid; Gal is galactose; and Xyl is xylose) (2).
The structural difference between various subclasses of proteoglycans emanates from the repeated extension of the linkage tetrasaccharide by two monosaccharides; e.g. GlcAβ3GalNAcβ4 in chondroitin sulfate proteoglycans (CSPGs), GlcAβ4GlcNAcα4 in heparan sulfate proteoglycans (HSPGs) and from the extent and positions of O- and N-sulfations, GlcA epimerization and from the actual chain length of the glycosaminoglycan. The GAG chains may also be covalently attached, or cross-linked, to other proteins at the nonreducing end of the chain through a C-terminal aspartic acid ester linkage. In this report we concentrate on the structural characterization of the human inter-α-trypsin inhibitor CSPG complex.
Chondroitin sulfate proteoglycans play a significant role in maintaining the structural integrity of most extracellular matrices. In addition to their structural role as matrix components, eukaryote CSPGs are known to be involved in more specialized functions such as signal transduction, morphogenesis and regulation of stem cell behavior and differentiation (3–9). In many cases, their biological activity is mediated by selective binding of protein ligands to distinct glycan structural variants (10, 11). Previous studies have made possible the analysis of the average fine structure of chondroitin sulfate (CS) chains facilitating the identification of discrete glycan domains likely involved in ligand interaction (12–16). The CSPG saccharide linkage region differs significantly from the structure of the rest of the glycan chain and its assembly has been shown to be essential for the regulation of the GAG biosynthesis (17–19).
The glycomics approach for CSPGs structural analysis involves the release of the GAG chains from the core proteins, digestion of the released chain into disaccharides followed by their subsequent analysis by e.g. ion exchange chromatography, nuclear magnetic resonance spectroscopy or mass spectrometry. However, in this work-flow the identities of the core proteins as well as the attachment sites are lost hindering the assignment of specific structures to particular proteoglycan isoforms. In order to obtain integrated glycan-protein information, we recently developed a glycoproteomics approach allowing site-specific analysis of CSPG linkage region glycopeptides (20). Samples from human urine, plasma and cerebrospinal fluid were trypsinized and subjected to strong anion exchange (SAX) chromatography in order to enrich for anionic GAG-substituted glycopeptides. The fractions were then treated with chondroitinase ABC to depolymerize CS-chains into free disaccharides and residual hexasaccharide substituted peptides comprising the tetrasaccharide linkage region (21). The resulting CS-glycopeptides were finally characterized by reversed phase nLC-MS/MS run in positive mode using higher-energy collisional dissociation (HCD). In that study, 13 novel CSPGs were successfully identified and site-specific information regarding 13 already established CSPGs was obtained. However, the fine-structure analysis of the CS glycopeptides and their substitution patterns was hampered by the relatively weak intensities of sulfated and/or phosphorylated fragment ions obtained under the HCD conditions used. This problem became especially obvious for bikunin, the major component of the inter-α-trypsin inhibitor (IαI) complex and the most abundant and heterogeneous CSPG of those samples.
Bikunin is a small acidic glycoprotein of about 40 kDa that is modified at Ser-10 by the attachment of one single CS chain. The linkage region of this GAG chain has been the target for several studies concluding a uniform galactose-4-sulfate tetrasaccharide structure (GlcAβ3Galβ3(4-O-SO3)Galβ4Xylβ-O-) in both human plasma and urinary samples (22, 23). The IαI complex is a unique macromolecular arrangement of structurally related proteins, i.e. heavy chains, covalently linked to the CS chain of bikunin, also called the light chain of the IαI complex (Fig. 1) (24–27). In humans, one bikunin molecule is typically cross-linked to both heavy chain 1 (H1) and heavy chain 2 (H2) in the IαI-complex, but only to heavy chain 3 (H3) in the pre-α-trypsin inhibitor complex (28). Free bikunin is mainly found in urine and also known as the urinary trypsin inhibitor (UTI) (29). The plasma and urinary levels of bikunin are often raised during pathological processes such as chronic and acute inflammations, infections, cancer, renal diseases and diabetes (30–36). During inflammation, the CS chain may undergo changes in size and sulfation that correlate with the severity of the inflammatory response (37, 38).
In the present work, we further developed our glycoproteomics approach to enable a detailed characterization and relative quantification of the IαI linkage and cross-linkage region glycopeptides derived from human plasma, urine and CSF samples. The use of alternating collisional energies proved to have a major impact on the fragmentation of CS glycopeptides and added valuable complementary structural information. The addition of sodium ions readily increased the stability of labile sulfate substituents upon HCD conditions allowing a more precise glycan characterization.
The CS linkage region of bikunin was found to display a much larger heterogeneity than previously realized. The linkage region modifications were found to include sulfate, phosphate, fucose and sialic acid substitutions with notable differences across body fluids. We were also able to specifically identify glycopeptides derived from the cross-linking regions of H1 and H2. The heavy chains were often simultaneously attached to the same CS-chain, in close proximity to each other and sometimes even to adjacent GalNAc residues. Taken together, this study demonstrates that the linkage region of human CSPGs displays a vast greater structural complexity than previously perceived. Given that specific modifications of the linkage region may function as a molecular regulator of CS biosynthesis, these findings could also assist in further elucidating the mechanisms of CS chain elongation and substitution.
EXPERIMENTAL PROCEDURES
Sample Preparation and Enrichment of CS-glycopeptides
Plasma (5 ml) was taken from a single blood donor and 100 μl of plasma was used for the glycopeptide analysis. Eight ml of morning urine was collected from a healthy male individual and was cleared from cell debris by centrifugation (3000 × g for 10 min). The CSF sample, collected during 5 days post operation, was kindly donated from a patient undergoing neurosurgery because of a benign condition. The CSF was centrifuged at 1800 × g for 10 min, separated into 2 ml aliquots and kept at −80 °C until use. The use of de-identified human samples for method development is in agreement with Swedish law and the study was permitted by the head of the Clinical Chemistry Laboratory, Sahlgrenska University Hospital (Dnr 797–540/15).
Preparation and enrichment of linkage region glycopeptides was conducted essentially as already published (20) although with some minor modifications. Briefly, plasma samples were treated with ProteoPrep Immunoaffinity Albumin and IgG Depletion Kit (PROTIA, Sigma-Aldrich, Saint Louis, MO) according to the manufacturer's specifications. Urine samples were mixed with SDS to a final concentration of 0.1% and run through a PD-10 column (GE Healthcare) using 0.1% SDS. The eluted samples were thereafter run through a second PD-10 column, equilibrated in dH2O, to remove the SDS. CSF, urine and depleted plasma samples (initial volumes of 2 ml, 8 ml, and 100 μl, respectively) were lyophilized and reduced with 10 mm DTT for 1h at 56 °C. Alkylation was conducted with 55 mm iodoacetamide for 45 min, in darkness at room temperature. Thereafter, the samples were digested overnight with sequence grade porcine trypsin (Promega). The trypsin-digested samples were diluted in 10 ml coupling buffer (50 mm sodium acetate, 200 mm NaCl, pH 4.0) and loaded onto a SAX spin column (Vivapure, Q Mini H). Samples were washed (50 mm Tris-HCl, 200 mm NaCl, pH 8.0) and the flow through and wash fractions were set aside. GAG-modified peptides were eluted in three steps in 50 mm Tris-HCl buffer pH 8 (0.4 m, 0.8 m and 1.6 m NaCl). The three fractions were collected, desalted, lyophilized, and subjected to chondroitinase ABC treatment (Sigma-Aldrich).
Depolymerization of the CS chains was conducted either for 1h at 37 °C to achieve partial degradation of the glycosaminoglycan or for complete degradation, overnight at 37 °C. The samples were then purified using C18 spin column (PierceTM Spin Columns, Thermo Fischer Scientific, Waltham, MA), according to the manufacturer's protocol, and lyophilized pending MS/MS analysis. The day of analysis the samples were reconstituted in 0.2% formic acid in water containing 5% acetonitrile. For nLC-MS/MS analysis with sodium adducts, stock solution of 5 m sodium acetate and 5 m formic acid in water was used for preparing a fresh 500 mm sodium formate solution directly pipetted into the MS vial.
β-elimination of O-linked Glycans
Samples were subjected to beta-elimination using methylamine vapor according to Mirgorodskaya et al. (39). Briefly, 300 μl 40% methylamine (aq) was added to a microcentrifuge tube and placed in a 35 ml Kimax tube, which had been flushed with nitrogen. A lyophilized CS glycopeptide sample in another microcentrifuge tube was added to the Kimax tube, capped and kept at 70 °C for 2 h. The beta- eliminated sample was then stored at −20 °C until analysis.
nLC-MS/MS Analysis
The samples were analyzed on a Q Exactive mass spectrometer coupled onto an Easy-nLC 1000 system (Thermo Fisher Scientific). Ions were injected into the mass spectrometer under a spray voltage of 1.6 kV in positive ion mode. MS precursor scans were performed at 70,000 resolution (at m/z 200), an Automatic Gain Control (AGC)-target value of 3 × 106 with a mass range of m/z 600–2000. MS2 spectra were generated by HCD of the six largest precursor peaks using an isolation window of m/z 2.0 at a normalized collision energy (NCE) of 20 and 30% using profile mode at a resolution of 35,000. A dynamic exclusion of 30 s was used. Glycopeptides (10 μl injection volume) were separated using an in-house constructed trap column and analytical column set up (45 × 0.075, 14 mm I.D and 200 × 0.050 mm I.D., respectively) packed with 3 μm Reprosil-Pur C18-AQ particles (Dr. Maisch GmbH, Ammerbuch, Germany). The following gradient was run at about 150 nL/min; 7–37% B-solvent (acetonitrile in 0.2% formic acid) over 60 min, 37–80% B over 5 min with a final hold at 80% B for 10 min.
