

Abstract
Post-translational modifications (PTMs) of histone proteins play a fundamental role in regulation of DNA-templated processes. There is also growing evidence that proteolytic cleavage of histone N-terminal tails, known as histone clipping, influences nucleosome dynamics and functional properties. Using top-down and middle-down protein analysis by mass spectrometry, we report histone H2B and H3 N-terminal tail clipping in human hepatocytes and demonstrate a relationship between clipping and co-existing PTMs of histone H3. Histones H2B and H3 undergo proteolytic processing in primary human hepatocytes and the hepatocellular carcinoma cell line HepG2/C3A when grown in spheroid (3D) culture, but not in a flat (2D) culture. Using tandem mass spectrometry we localized four different clipping sites in H3 and one clipping site in H2B. We show that in spheroid culture clipped H3 proteoforms are mainly represented by canonical histone H3, whereas in primary hepatocytes over 90% of clipped H3 correspond to the histone variant H3.3. Comprehensive analysis of histone H3 modifications revealed a series of PTMs, including K14me1, K27me2/K27me3, and K36me1/me2, which are differentially abundant in clipped and intact H3. Analysis of co-existing PTMs revealed negative crosstalk between H3K36 methylation and H3K23 acetylation in clipped H3. Our data provide the first evidence of histone clipping in human hepatocytes and demonstrate that clipped H3 carry distinct co-existing PTMs different from those in intact H3.
Chromatin is a highly dynamic structure that must respond to different stimuli in order to orchestrate all DNA-dependent processes. Post translational modifications (PTMs)1 of histones play a major role in regulation of chromatin functionality. Evidence is emerging that not only ”classical” histone PTMs, such as methylation, acetylation, and phosphorylation at distinct residues, but also proteolytic processing of nucleosome proteins, known as “histone clipping,” can be involved in regulation of key cellular processes, such as transcriptional regulation, cell differentiation, and senescence (1–7).
Clipping of the histone H3 N-terminal tail was reported to be associated with gene activation in yeast. Santos-Rosa et al. demonstrated a serine protease activity in S. cerevisiae that cleaves histone H3 after residue Ala21 (A21) during sporulation and stationary phase (1). H3 clipping took place specifically within the promoters of sporulation-induced genes following the induction of transcription and prior to histone eviction from these DNA regions. Prevention of H3 N-tail cleavage by amino acid substitution at the endoproteinase recognition site (H3 Q19A, L20A) abolished expression of these genes, indicating that H3 clipping is essential for productive transcription.
The biological significance of histone clipping in higher eukaryotes is not yet understood but also appears to be related to functional commitment by the cell. Duncan et al. demonstrated that histone H3 is proteolytically cleaved by the enzyme Cathepsin L1 (CTSL1) at several sites between residues A21 and S28 during mouse ESC differentiation (5). The “in vitro” proteolytic activity of CTSL1 was found to be dependent on the H3 N-tail PTM status. H3K27me2 increased H3 cleavage by CTSL1, whereas H3K23ac reduced H3 cleavage. H3 clipping was also demonstrated in human ESCs (6).
A recent study by Duarte et al. proposed a role for H3 clipping in cellular senescence (7). Histone variant H3.3 was found to be proteolytically processed by CTSL1 upon oncogene-induced and replicative senescence in human fibroblasts and melanocytes. Ectopic expression of H3.3 and particularly its clipped proteoform was sufficient to induce senescence in fibroblasts presumably via transcriptional silencing of cell cycle regulatory genes.
Although the mechanism of regulation of histone clipping remains unclear, several studies suggested that this process might be affected by canonical histone PTMs (1, 3, 5, 8). However, because of technical challenges in the characterization of co-existing histone modifications the relation between histone clipping and covalent histone PTMs has remained poorly defined. In the present study, we address this question by using a middle-down proteomic workflow optimized in our laboratory for efficient characterization of combinatorial histone modifications (9).
First, we demonstrate that the N-terminal tails of two core histones, H2B and H3, undergo proteolytic processing in human hepatocytes both in vitro in hepatocarcinoma cell line HepG2/C3A and in vivo in primary hepatocytes and liver tissue. We find that cell culture conditions have profound effect on this process. Histone clipping takes place in HepG2/3CA cell line cultivated as a spheroid 3D culture (i.e. when cells are at their metabolic equilibrium (10)) but not when grown in a flat 2D culture using conventional cell culture techniques (when cells are in exponential growth). By using middle- and top-down proteomic approaches optimized for histone analysis we localize four different H3 cleavage sites and identify the position of H2B clipping. Finally we provide a comprehensive analysis of the PTM status of clipped H3 proteoforms and show that clipped H3 contain distinct PTM patterns enriched in K3K36 mono- and dimethylation.
MATERIALS AND METHODS
Cell Culturing
Standard culture conditions (2D culture): the immortal human hepatocellular carcinoma cell line, HepG2/C3A, was grown in monolayer cultures in Dulbecco's modified Eagle medium (DMEM) as previously described (10).
Spheroid culture conditions (3D culture): the HepG2/C3A cell spheroids were prepared using AggreWell™ 400 plates (Stemcel Technologies cat. no. 27845) and grown in ProtoTissue™ bioreactors as previously described (10). Spheroids were collected at specific time points, washed 5 times with PBS, snap-frozen in liquid nitrogen and stored at −80 °C until analysis.
Primary Human Hepatocytes
Primary adult human hepatocytes were obtained from BioreclamationIVT (Brussels, Belgium)
Human Liver Tissue
Protein lysate of normal liver tissue was obtained from Acris Antibodies GmbH (Herford, Germany)
Euchromatin and Heterochromatin Extraction Through Partial MNase Digestion
Nuclei were isolated from the 21 day old C3A 3D culture followed by chromatin fractionation and immunoblot analysis as described (11). Briefly, nuclei were isolated by cell lysis with Nuclear Isolation Buffer (NIB, 15 mm Tris pH = 7.5, 15 mm NaCl, 60 mm KCl, 5 mm MgCl2, 1 mm CaCl2, 250 mm sucrose and “complete” protease inhibitor mixture (Roche)) supplemented with 0.3% Nonidet P-40. The resulting nuclei pellet was separated by centrifugation (600 rpm for 5 min at 4 °C) and washed twice with NIB. Nuclei were resuspended in NIB to a concentration of ∼107 nuclei/ml and chromatin was partially digested with MNase (Sigma 1.6 U/ml) at 37 °C for 10 min. Reaction was quenched by 1 mm EGTA on ice for 10 min. The sample was centrifuged at 1000 rpm for 5 min at 4 °C to generate the first supernatant (S1). The pellet was resuspended in 2 mm EDTA at pH = 7.2 in the same volume as S1 and incubated on ice for 10 min. The sample was then centrifuged at 12,000 rpm at 4 °C to yield a second supernatant (S2) and a pellet (P).
Immunoblot Analysis
Whole cell extracts (WCEs) from cell cultures and liver tissues were prepared by cell/tissue sonication (10 cycles of 2s ON/5s OFF) on ice in modified ice-cold RIPA buffer (50 mm Tris-Cl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 0.5% sodium deoxycholate, and 0.5% SDS supplemented with “complete” protease inhibitor mixture (Roche) and “PhosSTOP” phosphatase inhibitors (Roche)) following by centrifugation (14,000 rpm, 10 min, 4 °C) to remove cell debris. Equal amount of WCE from each sample was subjected to Western blotting with following antibodies: anti-H2B (1:1000) (Abcam, ab1790), anti-H3 (1:3000) (Abcam, ab1791) and Donkey Anti-Rabbit IgG H&L (HRP) (1:5000) (Abcam, ab16284), as described (12).
