

Abstract
Aldol additions to isobutyraldehyde and cyclohexanone with lithium enolates derived from acylated oxazolidinones (Evans enolates) are described. Previously characterized trisolvated dimeric enolates undergo rapid addition to isobutyraldehyde to give a 12:1 syn:syn selectivity in high yield along with small amounts of one anti isomer. The efficacy of the addition depends critically on aging effects and the reaction quench. Unsolvated tetrameric enolates that form on warming the solutions are unreactive toward isobutyraldehyde and undergo retroaldol reaction under forcing conditions. Additions to cyclohexanone are relatively slow but form a single isomeric adduct in >80% yield. The ketone-derived aldolates are robust. All attempts to control stereoselectivity by controlling aggregation failed. Rate studies of addition to cyclohexanone trace the lack of aggregation-dependent selectivities to a monomer-based mechanism. The synthetic implications and possible utility of lithium enolates in Evans aldol additions are discussed.
TOC image
Introduction
Enolates bearing chiral oxazolidinone auxiliaries—so-called Evans enolates—have taken their rightful place in the annals of organic synthesis.1 Since the original report by Evans, Bartroli, and Shih2 in 1981, more than 1600 patents mentioning Evans enolates have been filed. Curious gaps in the technology persist, however. Whereas alkylations of lithiated Evans enolates remain central to asymmetric synthesis,1,3 the corresponding lithium-based aldol additions of enormous potential application in polyketide syntheses have drifted into relative obscurity (eq 1).2,4,5 The problem began with the seminal 1981 Evans et al.2 paper reporting a nearly stereorandom aldol addition using lithium, which redirected the investigators to the highly selective boron variant. Evans and other aldol enthusiasts subsequently developed asymmetric aldol additions by using Lewis acidic counterions containing titanium, boron, tin, zinc, and magnesium while exploiting an incredible range of oxazolidinone auxiliaries.1
 |
(1) |
Despite the success of the alternatives, lithium-based Evans aldol additions appeared sporadically—a dozen or so cases in total.4,5 Some show low yields and poor selectivities. There is also an oddly high proportion of additions to ketones,5 which are widely believed to be poor substrates owing to the tendency of hindered aldolates to undergo retroaldol additions.6 Singer et al.5a at Pfizer developed a plant-scale synthesis exploiting the addition in eq 2. We consulted Singer about the details, and he noted emphatically and with a noticeable grimace, “everything mattered.”
 |
(2) |
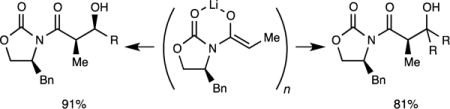
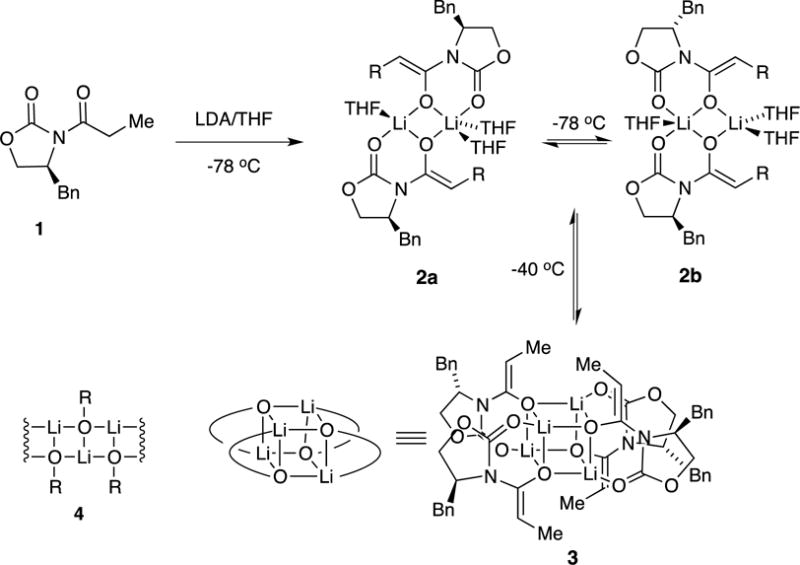
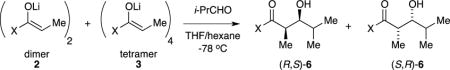
We describe herein studies of the aldol addition of lithiated Evans enolates.7 The first paper in this series laid the structural foundations (Scheme 1): spectroscopic and computational studies of several dozen structurally diverse enolates revealed isomeric dimers (2a and 2b), tetramers bearing D2d-symmetric cubic cores (3), and oligomers suspected to be ladders of various lengths (4).8 No monomeric enolates were detected under normal conditions.9 The distribution of aggregates depended on the choice of oxazolidinone auxiliary, steric demands of the substituent on the anionic enolate carbon, enolate and THF concentrations, and even the temperature of the enolization. On this last point, we noted in passing that enolizations of acylated oxazolidinones give dimeric enolates kinetically, and they equilibrate to various proportions of tetramer 3 only on warming. This equilibration proves to be key.10
Scheme 1.
We have studied two aldol additions (Scheme 2). The addition to isobutyraldehyde (i-PrCHO) to give syn adduct (R,S)-6 is not as selective as boron-based Evans aldol additions to generate (S,R)-6, but it is surprisingly effective. It is extremely sensitive to both quenching conditions and aging effects. Curiously, aggregation markedly influences reactivity yet has no effect on stereoselectivity. The totally stereoselective and high-yield addition to cyclohexanone is also influenced by aging effects but not destructively so. Rate studies delineate why the stereochemistries of the additions in Scheme 1 are insensitive to aggregation. Our work sheds light on why everything mattered for Singer et al.5a
Scheme 2.
Results
As is often the case, our narrative is by no means chronological; we became aware of aging effects and the impact of the acidic quench over time. Many experiments and computations became marginalized by this heightened understanding, but they retain merit and are archived in Supporting Information. Structural assignments for aggregates 2a, 2b, and 3 have been described in detail elsewhere8 and are not repeated here. We often refer to the highly fluctional dimers 2a and 2b collectively as 2.
Aggregate Aging
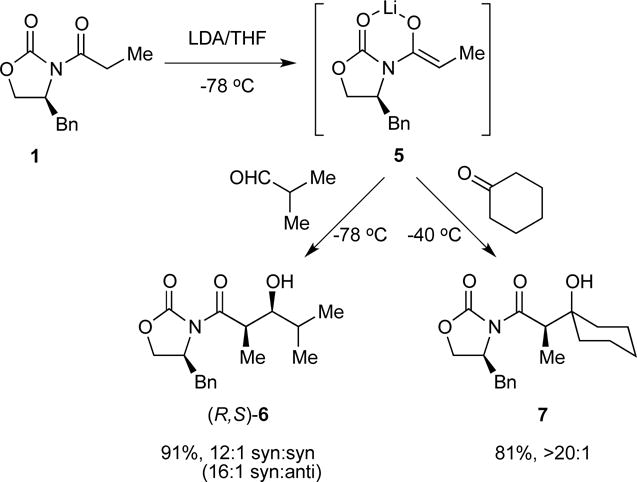
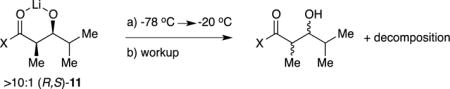
Substrate 1 and related oxazolidinones were enolized in tetrahydrofuran (THF) or THF/hexane mixtures at −78 °C with analytically pure, lithium chloride free lithium diisopropylamide (LDA), [6Li]LDA, or [6Li,15N]LDA.11 Previous studies showed that the metalation of oxazolidinone 1 with LDA in THF or THF/hexane mixtures at −78 °C proceeds according to Scheme 1.8 Intermediate mixed dimer 8 is also observable with excess LDA. The kinetically formed trisolvated dimers 2a and 2b are metastable in neat THF solution at −78 °C, isomerizing to tetramer 3 with an approximate half-life of 8–12 h at 0.10 M.12 In contrast to the tetramer–dimer equilibration, the dimers isomerize rapidly. Warming to −60 °C causes the rapid appearance of unsolvated tetramer 3 (half-life = 1.5–2.0 h), but warming to 0 °C with subsequent cooling results in no further changes. As implied by eq 3, tetramer 3 is favored by low THF concentrations, becoming the sole observable form at concentrations below 2.0 M THF. Moreover, low THF concentrations increase the rate at which the tetramer forms, consistent with a requisite THF dissociation. As we show below, tetramer formation has an enormous impact on the efficacy of the lithium-based aldol additions.
