

Abstract
Deubiquitinating enzymes (DUBs), cysteine or metallo- proteases that cleave ubiquitin chains or protein conjugates, are present in nearly every cellular compartment, with overlapping protein domain structure, localization, and functions. We discovered a cohort of DUBs that are involved in membrane trafficking (ubp4, ubp5, ubp9, ubp15, and sst2) and found that loss of all five of these DUBs but not loss of any combination of four, significantly impacted cell viability in the fission yeast Schizosaccharomyces pombe (1). Here, we delineate the collective and individual functions and activities of these five conserved DUBs using comparative proteomics, biochemistry, and microscopy. We find these five DUBs are degenerate rather than redundant at the levels of cell morphology, substrate selectivity, ubiquitin chain specificity, and cell viability under stress. These studies reveal the complexity of interplay among these enzymes, providing a foundation for understanding DUB biology and providing another example of how cells utilize degeneracy to improve survival.
Eukaryotic cells integrate signaling pathways to modulate their response to environmental changes, predominately through dynamic protein posttranslational modifications like ubiquitination (Ub'n) (2, 3). Cycles of Ub'n modulate protein stability, localization, and/or binding partners while maintaining cellular ubiquitin (Ub) homeostasis (3). Ub'n of substrate proteins is catalyzed by a linear sequence of enzymes (E1, E2, E3) and reversed by deubiquitinases (DUBs1). Ub chains can be formed through any of Ub's seven lysines (K6, K11, K27, K29, K33, K48, K63) or its N terminus (M1), generating a wide variety of Ub chain architectures that mediate specific cellular signals (4, 5). DUBs have been implicated in multiple essential cellular roles, including chromatin remodeling, DNA damage repair, kinase activation, endocytosis, ribosomal maturation, and immune responses (2, 3).
Surprisingly, while multiple Ub'n enzymes (E1, E2, and E3) are essential in yeast (6–8), only a single DUB is essential for viability of both budding and fission yeasts (6–11), suggesting that considerable functional overlap may exist in yeast under standard laboratory conditions. In contrast, in metazoans, knockdown or loss of individual DUBs often results in developmental defects or disease states (3, 12, 13). Consistent with this possibility, we previously found that loss of five DUBs (5DUB delete: ubp4Δ1 ubp5Δ ubp9Δ ubp15Δ sst2Δ) but not any combination of four intracellular membrane trafficking DUBs significantly impacted cell polarity, Ub conjugate accumulation, and viability in S. pombe (1). To begin to make sense of this functional overlap, here we dissected the shared and specific functions of these five DUBs on multiple levels, defining their contributions to cell polarity, Ub chain specificities, shared and specific putative substrates, and individual and combined effects of DUB loss on cell survival under stress. We find that this cohort of five DUBs is degenerate (different elements that have overlapping but not fully redundant roles), forming a robust functional module for maintenance of cell polarity and viability.
EXPERIMENTAL PROCEDURES
Phylogenetic Tree Generation
S. pombe and human DUB protein sequences (FASTA files) were downloaded from the UniprotKB database (September 2014) and analyzed in MEGA6 (14). The final image was colored in Adobe Illustrator (CS6).
Vector Construction
The Ub expression vector used for all large-scale purifications was constructed as follows. Ub (processed coding sequence) was amplified from a Xenopus Ub vector (15) by PCR using primers containing NdeI(5′)/XmaI(3′) and cloned into the pREP1 (16) vector using these sites. The His-biotin-His (HBH) tag (17) was amplified from the pFa6-HBH-kanR (KLG p3589) vector using primers containing NdeI restriction sites on both the 5′ and 3′ ends and cloned into the pREP1 vector containing Ub, yielding the pREP1-HBH-Ub construct (KLG p4954).
Strains and Yeast Methods
Strain construction and tetrad analysis were accomplished through standard methods. WT, DUB deletion strains, and endogenously tagged strains (Supplemental Table S1) were grown in rich YE media or Edinburgh minimal media (EMM) with appropriate supplements. For overexpression of Flag-Ub (KLG p3729) or HBH-Ub, strains were transformed with pREP1 expression vectors (containing a thiamine repressible promoter) using a standard sorbitol transformation procedure (18). Transformed strains were first grown in EMM containing thiamine to suppress expression and then in EMM lacking thiamine for 20–22 h (19). Cell pellets were frozen in a dry ice/ethanol bath. Pellets for large-scale experiments (for LC-MS/MS) were harvested from 8 liters of EMM (∼6,500 optical density pellets). Pellets for midscale experiments (for Western blots) were harvested from 1 liter of EMM (∼800 OD). To assay the response of various DUB deletion strains to stress (Fig. 5 and Supplemental Figs. S5 and S6), serial 10-fold dilutions of each strain were spotted onto EMM agar plates or YE agar plates in the absence or presence of the following drugs (from Sigma, except as noted): 25 μm Brefeldin A (Molecular Probes); 1 μg/ml bleomycin (Bleo); 0.5 mg/ml calcofluor; 10 μg/ml cycloheximide; 5 mm ethylene glycol tetraacetic acid; 5 mm hydroxyurea; 1 m KCl; 0.25 μm Latrunculin A (Cayman Chemical, Ann Arbor, MI); 10 μg/ml methyl benzimidazol-2-yl-carbamate; 0.01% methyl methanesulfonate; 0.005% sodium dodecyl sulfate (SDS, Fisher Scientific); 1.2 m sorbitol; 12.5 μg/ml thiabendazole; 100 μm CdCl2 (Fluka); 10 μg/ml canavanine, 1 mm H2O2. Plates were incubated at 32 °C for 2–6 days prior to colony imaging.
Fig. 5.