Histone Purification
Histones were acid extracted as described previously (13). One hundred micrograms of bulk histone mixture was separated by reverse-phase high performance liquid chromatography (RP-HPLC) as previously described (14). Twelve fractions were collected between 28–72 min of elution, lyophilized and stored at −80 °C until further analysis.
Chromatography and Mass Spectrometry
Top-down Protein Analysis
RP-HPLC fractions containing H2B were combined and fractions containing H3 were combined. Then three μg of protein sample (H2B or H3) from each pool were analyzed by our hybrid weak cation exchange/hydrophilic interaction liquid chromatography-tandem mass spectrometry (WCX/HILIC-MS/MS) method (9, 14). Protein samples were separated by using an UltiMate3000 (Dionex) HPLC system, equipped with a two-column setup, consisting of a reversed-phase trap column (3 cm, 100 μm i.d., 360 μm o.d., packed with ReproSil, Pur C18AQ 3 μm; Dr. Maisch) and a WCX/HILIC analytical column (18 cm, 75 μm i.d. 360 μm o.d., packed with PolycatA 1500 Å, 3 μm, PolyLC Inc). Solvent A and solvent B were composed as follows: solvent A 75% MeCN (Fisher Scientific), 20 mm propionic acid (Sigma) at pH 6; solvent B 25% MeCN and 0.3% formic acid (Sigma) at pH 2.5. Solvent C used for sample loading was composed of 0.1% formic acid. Proteins were eluted directly into an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) with a 70-min linear gradient of 50–90% buffer B at flow rate of 300 nL/min. MS was controlled by Xcalibur software (Thermo Fisher Scientific). The nanoelectrospray ion source (Thermo Fisher Scientific) was used with a spray voltage of 1.9 kV. The ion transfer tube temperature was 275 °C. MS survey scans were obtained for the m/z range of 700–1200 in the orbitrap with maximum ion injection time of 50 ms, auto gain control target 5 × 105, resolution 60,000 and 10 microscans per each full MS event. Four most abundant precursor ions with charge state in the range 9 to 21 from each MS scan were selected for ETD with 20 ms reaction time. MS/MS spectra were measured in the orbitrap with maximum ion injection time 500 ms, auto gain control target 2 × 105, mass resolution 30,000 and 8 microscans per MS/MS. Dynamic exclusion was set to a repeat count of one with a repeat duration of 30 s, exclusion duration 45 s, and exclusion m/z width 10.
Middle-down Protein Analysis
Acid-precipitated bulk histones were resuspended in 100 mm NH4HCO3. Then a 50-μg aliquot of histones was subjected to endoproteinase GluC digestion (Calbiochem, 1:20 enzyme to substrate ratio 1:20) performed for 8 h at room temperature. The resulting peptide mixture was analyzed in triplicate by the WCX/HILIC-MS/MS method adapted from our previous studies (9, 14) and described above in a previous section. Peptides were eluted directly into the ESI source of a LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) using a 180-min linear gradient of 40–80% buffer B at flow rate of 300 nL/min. The mass spectrometer was controlled by Xcalibur software (Thermo Fisher Scientific). A nanoelectrospray ion source (Proxeon, Odense, Denmark) was used with a ESI voltage of 2.2 kV. Capillary temperature was 270 °C. Dynamic exclusion (DE) was disabled during data acquisition. Data acquisition was performed in the Orbitrap for both precursor ions and product ions, with a mass resolution of 60,000 (full-width at half-height) for MS and 30,000 for MS/MS. Precursor charge states 1+, 2+, and 3+ were excluded. Isolation width was set at 2 m/z. The six most intense ions with MS signal higher than 5000 counts were isolated for fragmentation using ETD with an activation Q value of 0.25, activation time of 90 ms with charge state dependent ETD time enabled, and Supplemental activation. Three microscans were used for each MS/MS spectrum, and the AGC target was set to 2 × 105. Acquisition window was set at m/z 450–750, including charge states 5+ to 11+.
Data Analysis
Middle-down protein analysis. Raw files were processed and searched with Mascot (version 2.3.2, Matrix Science, London, UK) using the Proteome Discoverer (version 1.4.0.288, Thermo Fischer Scientific) data analysis package. Spectra were deconvoluted by using Xtract (Thermo Fisher Scientific) with the following parameters: S/N threshold was set to 0, resolution at 400 m/z was 30,000 and monoisotopic mass only was true. The following parameters were used in the Mascot search: MS mass tolerance: 2.1 Da, to include possible errors in isotopic recognition; MS/MS mass tolerance: 0.01 Da; semi GluC specificity with no missed cleavage allowed; mono- and dimethylation (KR), trimethylation (K), acetylation (K), and phosphorylation (ST) were set as variable modifications. MS/MS spectra were searched against histone human database, consisted of histone H3.1/H3.2 and H3.3 N-terminal sequences, covering residues 1–50, downloaded from Uniprot (updated September 2011). XML results file from Mascot were exported by ticking all the query level information and the start and end from the peptide match information (plus all the default settings). XML files were imported and processed using an in-house developed software Histone Coder (9) (see supplemental methods, supplemental Table S3). Histone Coder was used with a tolerance of 30 ppm and only c/z fragment ions allowed. Only PTMs with at least one site-determining ion before and after the assigned PTM site were accepted. Peptide quantification was performed using in-house developed software IsoScale (9) (see supplemental methods, supplemental Table S4). By using IsoScale we extracted the total ion intensity from MS/MS spectra of identified peptides. We used the output from Histone Coder after eliminating the matches that contain 0 (zeros) in the site determining ion cell, excluding all PTMs with ambiguous assignments. In addition, we loaded the .csv result file(s) from Mascot with all the query information included. The total ion intensity obtained from the Mascot result files was summed for all peptide-spectrum matches (PSMs) and divided by the sum of all peptides in all modified forms for each histone to obtain the relative abundance of each PTM form. Different clipped histone proteoform were considered as different proteins. IsoScale also calculated the fragment ion relative ratio (FIRR) of isobaric co-fragmented peptides and divided the total ion intensity of the given MS/MS spectrum between the two species (see supplemental Methods). The MS/MS tolerance to identify fragment ion for FIRR calculation was set to 0.05 Da. The software tools Histone Coder and isoScale are freely available (9).
The relative abundance of each individual modification was calculated as sum of relative abundances of all PTM forms containing this modification. Different histone proteoforms were considered as a different proteins and relative abundance of each individual modification was calculated for each H3 proteoform separately.
The “interplay score” was used to evaluate the likelihood of two histone marks to coexist on the same polypeptide as previously described (15). Interplay score was calculated by using the following equation: Interplayxy = log2 (Fxy/(Fx * Fy)), where xy is the coexistence frequency of the binary mark, Fx is the frequency or relative abundance of the mark x and Fy is the frequency of the mark y.
For a detailed description of the top-down protein data analysis, please see supplemental Methods.
Supplemental information
Supplemental information is present in two files: Supplemental data.pdf and Supplemental tables.xml. Supplemental data.pdf contains supplemental Figs. (supplemental Fig. S1–Fig. S6), supplemental Tables (supplemental Table S1 and Table S2) and supplemental Methods. Supplemental tables.xml contains supplemental Table S3 and supplemental Table S4.
RESULTS
Histones H2B and H3 Undergo Proteolytic Processing in a Hepatocarcinoma Cell Line Grown in 3D Culture
We first demonstrated the occurrence of histone clipping in the human hepatocarcinoma cell line HepG2/C3A (hereafter C3A). We used immunoblotting to probe whole-cell extracts (WCEs) of the C3A cell line grown in flat culture (2D) and as spheroids (3D). By using antibodies (Abs) specific for the C-terminal part of histone H3 we reproducibly observed two faster migrating H3 bands in WCE isolated from 3D culture, but not in C3A or HeLa grown in conventional 2D culture (Fig. 1A). To confirm the obtained results we analyzed histone fraction isolated from 3D culture using SDS-PAGE and liquid chromatography-tandem mass spectrometry (LC-MS/MS) after limited in-gel trypsin digestion. As expected the faster migrating H3 band contained peptides corresponding to histone H3. Interestingly, in addition to H3 this band also contained peptides derived from the histone H2B, suggesting that this protein might also be proteolytically processed (supplemental Fig. S1A). We confirmed the presence of clipped H2B in WCE from C3A cells by Western blot analysis using anti-H2B Ab. The additional low molecular weight H2B band was detected only in 3D culture, but not in C3A or Hela 2D cultures (Fig. 1A).