Automated Search Strategy for CS-glycopeptides Peptide Search Specifications
Mascot distiller (version 2.3.2.0, Matrix Science) was used to convert .raw spectra into singly protonated peak lists in .mgf format. Searches were performed against Homo sapiens (20,209 entries) in the UniProtKB/Swiss-Prot database (546,000 entries, 194,259,968 residues) using an in-house Mascot server (version 2.3.02). The following constraints were applied: MS tolerance, 10 ppm; MS/MS tolerance 0.1 Da; enzyme, trypsin or semitrypsin with 1 or 2 missed cleavages allowed; fixed carbamidomethyl modifications of Cys residues and variable Met oxidation.
Glycosaminoglycan Search Specifications
Variable modifications corresponding to CS-linkage regions on Ser residues were defined as their predicted chondroitinase ABC cleavage products: [ΔGlcAGalNAcGlcAGalGalXyl] without sulfate (C37H55NO30, 993.2809 Da), with one (C37H55NO33S, 1073.2377 Da) or two (C37H55NO36S2, 1153.1945 Da) sulfate groups attached. Loss of these masses for b- and y-ions including the arbitrarily assigned glycosylation site was also specified.
In separate searches, the allowed modification of ΔGlcAGalNAc and ΔGlcAGalNAcGlcAGalNAc were included. For these modifications, the neutral loss of ΔGlcAGalNAc and ΔGlcAGalNAcGlcAGalNAc, minus the mass of an acetyl group, were added. The acetyl shift (42.01 amu) was used because we experimentally observed fragmentation-generated acetylation of the y-ions including the C-terminal glycosylation site. Because the database employed (UniProtKB) does not contain known processing sites, and to potentially identify novel proteins cross-linked to the CS chain, these searches were performed using semitrypsin to allow for the identification of nontryptic C-terminal Asp of any cross-linking glycopeptide. For the beta-eliminated samples, the allowed modification of methylamine (–O, +NCH3, +13.0316 amu) on Ser and Thr residues was used in the Mascot search.
Manual Data Evaluation
All spectra with automatically proposed CS-glycopeptides from bikunin or any of the IαI heavy chains were manually evaluated. The final assignment was based on the following criteria: (1) deviation from the calculated monoisotopic mass of the precursor ions < 10 ppm, (2) the presence of ΔGlcAGalNAc-specific oxonium ions at m/z 362.11 or the GlcAGalNAc-specific oxonium ion at m/z 380.12; and the presence of oxonium ions at m/z 126.05, m/z 138.07, and m/z 186.08 originating from decomposition of the m/z 204.09 HexNAc B-ion (40), (3) stepwise glycosidic fragmentation confirming the sequence of the linkage or cross-linkage regions, (4) the presence of at least three peptide fragment ions originating from the intact C-terminal (y-ions) and N-terminal (b-ions), correct within ±0.01 amu, and consistent with the proposed amino acid sequence. LC-MS/MS files were additionally searched for the presence of CS glycopeptides by producing extracted ion chromatograms through filtering for the absolute presence of m/z 362.11–362.12 and m/z 380.12–380.13 at the MS2 level using the Xcalibur software (Thermo Scientific).
For estimation of relative amounts of the different bikunin glycoforms extracted ion chromatograms were prepared by tracing the first four isotopes of the [M+3H]3+ ions and then integrating the peaks using the Xcalibur software (Thermo Scientific). Relative abundances were reported as percentage values relative to all the observed glycoforms.
The raw mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD002707 (41, 42).
RESULTS
Enrichment and MS/MS Fragmentation Analysis of Bikunin Linkage Region Glycopeptides
The urine, the plasma, and the cerebrospinal fluid samples were all selected for study as they represent easily accessible human biological fluids traditionally used for diagnostic purposes. The amounts of each sample was fitted rather to what is clinically available than to what would give the most of information for each sample type. After initial purification of the urine and plasma samples, the work-up procedure was standardized as much as possible including lyophilization, reduction, trypsinization, stepwise ion exchange chromatography, desalting, enzymatic hydrolysis by chondroitinase ABC, purification by reversed phase chromatography and lyophilization of the enriched CS substituted glycopeptides. The glycopeptide fractions were then dissolved and subjected to positive mode nanoLC-MS/MS analyses very similar to a set up for standard proteomic analyses.
Analogous to our previous study (20), the analysis of all sample types revealed an intense CS-glycopeptide precursor ion (MS1 m/z 1094.44; 3+) in the 0.8 m-fraction that equated to the molecular mass of a tryptic bikunin peptide (1AVLPQEEEGSGGGQLVTEVTK21) carrying the CS hexasaccharide, including the linkage region, and two sulfate/phosphate (S/P) modifications (3280.2641 Da). However, the exact nature of this structural isomer could not directly be resolved because of the relatively weak intensities of sulfated and/or phosphorylated fragment ions. To further examine the nature of this ion we analyzed the total ion chromatograms of the fractions collected at 0.4, 0.8, and 1.6 m NaCl, respectively (Fig. 2A–2C). This revealed that the bikunin glycopeptide precursor ion (m/z 1094.44; 3+) eluted throughout all the three fractions, and was quantitatively the most dominating peak. The corresponding non- and monosulfated linkage region glycoforms at m/z 1041.13; 3+ and at m/z 1067.78; 3+, were mainly eluted at lower NaCl concentrations (Fig. 2A–2B).
Fig. 2.
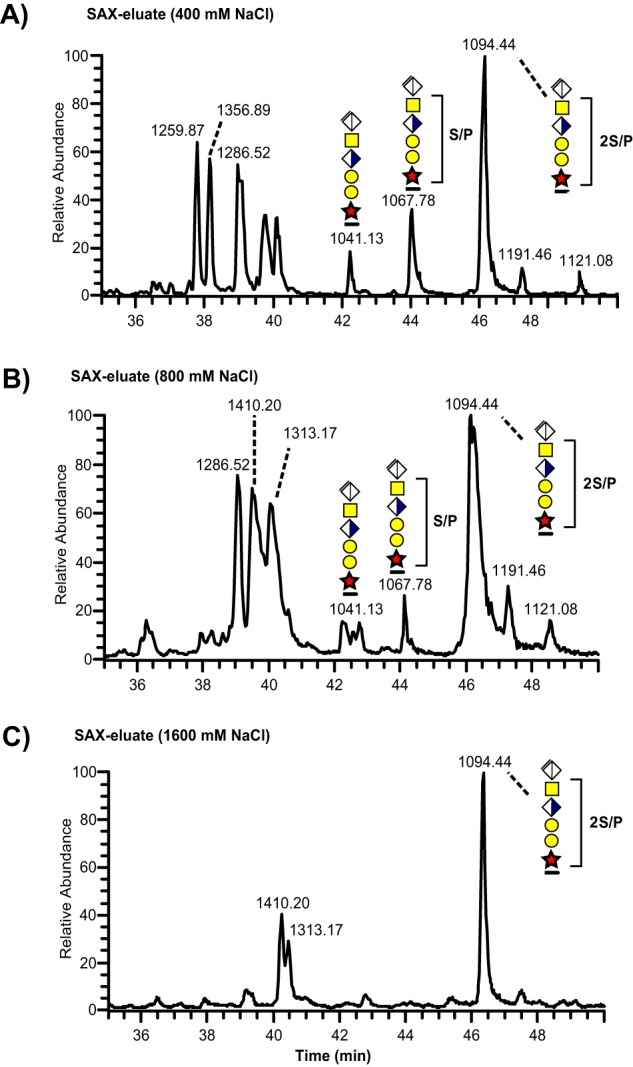
Total ion chromatograms of SAX-enriched CS-glycopeptides from a urine sample. Three fractions collected at (A) 400 mm, (B) 800 mm, and (C) 1600 mm NaCl were subjected to chondroitinase ABC hydrolysis and analyzed with nLC-MS/MS.
To investigate whether fragmentation at lower normalized collision energy (NCE) level would generate sulfated and/or phosphorylated fragment ions of higher intensities, the bikunin precursor ion (m/z 1094.44; 3+) was fragmented at the 20% NCE level (compared with our standard at 30%). The analysis resulted in abundant glycosidic fragmentation (Fig. 3A) into saccharide substituted peptide ions (Y-ions according to the nomenclature of Domon and Costello (40)) and poor peptide fragmentation. The presence of one sulfate group (SO3) at the sub-terminal GalNAc residue was indicated by a weak but significant [ΔGlcAGalNAc+HSO3]+ oxonium ion at m/z 442.07.We have previously determined that this is a sulfate group (79.9568 amu) as opposed to phosphate (79.9663 amu) based on the Orbitrap mass measurement at 35,000 resolution (20). The identity and location of the other S/P substitution could not be unambiguously determined as it readily dissociated upon fragmentation. See below regarding the assignment of sulfate identities using sodiated precursors.
Fig. 3.