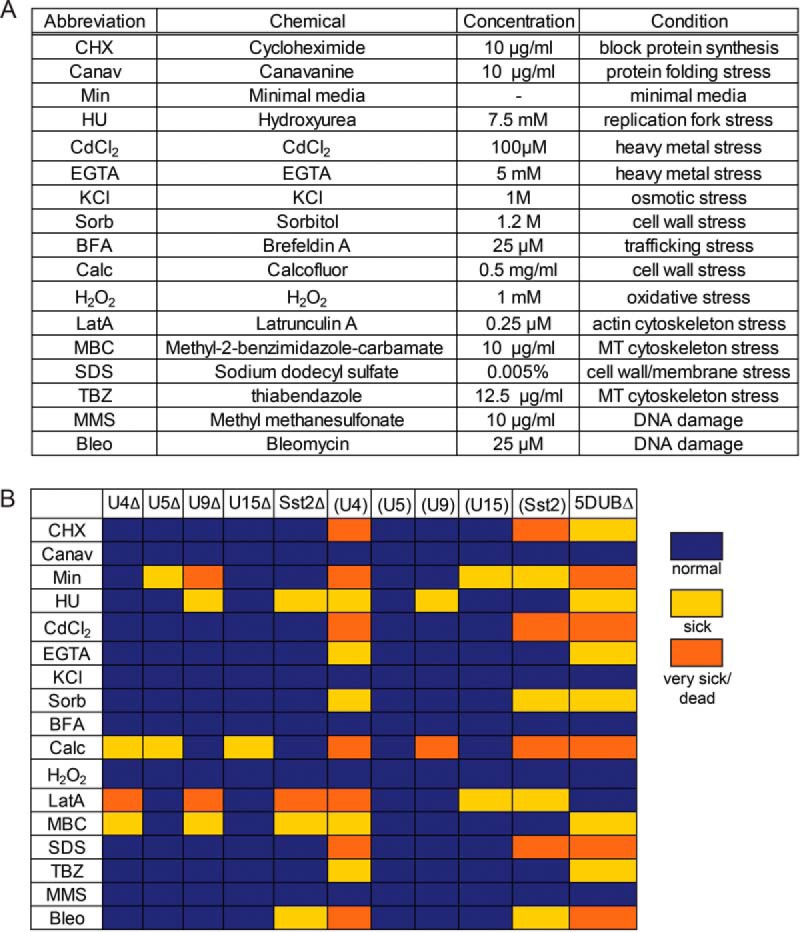
Sensitivities of S. pombe DUB deletion strains to various external stressors. (A) Table of stressors used. (B) Summary of sensitivities of DUB delete strains to stressors listed in part (A). Blue indicates normal growth under these conditions. Light orange indicates strain is sick under these conditions. Dark orange indicates strain is very sick or dead under these conditions. Strains are labeled as in Fig. 1.
HBH-Ub Purifications
Purifications were performed as previously described (17, 20) using equal amounts of cell pellets and affinity resin. For Western blots, 2X SDS gel loading buffer was added and samples were boiled prior to gel loading. For MS analysis, proteins were digested off streptavidin beads as described below.
LC-MS/MS Analysis
Streptavidin beads bound to HBH-ubiquitin were washed three times with Tris-urea buffer (100 mm Tris, pH 8.5, 8 m urea). Proteins were reduced with 3 mm Tris(2-carboxyethyl)phosphine hydrochloride, alkylated with 10 mm chloroacetamide, and digested with trypsin (0.4 μg of Trypsin Gold, Promega). The digest supernatant and washes were combined, concentrated, and desalted (Zebra spin column, Pierce). 2D-LC-MS/MS was performed as follows: Peptides were loaded onto 26-cm columns with a bomb pressure cell and then separated and analyzed by three-phase multidimensional protein identification technology on an linear trap quadrupole (LTQ) or Velos LTQ (Thermo Scientific, West Palm Beach, FL) coupled to a nanoHPLC (NanoAcquity; Waters Corporation). The NanoAcquity autosampler was used to inject 2 μl of varying concentrations of ammonium acetate (0, 10, 25, 50, 100, 200, 300, 400, 600, 800, 1,000, 5,000 mm) for 11 salt elution steps. Each injection was followed by elution of peptides with a 0–40% acetonitrile gradient (60 min) except the first and last injections, in which a 0–90% acetonitrile gradient was used. One full precursor MS scan (400–2,000 mass-to-charge ratio) and five tandem MS scans of the most abundant ions detected in the precursor MS scan under dynamic exclusion were performed. Ions with a neutral loss of 98 Da (singly charged), 49 Da (doubly charged), or 32.7 Da (triply charged) from the parent ions during MS2 were subjected to MS3 fragmentation.
RAW and processed LC-MS/MS files have been deposited in the PRIDE partner repository (ProteomeXchange Consortium) with the dataset identifier PXD001767. RAW files containing more than 20 peaks were converted to DTA files using Scansifter software (21) (v2.1.25). Each DTA file was searched using the SEQUEST algorithm (TurboSequest v.27 rev12) against the S. pombe protein database (created in May 2011 from pombase.org). Common contaminants were added and all sequences were reversed to estimate the false discovery rate, yielding 10,352 total entries. Variable modifications (C+57, M+16, K+42, K+114, [STY]+80), strict tryptic cleavage, <10 missed cleavages, fragment mass tolerance: 0.00 Da (this results in 0.5 Da tolerance in SEQUEST), and parent mass tolerance: 2.5 Da were allowed. Peptide identifications were assembled and filtered in Scaffold (v3.6.4, Proteome Software, Portland, OR) using the following criteria: minimum of 99% protein identification probability; minimum of three unique peptides; minimum of 95% peptide identification probability; minimum peptide length of five amino acids; minimum number of one tryptic terminus. These filtering criteria were used to achieve false discovery rates less than 1% (final false discovery rates were 0.7% and 0.1% at the protein and peptide levels, respectively). Control purifications (no HBH tag and unrelated HBH purifications) were performed; proteins identified with at least three unique peptides in all three purifications are listed in Supplemental Table S2.
Experiments were performed two or three separate times (biological replicates), and only proteins identified with at least three unique peptides in at least two biological replicates (six spectral count minimum) were included in our analysis (Supplemental Tables S3-S6). To compare DUB delete with WT, the total spectral counts for each LC-MS/MS experiment were normalized to total spectral counts for Ub (bait) and multiplied by 1,000 to generate “abundance.” The abundance was averaged across biological replicates and then the ratio of abundance in the DUB delete versus WT was calculated. Proteins with an abundance ratio greater than 2 were included as putative substrates (Supplemental Tables S4 and S6). Proteins missing from WT but present in the DUB delete were included as putative substrates (indicated by a ratio of “ND” in Supplemental Table S6). Proteins identified in WT but not in the DUB delete were not included in our substrate analysis but are listed in Supplemental Tables S3 and S5. Ub'n sites were filtered in Scaffold PTM v2.0 (Proteome Software, Portland OR) (Supplemental Table S7 and Supplemental Fig. S3) and only identifications with Ascores > 13 (22) are reported (1,200 di-gly modified peptides were identified in 494 proteins, which is ∼25% of all proteins identified in all HBH purifications). Gene ontology (GO) annotation categories for each substrate are listed in Supplemental Table S8.
Quantitative MS for Ub Chain Linkage Analysis
The analysis of all polyUb linkages in the yeast samples was performed as reported previously (23) with a few modifications. Briefly, JMP024 (24) was metabolically labeled with heavy lysine (Lys + 8.0142, Arg + 10.0083) and used as internal standard. Fission yeast (WT and 5DUB delete) were cultured in regular (light) media, harvested, and then each strain was separately mixed with an equal amount of the internal standard. Following mixing, yeast cells were lysed in lysis buffer (10 mm Tris, pH 8.0; 8 m urea, 0.02% SDS; 10 mm iodoacetamide; and Roche protease inhibitor mixture) with 0.5 mm glass beads (Biospec Products, Inc.). The sample was vortexed at the highest speed for 30 s with 30 s break on ice 10X, followed by cycles of sonication for a 1 s pulse and 30 s at 4 °C. The lysate was clarified by centrifugation at 21,000 g for 5 min.