Fig. 1.
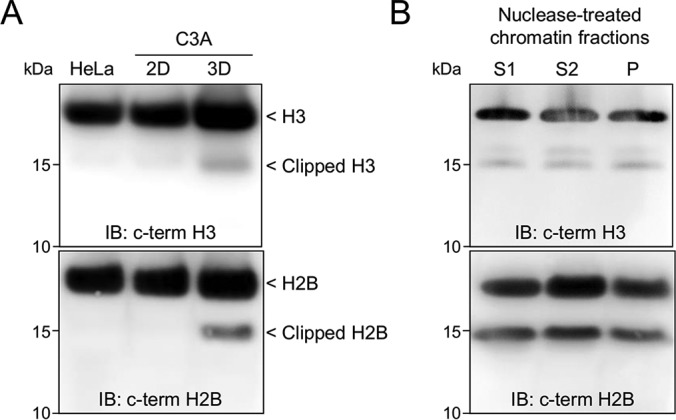
Histones H3 and H2B undergo proteolytic processing in 3D cell culture. A, C3A cell line cultured as a flat culture (2D) or as a spheroid culture (3D) and HeLa cell line (2D) were harvested and protein extracts were analyzed by immunoblotting(IB) using the antibodies indicated in each panel. B, Chromatin isolated from 21 days old 3D C3A cell culture was partially digested with MNase and subsequently fractionated into euchromatin (S1), heterochromatin (S2) and matrix-associated chromatin (P) subfractions. Resulting protein fractions were analyzed by immunoblotting.
To rule out the possibility that proteolysis of histone H3 occurred during sample preparation, we carried out a control experiment. Purified intact H3 from C3A cells grown in 2D culture was labeled with cysteine-reactive Tandem Mass Tag (TMT) reagent and added to 3D culture cell pellet with lysis buffer. Subsequently the cells were lysed and the resulting WCE was analyzed by Western blot using anti-TMT Ab (supplemental Fig. S2A). Uncontrolled degradation of TMT-tagged H3 by endogenous proteases during sample preparation would lead to multiple bands in the Western blot analysis. Thus the analysis revealed that histone H3 clipping did not occur during sample preparation (supplemental Fig. S2B). This procedure was not applicable to H2B because of the absence of cysteine residues in its sequence.
Clipped H2B and H3 Histone Forms are Associated with Eu- and Heterochromatin
To test whether clipped H2B and H3 are associated with distinct chromatin states, we evaluated the relative abundance of clipped histones in crude euchromatin and heterochromatin subfractions (11, 16, 17). Briefly, chromatin was isolated from 3D culture, partially digested with MNase and subsequently fractionated as described previously (11). The presence of clipped histones was examined by immunoblotting (Fig. 1B, supplemental Fig. S1B). We detected clipped H2B and H3 forms in both soluble (S1) and insoluble (S2) MNase-treated chromatin fractions, indicating that clipped histones are localized within both eu- and heterochromatin. Faster-migrating H2B and H3 bands were also observed in the final EDTA-insoluble chromatin pellet (P) containing matrix-associated chromatin (11).
Abundance of Clipped Histones Increases During C3A Cultivation as a 3D Culture
H3 clipping is involved in transcriptional silencing of cell cycle-promoting genes upon senescence in fibroblasts (7). Previously we demonstrated that C3A cells in 3D culture grow significantly slower than in classical 2D culture (18). The doubling time for the C3A cell line grown in 2D cultures is about 3 days, whereas in 3D it increases drastically to 17 days in 21-day old spheroids and to 60 days in 42-day old spheroids (18). To test whether histone clipping can be correlated with proliferation activity of the C3A cell line, we assessed the presence of clipped histones in 3D culture at different stages of cultivation (Fig. 2A). A signal from a faster migrating H3 band was first detected after 10 days of cultivation and increased up to day 15–24 (Fig. 2B). The similar trend was observed for histone H2B; the faster migrating H2B band was first observed after 10 days of C3A cultivation and increased in amount up to day 47 (Fig. 2B). To check whether histone clipping is associated with 3D culture or it occurs in cells growing by both cultivation techniques but gets diluted in 2D culture because of the faster cell doubling time we tested 2D culture cells exhibiting growth arrest caused by contact inhibition. C3A cells were grown in conventional 2D culture to confluence and then cultivated for 10 more days, prolonging the growth in arrested duplication state. Cells were collected at 1, 5, and 10 days post confluence and corresponding WCEs were probed with anti-H2B and anti-H3 Abs. No faster migrating histone band was detected in any of the post-confluence cultures (supplemental Fig. S3), demonstrating that histone clipping is not evenly accumulated in 2D culture, but is likely related to the functional differences between cells in 2D and 3D cultures. The possible contribution of cell death on proteolytic processing of histone proteins was eliminated by using TUNEL staining (data not shown), which confirmed that there are very few apoptotic cells (<3%) in the spheroids C3A culture, indicating that histone clipping is not caused by cell death-associated protein degradation.
Fig. 2.
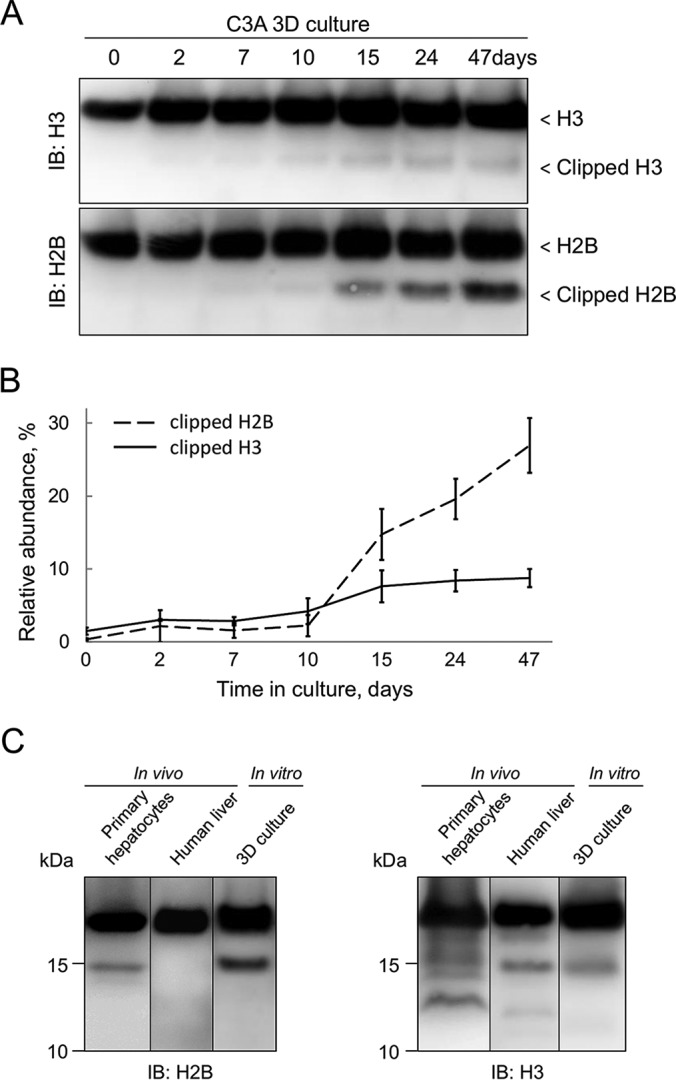
Clipped histones accumulate in 3D cell culture and are present in primary hepatocytes and liver tissue. A, C3A cell line was grown in 3D culture, harvested at the time points indicated on top of the panel and analyzed by immunoblotting. B, Semiquantitative analysis of clipped histone relative abundances by immunoblotting. Clipped H2B(H3) was normalized to total H2B(H3). Data are means ± s.d. (n = 3). C, WCEs from primary human hepatocytes, human liver bioptate and 3D culture were analyzed by immunoblotting with anti-H2B and anti-H3 Abs.