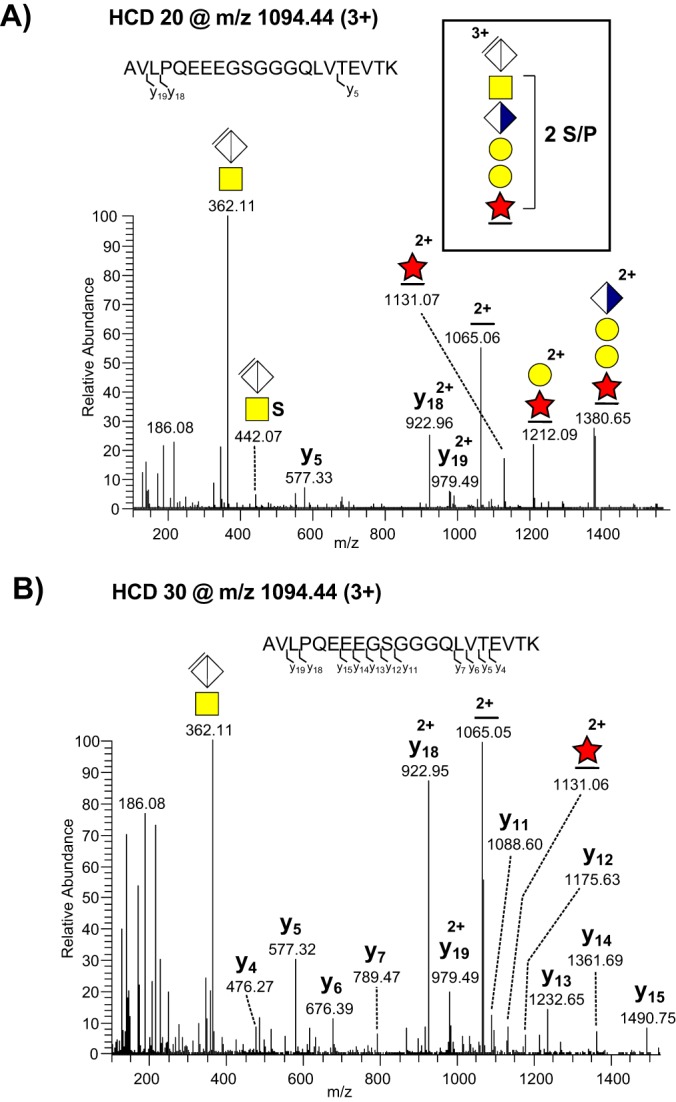
MS/MS fragmentation patterns recorded at different HCD levels from a triple charged bikunin CS-glycopeptide precursor ion at m/z 1094.44. (A) HCD performed at normalized collision energy of 20%; and (B) of 30%. Glycopeptide fragments as well as b- and y-ions are annotated. The schematic structure of the precursor ion is shown (boxed structure).
Collisional dissociation at the 30% NCE level yielded abundant peptide fragmentation at the expense of glycosidic cleavage and the assignment of substituents was similarly precluded by the absence of MS2 fragments with clear retention of S/P (Fig. 3B). We concluded that using different HCD fragmentation energies is an effective way of generating complementary structural information but S/P substituents are labile and readily abstracted regardless of energy level.
Sodium Adducts Stabilize Sulfate Modifications Under Positive Mode HCD Fragmentation
To investigate whether sodium ions could enhance the fragmentation stability of sulfate and phosphate groups, and thereby facilitate the characterization of their attachment site on the glycan of the linkage region, a buffer solution containing 10, 100, or 500 mm sodium was added to the sample vials prior to the LC-MS/MS analysis. A trapping precolumn was used to remove excess of sodium. At 500 mm the most abundant precursor ion for the bikunin CS-glycopeptide substituted with two sulfate/phosphate groups was observed at m/z 1116.41 [M+3Na]3+. The HCD spectrum (Fig. 4A) was now dominated by the presence of a series of sodiated oxonium ions e.g. [ΔGlcAGalNAc +Na]+ (m/z 384.09), [ΔGlcAGalNAc+SO3+2Na-H]+ (m/z 486.02), [ΔGlcAGalNAcGlcA+Na]+ (m/z 560.12) and [ΔGlcAGalNAcGlcA+SO3+2Na-H]+ (m/z 662.05) indicating stabilization of the saccharide sulfates by sodium complexation. The sodium ions seemed specifically bound to sulfate because an extra Na+ ion was added in going from unsulfated to monosulfated structures, shown by the shifts from m/z 384.09 to m/z 486.02 ions and from m/z 560.12 to m/z 662.05 ions. A significant peak corresponding to [GalNAc+SO3+2Na-H]+ at m/z 328.00 (Fig. 4B) showed that the sulfate was attached to the GalNAc, as opposed to GlcA, in line with previous studies (22, 23). Sulfate groups at sodiated glycopeptide fragments were also retained in MS2 (Y-fragments according to (40)). The other sulfate group was pinpointed to the outer Gal unit based on the presence of the [peptide+XylGalGal+SO3+2Na]2+ ion at m/z 1355.06 (Fig. 4C) and the [peptide+XylGalGal+SO3+3Na]3+ ion at m/z 911.04 (Fig. 4D) and the presence of a nonsulfated glycopeptide fragment (Fig. 4C) containing only the inner Gal (m/z 1223.07). Additionally, this fragmentation pattern excluded phosphorylation of Xyl for this particular precursor. The substitution assignment was further supported by the presence of an abundant oxonium ion corresponding to [ΔGlcAGalNAcGlcAGal+2SO3+3Na-H]+ at m/z 926.05 (Fig. 4D) pinpointing again to one sulfate group bound to the GalNAc and one to the outer Gal.
Fig. 4.

MS/MS fragmentation patterns recorded at the 20% HCD level from a sodiated bikunin CS-glycopeptide precursor ion, [M+3Na]3+ at m/z 1116.41. A, The complete HCD spectrum. B, the m/z 180–370 region; C, the m/z 1000–1500 region; and D, the m/z 900–1000 region. All major ions are annotated to charge, m/z, Na+ content and glycan structure.
In order to differentiate between sulfate and phosphate substitutions the mass resolution of the Orbitrap was used. The experimental mass accuracies of the observed oxonium ions were 5.9–8.1 ppm for an arbitrarily selected HCD spectrum (supplemental Table 1) demonstrating a high degree of mass accuracy for fragment ions at these low m/z values, and the correctness of the assigned ions. This included the [GalNAc+SO3 + 2Na-H]+ ion at m/z 328.0050, which was +7.6 ppm off, and the [ΔGlcAGalNAcGlcAGal+2SO3+3Na-2H]+ ion at m/z 926.0471 being +6.0 ppm off from the theoretical masses. When the theoretical structures, carrying sulfate, were changed to phosphate the experimental mass accuracies increased to 15.5–36.6 ppm convincingly demonstrating the identity of sulfate as opposed to phosphate. Taken together, the analysis of sodium adducts thus added structural information allowing the assignment of sulfate substitutions to specific monosaccharide units of the glycan chain.
The CS-linkage Region of Bikunin is Highly Heterogeneous and Can be Modified by Sulfate, Phosphate, Fucose, and Sialic Acid Substitutions
The distinction between sulfate and phosphate for the Xyl residue using exact fragment mass measurements has previously been demonstrated (20). The phosphorylation of the Xyl residue was conveniently characterized in a bikunin CS-glycopeptide containing one phosphate substitution (m/z 1067.78, Table I, supplemental Fig. S1 spectrum s3). In sharp contrast to saccharide sulfation, the Xyl phosphorylation of this CS-glycopeptide was stable upon HCD fragmentation at 20% NCE leaving distinct glycopeptide fragments at m/z 1171.04 [peptide+Xyl+PO3+3H]2+ and at m/z 1252.07 [peptide+XylGal+PO3+3H]2+. Those glycopeptide fragment ions were also clearly identified in the spectrum of the mono-phosphorylated mono-sulfated glycoform (supplemental Fig. S1 spectrum s19) co-eluting with the di-sulfated glycoform at 46–47 min (Fig. 2). We also managed to identify a glycoform carrying one phosphate attached to the Xyl residue, one sulfate group at the distal Gal residue and another sulfate group bound to the HexNAc (m/z 1121.08, Table I, supplemental Fig. S1 spectrum s4). The Xyl phosphorylation was detected only in urine samples but not in plasma or CSF.