 |
(3) |
Addition to i-PrCHO: in situ monitoring
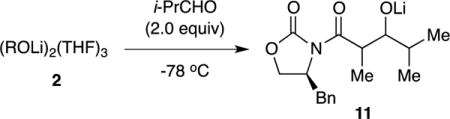
6Li NMR spectroscopic analysis of the aldol addition in eq 3 was limited because the aldolates appear as broad mounds. (Many lithium alkoxides do not form discrete aggregates in THF solution.13) The loss of starting materials, however, offered insights into the efficacy of the aldol addition. Enolizations at −78 °C without aging—kinetically controlled metalations—afford enolate dimers 2. The addition of 2.0 equiv of i-PrCHO consumes 2 in seconds (eq 4). Mixed dimer 8, present when a slight excess of LDA is used (as often occurs in synthetic applications), is also consumed immediately. We entertained the possibility that enolization is fast relative to the aldol addition and tried adding LDA last; that approach did not work. We eventually traced the fate of the LDA fragment in 8 to the formation of 1,2-adduct 9. Although the homoaggregated forms of 9 appear to be complex oligomers (probably ladders14), mixed adduct 10, formed by using 2.0 equiv of [6Li,15N]LDA, was characterized with 6Li, 13C, and 15N NMR spectroscopy,15 which made key 13C-15N and 6Li-15N couplings readily apparent.16
 |
(4) |
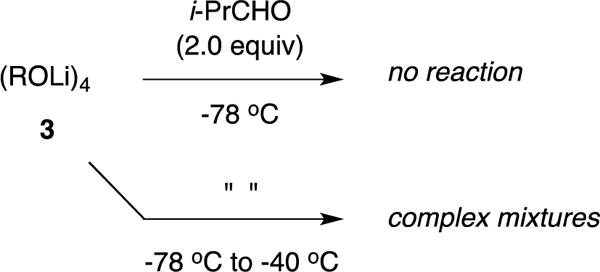
The addition of i-PrCHO to enolates that were generated at −78 °C, briefly aged at 0 °C, and recooled to −78 °C —solutions containing THF-concentration-dependent dimer–tetramer mixtures—show immediate dimer consumption and no detectable reaction of the tetramer over hours: tetramer 3 is unreactive (Scheme 3). Forcing the conversion of 3 by warming the solution to 0 °C affords broad mounds that provide no insight into what is achieved but was eventually traced to decomposition (vide infra).
Scheme 3.
The results of in situ IR spectroscopy qualitatively confirmed the outcomes observed with NMR spectroscopy. Injection of 0.95 equiv of i-PrCHO into unaged solutions at −78 °C caused immediate disappearance of the aldehyde absorbance envelope at 1710–1740 cm−1, whereas aged solutions containing exclusively tetramer showed no evidence of addition at −78 °C.
Addition to i-PrCHO: yield and selectivity
Those attempting to carry out Evans aldol additions with lithium enolates face several potential obstacles. In our hands, the aldol adducts were particularly sensitive to destruction during workup (more so than simple acylated oxazolidinones) primarily owing to the deacylation documented previously.17 The literature is replete with quenches of mild acids and buffers. Contrary to what we infer from these reports, however, deacylation is suppressed, and yields improve when aqueous HCl with decreasing pH is used: quenching with concentrated HCl affords the high yields shown in Scheme 2 with no detectable deacylation. Anticipating the obvious question, we note that HCl in saturated brine is not as effective.
The stereoselectivities and percent conversions were monitored by 1H NMR spectroscopic analysis of the crude extracts after acidic workup. Selected results illustrating the most salient features of the addition are shown in Table 1. A host of cross-checks showed that the isomer ratios were not distorted by the protocol. The stereochemistry was assigned by (1) determining the X–ray crystal structure of the known18 major syn isomer, (R,S)-6 (Supporting Information); (2) preparing the known18 minor syn isomer, (S,R)-6, via boron enolate based addition;19 (3) preparing the known20 minor anti isomer, (R,R)-6, via a titanium-catalyzed aldol addition;21 and (4) synthesizing (S,S)-6 from the opposite enantiomeric series (Supporting Information).22 The starting oxazolidinone 1 was easily monitored to assess percent conversion.
Table 1.
Condition-dependent percent conversion of oxazolidinone enolates 2 and 3 with added i-PrCHO at −78 °C.
| ||||
|---|---|---|---|---|
| [THF] (M)a | aging at 0 °C | estimatedb dimer:tetramer | percent conversion | selectivityc (R,S)-6:(S,R)-6 |
| 12.0 | no | >20:1 | >95% | 12:1 |
| 12.0 | yes | 1:1.4 | 33% | 12:1 |
| 0.20 | no | >20:1 | >95% | 12:1 |
| 0.20 | yes | 1:>20 | 2% | 12:1 |
Corresponds to neat tetrahydrofuran, which is 12.3 M without other additives.
Aggregate proportions derive from 6Li NMR spectroscopy.
Syn:anti = 16:1 owing to (R,R)-6.
The observed selectivities proved surprisingly impervious to the known (measurable)8 aggregate distribution: all reactions carried out at −78 °C afford approximately a 12:1 syn:syn selectivity with minor concentrations of the (R,R) anti isomer. The condition-dependent percent conversion is notable. Taken together, the results of product analyses and spectroscopic studies show that percent conversion and accompanying isolated yields correlate with the proportion of dimeric enolate, whereas stereoselectivity does not.