Histone Clipping Occurs in Human Primary Hepatocytes and Human Liver Tissue
Tumor-derived cell lines differ from both normal and tumor tissues (of the same origin) in many ways, including protein expression profiles (19). Much less is known about the epigenetic differences between those cell types, including differences in histone PTM abundance. To determine whether histone clipping is restricted to C3A cells and whether it also occurs in normal liver cells we analyzed human primary hepatocytes and liver tissue. Western blotting analysis confirmed the presence of clipped forms of both histones H3 and H2B in primary human hepatocytes (Fig. 2C). The immunoblot pattern for histone H2B was similar to those seen in hepatocarcinoma cell line, whereas for histone H3 more bands were detected in primary cells as compared with 3D culture. The similar pattern for H3 was observed in liver tissue lysate, however no faster migrating species for H2B were detected (Fig. 2C).
Identification of Histone Clipping Sites Using MS
To define the nature of the lower molecular weight histone proteoforms and determine the sequence localization of histone clipping sites we sequenced the faster migrating histone species using mass spectrometry (20). Histones were isolated from 3D culture and separated by reversed-phase liquid chromatography (RPLC) (13). The presence of clipped histones was determined by immunoblotting (supplemental Fig. S4C). Fractions containing faster migrating H2B and H3 species were analyzed using a top-down proteomics strategy, i.e. characterization of intact histones by using nanoliter flow LC interfaced to electrospray ionization tandem mass spectrometry (LC-MS/MS) (9, 21). We identified both the intact and clipped histone H2B proteoforms (supplemental Fig. S5, supplemental Table S1). The clipped H2B lacked 17 N-terminal residues demonstrating that proteolytic processing of H2B occurs between residues K17 and A18 (supplemental Fig. S5C).
Subsequently, we adopted a middle-down proteomics strategy optimized in our laboratory for the comprehensive characterization of H3 N-terminal tail PTM patterns for detailed analysis of clipped H3 (9, 14). Briefly, histones were isolated from 3D culture (or primary hepatocytes) and subjected to digestion by endoproteinase GluC to generate H3 N-terminal peptides covering amino acid residues 1–50. The peptide mixture was analyzed by LC-MS/MS using weak cation exchange/hydrophilic interaction liquid chromatography (WCX/HILIC) and high mass resolution ETD MS/MS. Two different H3 clipping sites at residues serine 10 (H3ΔS10) and glycine 12 (H3ΔG12) were localized (Fig. 3A, Fig. 3C) in H3 isolated from 3D culture and four different H3 clipping sites at residues lysine 9 (H3ΔK9), serine 10 (H3ΔS10), glycine G12 (H3ΔG12), and lysine 23 (H3ΔK23) were localized in H3 isolated from primary hepatocytes (Fig. 3B, supplemental Fig. S6). Clipped H3 proteoforms were identified for both H3.1/H3.2 and H3.3 variants: in 3D culture they were mainly represented by canonical histone H3, whereas in primary hepatocytes over 90% of clipped H3 correspond to the histone variant H3.3 (Table I, supplemental Fig. S6).
Fig. 3.

Determination of H3 and H2B clipping sites by mass spectrometry. A, Sequence of the intact histone H3 N-terminal tail peptide (black) with specified positions of two distinct H3 clipping sites localized by middle-down MS analysis of histones isolated from 3D culture: H3ΔS10 proteoform (red) and H3ΔG12 proteoform (green) are shown. B, Sequence of the intact histone H3.3 N-terminal tail peptide (black) with specified positions of four distinct H3.3 clipping sites localized by middle-down MS analysis of histones isolated from human primary hepatocytes: H3ΔK9 proteoform (purple), H3ΔS10 proteoform (red), H3ΔG12 proteoform (green), and H3ΔK23 proteoform (brown) are shown. C, Charge deconvoluted ETD-MS/MS spectra of intact H3 N-terminal tail peptide and two distinct clipped H3 N-terminal tail peptides (H3ΔS10 and H3ΔG12) obtained by middle-down MS/MS analysis of histones isolated from 3D culture. Matched z, z+1, z+2 type, and c type fragment ions are indicated by blue and red colors, respectively.
Table I.
N-terminal histone H3 tails identified by middle-down protein analysis by mass spectrometry. All LC-MS/MS files were processed by MASCOT and in-house developed software HistoneCoder to verify correct assignment of PTMs
| Protein name | Residues number | Modification state (top 5) | Theoretical mass, Da | Measured mass, Da | Delta mass, Da | Number of PSMs | Number of combinatorial PTMs | |
|---|---|---|---|---|---|---|---|---|
| H3.1/H3.2 | Intact H3 | 1–50 | 5 me | 5408.145 | 5408.176 | 0.031 | 2111 | 176 |
| 5 me +1 ac | 5450.156 | 5450.184 | 0.028 | |||||
| 3 me | 5380.114 | 5380.149 | 0.035 | |||||
| 4 me | 5394.130 | 5394.162 | 0.032 | |||||
| 3 me +1 ac | 5422.125 | 5422.155 | 0.030 | |||||
| H3ΔS10 | 11–50 | 2 me | 4238.446 | 4238.471 | 0.025 | 157 | 18 | |
| 3 me | 4252.462 | 4252.490 | 0.028 | |||||
| 3 me+1 ac | 4294.473 | 4294.502 | 0.029 | |||||
| 2 me+1 ac | 4280.457 | 4280.487 | 0.030 | |||||
| 4 me | 4266.478 | 4266.509 | 0.031 | |||||
| H3ΔG12 | 13–50 | 2 me | 4080.377 | 4080.404 | 0.027 | 574 | 31 | |
| 3 me | 4094.393 | 4094.421 | 0.028 | |||||
| 2 me+1 ac | 4122.388 | 4122.415 | 0.027 | |||||
| 3 me+1 ac | 4136.404 | 4136.429 | 0.025 | |||||
| 4 me | 4108.409 | 4108.436 | 0.027 | |||||
| H3.3 | Intact H3 | 1–50 | 5 me | 5424.141 | 5424.175 | 0.034 | 66 | 36 |
| 3 me | 5396.109 | 5396.15 | 0.041 | |||||
| 5 me +1 ac | 5466.151 | 5466.194 | 0.043 | |||||
| 4 me | 5410.125 | 5410.177 | 0.052 | |||||
| 3 me +1 ac | 5438.1200 | 5438.18 | 0.060 | |||||
| HΔS10 | 11–50 | 3 me | 4268.457 | 4268.511 | 0.054 | 4 | 4 | |
| 1 me + 1 ac | 4282.436 | 4282.492 | 0.056 | |||||
| H3ΔG12 | 13–50 | 2 me + 1 ac | 4138.383 | 4138.426 | 0.043 | 4 | 2 | |
| 3 me + 1 ac | 4152.398 | 4152.442 | 0.043 | |||||
We also observed truncated H3 N-tails starting at residue L20 (H3ΔQ19). The presence of such peptides can be explained by glutamine deamidation which results in the conversion of Gln19 to Glu19 (22). This, consequently, may lead to generation of artificial truncated H3 tails during histone digestion by endoproteinase GluC. We confirmed this hypothesis by analysis of Arg-C digested H3 using selected reaction monitoring (SRM) (supplemental Fig. S7).