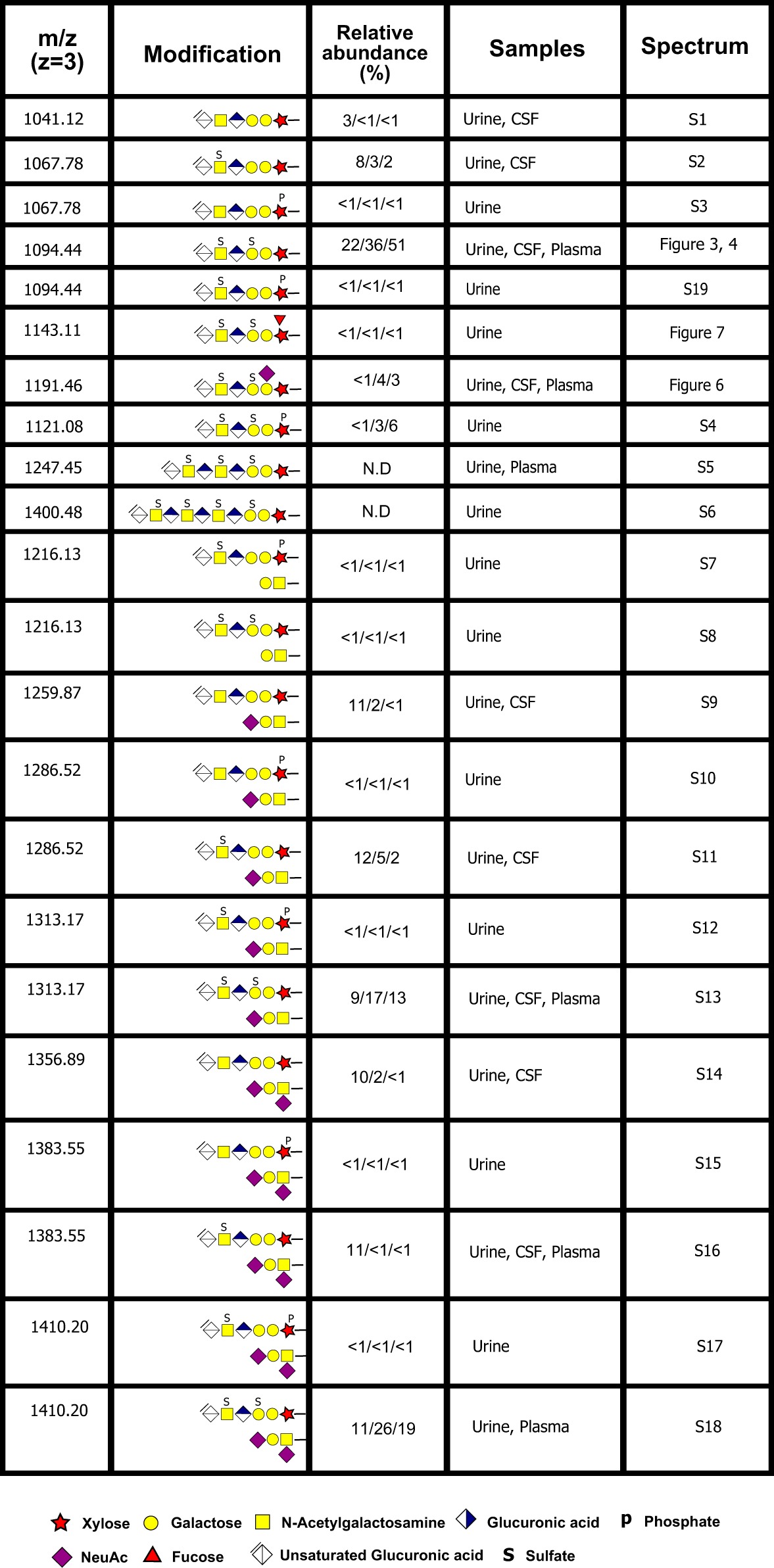
Table I.
Summary of all bikunin CS glycopeptides identified and analyzed in the present study
All glycopeptides are described from their monoisotopic m/z values of MS1 precursors, their glycoforms (symbols specified below the Table), their relative abundances in urine measured as percentage of all glycoforms in each eluted fraction from the SAX columns (corresponding to 0.4 M/ 0.8 M/ 1.6 M NaCl), their presence in urine, cerebrospinal fluid (CSF) and plasma and finally, reference is given to their individual spectra presented in Supplementary Fig. 1.
Novel modifications of the CS linkage region were also identified including bikunin CS-glycopeptides carrying a sialic acid (Table I, Fig. 5A–5C). The HCD spectrum showed the presence of two distinct oxonium ions at m/z 292.10 [Neu5Ac]+ and m/z 274.09 [Neu5Ac-H2O]+, demonstrating the presence of a Neu5Ac unit (Fig. 5A). The saccharide attachment site of the Neu5Ac was assigned to the inner Gal based on the presence of a [Peptide+XylGalNeu5Ac+2H]2+ fragment (m/z 1357.63) and no corresponding fragment lacking the Gal. This finding was also corroborated by close inspection of the corresponding spectrum of the sodiated compound (Fig. 5B) where HCD of the [M+3Na]3+ precursor (m/z 1213.44) showed that Neu5Ac was attached to the innermost Gal (m/z 1368.63 in Fig. 5C). As in the case of saccharide sulfation, Neu5Ac containing fragments were also partially protected from fragmentation because of sodium complexation e.g. for the sialic acid containing glycopeptide fragments at m/z 1187.12, m/z 1368.63 and m/z 1449.14 (Fig. 5C). This novel sialylation of the CS linkage region was found in all body fluids examined.
Fig. 5.
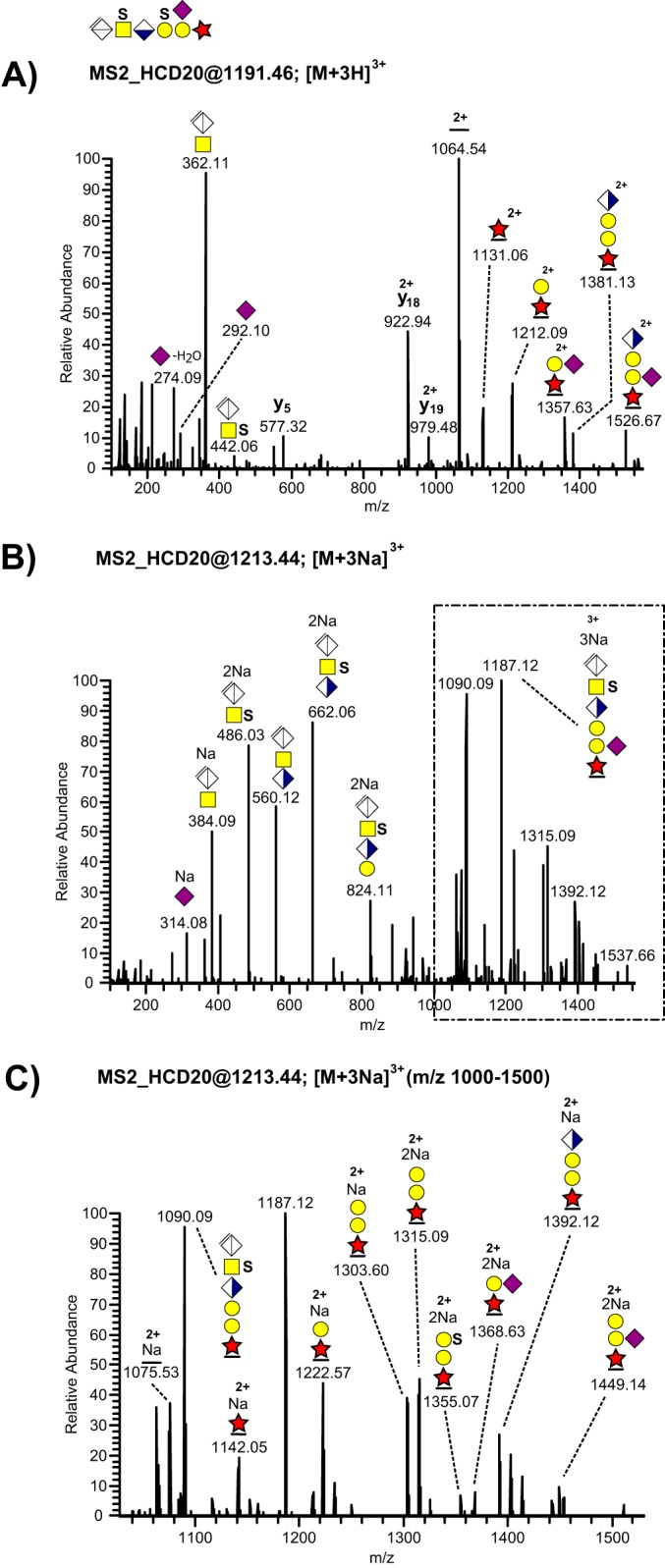
MS/MS fragmentation patterns recorded at the 20% HCD level from a disulfated and sialylated bikunin CS-glycopeptide precursor ion. A, The complete HCD spectrum of the [M+3H]3+ precursor ion at m/z 1191.46. B, The complete HCD spectrum of the [M+3Na]3+ precursor ion at m/z 1213.44 and (C) the expanded m/z 1000–1500 region of the sodiated precursor ion.
Similarly, we also detected fucose modification of the bikunin CS-linkage region (Table I, Fig. 6A-6C). The number and relative intensities of fucose-containing glycosidic fragments were also increased in the sodiated spectra (Fig. 6B–6C) compared with the protonated ones (Fig. 6A). The presence of an ion at m/z 1214.58; 2+ (Fig. 5C) allowed us to place this novel substitution at the innermost xylose residue showing resemblance with the well-characterized xylose phosphorylation. In contrast to sialylation, fucose modifications were only observed in urinary samples.
Fig. 6.