Aged samples containing predominantly tetrameric enolate 3 provide low conversion owing to the low reactivity of the tetramer. Forcing conditions (≥−40 °C) afford poor selectivities and debris traced to the retroaldol reaction. When a solution of aldolate prepared under optimum conditions to give high yields and 12:1 syn:syn selectivity is subsequently aged at −40 °C before quenching and workup, selectivity is lost and debris forms (eq 5). Thus, the aldolate product is incompatible with the temperatures required to elicit a tetramer-derived addition.
 |
(5) |
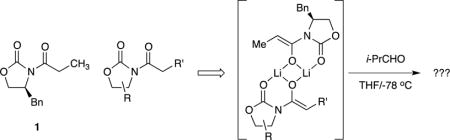
Heteroaggregation effects on selectivity
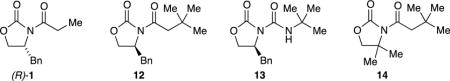
We generated mixtures of heteroaggregates (eq 6) as described in previous structural studies to assess the influence of pairing partners on the selectivity of aldol additions of (S)-5.8 Enolates (R)-5 and 12–14 are the most interesting of those assayed. Under both kinetic control, in which only dimers are formed, and aged conditions, the resulting distributions of homo- and heterodimers were shown by 6Li NMR spectroscopy are nearly statistical. (R)-5 is simply the antipode of the oxazolidinone denoted as 5, which affords a synthetic racemate at a 1:1 ratio. Hindered enolates such as 12 form exclusively homodimers in isolation and only homo- and heterodimers when mixed with 5. Anion 13 is structurally intriguing in that it could, in principle, heteroaggregate without undergoing a competitive aldol addition. Diastereoselectivities deriving from the oxazolidinone enolate 5 component remained invariant: heteroaggregation had no influence on the stereoselectivities. Hold that thought as we segue to studies of the mechanism of the aldol addition.
 |
(6) |
 |
Addition to ketones: yield, selectivity, and mechanism
Kinetically generated dimeric enolates 2 add to ketones such as cyclohexanone (Scheme 2) to form adduct 7 with total selectivity (>20:1), but the reaction stalls at approximately 30% conversion when carried out at equimolar concentrations.23 The addition was readily followed with IR spectroscopy by monitoring the disappearance of cyclohexanone (1715 cm−1) or the formation of the aldolate (1785 cm−1).24 Addition with 1:1 stoichiometry on unaged samples at −78 °C is sufficiently slow that dimer-to-tetramer aging is competitive, which appears to be the source of the incomplete conversion. Paradoxically, warming the samples to −40 °C to expedite the addition also accelerates the aging of the enolates to the less reactive tetrameric enolate. However, unlike additions to i-PrCHO, such forcing conditions are not destructive. Aldol addition at −40 °C affords high conversion (>98%), isolated yield (81%), and good selectivity (>20:1). Forcing conditions cause neither the loss of selectivity nor the decomposition expected for the retroaldol addition.
Rate studies were carried out under pseudo-first-order conditions with cyclohexanone at low concentrations (0.005 M). The absence of autocatalysis was demonstrated by completing a reaction, zeroing the baseline, and showing analogous rates with a second aliquot of ketone. The decays approximated first-order, but slight overlap of the absorbances of cyclohexanone and enolate 2 in tandem with dimer-to-tetramer aging effects distorted the data during the later half-lives. Monitoring initial rates of aldolate formation was expedient (1785 cm−1).
Plotting initial rates versus cyclohexanone concentration confirms a first-order ketone dependence (Figure 1). Plotting initial rates versus enolate concentration revealed a half-order dependence (rate = k′[enolate]n, such that n = 0.57 ± 0.06) on observable enolate dimer (Figure 2), which revealed that the aldol addition proceeds via reversible deaggregation and reaction via a fleeting monomeric intermediate.25
Figure 1.
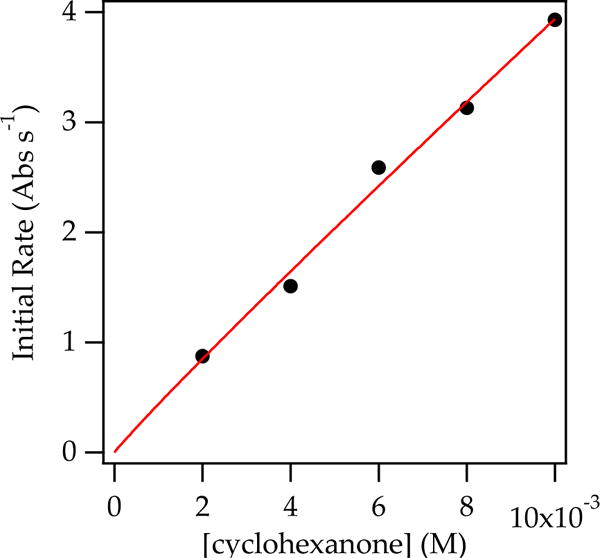
Plot of initial rate vs cyclohexanone concentration in neat tetrahydrofuran (THF) at −78 °C (0.10 M 5, neat THF,−78 °C). The data were fit to y = axn, such that n = 0.95 ± 06.
Figure 2.
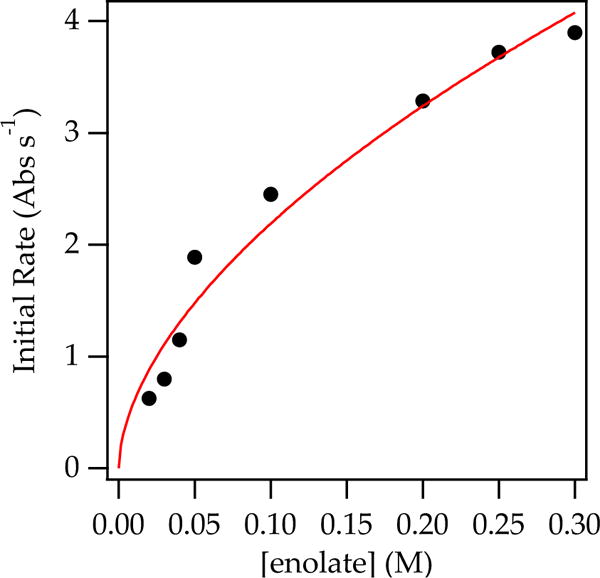
Plot of initial rate vs enolate 5 concentration at 0.0050 M cyclohexanone in neat THF at −78 °C. The data were fit to the following: y = axn, such that n = 0.57 ± 0.06.
The solvent order proved particularly interesting in the context of the previous structural assignments as isomeric trisolvated dimers 2. The uneven per-lithium solvation number—which was firmly established experimentally and computationally but disquieting nonetheless8—mandated that the reaction order in THF be a variant of a half-integer order (± 0.5, ± 1.5, and so forth).26 Indeed, a plot of initial rate versus THF concentration showed a distinct positive dependence that fits the following: initial rate = k′[THF]n, such that n = 0.56 ± 0.05 (Figure 3). Thus, the half-order in THF also confirms that the trisolvation state assignment is correct. As an aside, cursory examination of the rates of aged samples in which the sole observable form is tetramer reveals a 0.37 ± 0.05 order in enolate, consistent with a more sluggish tetramer-to-monomer conversion.
Figure 3.
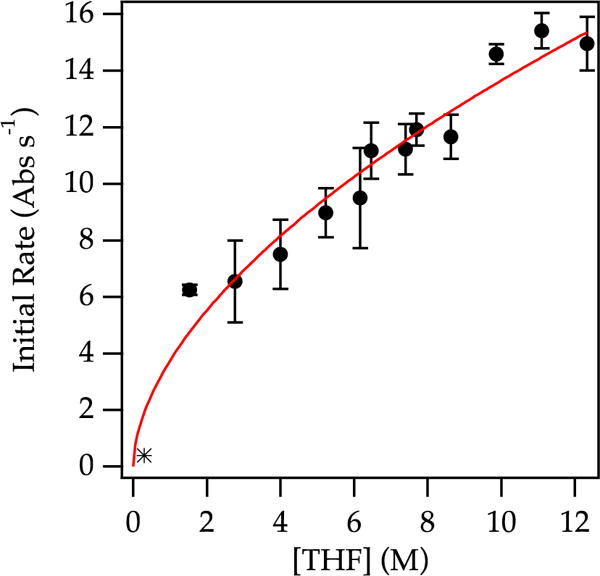
Plot of initial rate vs THF concentration using 0.10 M enolate and 0.0050 M cyclohexanone in toluene at −78 °C. The data were fit to the following: y = axn, such that n = 0.56 ± 0.05.