Clipped H3 Proteoforms have Specific PTM Signatures
We further characterized clipped H3 proteoforms with respect to their PTMs and compared the relative abundance of modifications in intact and clipped H3 tails. Using the middle-down proteomics strategy we mapped PTMs in intact H3 proteoforms: K4me1, R8me1, R26me1, R17me1/me2, K9me1/me2/me3/ac, K14me1/me2/me3/ac, K18me1/ac, K23me1/me2/me3/ac, K27me1/me2/me3/ac, and K36me1/me2/me3. Clipped H3 proteoforms were found to be modified at different lysine residues, including K14me1/ac, K18me2/me3/ac, K23me2/me3/ac, K27me1/me2/me3/ac, and K36me1/me2/me3. We mapped 212 unique combinatorial PTMs on intact H3 N-terminal tails and 55 combinatorial PTMs on two different clipped H3 N-terminal tails (Table I). IsoScale, an in-house developed software, was used for quantification of relative PTM abundance (9). The most frequently observed co-existing PTMs in intact H3 N-terminal tail were K9me2K27me3, K9me1K27me2, K9me1K14me1K27me2, K9me2K14acK27me3, K9me1K14me1K23acK27me2, K23acK27me2, K23acK27me3 (Fig. 4B). The most abundant single marks were K27me2, K27me3, K23ac, K14me1, K14ac, K9me1, and K9me2 (supplemental Table S2). The most frequently observed co-existing PTMs in clipped H3 N-terminal tails were K14acK27me2, K23acK27me2, K14acK27me3, K27me2K36me2, K18acK27me3, K27me2K36me1, and K23acK27me3 (in H3ΔS10) and K23acK27me2, K23acK27me3, K27me2K36me2, K27me2K36me1, K27me3K36me1, K14acK27me2, and K27me1K36me2 (in H3ΔG12). The abundance of some combinatorial PTMs, including K23acK27me2/me3, was similar between intact and clipped H3, whereas some combinatorial PTMs, including K27me2K36me1/me2, were enriched in clipped H3 proteoforms (Fig. 4C). To further explore the difference between intact and clipped H3 we compared the relative abundance of individual H3 PTMs. We found that clipped histone H3 N-terminal tails contain PTMs with significantly different frequency as compared with intact tails (Table II, Fig. 4A). One of the most dramatic differences was observed for K14me1 and K27me3, which were significantly lower in abundance in clipped H3 (t test p value <0.005). At the same time K36me1/K36me2 and K27me2 abundance was increased in clipped H3 tails (p value <0.05), (Table II, Fig. 4A). Although two clipped H3 proteoforms demonstrated similar PTM profiles, K14ac was found to be enriched in H3ΔS10 as compared with H3ΔG12. Other marks exhibiting significantly different relative abundance between intact and clipped H3 peptides were K27me1, K27ac, and K36me3, however, because of their low abundance these marks were excluded from further analysis.
Fig. 4.

Clipped histone H3 proteoforms contain PTMs with significantly different relative abundance as compared with intact H3. A, Relative abundances of the ten most abundant individual PTMs in the intact and clipped H3 proteoforms identified by middle-down MS/MS analysis. The relative abundance of each individual modification was calculated as the sum of relative abundances of all distinct PTM peptide forms containing this particular modification. Different H3 proteoforms are indicated by color in the top of the panel. The same colors for different H3 proteoforms are used in bar plot (C). Data are means ± s.d. (n = 6). Asterisk indicates differentially abundant PTMs (* (p < 0.05), ** (p < 0.005), and *** (p < 0.0005)). B, Relative abundances of the seven most frequently observed combinatorial marks identified in N-terminal tail of intact H3. C, Relative abundances of the nine most frequently observed combinatorial marks identified in N-terminal tails of clipped H3ΔS10 and H3ΔG12 proteoforms. The frequencies of these combinatorial marks in intact H3 were calculated as a sum of the frequency of the combinatorial PTM in intact H3 and frequencies of all combinatorial PTMs containing this PTM pair in combination with any of modifications of K4 and K9 (e.g. the frequency of K14acK27me2 was calculated as a sum of frequencies of K14acK27me2, K9me1K14acK27me2, K4me1K9me1K14acK27me2, and K9me3K14acK27me2).
Table II.
Comparison of relative PTM abundances of intact and clipped histone H3 proteoforms. Statistically significant difference is indicated by * (p < 0.05), ** (p < 0.005), and *** (p < 0.0005). No significant difference is indicated by n.s. Marks that are differentially abundant (p < 0.05) between intact and clipped H3 proteoforms are indicated in bold
| Relative PTM abundance, % |
t-test (H3ΔS10 vs Intact H3) | t-test (H3ΔG12 vs Intact H3) | |||
|---|---|---|---|---|---|
| Intact H3 | H3ΔS10 | H3ΔG12 | |||
| K14me1 | 17.5 | 0 | 0.1 | *** | *** |
| K14ac | 8.6 | 17.2 | 6.1 | n.s. | n.s. |
| K18ac | 1.3 | 7.9 | 0.5 | n.s. | n.s. |
| K23ac | 20.4 | 11.8 | 16.5 | n.s. | n.s. |
| K27me1 | 4.7 | 1.2 | 6.7 | ** | n.s. |
| K27me2 | 41.8 | 58.3 | 58.7 | ** | *** |
| K27me3 | 48.0 | 38.0 | 33.8 | ** | *** |
| K27ac | 2.7 | 1.0 | 0 | n.s. | ** |
| K36me1 | 1.8 | 5.9 | 9.9 | * | *** |
| K36me2 | 2.1 | 8.2 | 9.1 | ** | *** |
| K36me3 | 4.4 | 0.9 | 3.5 | ** | n.s. |
PTMs Interplay Within Intact and Clipped Histone H3
Next, using the data obtained by the middle-down proteomics analysis of residues 1–50 of the histone H3 tail we investigated the interplay between the different PTMs within intact and clipped histone H3. We calculated the “interplay score” of binary PTMs by comparing observed PTM pair frequencies with theoretical PTM pair frequencies (15). The “observed” co-occurrence frequencies of each co-existing PTM pairs were calculated based on the frequencies of peptides with combinatorial PTMs containing these PTM pairs. Theoretical PTM pair co-occurrence frequencies were calculated based on the single PTM relative abundances on the assumption that they are independent (14). Briefly, such interplay score considers how often two PTMs should co-exist on the same protein based on their relative abundance and compares this value with the experimental value (15). We examined which PTMs are unlikely to co-exist (negative interplay) and which are most likely co-dependent (positive interplay) for intact and clipped H3 N-terminal tails. The most abundant PTMs including K14ac, K23ac, K27me2/me3, and K36me1/me2 were examined (Fig. 5A). We found that K27me3 rarely co-exist with K36me1/me2 (except for the H3ΔG12 tail). This result conforms to previous studies suggesting antagonism between high methylation states of H3K27 and H3K36 (23, 24). K14ac and K23ac were found to be mutually exclusive by interplay analysis, in agreement with a previous report (15). Interestingly, the calculated interplay scores for some of the H3 PTM pairs were different for intact and clipped H3 tails. For instance, K36me1/me2 was found with a negative interplay score with K23ac on clipped H3 tails, whereas this was not so pronounced in intact H3 tails (Fig. 5A, Fig. 6).
Fig. 5.