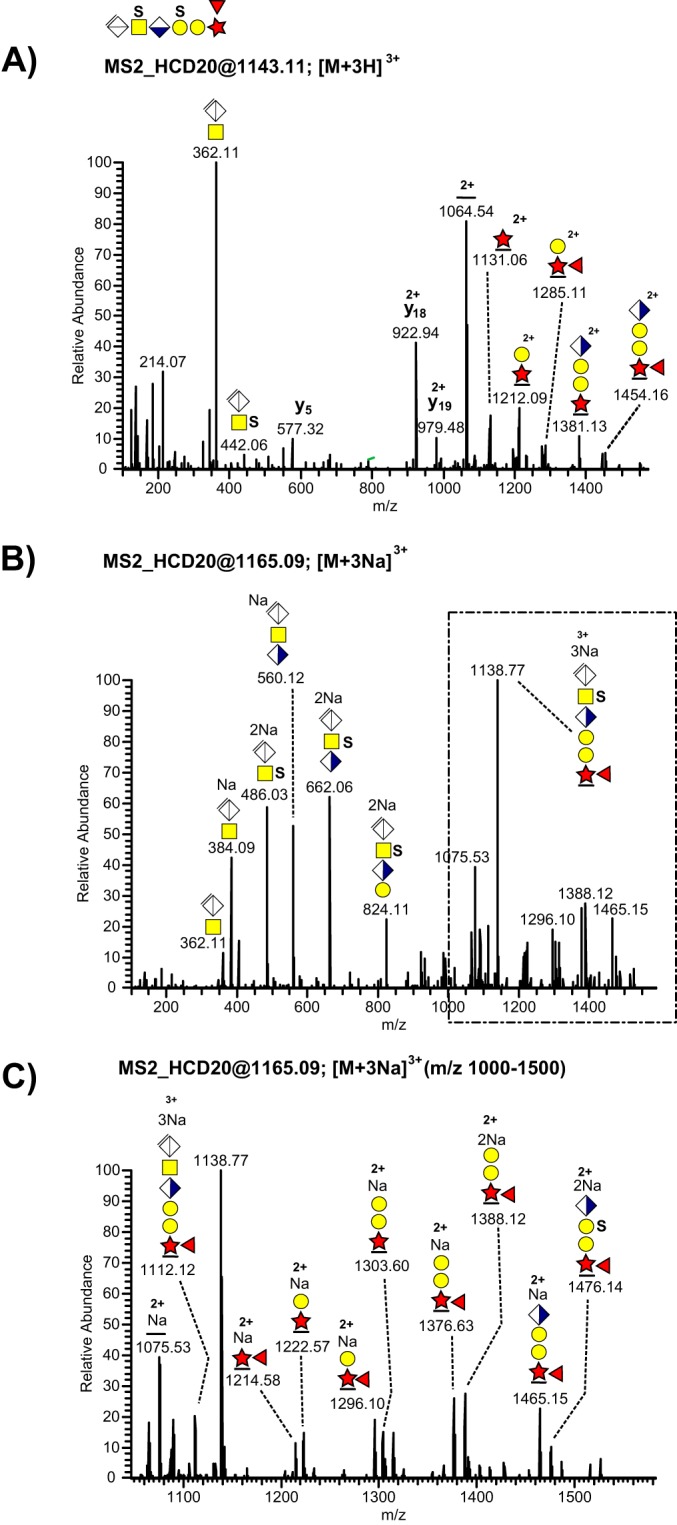
MS/MS fragmentation patterns recorded at the 20% HCD level from a disulfated and fucosylated bikunin CS-glycopeptide precursor ion. A, The complete HCD spectrum of the [M+3H]3+ precursor ion at m/z 1143.11. B, The complete HCD spectrum of the [M+3Na]3+ precursor ion at m/z 1165.09, and C, the expanded m/z 1000–1500 region of the sodiated precursor ion.
Given the high degree of heterogeneity of the CS-linkage region of urinary bikunin we prepared extracted ion chromatograms for all identified glycoforms in these samples and calculated their relative abundance for each of the three SAX fractions (Table I, supplemental Fig. S2A). Glycoforms containing the unsubstituted hexasaccharide linkage region and glycoforms containing the linkage region with only one sulfate or one phosphate group (i.e. 6-mer and S/P glycoforms) were less abundant, eluted mainly in the low salt fractions and contributed to 24%, 3 and 1%, and 33%, 8 and 6% of the three fractions, respectively. Conversely, glycoforms containing the linkage hexasaccharide substituted with two sulfates or one sulfate and one phosphate group (SS or SP forms) were more abundant, eluted mainly in the high salt fractions, and contributed to 44%, 85 and 88% of the three fractions. The glycoform containing two sulfates and one phosphate group was less than 1% in the low salt fraction but contributed to 3 and 6% of the glycoforms of the last two fractions.
The Identification of Extended CS Chains and CS-glycopeptides Additionally Modified with Core 1 O-glycans
In addition to the common modifying hexasaccharide we also found extended linkage region glycopeptides carrying one or two additional GlcAGalNAc units (Table I, supplemental Fig. S1 spectra s5–s6). These longer structures, carrying 8 and 10 monosaccharides, were only observed in the samples treated with chondroitinase ABC for 1 h but were absent in the samples digested overnight. For these glycopeptides, the diagnostic [ΔGlcAGalNAc]+ oxonium ion at m/z 362.11 was always accompanied by its nondehydrated counterpart [GlcAGalNAc]+ at m/z 380.12 arising from an internal GlcAGalNAc fragment. Both the octameric and decameric structures were substituted with three and four sulfate groups, respectively, suggesting that at least 2 or 3 of the innermost GlcAGalNAc disaccharides of the elongated CS structure contained sulfation. Furthermore, we detected several glycopeptides carrying non-, mono- or disialylated core 1 O-linked structures in addition to the CS-hexasaccharide (Table I, supplemental Fig. S1 spectra s7–s18).
As is shown in Fig. 7 all major peaks in the total ion chromatograms of urinary samples could be attributed to different glycoforms of bikunin CS-linkage region glycopeptides. Glycoforms containing the additional mucin type O-glycan were highly abundant making up for 66%, 54 and 39% of the different glycoforms in the three salt fractions (supplemental Fig. S2B) and thus contributed significantly to the chromatographic profile (Fig. 7). There are two candidate Thr residues (Thr-17, Thr-20) close to the CS modified Ser-10. To allow pinpointing of the O-glycosylation sites a urine CS glycopeptide sample (0.8 m fraction) was subjected to beta elimination using methylamine. This demonstrated that both Ser-10 and Thr-17 but not Thr-20 were O-glycosylated (supplemental Fig. S3A and S3B).
Fig. 7.
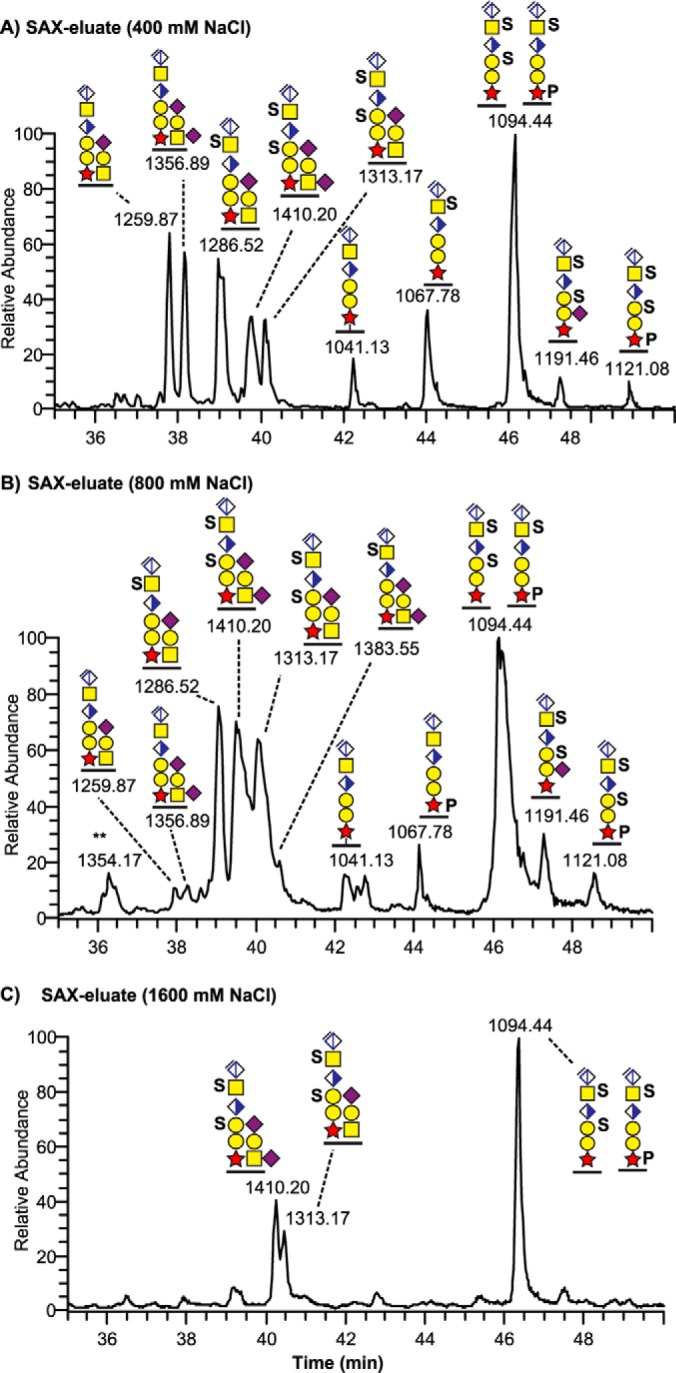
Total ion chromatograms of three fractions of a SAX purified and chondroitinase ABC treated urine sample with all major peaks annotated as different glycoforms of bikunin. The first fraction was eluted with 400 mm NaCl (top), the second fraction with 800 mm NaCl (middle), and the last fraction with 1600 mm NaCl (bottom).