Samples were loaded on an 8% SDS gel and stained with Coomassie blue G250. The gel lane was excised for in-gel digestion by trypsin (12.5 ng/μl) at 37 °C overnight. The resulting peptides were extracted with a buffer of 5% formic acid and 50% acetonitrile at room temperature, dried, and reconstituted in 1x IAP buffer (50 mm MOPS/NaOH, pH7.2; 10 mm Na2HPO4; 50 mm NaCl). Then the peptide samples were transferred to K-ε-GG antibody coupled to protein A agarose beads (Cell Signaling Technology, Inc.), and the mixture was incubated for 1.5 h at 4 °C with gentle shaking. The antibody beads were washed three times with 1X IAP (Cell Signaling Technology, Inc.) buffer containing 0.15% sodium deoxycholate at 4 °C. Tryptic diGly-containing peptides were eluted from beads by incubating with 0.15% TFA at room temperature for 10 min.
Eluted peptides were desalted using StageTips (Thermo Fisher Scientific, Inc.) and analyzed by LC-MS/MS on a Thermo Q-Exactive mass spectrometer. Peptides were loaded onto a 75 μm inner diameter × 10 cm PicoFrit capillary column packed with C18 resins (2.7 μm HALO beads) (New Objective, Inc.) by the autosampler and eluted with a 40 min gradient with 6–40% of buffer B (buffer A, 0.2% formic acid; buffer B, 0.2% formic acid and 70% acetonitrile; flow rate: ca. 400 nl/min). The eluted peptides were detected in a precursor MS scan by Q Exactive (400–900 m/z, 140,000 resolution at m/z 200, 1 μscan, and 1 × 106 for automatic gain control), followed by targeted MS/MS scans of Ub linkage-specific GG peptide ions (isolation width of 2 m/z, 1 μscan, target value of 500,000 for automatic gain control; see Supplemental Table S9). The Ub-specific diGly-containing peptides from fission yeast and budding yeast were eluted together and separated in the mass spectrometer due to mass difference, enabling relative quantification.
The quantitation of polyUb linkages in WT and DUB mutant yeast cells was carried out following a previously reported protocol (23). The intensities of the tryptic peptides (GG-signature peptides) derived from Ub were manually analyzed by ion chromatograms using Xcalibur v2.2.0 software (Thermo Fisher Scientific, Inc.) and used to estimate the Ub linkage changes between WT and 5DUB delete. The peak height of each precursor was calculated using Genesis peak algorithm with a mass tolerance of 20 ppm. The relative abundance of each peptide was obtained by calculating the ratio of peak height of the light form of each peptide and the corresponding peak height of the heavy form (internal standard).
Biochemistry
HBH purifications and GST pull-down experiments analyzed by SDS-PAGE on 4–12% Tris-Glycine gels (Life Technologies) were blotted on Immobilon P (Millipore) and probed with the following primary antibodies: anti-Flag (Sigma M2 monoclonal antibody) and secondary antibodies/conjugates: streptavidin (Li-COR, 680 nm or 800 nm conjugates), goat-anti rabbit or goat anti-mouse 680 nm or 800 nm conjugates (Life Technologies). Membranes were imaged on a Li-COR instrument using Odyssey software.
Di-Ub cleavage assays were performed using M1, K6, K11, K27, K29, K33, K48, and K63 linkages. For each reaction, lysates of strains containing TAP-tagged DUBs were made from 40 OD pellets in native lysis buffer (6 mm Na2HPO4, 4 mm NaH2PO4·H2O, 1% Nonidet P-40, 300 mm NaCl, 2 mm EDTA, 50 mm NaF, 0.1 mm Na3VO4 plus Roche complete protease mixture, 1 μm benzamidine (Sigma) and 0.1 mm diisopropyl fluorophosphate (Sigma)). Cleared lysates were incubated with 10 μl slurry of M280 tosylactivated dynabeads (Life Technologies) coated with rabbit IgG (MP Biomedicals) to isolate DUB-TAPs. After washing with lysis buffer, the dynabeads were equilibrated with reaction buffer (20 mm HEPES-KOH [pH 8], 20 mm NaCl, 0.1 mg/ml BSA 0.5 mm EDTA, and 10 mm DTT). Each cleavage reaction was started by adding 1 μg of a di-Ub chain (Boston Biochem, Boston, MA, USA) in reaction buffer to the dynabeads containing the DUB-TAPs complexes and incubating with shaking at 30 °C for 1–18 h. Negative controls using strains lacking DUB-TAPs were included for each set of reactions to control for proteases nonspecifically bound to the beads.
Microscopy
All images of S. pombe cells were acquired using either: (1) a spinning disk confocal microscope (Ultraview LCI; PerkinElmer), which is equipped with a Zeiss Axiovert 200m microscope, 100X NA 1.40 PlanApo oil immersion objective, a 488-nm argon ion laser (GFP), a 594-nm helium neon laser (RFP, mCherry), a charge-coupled device camera (Orca-ER; Hamamatsu Phototonics), and Metamorph 7.1 software (MDS Analytical Technologies; Molecular Devices) or (2) a Personal DeltaVision microscope system (Applied Precision), which includes an Olympus IX71 microscope, 60X NA 1.42 PlanApo and 100X NA 1.40 UPlanSApo objectives, fixed- and live-cell filter wheels, a Photometrics CoolSnap HQ2 camera, and softWoRx imaging software. Images were processed in Image J (Fiji) (25), vacuolar volume was calculated using the 3D object counter plugin (26), and final figures were assembled in Adobe Creative Suite (CS6).
RESULTS
These five membrane trafficking DUBs—Ubp4, Ubp5, Ubp9, Ubp15, and Sst2—span two of the four Ub protease domain families found in S. pombe—Ub-specific protease (USP: Ubp4, Ubp5, Ubp9, and Ubp15) and Jab1/MPN domain-associated metallo-isopeptidase (JAMM: Sst2) (Fig. 1A). Although four of the five enzymes contain USP domains, they differ in sequence, metal binding capacity (Ubp5 and Ubp15 lack a functional metal binding site), and domain architecture (Fig. 1A) (1, 27). While there is significant overlap of cellular localization of these membrane trafficking DUBs, each enzyme has a unique localization pattern under standard laboratory growth conditions (1).
Fig. 1.