In total, the rate data are consistent with the idealized27 rate law in eq 7 and the generic mechanism delineated in eqs 8 and 9.
| (7) |
| (8) |
| (9) |
Computations
The reaction coordinate was examined with density functional theory (DFT) computations at the B3LYP/6–31G(d) level of theory with single-point calculations at the MP2 level of theory.28 Each transition structure showed the anticipated single imaginary frequency corresponding to the reaction coordinate. Intrinsic reaction coordinate (IRC) calculations29 probed the minima bracketing the transition structures. The supporting information includes probes of various solvation states and conformational isomers of the transition structures. We have distilled the results to the lowest energy transition structures of central importance to the issues at hand.30
The cornerstone for the results is the previously reported stability of the relative aggregation states summarized in Scheme 4, which illustrates the small predicted energy separations of germane aggregates.8 The lowest energy transition structures stemming from the aggregates in Scheme 4 as well as the post-transition-structure minima are shown in Scheme 5. Comparing the relative energies presented a challenge in standard states. We benchmarked the computed activation free energies on a per-aldehyde basis to the most stable trisolvated dimer (eqs 10–13). As demonstrated by the energies, the computations do not capture the preference for monomer-derived reactivity (vide infra).
| (10) |
| (11) |
| (12) |
| (13) |
Scheme 4.
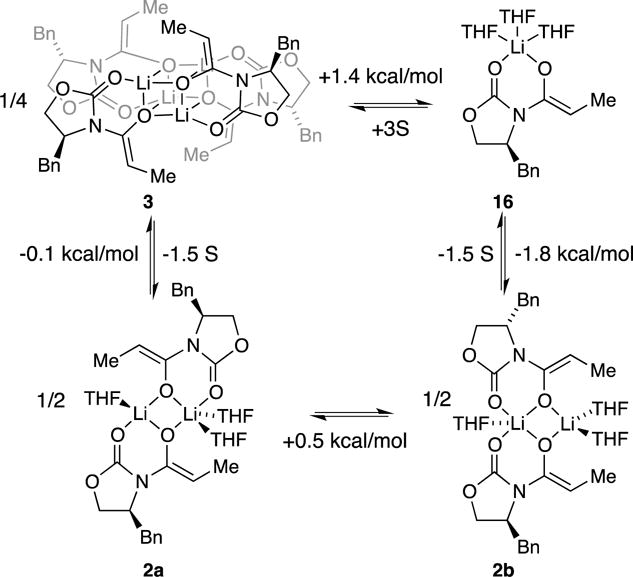
Scheme 5.
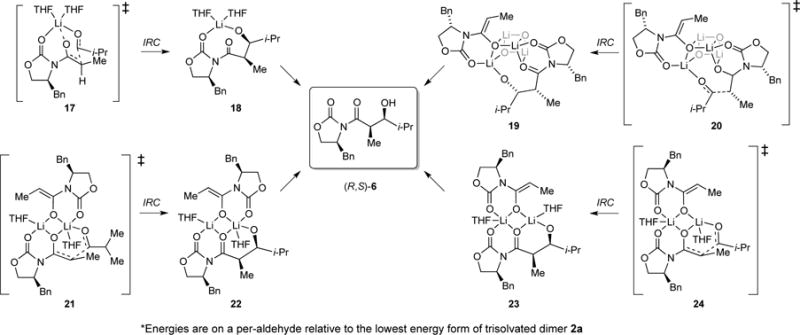
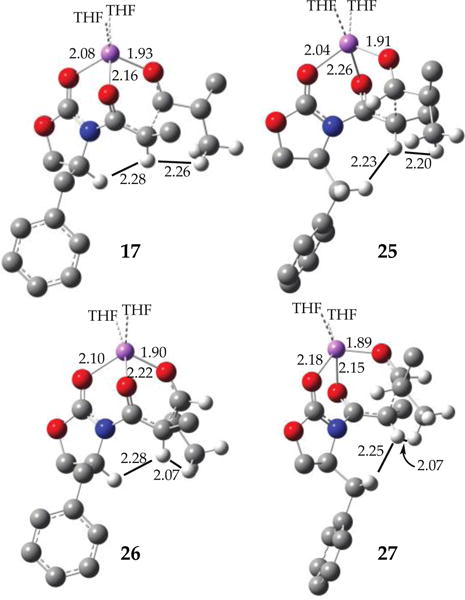
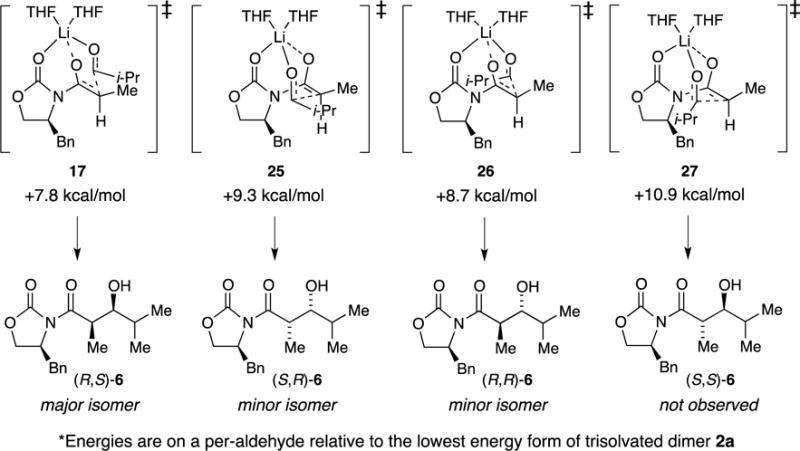
The most pressing issue seems to pertain to stereocontrol: all aggregation states predict the formation of the observed syn adduct (R,S)-6. In light of the rate studies, we focused on the four Zimmerman-Traxler31 diastereomeric transition structures depicted in Scheme 6 and as artists’ renditions with energies in Scheme 7. IRC calculations show the O–Li bond of the propionamide carbonyl cleaving at the transition structure with an affiliated rotation about the N–C bond. The relative energies of 25 and 27 are surprisingly close, but a minor canting of the i-Pr moiety in 27 appears to alleviate serious interaction. Transition states without coordination of the carbamate carbonyl or those retaining the tripodal contact through to the product aldolate are viable but higher-energy (vide infra).
Scheme 6.
Scheme 7.
Discussion
The results of structural, stereochemical, and mechanistic studies illustrate why lithiated Evans enolates are rarely used in synthesis and suggest ways to overcome some of the limitations of these compounds. Key aging effects are summarized in Scheme 8. Omitted structural details including THF ligands appear in Scheme 1.
Scheme 8.
Aggregate aging
Reich has shown that when kinetically formed mixtures of enolate aggregates react with aldehydes, the lower aggregates are decidedly more reactive.8,10,32 That is not to say, however, that deaggregation necessarily occurs before aldol addition. The results of our previous studies showed that enolization with LDA forms isomeric trisolvated dimers 2a/b, which are kinetically stable at −78 °C.8 On warming to −40 °C (or standing for hours at −78 °C), the dimers equilibrate to unsolvated tetramer 3 (see Scheme 1). Low THF concentrations in THF/toluene mixtures favor the unsolvated tetramers at equilibrium and facilitate the dimer-to-tetramer conversion.