Analysis of Heavy Chain CS Cross-linking Glycopeptides
By tracing the signal of the diagnostic [ΔGlcAGalNAc]+ oxonium ion at m/z 362.11 in plasma samples we observed a series of early eluting glycopeptide peaks which were selected for MS2 fragmentation and which were not related to the amino acid sequence of bikunin (Fig. 8A–8E). These peaks did not contain any fragment ions indicating a GalNAcGalGalXyl linkage region but only modifications corresponding to ΔGlcAGalNAc or ΔGlcAGalNAcGlcAGalNAc units. We reasoned that chondroitinase ABC hydrolysis of the IαI-complex would render not only CS-glycopeptides derived from the bikunin linkage region but possibly also fragments derived from the cross-linking regions of heavy chains present in the complex.
Fig. 8.

MS/MS fragmentation spectra of H1 and H1+H2 Heavy chain peptides cross-linked to CS-chains hydrolyzed by chondroitinase ABC. The H1 C-terminal tryptic and semitryptic peptides LPDRVTGVDTD and VTGVDTD were identified cross-linked to ΔGlcAGalNAc at m/z 774.85 (A), to ΔGlcAGalNAcGlcAGalNAc at m/z 964.41 (B), to ΔGlcAGalNAcGlcAGalNAc at m/z 723.77 (C) and together with the C-terminal peptide VEND of H2 cross-linked to ΔGlcAGalNAcGlcAGalNAc at m/z 952.36 (D) and cross-linked to ΔGlcAGalNAcGlcAGalNAcGlcAGalNAc at m/z 1142.42 (E). Schematic structures of the corresponding precursor ions are shown in the box.
As is illustrated in Fig. 8A, an ion at m/z 774.85; 2+ from such an early eluting peak was unambiguously determined as a tryptic peptide (LPDRVTGVDTD) of the C terminus of heavy chain 1 (H1) carrying one ΔGlcAGalNAc modification. Earlier structural studies of the IαI-complex provided compelling evidence for the existence of an unusual ester bond between internal GalNAc units of the CS-chain and the α-carboxyl of the proteolytically produced C-terminal Asp of the complexed heavy chains (Fig. 1) (24). Both the deglycosylated peptide and the y2 fragment ions were found carrying an extra mass of +42.01 amu, as indicated by fragment ions at m/z 615.30 (2+) and m/z 277.10 (Fig. 8A). This mass shift was tentatively interpreted as an additional acetyl group derived from cross-ring fragmentation of the ester linked GalNAc. This extra mass was not observed for the b- ion series (b8 - b10, Fig. 8A and 8B) pointing to the peptide C terminus as the potential site of modification. A similar fragmentation signature was consistently observed in the HCD spectra of a glycopeptide carrying one additional GlcAGalNAc unit (Fig. 8B) and of glycopeptides derived from the same region, but with alternative proteolytic cleavage (VTGVDTD, Fig. 8C). This +42.01 amu finding gave us further support for the presence of a C-terminal Asp to GalNAc bond in the CS cross-linkage region of H1.
We also found mass spectrometric evidence indicating that H1 (VTGVDTD) and the corresponding tryptic peptide from heavy chain 2 (H2) (VEND) were simultaneously bound to two (Fig. 8D) and three (Fig. 8E) CS-disaccharide units, respectively, showing that these peptides can be situated close and even on adjacent GalNAc units of the CS chain. The deglycosylated peptide fragment ion of H2 (VEND, m/z 518.21, Fig. 8D) was observed carrying an additional mass of +42.01 amu suggesting the same type of ester bond and fragmentation behavior as for H1. The presence of a prominent oxonium ion at m/z 362.11 (Fig. 8B–8C) is typical for an enzymatically hydrolyzed extended CS-chain but its total absence in Fig. 8A and 8D indicates that the GalNAc unit of the ΔGlcAGalNAc disaccharide and the two GalNAc units of the ΔGlcAGalNAcGlcAGalNAc tetrasaccharide are all cross-linked to the H1 and H1 + H2 peptides, respectively (Fig. 8D). Analogously, the outermost GalNAc of the ΔGlcAGalNAcGlcAGalNAcGlcAGalNAc hexasaccharide was likely linked to H1 because no m/z 362.11 was observed upon fragmentation of the ion at m/z 1142.42 (2+) (Fig. 8E). The cross-linked VTGVDTD + VEND CS-glycopeptides were also identified in CSF but were scarce in urine, confirming the current notion that UTI is essentially deployed of heavy chains. Finally, we also detected similar H1 cross-linking glycopeptides displaying abundant oxonium ions at m/z 380.12 but not at m/z 362.11, indicating terminal GlcAGalNAc structure at the very end of the CS glycan chain (supplemental Fig. S4A–S4D).
DISCUSSION
A complete determination of the structural details of CS chains is a laborious task given their polydisperse nature with respect to sulfation patterns, length of the chain and variations in terms of core proteins and occupancy of specific attachment sites. Owing to their different chemical nature, the CS-chain(s) and corresponding core proteins are usually separated from each other during structural analysis. As a complementary approach, glycoproteomics has become an alternative analytical platform where glycopeptides, obtained from the protease digestion of glycoproteins, can be characterized in a bottom-up fashion (1, 43, 44). The main advantage of this approach is the prospect of simultaneously deducing the glycan structure and its specific protein attachment site.
Recently, we reported a glycoproteomics protocol for enrichment and analysis of proteoglycan linkage region glycopeptides from complex samples (20). However, proper analysis of the glycan microheterogenity remained unaddressed because of the intrinsic fragility of linkage regions substituents upon HCD fragmentation. In this study we decided to focus on the IαI complexes because these proteins are highly abundant in human body fluids. We analyzed human plasma, urine and CSF samples and optimized the analytical protocol by taking advantage of the complementary information obtained from alternating HCD-fragmentation energies as well as the analysis of sodiated molecular ions in positive mode. We also targeted the bioinformatics searches to include glycopeptides not only derived from the bikunin linkage region but also from the cross-linkage regions of the heavy chains. Switching HCD fragmentation energies proved to be a useful way to obtain complementary structural information. Dissociation of glycopeptides at low collisional energy (NCE 20%) tended to yield a more pronounced glycosidic fragmentation which facilitated sugar composition analysis at the expense of poorer peptide characterization. Increased energy resulted in the opposite effect, providing a higher coverage of the peptide sequence in terms of b- and y- peptide ions but removing most of the glycans. However, the site-specific assignment of substituents, especially sulfates, was still difficult to fulfill given their intrinsic fragility upon fragmentation.
Traditionally, the MS analysis of sulfated glycans has been conducted using negative mode ionization or the addition of ion-pairing reagents to avoid sulfate losses. Further, it has been demonstrated that sulfopeptides complexed to sodium ions were protected from fragmentation using positive mode electron capture/transfer dissociation (ECD/ETD)(45). Here, we explored if sodium ion complexation could be a feasible way of enhancing the stability of sulfate glycan substituents in positive mode MS/MS. The results presented show that the effect of sodium ions upon fragmentation of linkage region glycopeptide was two-folded. First a general increase in the number and relative intensities of oxonium ions and glycopeptide fragments was observed, suggesting that sodium ion complexation promotes better sugar ionization and a more gentle glycosidic fragmentation than protonation. Second, a higher retention of sulfate substituents allowed for pinpointing of substitution sites on the CS linkage oligosaccharide. In order to distinguish between sulfation and phosphorylation of the CS hexasaccharide we used the high mass resolving power of the Q-Exactive Orbitrap at the MS2 level and determined that sulfates, as opposed to phosphates, were attached to the GalNAc and to the outer Gal of the CS hexasaccharide. We have previously showed that the Xyl residue could become phosphorylated, and in the present study we observed a higher stability of phosphate bound to Xyl versus sulfate bound to Gal under nonsodium HCD conditions, which may be used in future studies to distinguish between the two substitutions. In line with this observation, analyses in positive mode MS/MS showed that synthetic sulfopeptides were more prone to desulfation as opposed to the corresponding phosphopeptides (46).
In contrast to the expected uniformity of the proteoglycan linkage region of bikunin, we documented a large structural heterogeneity in our samples. In addition to sulfation, we observed the presence of fucose and phosphate on the CS linkage region of UTI but not in CSF or plasma samples. Because xylose phosphorylation is thought to act as a transitional switch that regulates CS-biosynthesis (19, 47), such information may be of value for understanding the structural differences between body fluids. As our analysis was restricted to linkage region glycopeptides we could not assess whether full-length CS- chains carrying phosphate or fucose modifications actually differed in any respect from those only substituted with sulfates. However, most of the nonsulfated glycoforms eluted from the SAX-column at low salt concentrations, suggesting that these chains may be of shorter length and/or carrying fewer sulfate groups. Interestingly, a relationship between modifications at the linkage region as well as the number and character of full-length CS-chains has previously been suggested (47). The results presented in this study give further support to that notion. An additional novel sialic acid substitution was also determined in all three body fluids. Similar to fucosylated glycoforms, sialic acid substitutions were present together with sulfation adding further negative charge and structural complexity to the combinatorial possibilities of CS-linkage regions.