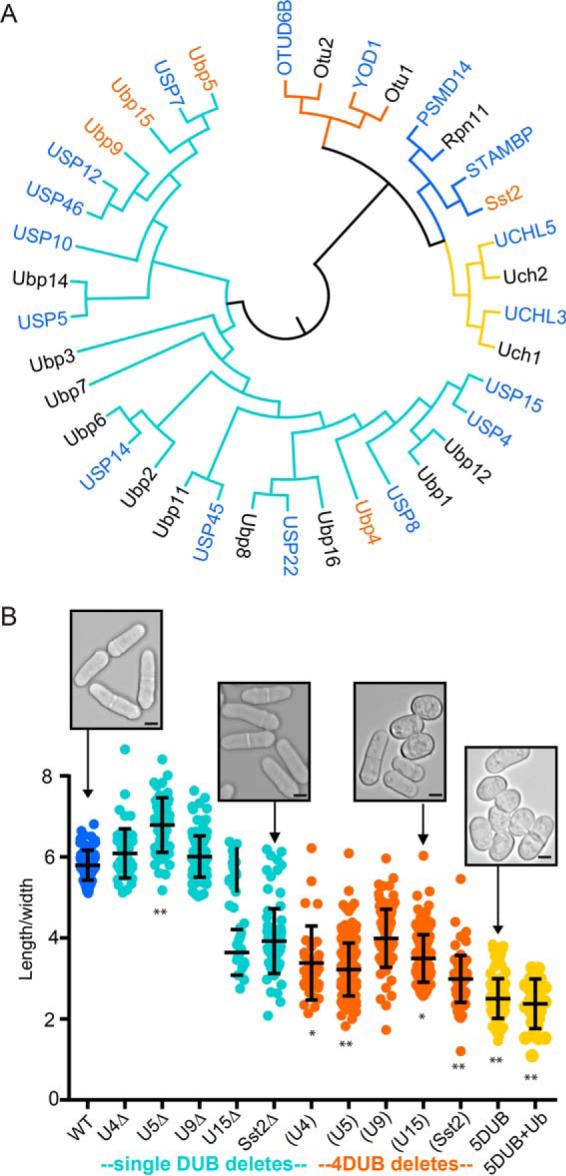
S. pombe membrane trafficking DUB degeneracy. (A) Phylogenetic tree of S. pombe DUBs (four enzyme classes, lowercase font) and their human homologs (if present, uppercase blue font). The cohort of five DUBs involved in trafficking are highlighted in orange font. Branches are color-coded to indicate different catalytic domain families (cyan—USP, dark orange—OTU, medium blue—JAMM, light orange—UCH). DUB relationships, but not sequence distances, are indicated. (B) Plot of cell aspect ratios at septation (cell length/cell width ratio) for the indicated strains measured using Image J from DIC images (each point represents one measurement (n > 30); the average and standard deviations are shown as a solid bar and lines). Abbreviations: U4Δ = ubp4Δ1, U5Δ = ubp5Δ, U9Δ = ubp9Δ, U15Δ = ubp15Δ, (U4) = ubp5Δ ubp9Δ ubp15Δ sst2Δ, (U5) = ubp4Δ1 ubp9Δubp15Δ sst2Δ, (U9) = ubp4Δ1 ubp5Δ ubp15Δ sst2Δ, (U15) = ubp4Δ1 ubp5Δ ubp9Δ sst2Δ, (Sst2) = ubp4Δ1 ubp5Δ ubp9Δ ubp15Δ, 5DUB = ubp4Δ1 ubp5Δubp9Δubp15Δ sst2Δ. Asterisks indicated a significant difference between WT and the DUB delete strains (one way ANOVA performed in GraphPad PRISM6). (*) = p < .05 and (**) = p < .0001. Inset images are DIC images from the strains indicated by arrows (scale bars = 4 μm).
Membrane trafficking is intimately linked to cell polarity. The fission yeast S. pombe exhibits a stereotypical rod shape that is maintained by polarized growth throughout the cell cycle (28) and is highly correlated with actin cytoskeleton organization (29, 30) and membrane trafficking (31, 32). Loss of cell polarity is easily detected by light microscopy of live cells (33), and the ratio of cell length divided by cell width at septation is a simple way to quantitate changes in cell polarity and morphology (apolar cells become misshapen or round). The ratio of cell length/width at septation varies among a series of single and multiple membrane trafficking DUB deletion strains (Fig. 1B). Loss of any one of these DUBs results in subtle and distinct changes in cell morphology (Fig. 1B, single deletes). Of the single deletions, only ubp5Δ cells have a statistically significant change in cell morphology, but a larger range in length/width variation is apparent for all the single deletions. The 4DUB delete strains each maintain one of these membrane trafficking DUBs (indicated in parentheses), and all but one exhibit significant and variable changes in cell morphology (Fig 1B and inset images). These morphological changes could potentially be due to depletion of the cellular Ub pool, as we previously showed that the 5DUB delete accumulates Ub conjugates (1). However, we found that the 5DUB delete strain exhibits severe cell morphology/polarity defects even when the Ub pool has been replenished ectopically, indicating that the cell polarity defects are a consequence of DUB loss and not Ub depletion (Fig. 1B). Confirming significant defects in intracellular transport, 5DUB delete cells exhibit other changes in cell morphology (Supplemental Fig. S1), including fragmented, smaller vacuoles, a phenotype reminiscent of a Cdc42 mutant deficient in membrane trafficking, endocytosis, and cell polarity (32).
To understand the overlapping and unique functions of these membrane trafficking DUBs, we defined putative substrates of each DUB using denaturing ubiquitin–conjugate purifications and comparative MS-based proteomics of the ubiquitinome (Fig. 2A, Supplemental Tables S3 and S4), as we have done previously (20). Briefly, we overexpressed N-terminally His-biotin-His (HBH) tagged Ub using the nmt1 promoter in DUB delete and WT cells, performed duplicate denaturing purifications, and used 2D-LC-MS/MS to identify ubiquitinated (Ub'd) proteins (Fig. 2A). We identified over 2,000 Ub'd proteins in total (Supplemental Table S3), many of which have not previously been reported as ubiquitin conjugates in yeast (17, 34, 35). These two-step denaturing purifications result in low nonspecific background ((17) and Supplemental Table S2), and our relatively stringent filtering (three distinct peptides in at least two biological replicates) further reduces false positive results. Comparison of protein abundance in WT versus the DUB deletions yielded a list of putative substrates (proteins enriched in the DUB deletion strains) for each of the membrane trafficking DUBs (Fig. 2B and Supplemental Table S4), some of which were unique to specific strains (colored bars in Fig. 2B). We biochemically confirmed the Ub'n status of a subset of putative substrates in WT and DUB delete backgrounds (Supplemental Fig. S2) using denaturing HBH purifications and Western blots as detailed in Experimental Procedures. Every protein tested was found to be a bona fide ubiquitin conjugate, validating our strategy. There was significant overlap in putative substrates affected by the DUB deletions, with the majority of proteins identified as putative substrates of at least two of these DUBs (Fig. 2C, each line represents a substrate and shading in the DUB delete column indicates that substrate is a putative target of the deleted DUB).
Fig. 2.

Comparative proteomics reveals unique and shared putative substrates among five S. pombe DUBs. (A) Schema of comparative proteomics strategy (see Experimental Procedures for details). (B) Graph of numbers of putative substrates identified for each DUB using the indicated deletion strains (intersection of two independent experiments). Colored bars indicate substrates unique to that strain (DUB), and black bars indicate substrates shared with at least one other DUB. (C) Graph showing the complex relationships between the putative DUB substrates. Each row represents a substrate and the colored bars indicate which DUBs potentially target each substrate. Bar and column titles for the strains are as in Fig. 1.