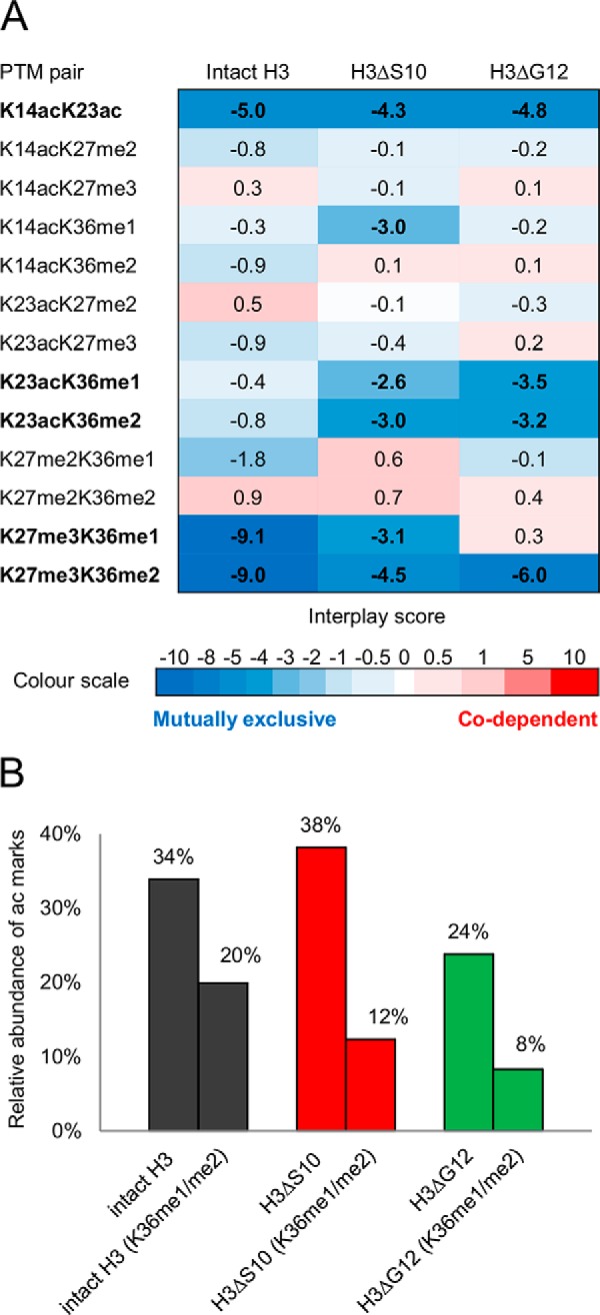
Analysis of co-existing histone H3 PTMs reveal mutually exclusive modifications in intact and clipped H3 proteoforms. A, Heat map of interplay scores for H3 N-tail PTM binary combinations. Positive and negative interplay is represented in red and blue, respectively. PTM pairs with negative (positive) interplay values co-occur less (more) frequently than if they were independent. A negative interplay score corresponds therefore to mutual exclusion indicating competition between the two marks whereas positive interplay reveals enhanced, correlated crosstalk. PTMs which appear to be mutually exclusive are indicated in bold. B, The total relative abundance of acetylation marks in peptides carrying K36me1 or K36me2 and the total relative abundance of acetylation marks calculated based on all identified peptides. Histone H3 containing K36me1 or K36me2 demonstrate hypoacetylated status.
Fig. 6.
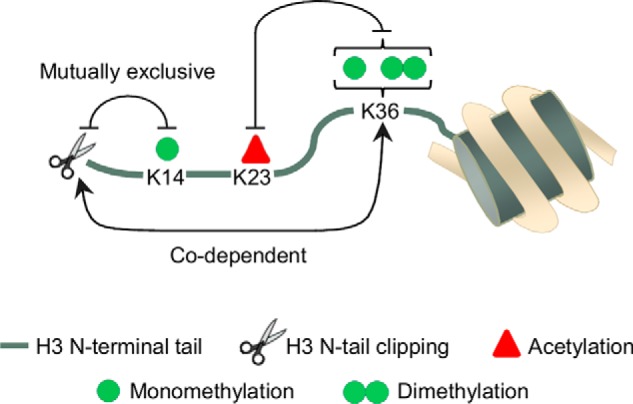
PTM crosstalk in the clipped histone H3. Schematic representation of PTM crosstalk in clipped H3. Positive crosstalk is indicated by an arrowhead line and a negative crosstalk is indicated by a flat head line. Histone H3 clipping positively correlates with mono- and dimethylation of K36 and negatively correlates with methylation of K14. K36 methylation and K23 acetylation are mutually exclusive in clipped H3.
To further explore the relation between H3K36 methylation and H3 N-tail acetylation status we compared the total relative abundance of acetylation marks in peptides carrying K36me1 or K36me2 with the total relative abundance of acetylation calculated based on all identified acetylated peptides. Both intact and clipped H3 tails containing K36me1 or K36me2 exhibited a hypo-acetylated status, which was especially pronounced in clipped H3 proteoforms (2.8-fold decrease (H3ΔG12) and 3-fold decrease (H3ΔS10) versus 1.7-fold decrease (intact H3)) (Fig. 5B). Collectively, our results demonstrate that clipped H3 forms contain distinct PTM patterns that are different from those in intact H3.
DISCUSSION
Histone Clipping Activity in Human Hepatocytes
A number of studies demonstrated that clipping of the N-terminal tail of histone H3 takes place in various organisms, including Tetrahymena thermophila, Saccharomyces cerevisiae, chicken, mouse, and human (1, 2, 5–7, 25, 26). Although this phenomenon is likely associated with transcriptional activation in yeast, the significance of H3 clipping in higher eukaryotes remains poorly understood (1–4). Here, we demonstrate that site-specific clipping of core histones H2B and H3 takes place in human hepatocytes both in vitro and in vivo. Histone clipping was induced during the cultivation of human hepatocarcinoma cell line, HepG2/C3A, in 3D culture but not in 2D culture. There is growing evidence that cell culture techniques can have a profound effect on cell metabolism (27, 28). In particular, cells cultured in 3D spheroids more closely mimic original tissues compared with those grown using traditional 2D cultures (29). Recently Duarte et al. proposed a role for histone H3.3 clipping in down-regulation of cell cycle-promoting genes during senescence in human fibroblasts (30). Previously we demonstrated that proliferation rate of hepatocarcinoma cells constitutively decreases during the cultivation in 3D culture and after 21 days of growth the majority of cells are either arrested in G1 or have entered Go phase (31). We now show that the abundance of clipped H2B and H3 consistently increases in C3A cells during their cultivation in 3D culture. By using middle-down MS analysis of histones isolated from human primary hepatocytes we identified four different clipped H3 proteoforms—namely H3ΔK9, H3ΔS10, H3ΔG12, and H3ΔK23—and demonstrate that clipping preferentially occurs in histone variant H3.3 at the same regions within N-terminal tail (between lysine 9 and glycine 12 and after lysine 23) as was observed in senescent fibroblasts (30). Although the role of histone clipping in hepatocytes remains undefined, we hypothesize that it might be involved in regulation of cell-cycle progression as was previously proposed for human fibroblasts.