In CSF and plasma samples we were also able to identify abundant glycopeptides derived from the cross-linkage region of H1 and H2. The presence of these ions was, on the other hand, rather scarce in urinary samples suggesting that UTI is mostly deployed of heavy chains. It is well known that enzymatic degradation of the IαI complex rapidly leads to the release of bikunin from the heavy chains but separation of the heavy chains from each other is more difficult to achieve (27). Based on this finding it has been suggested that the heavy chains may be located close to each other. Here we present mass spectrometric evidence for C-terminal peptides from H1 and H2 being simultaneously attached to the same CS-stretch. The peptides were indeed spatially close to each other and in some cases even at adjacent GalNAc residues. The C-terminal domains of H1 and H2 are known to be intrinsically disordered explaining the close arrangement of the heavy chains onto the CS-chain of bikunin without leading to steric hindrances or clashes (48).
According to a previous publication, the number of theoretical structures of bikunin is 43 million (49) because of the combination of sulfate and chain-length heterogeneities. In contrast to that and based on an elegant top-down MS approach, the same authors showed that the CS-chain of bikunin has a defined sequence and actually displays a discrete number of full-length structures. Interestingly, only sulfation of the second Gal was identified as a variation of the linkage region. In the present study, galactose sulfation was also found to be the most abundant substitution of the bikunin CS linkage tetrasaccharide but several additional structural variants including the phosphate, sialic acid and fucose containing glycoforms (Table I) were also detected and sequenced.
Although the pathophysiological relevance of bikunin and the IαI-complex has been experimentally demonstrated in numerous studies, the structure-function relationships of these processes still remain largely unexplored. It is reasonable to assume that these molecules display substantial structural diversity to accommodate divergent physiological functions. Additionally, the biosynthesis of the CS- chain must be relatively dynamic so the required structure will be synthesized at any given condition. As modifications of the CS linkage region are known to modulate GAG biosynthesis we propose that the methods employed in this study may help to explore such relationships in relevant systems.
In conclusion, in this work we developed a method for fine-structural analysis of CS-hexasaccharide linkage regions focusing on the IαI-complex derived from human body fluids. Our results suggest that the structural heterogeneity of bikunin, and the related IαI complexes, is much greater than previously realized. We also provide evidence for novel combinatorial possibilities at the linkage regions of human CS-proteoglycans. However, if these novel modifications are specific to the CS chain of bikunin or reflect a more general phenomenon of CSPGs needs further investigation.
Acknowledgments
The Proteomics Core Facility, Sahlgrenska Academy is acknowledged for running all the MS-analyses.
Footnotes
Author contributions: A.G., J.N., F.N., C.S., and G.L. designed research; A.G., J.N., F.N., C.S., and G.L. performed research; A.G., J.N., F.N., C.S., and G.L. contributed new reagents or analytic tools; A.G., J.N., F.N., and G.L. analyzed data; A.G., J.N., F.N., and G.L. wrote the paper.
* The work was supported by grants from the Swedish Research Council (8266), by governmental grants to the Sahlgrenska University Hospital and by donations from the IngaBritt and Arne Lundberg Foundation.
 This article contains supplemental Figs. S1 to S4 and Table S1.
This article contains supplemental Figs. S1 to S4 and Table S1.
1 The abbreviations used are:
- List of Abbreviations: GAG
- glycosaminoglycan
- CS
- chondroitin sulfate
- CSF
- cerebrospinal fluid
- CSPG
- chondroitin sulfate proteoglycan
- Fuc
- fucose
- ΔGlcA
- unsaturated glucuronic acid
- Gal
- galactose
- GlcA
- glucuronic acid
- GalNAc
- N-acetylgalactosamine
- GlcNAc
- N-acetylglucosamine
- HCD
- higher-energy collisional dissociation
- H1
- inter-alpha-trypsin inhibitor heavy chain 1 (UniProtKB P19827)
- H2
- inter-alpha-trypsin inhibitor heavy chain 2 (UniProtKB P19823)
- HexNAc
- N-acetylhexosamine
- HSPG
- heparan sulfate proteoglycan
- IαI
- inter-alpha-trypsin inhibitor
- NCE
- normalized collision energy
- Neu5Ac
- N-acetylneuraminic acid
- PG
- proteoglycan
- ppm
- parts per million
- SAX
- strong anion exchange
- UTI
- urinary trypsin inhibitor, bikunin, inter-alpha-trypsin inhibitor light chain (UniProtKB P02760)
- Xyl
- xylose.
REFERENCES
- 1.Thaysen-Andersen M., and Packer N. H. (2014) Advances in LC-MS/MS-based glycoproteomics: Getting closer to system-wide site-specific mapping of the N- and O-glycoproteome. Biochim Biophys Acta-Proteins Proteom 1844, 1437–1452 [DOI] [PubMed] [Google Scholar]
- 2.Esko J. D., Kimata K., and Lindahl U. (2009) Proteoglycans and Sulfated Glycosaminoglycans. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E. eds.), 2nd Ed., Cold Spring Harbor (NY) pp 229–248 [PubMed] [Google Scholar]
- 3.Pickford C. E., Holley R. J., Rushton G., Stavridis M. P., Ward C. M., and Merry C. L. (2011) Specific glycosaminoglycans modulate neural specification of mouse embryonic stem cells. Stem Cells 29, 629–640 [DOI] [PubMed] [Google Scholar]
- 4.Izumikawa T., Sato B., and Kitagawa H. (2014) Chondroitin sulfate is indispensable for pluripotency and differentiation of mouse embryonic stem cells. Sci. Reports 4, 3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan C. K., Rolle M. W., Potter-Perigo S., Braun K. R., Van Biber B. P., Laflamme M. A., Murry C. E., and Wight T. N. (2010) Differentiation of cardiomyocytes from human embryonic stem cells is accompanied by changes in the extracellular matrix production of versican and hyaluronan. J. Cell. Biochem. 111, 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang H. Y., Olson S. K., Esko J. D., and Horvitz H. R. (2003) Caenorhabditis elegans early embryogenesis and vulval morphogenesis require chondroitin biosynthesis. Nature 423, 439–443 [DOI] [PubMed] [Google Scholar]
- 7.Mizuguchi S., Uyama T., Kitagawa H., Nomura K. H., Dejima K., Gengyo-Ando K., Mitani S., Sugahara K., and Nomura K. (2003) Chondroitin proteoglycans are involved in cell division of Caenorhabditis elegans. Nature 423, 443–448 [DOI] [PubMed] [Google Scholar]
- 8.Olson S. K., Bishop J. R., Yates J. R., Oegema K., and Esko J. D. (2006) Identification of novel chondroitin proteoglycans in Caenorhabditis elegans: embryonic cell division depends on CPG-1 and CPG-2. J. Cell Biol. 173, 985–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izumikawa T., Kanagawa N., Watamoto Y., Okada M., Saeki M., Sakano M., Sugahara K., Sugihara K., Asano M., and Kitagawa H. (2010) Impairment of embryonic cell division and glycosaminoglycan biosynthesis in glucuronyltransferase-I-deficient mice. J. Biol. Chem. 285, 12190–12196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawashima H., Atarashi K., Hirose M., Hirose J., Yamada S., Sugahara K., and Miyasaka M. (2002) Oversulfated chondroitin/dermatan sulfates containing GlcAbeta1/IdoAalpha1–3GalNAc(4,6-O-disulfate) interact with L- and P-selectin and chemokines. J. Biol. Chem. 277, 12921–12930 [DOI] [PubMed] [Google Scholar]
- 11.Zhou Z. H., Karnaukhova E., Rajabi M., Reeder K., Chen T., Dhawan S., and Kozlowski S. (2014) Oversulfated chondroitin sulfate binds to chemokines and inhibits stromal cell-derived factor-1 mediated signaling in activated T cells. PloS One 9, e94402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaia J. (2013) Glycosaminoglycan Glycomics Using Mass Spectrometry. Mol. Cell. Proteomics 12, 885–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gill V. L., Aich U., Rao S., Pohl C., and Zaia J. (2013) Disaccharide Analysis of Glycosaminoglycans Using Hydrophilic Interaction Chromatography and Mass Spectrometry. Anal. Chem. 85, 1138–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staples G. O., Bowman M. J., Costello C. E., Hitchcock A. M., Lau J. M., Leymarie N., Miller C., Naimy H., Shi X. F., and Zaia J. (2009) A chip-based amide-HILIC LC/MS platform for glycosaminoglycan glycomics profiling. Proteomics 9, 686–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oguma T., Tomatsu S., Montano A. M., and Okazaki O. (2007) Analytical method for the determination of disaccharides derived from keratan, heparan, and dermatan sulfates in human serum and plasma by high-performance liquid chromatography/turbo ionspray ionization tandem mass spectrometry. Anal. Biochem. 368, 79–86 [DOI] [PubMed] [Google Scholar]