Because cell viability drastically changes upon deletion of five as opposed to any combination of four of these membrane trafficking DUBs (1), we hypothesized that there were potentially substrates shared by all five of these membrane trafficking DUBs that are presumably determinants of cell viability. Comparative analysis of the ubiquitinomes of the 5DUB delete cells and WT cells revealed just over 200 putative substrates shared among these DUBs (increased in abundance by at least twofold in 5DUB compared with WT; see Experimental Procedures for details) (Fig. 3A and Supplemental Tables S5 and S6). Approximately 60% of the 5DUB putative substrates were not detected in purifications from WT cells, confirming accumulation of Ub'd proteins in the 5DUB delete (Fig. 3A, green circle in lower panel). A bit surprisingly, the majority of the putative 5DUB substrates were not identified as putative substrates in any of the single DUB deletions (Fig. 3B), indicating significant overlap of substrate specificity of these DUBs. Thus, these membrane trafficking DUBs potentially have both unique and overlapping substrates, a key characteristic of degeneracy. Moreover, we identified ∼1,200 Ub'n sites in our combined datasets without di-Gly remnant enrichment (Supplemental Table S7 and Supplemental Fig. S3).
Fig. 3.
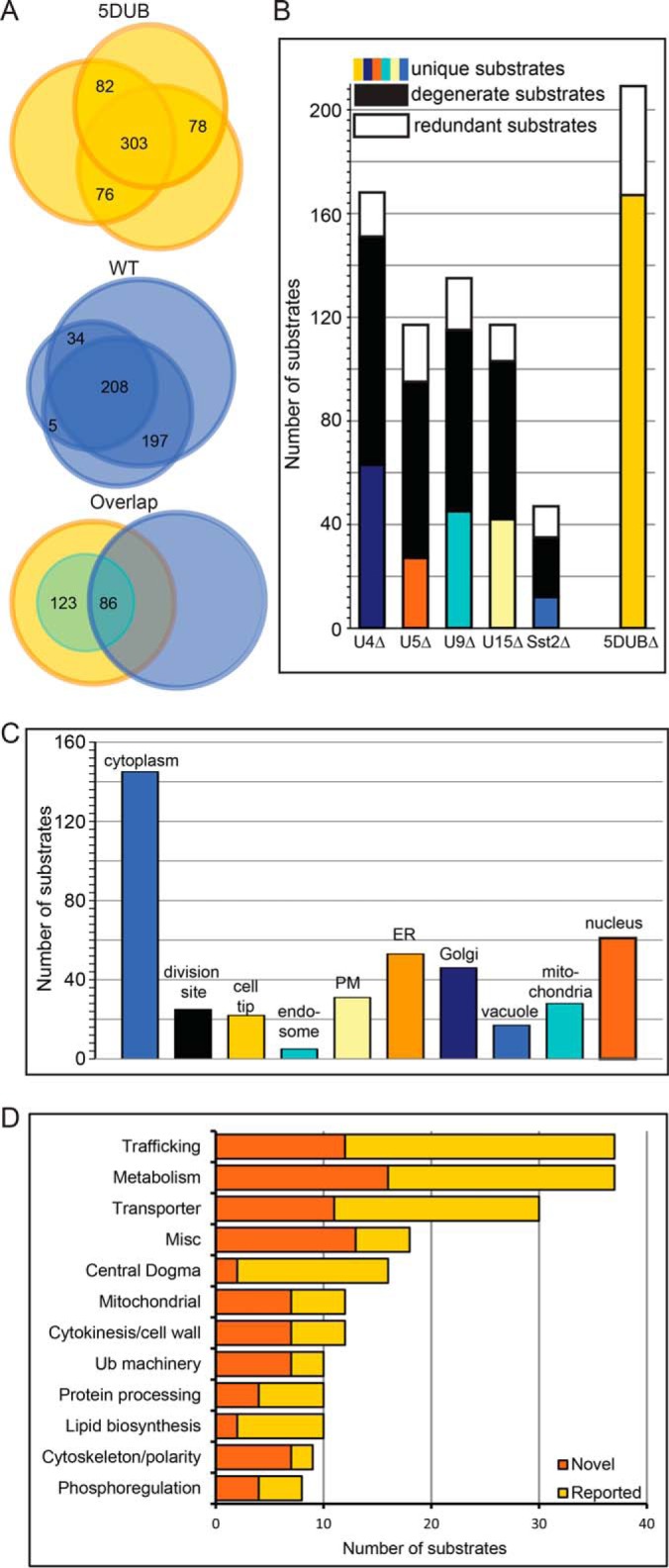
Comparative proteomics of wildtype (WT) and the 5DUB delete S. pombe strains reveals the degeneracy of these 5 DUBs. (A) Venn diagrams indicating the overlap of proteins identified in three independent experiments for the 5DUBΔ (upper) and WT (middle) strains. Bottom panel shows overlap of proteins identified in both strains and the small inset circle represents the putative substrates. (B) Bar graph showing numbers of substrates identified for each DUB using the indicated strains. Colored bars indicate substrates unique to specific strains, black bars indicate substrates shared with at least one other DUB, and white bars indicate substrates identified in at least one of the single deletions in addition to the 5DUB delete. (C) Cellular localization of the putative DUB substrates. Note that many substrates are present in multiple cellular locations. (D) Bar graph indicating the number of substrates for each gene ontology category (Pombase (50)) for the shared substrates of the five membrane trafficking DUBs. The dark orange bars represent ubiquitinated substrates that have not been reported as ubiquitinated, and the light orange bars represent substrates whose budding yeast orthologs have been reported as ubiquitinated proteins (17, 34, 35) (see Supplemental Table S6).
Shared putative substrates of the five membrane trafficking DUBs are found in virtually every cellular locale but are especially abundant in membrane trafficking compartments (ER, Golgi, cell division site, membrane, Fig. 3C). In line with their enriched localization in membrane compartments, gene ontology (GO) analysis of the shared putative substrates revealed their involvement in an array of essential membrane-centric cellular processes, including transport, cytokinesis, lipid metabolism, and protein trafficking (Fig. 3D, and Supplemental Table S8). The fact that many (34%) of the putative substrates are transporters or known players in membrane traffic further validates our approach to substrate discovery (Fig. 3D, Trafficking and Transporter clusters). Transporters are known to be regulated by ubiquitination (36), and a complex web of ligases, adaptors, and DUBs dictate their ultimate fate (37, 38). In addition, many novel putative substrates (e.g. the 14–3-3 protein Rad24) were identified (Supplemental Table S6). Future work will define the role of Ub'n in their function.
Interestingly, many of the candidate 5DUB substrates are involved in processes related to cytokinesis, including lipid metabolism, cell wall metabolism, and cytoskeletal regulation and polarity (Fig. 3D). For instance, multiple players in cell polarity (e.g. Cdc42 and Tea1) were identified as putative substrates (Fig. 3D and Supplemental Tables S6 and S8). In line with this cohort of DUBs targeting polarity and cytokinetic proteins, the 5DUB delete phenotype includes defects in abscission, septation, and polarized growth. (Fig. 1B and Supplemental Fig. S1).