Histone modifications are known to play an important role in the regulation of many chromatin-associated processes. Since Allis and colleagues have proposed the “histone code hypothesis” in 2001, many findings provided strong evidence that histone modifications can act in a cooperative manner (32–34). Different combinations of histone PTMs lead to different functional outcomes, providing a strong and flexible platform for the regulation of key cellular processes. Previously, Duncan et al. found that different H3 PTMs can modulate proteolytic activity of the Cathepsin L enzyme, which was found to be responsible for histone H3 clipping during mouse ESCs differentiation, in vitro (5). The combinatorial PTM status of cellular clipped histones, however, remains poorly defined. By using an optimized middle-down MS platform that is highly efficient for the characterization of histone PTM isoforms we carried out for the first time a comprehensive comparison of histone PTMs between intact and clipped H3 N-terminal tails isolated from the C3A cell line grown in 3D culture. First, by using MS and MS/MS we identified two different clipped proteoforms of H3, namely H3ΔS10 and H3ΔG12. Furthermore we show that the relative abundance of distinct H3 PTMs, such as K14me1, K27me2, K27me3, K36me1, and K36me2, is significantly different between clipped and intact H3 N-terminal tails. H3ΔS10 and H3ΔG12 forms were found to contain extremely low levels of K14me1, suggesting a strong negative correlation between the H3K14 methylation and clipping of H3. This finding supports the previously reported idea that H3 clipping can be regulated by other H3 PTMs (5). However which of these events occurs first is not known. Thus, there are several possible explanations of the relationship between H3 clipping and H3 lysine 14 methylation. First, H3K14me1, as a mark near the H3 clipping site, can decrease the efficiency of the cleavage site recognition by the “clipping protease,” thus, preventing H3 processing. Alternatively, H3K14me1 can be specifically demethylated in clipped H3. It is also possible that these events do not have a direct link and H3 clipping just occurs in chromatin regions which have low level of H3K14 methylation. These questions need to be addressed in future studies.
We found that clipped H3 proteoforms H3ΔS10 and H3ΔG12 are highly enriched in both H3K36me1 and H3K36me2 as compared with intact H3. Histone H3 K36 methylation is known as an active mark which has several well established roles in different nuclear processes, including transcription, DNA repair and regulation of Polycomb repressive complex 2 (PRC2) (23, 35–37).
Numerous studies demonstrated that H3K36 methylation is associated with transcribed chromatin in various organisms (38–40). In human TKO2 myeloma cell line H3K36me2 was enriched near the transcription start sites (TSSs) and the level of H3K36me2 correlated with gene activity (41). In yeast co-transcriptional dimethylation of H3K36, catalyzed by RNA polymerase II (pol II)-associated methyltransferase Set2, recruits Rpd3S deacetylase complex and consequently prevents aberrant transcriptional initiation within coding sequences by maintaining the hypo-acetylated state of transcribed chromatin (42, 43). Our data reveal that clipped H3 proteoforms are over 4 times enriched in K36me2 as compared with intact H3. Furthermore, H3 tails methylated at K36 exhibit a low acetylation level primarily because of the negative crosstalk between K36me1/me2 and H3K23ac. Taken together, these findings demonstrate that clipped H3 proteoforms have a PTMs pattern similar to those known to occur in H3 located in transcribed chromatin regions. Although the role of histone clipping in transcriptional regulation still needs to be explored previous studies suggested that this irreversible modification can act both as a transcriptional activator and repressor (1, 30, 44, 45). Here we provide firs time comprehensive characterization of H3 clipping sites in human cells. We mapped four different cleavage sites within the H3 N-terminal tail: after lysine 9 (H3ΔK9), serine 10 (H3ΔS10), glycine 12 (H3ΔG12), and lysine 23 (H3Δ23). This result can be used for generation of antibodies specific to distinct clipped H3 proteoforms and further characterization of proteolytic processing of histone H3 and its genomic distribution.
Although proteolytic processing of histone H3 has been demonstrated in several species there was no prior evidence of clipping of H2B, except for nickel-induced H2B truncation (46). We here provide the first evidence of H2B clipping as a part of normal cellular functionality. Using top-down MS we identified two distinct intact H2B variants, H2B type 1-C/E/F/G/I and H2B type 2-E. The clipped proteoform lacking 17 N-terminal residues was identified only for H2B type 1-C/E/F/G/I, suggesting that H2B clipping might be variant-specific.
In summary, our study provides the first observation of the proteolytic processing of histones H2B and H3 in human hepatocytes and the first detailed PTM characterization of clipped H3, including co-existing PTMs. Our data highlight the relationship between H3 clipping and canonical H3 PTMs and provide a basis for further investigation of this phenomenon.
Supplementary Material
Acknowledgments
We thank Dr. Ove Schaffalitzky de Muckadell for providing the human liver biopsy sample.
Footnotes
Author contributions: A.T., K.W., S.J.F., A.R., and O.N.J. designed research; A.T., K.W., and S.S. performed research; A.T. and S.S. analyzed data; A.T. wrote the paper; K.W., S.J.F., and A.R. edited the manuscript; O.N.J. edited the manuscript, supervised the project.
* This work was supported by the VILLUM Center for Bioanalytical Sciences (VILLUM Foundation) and a generous grant to the Center for Epigenetics from the Danish National Research Foundation (DNRF #82). SJF and KW were supported by MC2 Biotek.
 This article contains supplemental Figs. S1 to S6, Tables S1 to S4 and Methods.
This article contains supplemental Figs. S1 to S6, Tables S1 to S4 and Methods.
1 The abbreviations used are:
- List of Abbreviations: DMEM
- Dulbecco's modified Eagle medium
- ETD
- electron transfer dissociation
- GDH
- glutamate dehydrogenase
- hESC
- human embryonic stem cell
- mESC
- mouse embryonic stem cell
- MNase
- micrococcal nuclease
- MS/MS
- tandem mass spectrometry
- PTM
- post translational modification
- PSM
- peptide spectrum match
- RP-HPLC
- reversed-phase high performance liquid chromatography
- SRM
- selected reaction monitoring
- TMT
- tandem mass tag
- WCE
- whole cell extract
- WCX/HILIC
- weak cation exchange/hydrophilic interaction liquid chromatography.
REFERENCES
- 1.Santos-Rosa H., Kirmizis A., Nelson C., Bartke T., Saksouk N., Cote J., and Kouzarides T. (2009) Histone H3 tail clipping regulates gene expression. Nat. Struct. Mol. Biol. 16, 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azad G. K., and Tomar R. S. (2014) Proteolytic clipping of histone tails: the emerging role of histone proteases in regulation of various biological processes. Mol. Biol. Rep. 41, 2717–2730 [DOI] [PubMed] [Google Scholar]
- 3.Zhou P., Wu E., Alam H. B., and Li Y. (2014) Histone Cleavage as a Mechanism for Epigenetic Regulation: Current Insights and Perspectives. Current Mol. Med. 14, 1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhaenens M., Glibert P., Meert P., Vossaert L., and Deforce D. (2015) Histone proteolysis: A proposal for categorization into ‘clipping’ and ‘degradation’. Bioessays 37, 70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duncan E. M., Muratore-Schroeder T. L., Cook R. G., Garcia B. A., Shabanowitz J., Hunt D. F., and Allis C. D. (2008) Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135, 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vossaert L., Meert P., Scheerlinck E., Glibert P., Van Roy N., Heindryckx B., De Sutter P., Dhaenens M., and Deforce D. (2014) Identification of histone H3 clipping activity in human embryonic stem cells. Stem Cell Res. 13, 123–134 [DOI] [PubMed] [Google Scholar]
- 7.Duarte L. F., Young A. R., Wang Z., Wu H. A., Panda T., Kou Y., Kapoor A., Hasson D., Mills N. R., Ma'ayan A., Narita M., and Bernstein E. (2014) Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 5, 5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams-Cioaba M. A., Krupa J. C., Xu C., Mort J. S., and Min J. (2011) Structural basis for the recognition and cleavage of histone H3 by cathepsin L. Nat. Commun. 2, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sidoli S., Schwammle V., Ruminowicz C., Hansen T. A., Wu X., Helin K., and Jensen O. N. (2014) Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 14, 2200–2211 [DOI] [PubMed] [Google Scholar]