Aggregate-dependent reactivity
The outcome of an aldol addition depends markedly on the dimer–tetramer distribution. Under kinetically controlled enolization in which only dimers form, aldol (R,S)-6 can be isolated in a credible 12:1 syn:syn selectivity (with lower levels of anti addition) and 91% combined isolated yield (Scheme 2). The result is contingent on an optimized quench—concentrated HCl—to preclude deacylation. If the solution is allowed to age to form tetramer 3, the aldol addition fails (Table 1). This outcome is consistent with Reich’s observations that metastable dimers are more reactive than tetramers.7,10,32 Attempts to force a tetramer-derived aldol addition, however, lead to debris and stereochemical erosion (Scheme 3) owing to the penchant of the aldolate to undergo retroaldol addition (eq 5).6
Aggregate-independent selectivity
We were determined to show that the stereochemistry of the additions could be controlled and influenced by controlling the aggregation state of the enolate. We explored this hypothesis by preparing assorted heterodimeric enolates and carrying out subsequent aldol additions (eq 6). Structural variations within the partnered oxazolidinone enolates were examined. We knew from the results of 6Li NMR spectroscopic studies8 that these reactions form heterodimers. Some pairs had interesting flairs. By attempting to alter the diastereoselectivity using a 1:1 mixture of two enantiomers of 5—a synthetic racemate—we likely carried out the first (and only) stereochemical study of an Evans aldol addition using racemic starting material. Other pairing partners such as 14 lack chirality while retaining potentially biasing substituents. Anion 13 affords an Evans enolate surrogate neutered by replacing the carbanionic carbon with a nitrogen. None—not one—afforded a significant change in the stereochemistry.
Mechanism
There is an interesting circularity to our inability to control the stereoselectivity of an organolithium reaction by controlling aggregation state: our first mechanistic organolithium paper in 1986 describes a similar failure to alter the stereochemistry of lithiated hydrazone alkylations through aggregation control.33 These two rate studies spanning three decades provided the same outcome: changes in aggregate structures fail to control selectivity because the reaction proceeds via monomers. In the case of adding enolate 5 to cyclohexanone (Scheme 2), a rate law (eq 7) in conjunction with the assigned reactants as trisolvated dimers provided the generic mechanism involving a disolvated monomer intermediate (eqs 8 and 9). Despite speculation34 and hard evidence32 that the aldol addition occurs from aggregates, we found no support for aggregate-based Evans aldol additions. Of course, generalizing our results to other aldol additions is ill-advised.
Computations
The results of DFT computations, although at times quantitatively in conflict with experimental observations, proved helpful. The large cache of results are archived in Supporting Information. Scheme 4 shows that the computations captured the essence of the energetic equivalence of isomeric dimers 2a and 2b and tetramer 3.8 The most stable transition structures representing the four aggregate forms are illustrated in Scheme 5, with affiliated energies in eqs 10–13—all of which predict the formation of (R,S)-6 as the major product. The per-aldehyde energies normalized to a single trisolvated dimer ground state, however, did not reflect the experimental results well, with the experimentally implicated monomer-based transition structure 17 garnering the highest energy and the tetramer-based variant 20 the lowest.
Despite the failure of the admittedly non-isodesmic comparisons,35 the computations provoke thought. IRC calculations29 of monomer-based transition structure 17 have revealed the carbonyl of the propionate undergoing an Li–O scission bond to adduct 18 as the first-formed minimum. Transition structure 28, retaining this interaction, as well as transition structure 29, showing scission of the oxazolidinone carbonyl Li–O bond, are viable but higher in energy. The dimer-based transition structures show similarities to those calculated by Cossío and co-workers30a for simple aldol condensations. Owing to the stereochemical complexity, a number of isomeric forms occur (Supporting Information). Tetramer-based transition structure 20 undergoes addition involving a scission of an O–Li bond in the cube to give partially fragmented cube 19 as the immediate minimum. The stability of 20 seems to find origins in precursor complex 30 that, for reasons that are not obvious to us, derives significant stabilization via aldehyde complexation that carries through to the transition structure.
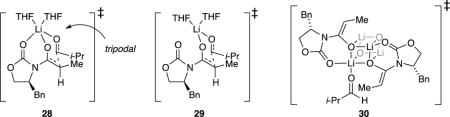 |
Focusing on the experimentally implicated monomer-based transition structures, we found that the computations mimicked experimental outcomes nicely (Scheme 6), although the numbers should not be taken literally. If viewed from the right angle, one is struck at how closely the six-centered transition structure mimics the idealized chair first proffered by Zimmerman and Traxler.31 The most stable transition structure, 17, corresponds to the experimentally observed syn isomer. Probably the most vexing result is that transition structure 27 corresponding to synfacial addition to the benzylic group with anti orientation is not wildly unstable. Squinting at three-dimensional depictions in Scheme 6 (or using the more tractable interactive capabilities of a computer) reveals a notable absence of consequential interactions between any portions of i-PrCHO and the benzylic CH2 protons. As redrawn and labeled below, the notable H–H van der Waals interactions appear to be those of the benzylic CH2 or aldehyde CH3 moieties with the enolate vinyl proton in concert with a significant (27–33°) rotation about the N–C(=O) bond. In short, preferred attack antarafacially to the benzylic group (25) rotates the N–C(=O) bond and moves the vinyl hydrogen away from the benzylic protons, whereas attack synfacially (25) rotates the vinyl hydrogen toward one of the benzylic protons. Also notable is an even greater elongation of the propionamide O–Li bond, which is in the process of cleaving en route to the aldolate.
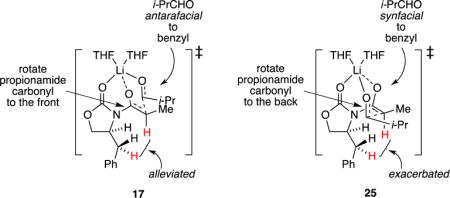 |
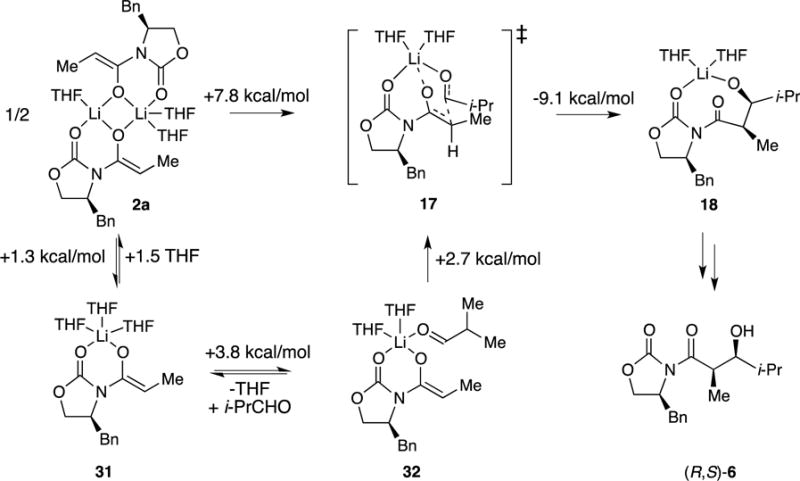
We summarize the reaction coordinate for the conversion of dimer 2a to give (R,S)-6 to provide a fairly coherent narrative in Scheme 9. Some may find the preferred five-coordinate monomers 31 and 32 disquieting relative to their more conventional four-coordinated analogs, but evidence has amassed over the years that the limit of four-coordinate, tetrahedral lithium does not stand up to scrutiny.36
Scheme 9.