The results described above along with previous work (1) show that these five membrane trafficking DUBs have overlapping cellular localization and substrate selectivity. To clarify the specificity of these DUBs at a mechanistic level, we defined their Ub chain specificities in vitro and in vivo. In vitro assays were performed with di-Ub conjugates of each chain type (M1, K6, K11, K27, K29, K33, K48, and K63) and TAP-tagged DUBs (including their associated partners) (Supplemental Fig. S4A), using silver stained SDS-PAGE gels to detect cleaved Ub (Supplemental Fig. S4B). Negative control assays with untagged strains showed no di-Ub cleavage (Supplemental Fig. S4C). The cellular abundance of these five DUBs is variable (Supplemental Fig. S4A) (39, 40), so the activity of each DUB toward each chain type was normalized to its preferred chain type (bright orange in Fig. 4A) for comparison (see Supplemental Fig. S4D for data prior to normalization). Ubp4 predominately targets K11, then K48, and finally K6 and K63 Ub chains to some extent (Fig. 4A). In contrast, Ubp5 prefers K63, then K11 followed by K6, K27, K33, and K48 linkages. Ubp9 shares significant overlap in chain specificity with Ubp4 but can also target K33 Ub chains. Although Sst2 prefers K63 linkages, as has previously been reported for its ortholog AMSH (41, 42), Sst2 also targets K11 and to a lesser extent K6, K27, K33, and K48 Ub chain linkage types. Ubp15 prefers K11 linkages but also targets K63 and many other chain types to a lesser extent. Thus, this cohort of DUBs targets multiple chain types with varying degrees of selectivity (Fig. 4A and Supplemental Fig. S4D).
Fig. 4.
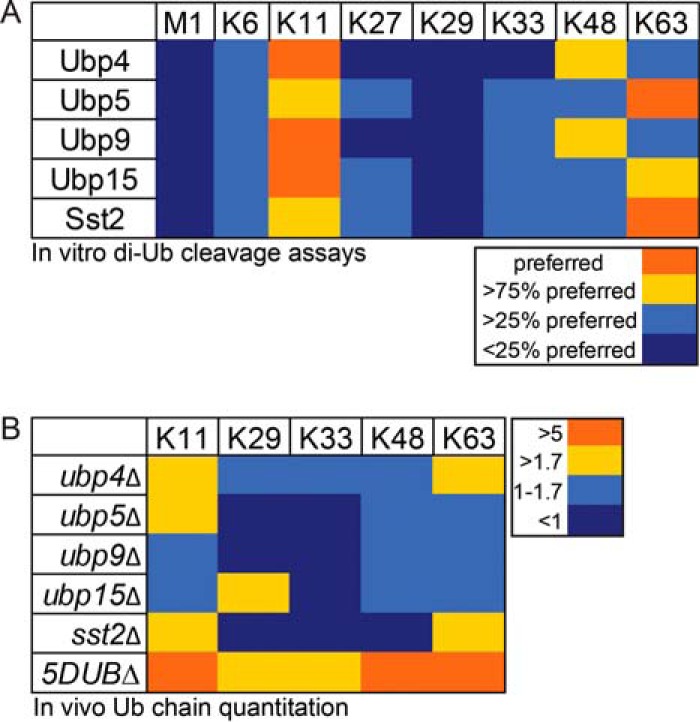
Ub chain specificity of 5 S. pombe membrane trafficking DUBs. (A) In vitro di-Ub cleavage reactions (see Experimental Procedures for details). Cleavage efficiencies were calculated as the ratio of cleaved/uncleaved x100. Preferred chains (bright orange) were cleaved most efficiently (i.e. 100% normalized cleavage efficiency), and the other colors are as indicated with lower cleavage efficiency (see Supplemental Fig. S4). These results are the average of two independent experiments. (B) In vivo detection of Ub chains using quantitative mass spectrometry (see Experimental Procedures and Supplemental Table 9 for details). Numbers indicate fold increases in detected chain types in the DUB delete (indicated) strains compared with WT S. pombe cells (ratios are average of three experiments).
We then performed quantitative targeted MS measurements of specific Ub linkages found in vivo in various DUB deletion strains. The in vivo results largely recapitulate the in vitro results (Fig. 4B). For example, sst2Δ shows an accumulation of K11 and K63 chains (Fig. 4B). Collectively, these five DUBs target K11, K48, and K63 linkages (Fig. 4B, bottom row), correlating with the most highly targeted chain types in the in vitro di-Ub cleavage assays (Fig. 4A). Surprisingly, most of the DUBs in this cohort target K11-linked Ub chains (Fig. 4), which have been implicated in proteasomal degradation (43, 44) and as mixed chains in endocytosis and NF-κB signaling (45, 46). We can infer from these results that K11 chain linkages are common in membrane trafficking and likely have many as yet unknown functions in cellular signaling. Although we cannot differentiate between homotypic and mixed Ub chains using this in vivo method, the in vitro experiments show that mixed chains are not a prerequisite for activity of these DUBs (Fig. 4A). Taken together, both in vitro and in vivo analyses highlight overlapping Ub chain specificities of this cohort of DUBs and reveal another level of degeneracy.
In light of the many levels of degeneracy described above, we sought to test the essentiality of each DUB in this cohort under stress conditions. We predicted that exposure of the cells to external stressors would reveal conditional essentialities, illustrating the unique contributions of each DUB to cell survival. The collective and individual contributions of each DUB to cell survival were assayed in response to a battery of external stressors (Fig. 5A). Log phase cells were spotted in serial dilutions on each type of stressor plate and incubated at 32 °C for 2–6 days prior to imaging and interpretation (Supplemental Figs. S5 and S6). As predicted, the DUB deletions varied in their ability to grow under different stress conditions (Fig. 5B). For instance, ubp5Δ growth is only sensitive to minimal media and calcofluor (a chemical that perturbs cell wall formation), but ubp9Δ growth was significantly reduced by multiple stressors (Fig. 5B). The nonadditive, unique patterns of growth sensitivity for each strain reveal how the phenotypic landscape is altered by the presence of different combinations of these five DUBs. That is, the specific and overlapping roles of each DUB are revealed by these different cellular environments where they become essential for viability. It is interesting to note that while Latrunculin A affects growth of multiple single and quadruple DUB delete strains, it appears to have little or no effect on the growth of the 5DUB delete, suggesting that the actin cytoskeleton is already disrupted in this mutant, and thus Latrunculin A has no further effect. Indeed phalloidin staining of the 5DUB delete revealed the extent of actin cytoskeletal disorganization, in line with the loss of polarity in this mutant (Supplemental Fig. S1). As predicted, we found that the degeneracy of this cohort of enzymes under standard laboratory conditions masks their individual contributions to cellular function and viability.