- 16.Beaudet J. M., Mansur L., Joo E. J., Kamhi E., Yang B., Clausen T. M., Salanti A., Zhang F., and Linhardt R. J. (2014) Characterization of human placental glycosaminoglycans and regional binding to VAR2CSA in malaria infected erythrocytes. Glycoconjugate J. 31, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gulberti S., Lattard V., Fondeur M., Jacquinet J. C., Mulliert G., Netter P., Magdalou J., Ouzzine M., and Fournel-Gigleux S. (2005) Phosphorylation and sulfation of oligosaccharide substrates critically influence the activity of human beta1,4-galactosyltransferase 7 (GalT-I) and beta1,3-glucuronosyltransferase I (GlcAT-I) involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 280, 1417–1425 [DOI] [PubMed] [Google Scholar]
- 18.Tone Y., Pedersen L. C., Yamamoto T., Izumikawa T., Kitagawa H., Nishihara J., Tamura J., Negishi M., and Sugahara K. (2008) 2-o-phosphorylation of xylose and 6-o-sulfation of galactose in the protein linkage region of glycosaminoglycans influence the glucuronyltransferase-I activity involved in the linkage region synthesis. J. Biol. Chem. 283, 16801–16807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen J., Xiao J., Rahdar M., Choudhury B. P., Cui J., Taylor G. S., Esko J. D., and Dixon J. E. (2014) Xylose phosphorylation functions as a molecular switch to regulate proteoglycan biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 111, 15723–15728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noborn F., Gomez Toledo A., Sihlbom C., Lengqvist J., Fries E., Kjellen L., Nilsson J., and Larson G. (2015) Identification of chondroitin sulfate linkage region glycopeptides reveals prohormones as a novel class of proteoglycans. Mol. Cell. Proteomics 14, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugahara K., Yamashina I., De Waard P., Van Halbeek H., and Vliegenthart J. F. (1988) Structural studies on sulfated glycopeptides from the carbohydrate-protein linkage region of chondroitin 4-sulfate proteoglycans of swarm rat chondrosarcoma. Demonstration of the structure Gal(4-O-sulfate)beta 1–3Gal beta 1–4XYL beta 1-O-Ser. J. Biol. Chem. 263, 10168–10174 [PubMed] [Google Scholar]
- 22.Yamada S., Oyama M., Kinugasa H., Nakagawa T., Kawasaki T., Nagasawa S., Khoo K. H., Morris H. R., Dell A., and Sugahara K. (1995) The sulphated carbohydrate-protein linkage region isolated from chondroitin 4-sulphate chains of inter-alpha-trypsin inhibitor in human plasma. Glycobiology 5, 335–341 [DOI] [PubMed] [Google Scholar]
- 23.Yamada S., Oyama M., Yuki Y., Kato K., and Sugahara K. (1995) The uniform galactose 4-sulfate structure in the carbohydrate-protein linkage region of human urinary trypsin inhibitor. Eur. J. Biochem. / FEBS 233, 687–693 [DOI] [PubMed] [Google Scholar]
- 24.Enghild J. J., Salvesen G., Thogersen I. B., Valnickova Z., Pizzo S. V., and Hefta S. A. (1993) Presence of the protein-glycosaminoglycan-protein covalent cross-link in the inter-alpha-inhibitor-related proteinase inhibitor heavy chain 2/bikunin. J. Biol. Chem. 268, 8711–8716 [PubMed] [Google Scholar]
- 25.Jessen T. E., Faarvang K. L., and Ploug M. (1988) Carbohydrate as Covalent Crosslink in Human Inter-Alpha-Trypsin Inhibitor - a Novel Plasma-Protein Structure. FEBS Lett. 230, 195–200 [DOI] [PubMed] [Google Scholar]
- 26.Morelle W., Capon C., Balduyck M., Sautiere P., Kouach M., Michalski C., Fournet B., and Mizon J. (1994) Chondroitin sulphate covalently cross-links the three polypeptide chains of inter-alpha-trypsin inhibitor. Eur. J. Biochem. / FEBS 221, 881–888 [DOI] [PubMed] [Google Scholar]
- 27.Enghild J. J., Thogersen I. B., Cheng F., Fransson L. A., Roepstorff P., and Rahbek-Nielsen H. (1999) Organization of the inter-alpha-inhibitor heavy chains on the chondroitin sulfate originating from Ser(10) of bikunin: posttranslational modification of IalphaI-derived bikunin. Biochemistry 38, 11804–11813 [DOI] [PubMed] [Google Scholar]
- 28.Enghild J. J., Thogersen I. B., Pizzo S. V., and Salvesen G. (1989) Analysis of inter-alpha-trypsin inhibitor and a novel trypsin inhibitor, pre-alpha-trypsin inhibitor, from human plasma. Polypeptide chain stoichiometry and assembly by glycan. J. Biol. Chem. 264, 15975–15981 [PubMed] [Google Scholar]
- 29.Salier J. P., Rouet P., Raguenez G., and Daveau M. (1996) The inter-alpha-inhibitor family: from structure to regulation. Biochem. J. 315 (Pt 1), 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balduyck M., Albani D., Jourdain M., Mizon C., Tournoys A., Drobecq H., Fourrier F., and Mizon J. (2000) Inflammation-induced systemic proteolysis of inter-alpha-inhibitor in plasma from patients with sepsis. J. Lab. Clin. Med. 135, 188–198 [DOI] [PubMed] [Google Scholar]
- 31.Bayraktar E., Igci M., Erturhan S., Igci Y. Z., Karakok M., Gogebakan B., Ulasli M., Cakmak E. A., and Arslan A. (2014) Reduced gene expression of bikunin as a prognostic marker for renal cell carcinoma. Exp. Oncol. 36, 107–111 [PubMed] [Google Scholar]
- 32.Daveau M., Rouet P., Scotte M., Faye L., Hiron M., Lebreton J. P., and Salier J. P. (1993) Human inter-alpha-inhibitor family in inflammation: simultaneous synthesis of positive and negative acute-phase proteins. Biochem. J. 292, 485–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lepedda A., Fancellu L., Zinellu E., De Muro P., Nieddu G., Deiana G. A., Canu P., Concolino D., Sestito S., Formato M., and Sechi G. (2013) Urine bikunin as a marker of renal impairment in Fabry's disease. Biomed. Res. Int. Art. ID. 205948, 9 pages [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lepedda A. J., Nieddu G., Rocchiccioli S., Fresu P., De Muro P., and Formato M. (2013) Development of a method for urine bikunin/urinary trypsin inhibitor (UTI) quantitation and structural characterization: Application to type 1 and type 2 diabetes. Electrophoresis 34, 3227–3233 [DOI] [PubMed] [Google Scholar]
- 35.Seifert G., Seifert M., and Wittel U. A. (2013) Searching for the common denominator of systemic inflammation and tumor invasion in the cases of bikunin and the inter-alpha-trypsin inhibitor heavy chains. J. Surg. Res. 183, 983. [DOI] [PubMed] [Google Scholar]
- 36.Tsui K. H., Tang P., Lin C. Y., Chang P. L., Chang C. H., and Yung B. Y. M. (2010) Bikunin Loss in Urine as Useful Marker for Bladder Carcinoma. J. Urol. 183, 339–344 [DOI] [PubMed] [Google Scholar]
- 37.Capon C., Mizon C., Lemoine J., Rodie-Talbere P., and Mizon J. (2003) In acute inflammation, the chondroitin-4 sulphate carried by bikunin is not only longer; it is also undersulphated. Biochimie 85, 101–107 [DOI] [PubMed] [Google Scholar]
- 38.Mizon C., Mairie C., Balduyck M., Hachulla E., and Mizon J. (2001) The chondroitin sulfate chain of bikunin-containing proteins in the inter-alpha-inhibitor family increases in size in inflammatory diseases. Eur. J. Biochem. 268, 2717–2724 [DOI] [PubMed] [Google Scholar]
- 39.Mirgorodskaya E., Hassan H., Clausen H., and Roepstorff P. (2001) Mass spectrometric determination of O-glycosylation sites using beta-elimination and partial acid hydrolysis. Anal. Chem. 73, 1263–1269 [DOI] [PubMed] [Google Scholar]
- 40.Domon B., and Costello C. E. (1988) A Systematic Nomenclature for Carbohydrate Fragmentations in Fab-Ms Ms Spectra of Glycoconjugates. Glycoconjugate J. 5, 397–409 [Google Scholar]
- 41.Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., and Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nilsson J., Halim A., Grahn A., and Larson G. (2013) Targeting the glycoproteome. Glycoconjugate J. 30, 119–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan S., Chen R., Aebersold R., and Brentnall T. A. (2011) Mass Spectrometry Based Glycoproteomics-From a Proteomics Perspective. Mol. Cell. Proteomics 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medzihradszky K. F., Guan S., Maltby D. A., and Burlingame A. L. (2007) Sulfopeptide fragmentation in electron-capture and electron-transfer dissociation. J. Am. Soc. Mass Spectrom. 18, 1617–1624 [DOI] [PubMed] [Google Scholar]
- 46.Medzihradszky K. F., Darula Z., Perlson E., Fainzilber M., Chalkley R. J., Ball H., Greenbaum D., Bogyo M., Tyson D. R., Bradshaw R. A., and Burlingame A. L. (2004) O-sulfonation of serine and threonine: mass spectrometric detection and characterization of a new posttranslational modification in diverse proteins throughout the eukaryotes. Mol. Cell. Proteomics 3, 429–440 [DOI] [PubMed] [Google Scholar]
- 47.Koike T., Izumikawa T., Tamura J., and Kitagawa H. (2009) FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 421, 157–162 [DOI] [PubMed] [Google Scholar]
- 48.Blom A. M., Morgelin M., Oyen M., Jarvet J., and Fries E. (1999) Structural characterization of inter-alpha-inhibitor. Evidence for an extended shape. J. Biol. Chem. 274, 298–304 [DOI] [PubMed] [Google Scholar]
- 49.Ly M., Leach F. E., Laremore T. N., Toida T., Amster I. J., and Linhardt R. J. (2011) The proteoglycan bikunin has a defined sequence. Nat. Chem. Biol. 7, 827–833 [DOI] [PMC free article] [PubMed] [Google Scholar]