Synthetic implications
Our results yield a number of interesting implications that are specific to the Evans chemistry or have even broader interest. We noted at the outset, for example, that ketones are unusually well-represented in the small group of lithium-based Evans aldol additions.5 The Pfizer team that took the aldol in eq 4 to a plant-scale production level5a used several tricks that we suspect circumvented the deleterious aging effects: they (1) carried out the aldol addition in a large number of smaller batches, and (2) generated the unhindered (rapidly enolized?) acetate enolate in the presence of the hindered (slowly enolized?) ketone, which may have allowed fleeting lower aggregates to be captured before association into higher aggregates. (We are currently examining some of these hypotheses.) That said, additions to ketones are less susceptible to the problems of aging: the aldol addition to cyclohexanone shown in Scheme 2 gives a credible 81% isolated yield if the enolate solution is allowed to age to give the less reactive tetramer 3. The ketone adduct (the aldolate of 7) is less sensitive than aldehyde adduct 11 to destructive retroaldol addition, which seems to contradict the widely held notions about ketone-based aldolates.6 We are reluctant to conclude that the Evans enolates are exceptional and wonder what these potentially dogmatic notions are based on. Given that aldol additions to ketones are still considered unusual, the small handful of oxazolidinone-based additions to ketones are probably worthy of more attention, especially if the low isolated yields derive from reparable quenching problems.
The aldehydes should not be discounted either. The 12:1 syn:syn selectivity contaminated by a small amount of anti isomer may be economically viable. Would a plant-scale boron-based aldol necessarily be economically more viable? The answer to that question depends on the details of handling and purification. The compelling part to us, however, stems from the insights provided by the computations demonstrating that the central interaction dictating facial selectivity appears to involve the interaction of the enolate vinyl hydrogen with one of the two benzylic hydrogens on the oxazolidinone substituent. We briefly examined oxazolidinones bearing isopropyl (from valine), phenyl, and diphenylmethyl moieties trolling for better selectivities and we found none. In retrospect, none appears to be optimally suited to improve selectivity based on the computational model. We cannot help but wonder whether there are oxazolidinones engineered to magnify the interaction with the enolate hydrogen atom (and whether they could be prepared economically enough to justify an improvement).
We plan to examine alkylations of Evans enolates, but we suspect that the requisite warmer temperatures foreshadow deaggregation. Do lithium enolate tetramers ever react directly without deaggregation? There is precious little direct data with which to address this question,7 but Reich shows that tetramers, although less reactive than dimers, react directly without dissociation.32 In a very clever experiment, Flowers and co-workers10f correlated the product distribution of an oxidative enolate coupling with the distribution of homo- and heterotetramers.
As a final note, it is common practice to use a slight excess of LDA as a margin of error, and its fate is worth considering. LDA forms 1,2-adduct 9 with i-PrCHO. Although the homoaggregated adducts are spectroscopically intractable, the mixed dimer with LDA (10) is readily characterized. Adduct 9 is akin to that exploited to exact ortholithiations of aryl aldehydes,37 and 1,2-adducts are surely intermediates in lithium hexamethyldisilazide additions used to form imines.38 There is additional evidence of 1,2-addition by lithium amides,39 but to observe it with LDA remains somewhat sobering. Are such adducts necessarily precluded if the electrophile is a ketone rather than a less hindered aldehyde? In a word, no. Such 1,2-adducts to ketones have been documented.16 Of course, none of this matters much unless the aldehyde is the precious component, at which point excess LDA will cause a loss in yield. It makes us wonder, however, what fleeting intermediates go undetected during enolizations.40
Conclusion
We believe that the lithium-based Evans aldol addition is potentially more useful than many realize, especially for potentially challenging additions to ketones. The inherently lower selectivities of lithium-based Evans aldols compared with those of their dibutylboron triflate variants are unappealing in academic laboratories (ketone additions excepted): an increase in per-unit cost to obtain spectacular selectivities is a minor sacrifice. In an industrial setting, however, the economics of large-scale reactions that include purifications relying on recrystallization leave lead to altogether different calculations. Industrial practitioners working on large scales should be aware, however, that they could fall prey to non-equilibrium kinetics in which solutions of highly reactive dimeric enolates age to become much less reactive tetrameric enolates. Enolizations in the presence of electrophiles possibly with a more judicious choice of hindered lithium amide base, should be considered.
Experimental
Reagents and Solvents
THF and toluene were distilled from solutions containing sodium benzophenone ketyl. The toluene stills contained approximately 1% tetraglyme to dissolve the ketyl. LDA, [6Li]LDA, and [6Li,15N]LDA were prepared as described previously.11 Solutions of LDA were titrated for active base with a literature method.41 Air- and moisture-sensitive materials were manipulated under argon with standard glove box, vacuum line, and syringe techniques. Oxazolidinone 1 was purchased.
NMR Spectroscopy
Individual stock solutions of substrates and LDA were prepared at room temperature. An NMR tube under vacuum was flame-dried on a Schlenk line and allowed to return to room temperature, then backfilled with argon and placed in a −78 °C dry ice/acetone bath. The appropriate amounts of oxazolidinone and LDA (1.1 equiv) were added sequentially via syringe. The tube was sealed under partial vacuum, vortexed three times on a vortex mixer for 5 s with cooling between each vortexing. Equilibrated samples could be stored overnight in a −86 °C freezer. Each sample routinely contained 0.10 M total enolate with a 0.050 M excess of LDA. (Excess base forms mixed dimers 8 with the resulting enolate, which was characterized with 6Li and 15N NMR spectroscopy.8) Standard 6Li and 13C NMR spectra were recorded on a 500 MHz spectrometer at 73.57 and 125.79, respectively. The 6Li and 13C resonances are referenced to 0.30 M [6Li]LiCl/MeOH at −90 °C (0.0 ppm) and the CH2O resonance of THF at −90 °C.
IR spectroscopic analyses
IR spectra were recorded with an in situ IR spectrometer fitted with a 30-bounce, silicon-tipped probe. The spectra were acquired in 16 scans at a gain of 1 and a resolution of 4 cm−1. A representative reaction was carried out as follows: The IR probe was inserted through a nylon adapter and O-ring seal into an oven-dried, cylindrical flask fitted with a magnetic stir bar and a T-joint. The T-joint was capped with a septum for injections and a nitrogen line. After evacuation under full vacuum, heating, and flushing with nitrogen, the flask was charged with LDA (51 mg, 0.475 mmol) in THF, cooled in a dry ice–acetone bath prepared with fresh acetone, and charged with 1 (117 mg, 0.50 mmol) in THF. The total volume was brought to 4.9 mL with THF. After recording a background spectrum, we added cyclohexanone (0.05 mmol) with stirring. IR spectra were recorded every 15 s with monitoring of the absorbance at 1785 cm−1 over the course of the reaction.