- 10.Wrzesinski K., and Fey S. J. (2013) After trypsinisation, 3D spheroids of C3A hepatocytes need 18 days to re-establish similar levels of key physiological functions to those seen in the liver. Toxicol. Res. 2, 123–135 [Google Scholar]
- 11.Torrente M. P., Zee B. M., Young N. L., Baliban R. C., LeRoy G., Floudas C. A., Hake S. B., and Garcia B. A. (2011) Proteomic interrogation of human chromatin. PLoS ONE 6, e24747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baird L., Lleres D., Swift S., and Dinkova-Kostova A. T. (2013) Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. U.S.A. 110, 15259–15264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shechter D., Dormann H. L., Allis C. D., and Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 14.Jung H. R., Sidoli S., Haldbo S., Sprenger R. R., Schwammle V., Pasini D., Helin K., and Jensen O. N. (2013) Precision mapping of coexisting modifications in histone H3 tails from embryonic stem cells by ETD-MS/MS. Anal. Chem. 85, 8232–8239 [DOI] [PubMed] [Google Scholar]
- 15.Schwammle V., Aspalter C. M., Sidoli S., and Jensen O. N. (2014) Large scale analysis of co-existing post-translational modifications in histone tails reveals global fine structure of cross-talk. Mol. Cell. Proteomics 13, 1855–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmiedeberg L., Weisshart K., Diekmann S., Meyer Zu Hoerste G., and Hemmerich P. (2004) High- and low-mobility populations of HP1 in heterochromatin of mammalian cells. Mol. Biol. Cell 15, 2819–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henikoff S., Henikoff J. G., Sakai A., Loeb G. B., and Ahmad K. (2009) Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 19, 460–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wrzesinski K., Magnone M. C., Hansen L. V., Kruse M. E., Bergauer T., Bobadilla M., Gubler M., Mizrahi J., Zhang K. L., Andreasen C. M., Joensen K. E., Andersen S. M., Olesen J. B., de Muckadell O. B. S., and Fey S. J. (2013) HepG2/C3A 3D spheroids exhibit stable physiological functionality for at least 24 days after recovering from trypsinisation. Toxicol. Res. 2, 163–172 [Google Scholar]
- 19.Sandberg R., and Ernberg I. (2005) Assessment of tumor characteristic gene expression in cell lines using a tissue similarity index (TSI). Proc. Natl. Acad. Sci. U.S.A. 102, 2052–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidoli S., Cheng L., and Jensen O. N. (2012) Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteomics 75, 3419–3433 [DOI] [PubMed] [Google Scholar]
- 21.Molden R. C., and Garcia B. A. (2014) Middle-Down and Top-Down Mass Spectrometric Analysis of Co-occurring Histone Modifications. Curr. Protoc. Protein Sci. 77, 23 27 21,–23 27 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson N. E., Robinson Z. W., Robinson B. R., Robinson A. L., Robinson J. A., Robinson M. L., and Robinson A. B. (2004) Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pept. Res. 63, 426–436 [DOI] [PubMed] [Google Scholar]
- 23.Yuan W., Xu M., Huang C., Liu N., Chen S., and Zhu B. (2011) H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y., Sweet S. M., Popovic R., Martinez-Garcia E., Tipton J. D., Thomas P. M., Licht J. D., and Kelleher N. L. (2012) Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. U.S.A. 109, 13549–13554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allis C. D., Bowen J. K., Abraham G. N., Glover C. V., and Gorovsky M. A. (1980) Proteolytic processing of histone H3 in chromatin: a physiologically regulated event in Tetrahymena micronuclei. Cell 20, 55–64 [DOI] [PubMed] [Google Scholar]
- 26.Mandal P., Azad G. K., and Tomar R. S. (2012) Identification of a novel histone H3 specific protease activity in nuclei of chicken liver. Biochem. Biophys. Res. Commun. 421, 261–267 [DOI] [PubMed] [Google Scholar]
- 27.Wu Y. M., Tang J., Zhao P., Chen Z. N., and Jiang J. L. (2009) Morphological changes and molecular expressions of hepatocellular carcinoma cells in three-dimensional culture model. Exp. Mol. Pathol. 87, 133–140 [DOI] [PubMed] [Google Scholar]
- 28.Frith J. E., Thomson B., and Genever P. G. (2010) Dynamic three-dimensional culture methods enhance mesenchymal stem cell properties and increase therapeutic potential. Tissue Eng. Part C Methods 16, 735–749 [DOI] [PubMed] [Google Scholar]
- 29.Pampaloni F., Reynaud E. G., and Stelzer E. H. (2007) The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 8, 839–845 [DOI] [PubMed] [Google Scholar]
- 30.Duarte L. F., Young A. R. J., Wang Z. C., Wu H. A., Panda T., Kou Y., Kapoor A., Hasson D., Mills N. R., Ma'ayan A., Narita M., and Bernstein E. (2014) Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wrzesinski K., Rogowska-Wrzesinska A., Kanlaya R., Borkowski K., Schwammle V., Dai J., Joensen K. E., Wojdyla K., Carvalho V. B., and Fey S. J. (2014) The Cultural Divide: Exponential Growth in Classical 2D and Metabolic Equilibrium in 3D Environments. PLoS ONE 9, e106973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenuwein T., and Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 33.Bannister A. J., and Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rando O. J. (2012) Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr. Opin. Genet. Dev. 22, 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner E. J., and Carpenter P. B. (2012) Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keogh M. C., Kurdistani S. K., Morris S. A., Ahn S. H., Podolny V., Collins S. R., Schuldiner M., Chin K., Punna T., Thompson N. J., Boone C., Emili A., Weissman J. S., Hughes T. R., Strahl B. D., Grunstein M., Greenblatt J. F., Buratowski S., and Krogan N. J. (2005) Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123, 593–605 [DOI] [PubMed] [Google Scholar]
- 37.Fnu S., Williamson E. A., De Haro L. P., Brenneman M., Wray J., Shaheen M., Radhakrishnan K., Lee S. H., Nickoloff J. A., and Hromas R. (2011) Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. U.S.A. 108, 540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kizer K. O., Phatnani H. P., Shibata Y., Hall H., Greenleaf A. L., and Strahl B. D. (2005) A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 25, 3305–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao F., and Natarajan R. (2005) Mapping global histone methylation patterns in the coding regions of human genes. Mol. Cell. Biol. 25, 4650–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris S. A., Shibata Y., Noma K., Tsukamoto Y., Warren E., Temple B., Grewal S. I., and Strahl B. D. (2005) Histone H3 K36 methylation is associated with transcription elongation in Schizosaccharomyces pombe. Eukaryot. Cell 4, 1446–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo A. J., Cheung P., Chen K., Zee B. M., Kioi M., Lauring J., Xi Y., Park B. H., Shi X., Garcia B. A., Li W., and Gozani O. (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B., Jackson J., Simon M. D., Fleharty B., Gogol M., Seidel C., Workman J. L., and Shilatifard A. (2009) Histone H3 Lysine 36 Dimethylation (H3K36me2) Is Sufficient to Recruit the Rpd3s Histone Deacetylase Complex and to Repress Spurious Transcription. J. Biol. Chem. 284, 7970–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carrozza M. J., Li B., Florens L., Suganuma T., Swanson S. K., Lee K. K., Shia W. J., Anderson S., Yates J., Washburn M. P., and Workman J. L. (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 [DOI] [PubMed] [Google Scholar]
- 44.Huh J. W., Wu J., Lee C. H., Yun M., Gilada D., Brautigam C. A., and Li B. (2012) Multivalent di-nucleosome recognition enables the Rpd3S histone deacetylase complex to tolerate decreased H3K36 methylation levels. EMBO J. 31, 3564–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Protacio R. U., Li G., Lowary P. T., and Widom J. (2000) Effects of histone tail domains on the rate of transcriptional elongation through a nucleosome. Mol. Cell. Biol. 20, 8866–8878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karaczyn A. A., Golebiowski F., and Kasprzak K. S. (2005) Truncation, deamidation, and oxidation of histone H2B in cells cultured with nickel(II). Chem. Res. Toxicol. 18, 1934–1942 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.