DISCUSSION
Through cell biological, biochemical, and proteomic experiments, we have dissected the individual and collective contributions of five membrane trafficking DUBs. Although these membrane trafficking DUBs appear redundant at the level of cell viability (1), they are degenerate on multiple levels including cellular localization pattern, effect on cell polarity (Fig. 1B), substrate specificity (Figs. 2 and 3), Ub chain specificity (Fig. 4), and cell viability in the face of external stress (Fig. 5B). Thus, although yeast DUBs appear to have significant functional overlap when cultured in rich media, exposure to different environments reveals their individual functions. From this perspective, yeast DUBs have specific functions, similar to metazoans, but these functions are simply masked by growth under standard conditions. The architecture of the degeneracy (unique and overlapping functions by diverse structures) of these five DUBs is revealed by their substrate network (Supplemental Fig. S7), which hints at the circuitry underlying their functional overlap in mediating cell polarity and viability (each node is a substrate or DUB and lines connecting DUBs to putative substrates are color-coded to indicate which DUB targets the substrate). This network contains each DUB and all the potential DUB substrates as nodes (dots) that are connected by edges that indicate which DUB targets that specific substrate. It is likely that the edges and nodes in this network would shift and change in the face of external stressors, mediating cell survival by modulating the cellular phenotype. We conclude that, collectively, this cohort of DUBs forms a degenerate module that mediates cell polarity and survival, and, individually, each DUB has unique contributions to cell survival in different cellular environments (stresses).
Adding further layers of complexity to the modes of degeneracy and diversity discussed above, this cohort of membrane trafficking DUBs is regulated by protein binding partners and posttranslational modifications. Ubp4, Ubp5, and Ubp9 each have unique protein binding partners that impact their cellular localization and/or activity (1) and AMSH, the human ortholog of Sst2, also has partners that impact its activity (42, 47, 48). Furthermore, all five of these DUBs and most of their partners are phosphorylated (49), indicating another layer of regulation. It will be interesting to explore the signaling circuits that impact DUB function and whether they are modulated by cellular stressors.
Finally, we note that this is the first report of a systemwide study of ubiquitination in S. pombe, providing a valuable resource to multiple research communities. Our strategy yielded 2,000 potentially ubiquitinated proteins, comprised of both well-characterized (e.g. histone H2B Htb1), and uncharacterized (e.g. the 14–3-3 protein Rad24) ubiquitin conjugates. Based on our previous success in DUB substrate identification using this straightforward, inexpensive approach (20), we chose not to use di-gly enrichment methods prior to MS analysis. Validating our approach, every substrate assayed for Ub'n was indeed ubiquitinated and exhibited an increase in ubiquitination in the DUB delete strain (Supplemental Fig. S2). Additionally, we identified 1,200 Ub'n sites in 494 proteins (25%) (Supplemental Table S7 and Supplemental Fig. S3), providing site-specific information for many putative substrates of the cellular Ub'n machinery.
Acknowledgments
We thank Ilektra Kouranti for plasmid construction and preliminary experiments, Jianqiu Wang, Malwina Huzarska, and Ping Liang for performing LC-MS experiments, and Paul Randazzo for providing the pGEX5T-GGA3 construct. We also thank members of the Gould lab for critically reading and editing the manuscript.
Footnotes
Author contributions: J.R.B., Y.Y., J.P., and K.L.G. designed the research; J.R.B., J.C., and Y.Y. performed the research; J.R.B. and Y.Y. analyzed data; and J.R.B. and K.L.G. wrote the paper.
* J.P. is supported American Lebanese Syrian Associated Charities (ALSAC). The authors declare that they have no conflict of interest.
 This article contains supplemental material Supplemental Tables S1-S9 and Supplemental Figs. S1-S7.
This article contains supplemental material Supplemental Tables S1-S9 and Supplemental Figs. S1-S7.
1 The abbreviations used are:
- DUB
- deubiquitinase
- Ub
- ubiquitin
- Ub'n
- ubiquitination
- OD
- optical density
- GO
- Gene ontology
- MS
- mass spectrometry
- LC
- liquid chromatography
- EMM
- Edinburgh Minimal Medium
- AMSH
- Associated molecule with the SH3 domain of STAM
- ANOVA
- analysis of variance
- DIC
- Differential interference contrast.
REFERENCES
- 1.Kouranti I., McLean J. R., Feoktistova A., Liang P., Johnson A. E., Roberts-Galbraith R. H., and Gould K. L. (2010) A global census of fission yeast deubiquitinating enzyme localization and interaction networks reveals distinct compartmentalization profiles and overlapping functions in endocytosis and polarity. PLoS Biol. 8, e1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reyes-Turcu F. E., Ventii K. H., and Wilkinson K. D. (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clague M. J., Barsukov I., Coulson J. M., Liu H., Rigden D. J., and Urbé S. (2013) Deubiquitylases from genes to organism. Physiol. Rev. 93, 1289–1315 [DOI] [PubMed] [Google Scholar]
- 4.Ikeda F., and Dikic I. (2008) Atypical ubiquitin chains: New molecular signals. Protein modifications: Beyond the usual suspects' review series. EMBO Rep. 9, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–29 [DOI] [PubMed] [Google Scholar]
- 6.Kim D. U., Hayles J., Kim D., Wood V., Park H. O., Won M., Yoo H. S., Duhig T., Nam M., Palmer G., Han S., Jeffery L., Baek S. T., Lee H., Shim Y. S., Lee M., Kim L., Heo K. S., Noh E. J., Lee A. R., Jang Y. J., Chung K. S., Choi S. J., Park J. Y., Park Y., Kim H. M., Park S. K., Park H. J., Kang E. J., Kim H. B., Kang H. S., Park H. M., Kim K., Song K., Song K. B., Nurse P., Hoe K. L., Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28, 617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayles J., Wood V., Jeffery L., Hoe K. L., Kim D. U., Park H. O., Salas-Pino S., Heichinger C., and Nurse P. (2013) A genome-wide resource of cell cycle and cell shape genes of fission yeast. Open Biol. 3, 130053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Güldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S.L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature, 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 9.Amerik A. Y., Li S. J., and Hochstrasser M. (2000) Analysis of the deubiquitinating enzymes of the yeast Saccharomyces cerevisiae. Biol. Chem. 381, 981–992 [DOI] [PubMed] [Google Scholar]
- 10.Shimanuki M., Saka Y., Yanagida M., and Toda T. (1995) A novel essential fission yeast gene pad1+ positively regulates pap1(+)-dependent transcription and is implicated in the maintenance of chromosome structure. J. Cell Sci. 108, p. 569–579 [DOI] [PubMed] [Google Scholar]
- 11.Penney M., Wilkinson C., Wallace M., Javerzat J. P., Ferrell K., Seeger M., Dubiel W., McKay S., Allshire R., and Gordon C. (1998) The Pad1+ gene encodes a subunit of the 26 S proteasome in fission yeast. J. Biol. Chem. 273, 23938–23945 [DOI] [PubMed] [Google Scholar]