Preparative scale addition to i-PrCHO
LDA was prepared in situ via the addition of 0.91 mL n-butyllithium (9.6 mmol) to a solution of 1.44 mL diisopropylamine (10.3 mmol) freshly distilled from sodium benzophenone ketyl in 100 mL THF in a dry ice–acetone bath prepared with fresh acetone. This solution was warmed with an ice water bath for 1 h. The solution was recooled with a dry ice–acetone bath, and a solution of 1 in THF (2.0 g, 8.6 mmol) was added over 5 min. This solution was stirred for 30 min, and 1.57 mL of i-PrCHO (17.1 mmol) was added. After 30 min of stirring, the reaction was quenched with 10 mL concentrated HCl. The solution was warmed to room temperature and extracted with three 100 mL portions of diethyl ether. The organic extracts were washed with 100 mL phosphate buffer (pH 7), dried over sodium sulfate, and concentrated in vacuo. The crude product was analyzed with 1H and 13C NMR. After flash chromatography with 27% ethyl acetate in hexanes, 2.365 g of product isomer was obtained (91% combined yield). Analytically pure (R,S)-6 was obtained via recrystallization from the mixture of products with ethyl acetate in hexanes at −20 °C and is a literature compound.18 The recovered crystals were subjected to X-ray crystallographic analysis (Supporting Information). (R,S)-6: 1H in CDCl3 (600 MHz): 7.27 ppm (m, 5H); 4.70 ppm (ddt, J = 9.8, 7.7, 3.3 Hz, 1H); 4.20 ppm (m, 2H); 4.03 ppm (qd, J = 7.0, 2.7 Hz, 1H); 3.60 ppm (dd, J = 8.6, 2.7 Hz, 1H); 3.31 ppm (dd, J = 13.4, 3.4 Hz, 1H); 2.77 ppm (dd, J = 13.4, 9.7 Hz, 1H); 2.59 ppm (s, 1H); 1.73 ppm (dhept, J = 8.5, 6.6 Hz, 1H); 1.19 ppm (d, J = 7.0 Hz, 3H); 1.06 ppm (d, J = 6.6 Hz, 3H); 0.95 ppm (d, J = 6.8 Hz, 3H). 13C in CDCl3 (126 MHz): 177.5, 153.0, 135.2, 129.4, 129.0, 127.4, 76.8, 66.2, 55.2, 39.7, 38.0, 31.1, 19.3, 18.9, 9.7. Dart-MS m/z: calculated for C17H24NO4 (M+H)+ 306.16998, found 306.16981. The minor isomers were observed using NMR spectroscopy with comparison to independently prepared authentic samples of (S,R)-6:18,19 1H NMR in CDCl3 (600 MHz): 7.29 ppm (m, 5H); 4.70 ppm (dddd, J = 9.5, 7.6, 3.4, 2.8 Hz, 1H); 4.22 ppm (m, 1H); 3.96 ppm (qd, J = 7.1, 2.5 Hz, 1H); 3.54 ppm (dd, J = 8.6, 2.5 Hz, 1H); 3.26 ppm (dd, J = 13.4, 3.4 Hz, 1H); 2.79 ppm (dd, J = 13.4, 9.5 Hz, 1H); 1.73 ppm (dhept, J = 8.7, 6.7 Hz, 1H); 1.24 ppm (d, J = 7.1 Hz, 3H); 1.04 ppm (d, J = 6.6 Hz, 3H); 0.91 ppm (d, J = 6.8 Hz, 3H). 13C NMR in CDCl3 (126 MHz): 177.8, 152.9, 135.1, 129.4, 129.0, 127.4, 76.7, 66.2, 55.2, 39.7, 37.8, 30.8, 19.3, 18.9, 9.9. Dart-MS m/z: calculated for C17H24NO4 (M+H)+ 306.16998, found 306.16986. (R,R)-6:20,21 1H NMR in CDCl3 (600 MHz): 7.28 ppm (m, 5H); 4.68 ppm (ddt, J = 10.3, 7.1, 3.2 Hz, 1H); 4.18 ppm (m, 2H); 4.05 ppm (p, J = 7.0 Hz, 1H); 3.51 ppm (s, 1H); 3.34 ppm (dd, J = 13.5, 3.5 Hz, 1H); 2.75 ppm (dd, J = 13.5, 9.7 Hz, 1H); 1.82 ppm (heptd, J = 6.9, 4.4, 1H); 1.21 ppm (d, J = 6.9 Hz, 3H); 1.00 ppm (d, J = 6.9 Hz, 3H); 0.97 ppm (d, J = 6.8 Hz, 3H). 13C NMR in CDCl3 (126 MHz): 177.4, 153.7, 135.3, 129.4, 129.0, 127.3, 79.6, 66.1, 55.6, 40.4, 37.8, 30.7, 19.9, 15.7, 15.0. Dart-MS m/z: calculated for C17H24NO4 (M+H)+ 306.16998, found 306.16982. Isomer (S,S)-622 was not observed on comparison with an authentic sample: 1H NMR in CDCl3 (600 MHz): 7.29 (m, 5H), 4.72 (ddt, J = 9.3, 7.7, 3.1 Hz, 1H), 4.20 (m, 2H), 3.98 (p, J = 7.0 Hz, 1H), 3.53 (q, J = 6.2 Hz, 1H), 3.25 (dd, J = 13.4, 3.4 Hz, 1H), 2.79 (dd, J = 13.4, 9.4 Hz, 1H), 2.48 (m, 1H), 1.82 (pd, J = 6.8, 4.6 Hz, 1H), 1.26 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.8 Hz, 3H). 13C NMR in CDCl3: δ 177.4, 153.5, 135.2, 129.6, 129.1, 127.6, 78.9, 66.3, 55.3, 40.8, 38.0, 30.6, 20.2, 15.9, 15.3. Dart-MS m/z: calculated for C17H24NO4 (M+H)+ 306.16998, found 306.16913.
Preparative scale addition to cyclohexanone
LDA was prepared in situ via the addition of 0.16 mL n-butyllithium (1.6 mmol) to a solution of 0.25 mL diisopropylamine (1.8 mmol) freshly distilled from sodium benzophenone ketyl in 20 mL THF in a dry ice–acetone bath prepared with fresh acetone. This solution was warmed with an ice water bath for 1 h. The solution was recooled with a dry ice–acetone bath, and a solution of 1 in THF (349 mg, 1.5 mmol) was added over 5 min. This solution was stirred for 30 min, and 0.50 mL of cyclohexanone (4.8 mmol) was added. The reaction was warmed to −40 °C in a dry ice–acetonitrile bath. After 30 min of stirring, the reaction was quenched with 2 mL concentrated HCl. The solution was warmed to room temperature and extracted with three 20 mL portions of diethyl ether. The organic extracts were washed with 20 mL phosphate buffer (pH 7), dried over sodium sulfate, and concentrated in vacuo. The crude product was analyzed with 1H and 13C NMR. After flash chromatography with 30% ethyl acetate in hexanes, 398 mg (1.2 mmol, 80%) of adduct 7 was isolated. Recrystallization with ethyl acetate in hexanes at −20 C afforded crystals were for an X-ray crystallographic analysis. 7: 1H NMR in CDCl3: 7.49 ppm (m, 5H); 4.90 ppm (ddt, J = 10.2, 6.8, 3.5 Hz, 1H); 4.37 ppm (m, 2H); 4.19 ppm (q, J = 7.0 Hz, 1H); 3.57 ppm (dd, J = 13.3, 3.5 Hz, 1H); 3.45 ppm (d, J = 1.8 Hz, 1H); 2.92 ppm (dd, J = 13.3, 10.0 Hz, 1H); 1.77 ppm (m, 8H); 1.53 ppm (m, 2H); 1.43 ppm (d, J = 7.0 Hz, 3H) 13C NMR in CDCl3: 178.0, 153.4, 135.2, 129.4, 129.1, 129.0, 128.5, 127.4, 126.5, 72.6, 65.9, 55.5, 38.1, 37.2, 33.5, 25.7, 21.8, 21.5, 12.2. Dart-MS m/z: 332.18561
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM077167) for support.