- 12.Tsou W. L., Sheedlo M. J., Morrow M. E., Blount J. R., McGregor K. M., Das C., and Todi S. V. (2012) Systematic analysis of the physiological importance of deubiquitinating enzymes. PLoS ONE 7, e43112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCloskey R. J., and Kemphues K. J. (2012) Deubiquitylation machinery is required for embryonic polarity in Caenorhabditis elegans. PLoS Genet. 8, e1003092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamura K., Stecher G., Peterson D., Filipski A., and Kumar S. (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shih S. C., Katzmann D. J., Schnell J. D., Sutanto M., Emr S. D., and Hicke L. (2002) Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat. Cell Biol. 4, 389–393 [DOI] [PubMed] [Google Scholar]
- 16.Maundrell K., (1993) Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123, 127–130 [DOI] [PubMed] [Google Scholar]
- 17.Tagwerker C., Flick K., Cui M., Guerrero C., Dou Y., Auer B., Baldi P., Huang L., and Kaiser P. (2006) A tandem affinity tag for two-step purification under fully denaturing conditions: Application in ubiquitin profiling and protein complex identification combined with in vivocross-linking. Mol. Cell. Proteomics 5, 737–748 [DOI] [PubMed] [Google Scholar]
- 18.Prentice H. L. (1992) High efficiency transformation of Schizosaccharomyces pombe by electroporation. Nucleic Acids Res. 20, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno S., Klar A., and Nurse P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823 [DOI] [PubMed] [Google Scholar]
- 20.Elmore Z. C., Beckley J. R., Chen J. S., and Gould K. L. (2014) Histone H2B ubiquitination promotes the function of the anaphase-promoting complex/cyclosome in Schizosaccharomyces pombe. G3 4, 1529–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Z. Q., Tabb D. L., Burden J., Chambers M. C., Cox M. B., Cantrell M. J., Ham A. J., Litton M. D., Oreto M. R., Schultz W. C., Sobecki S. M., Tsui T. Y., Wernke G. R., and Liebler D. C. (2011) Supporting tool suite for production proteomics. Bioinformatics 27, 3214–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beausoleil S. A., Villén J., Gerber S. A., Rush J., and Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 23.Na C. H., Jones D. R., Yang Y., Wang X., Xu Y., and Peng J. (2012) Synaptic protein ubiquitination in rat brain revealed by antibody-based ubiquitome analysis. J. Proteome Res. 11, 4722–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., and Peng J. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolte S., and Cordelieres F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microscopy 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 27.Ye Y., Scheel H., Hofmann K., and Komander D. (2009) Dissection of USP catalytic domains reveals five common insertion points. Mol. Biosyst. 5, 1797–1808 [DOI] [PubMed] [Google Scholar]
- 28.Streiblová E., and Wolf A. (1972) Cell wall growth during the cell cycle of Schizosaccharomyces pombe. Z. Allg. Mikrobiol. 12, 673–684 [PubMed] [Google Scholar]
- 29.Marks J., Hagan I. M., and Hyams J. S. (1986) Growth polarity and cytokinesis in fission yeast: the role of the cytoskeleton. J. Cell Sci. Suppl. 5, 229–241 [DOI] [PubMed] [Google Scholar]
- 30.Arai R., Mabuchi I. (2002) F-actin ring formation and the role of F-actin cables in the fission yeast Schizosaccharomyces pombe. J Cell Sci. 115, 887–98 [DOI] [PubMed] [Google Scholar]
- 31.Kanbe T., Kobayashi I., and Tanaka K. (1989) Dynamics of cytoplasmic organelles in the cell cycle of the fission yeast Schizosaccharomyces pombe: Three-dimensional reconstruction from serial sections. J. Cell Sci. 94, 647–656 [DOI] [PubMed] [Google Scholar]
- 32.Estravís M., Rincón S. A., Santos B., and Pérez P. (2011) Cdc42 regulates multiple membrane traffic events in fission yeast. Traffic 12, 1744–58 [DOI] [PubMed] [Google Scholar]
- 33.Snell V., and Nurse P. (1994) Genetic analysis of cell morphogenesis in fission yeast—A role for casein kinase II in the establishment of polarized growth. EMBO J. 13, 2066–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., and Gygi S. P. (2003) A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 [DOI] [PubMed] [Google Scholar]
- 35.Hitchcock A. L., Auld K., Gygi S. P., and Silver P. A. (2003) A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. Proc. Natl. Acad. Sci. U.S.A. 100, 12735–12740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonifacino J. S., and Weissman A. M. (1998) Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol. 14, 19–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin C. H., MacGurn J. A., Chu T., Stefan C. J., and Emr S. D. (2008) Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 135, 714–725 [DOI] [PubMed] [Google Scholar]
- 38.MacGurn J. A., Hsu P. C., Smolka M. B., and Emr S. D. (2011) TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147, 1104–1117 [DOI] [PubMed] [Google Scholar]
- 39.Carpy A., Krug K., Graf S., Koch A., Popic S., Hauf S., and Macek B. (2014) Absolute proteome and phosphoproteome dynamics during the cell cycle of Schizosaccharomyces pombe (fission yeast). Mol. Cell. Proteomics 13, 1925–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marguerat S., Schmidt A., Codlin S., Chen W., Aebersold R., and Bähler J. (2012) Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151, 671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCullough J., Clague M. J., and Urbé S. (2004) AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell Biol. 166, 487–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCullough J., Row P. E., Lorenzo O., Doherty M., Beynon R., Clague M. J., and Urbé S. (2006) Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr. Biol. 16, 160–165 [DOI] [PubMed] [Google Scholar]
- 43.Matsumoto M. L., Wickliffe K. E., Dong K. C., Yu C., Bosanac I., Bustos D., Phu L., Kirkpatrick D. S., Hymowitz S. G., Rape M., Kelley R. F., and Dixit V. M. (2010) K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol. Cell 39, 477–484 [DOI] [PubMed] [Google Scholar]
- 44.Meyer H. J., and Rape M. (2014) Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boname J. M., Thomas M., Stagg H. R., Xu P., Peng J., and Lehner P. J. (2010) Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 11, 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dynek J. N., Goncharov T., Dueber E. C., Fedorova A. V., Izrael-Tomasevic A., Phu L., Helgason E., Fairbrother W. J., Deshayes K., Kirkpatrick D. S., and Vucic D. (2010) c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29, 4198–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Y. M., Boucrot E., Villén J., Affar el B., Gygi S. P., Göttlinger H. G., Kirchhausen T. (2007) Targeting of AMSH to endosomes is required for epidermal growth factor receptor degradation. J. Biol. Chem. 282, 9805–9812 [DOI] [PubMed] [Google Scholar]
- 48.Zamborlini A., Usami Y., Radoshitzky S. R., Popova E., Palu G., and Göttlinger H. (2006) Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc. Natl. Acad. Sci. U.S.A. 103, p. 19140–19145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLean J. R., Kouranti I., and Gould K. L. (2011) Survey of the phosphorylation status of the Schizosaccharomyces pombe deubiquitinating enzyme (DUB) family. J. Proteome Res. 10, 1208–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wood V., Harris M. A., McDowall M. D., Rutherford K., Vaughan B. W., Staines D. M., Aslett M., Lock A., Bähler J., Kersey P. J., and Oliver S. G. (2012) PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Res. 40, D695-D699 [DOI] [PMC free article] [PubMed] [Google Scholar]