Footnotes
Supporting Information: Spectroscopic, kinetic, and computational data and authors for reference 28. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References and Footnotes
- 1.(a) Ager DJ, Prakash I, Schaad DR. Chem Rev. 1996;96:835. doi: 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]; (b) Wu G, Huang M. Chem Rev. 2006;106:2596. doi: 10.1021/cr040694k. [DOI] [PubMed] [Google Scholar]; (c) Farina V, Reeves JT, Senanayake CH, Song JJ. Chem Rev. 2006;106:2734. doi: 10.1021/cr040700c. [DOI] [PubMed] [Google Scholar]; (d) Evans DA, Shaw JT. Ĺactualité Chimique. 2003;35 [Google Scholar]; (e) Lin G-Q, Li Y-M, Chan ASC. Principles and Applications of Asymmetric Synthesis. Vol. 2001. Wiley & Sons; New York: p. 135. [Google Scholar]; (f) Palomo C, Oiarbide M, Garcia JM. Chem Soc Rev. 2004;33:65. doi: 10.1039/b202901d. [DOI] [PubMed] [Google Scholar]; (g) Franklin AS, Paterson I. Contemp Org Syn. 1994;1:317. [Google Scholar]; (h) Baiget J, Cosp A, Galvez E, Gomez-Pinal L, Romea P, Urpi F. Tetrahedron. 2008:5637. [Google Scholar]; (i) Velazques F, Olivio H. Curr Org Chem. 2002;6:303. [Google Scholar]; (j) Mahrwald R, editor. Modern Aldol Reactions, Vols 1 and 2. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 2.Evans DA, Bartroli J, Shih TL. J Am Chem Soc. 1981;103:2127. [Google Scholar]
- 3.Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]
- 4.(a) Sobahi TR. Oriental J Chem. 2004;20:17. [Google Scholar]; (b) Bonner MP, Thornton ER. J Am Chem Soc. 1991;113:1299. [Google Scholar]; (c) Pridgen LN, Abdel-Magid AF, Lantos I, Shilcrat S, Eggleston DS. J Org Chem. 1993;58:5107. [Google Scholar]; (d) Abdel-Magid A, Pridgen LN, Eggleston DS, Lantos I. J Am Chem Soc. 1986;108:4595. [Google Scholar]; (e) Pridgen LN, Abdel-Magid A, Lantos I. Tetrahedron Lett. 1989;30:5539. [Google Scholar]; (f) Banks MR, Blake AJ, Cadogan JIG, Dawson IM, Gaur S, Gosney I, Gould RO, Grant KJ, Hodgson PKG. J Chem Soc, Chem Commun. 1993:1146. [Google Scholar]; (g) Xue C, Voss ME, Nelson DJ, Duan JJW, Cherney RJ, Jacobson IC, He X, Roderick J, Chen L, Corbett RL, Wang L, Meyer DT, Kennedy K, DeGrado WF, Hardman KD, Teleha CA, Jaffee BD, Liu R, Copeland RA, Covington MB, Christ DD, Trzaskos JM, Newton RC, Magolda RL, Wexler RR, Decicco CP. J Med Chem. 2001;44:2636. doi: 10.1021/jm010127e. [DOI] [PubMed] [Google Scholar]; (h) Jacobsen IC, Reddy GP. Tetrahdron Lett. 1996;37:8263. [Google Scholar]; (i) Doi T, Iijima Y, Shin-a K, Ganesan A, Takahashi T. Tetrahedron Lett. 2006;47:1177. [Google Scholar]
- 5.(a) Singer RA, Ragan JA, Bowles P, Chisowa E, Conway BG, Cordi EM, Leeman KR, Letendre LJ, Sieser JE, Sluggett GW, Stanchina CL, Strohmeyer H, Blunt J, Taylor S, Byrne C, Lynch D, Mullane S, O’Sullivan MM, Whelan M. Org Process Res Dev. 2014;18:26. [Google Scholar]; (b) Fürstner A, Bouchez LC, Morency L, Funel J, Liepins V, Porée F, Gilmour R, Laurich D, Beaufils F, Tamiya M. Chem Euro J. 2009;15:3983. doi: 10.1002/chem.200802067. [DOI] [PubMed] [Google Scholar]; (c) Theurer M, Fischer P, Baro A, Nguyen GS, Kourist R, Bornscheuer U, Laschat S. Tetrahedron. 2010;66:3814. [Google Scholar]; (d) Peters R, Althaus M, Diolez C, Rolland A, Manginot E, Veyrat M. J Org Chem. 2006;71:7583. doi: 10.1021/jo060928v. [DOI] [PubMed] [Google Scholar]; (e) Liu W, Sheppeck JE, II, Colby DA, Huang H, Nairn AC, Chamberlin AR. Bioorg Med Chem Lett. 2003;13:1597. doi: 10.1016/s0960-894x(03)00105-7. [DOI] [PubMed] [Google Scholar]; (f) Ojida A, Yamano T, Taya N, Tasaka A. Org Lett. 2002;4:3051. doi: 10.1021/ol0263189. [DOI] [PubMed] [Google Scholar]; (g) Bartroli J, Turmo E, Belloc J, Forn J. J Org Chem. 1995;60:3000. [Google Scholar]
- 6.March J. Advanced Organic Chemistry. 4th. Wiley; New York: 1980. p. 959. [Google Scholar]
- 7.(a) Reich HJ. Chem Rev. 2013;113:7130. doi: 10.1021/cr400187u. [DOI] [PubMed] [Google Scholar]; (b) Reich HJ. J Org Chem. 2012;77:5471. doi: 10.1021/jo3005155. [DOI] [PubMed] [Google Scholar]; (c) Seebach D. Angew Chem, Int Ed Engl. 1988;27:1624. [Google Scholar]; (d) Braun M. Helv Chim Acta. 2015;98:1. [Google Scholar]; (e) Setzer WN, Schleyer PvR. Adv Organomet Chem. 1985;24:353. [Google Scholar]; (f) Williard PG. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 1. Pergamon; New York: 1991. Chapter 1.1. [Google Scholar]; (g) Kim Y-J, Streitwieser A. Org Lett. 2002;4:573. doi: 10.1021/ol017175d. [DOI] [PubMed] [Google Scholar]; (h) Jackman LM, Lange BC. Tetrahedron. 1977;33:2737. [Google Scholar]; (i) Li D, Keresztes I, Hopson R, Williard PG. Acc Chem Res. 2009;42:270. doi: 10.1021/ar800127e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zune C, Jerome R. Prog Polymer Sci. 1999;24:631. [Google Scholar]; (k) Baskaran D. Prog Polym Sci. 2003;28:521. [Google Scholar]
- 8.Tallmadge EH, Collum DB. J Am Chem Soc. 2015;137:13087. doi: 10.1021/jacs.5b08207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Highly hindered enolates in toluene with N,N,N′,N′-tetramethylcyclohexanediamine afforded monomer, illustrating that they are thermochemically reasonable as fleeting intermediates.8
- 10.For examples of reactions that are fast relative to the rates of aggregate–aggregate exchanges see:; (a) McGarrity JF, Ogle CA. J Am Chem Soc. 1985;107:1810. [Google Scholar]; (b) Jones AC, Sanders AW, Bevan MJ, Reich HJ. J Am Chem Soc. 2007;129:3492. doi: 10.1021/ja0689334. [DOI] [PubMed] [Google Scholar]; (c) Thompson A, Corley EG, Huntington MF, Grabowski EJJ, Remenar JF, Collum DB. J Am Chem Soc. 1998;120:2028. [Google Scholar]; (d) Jones AC, Sanders AW, Sikorski WH, Jansen KL, Reich HJ. J Am Chem Soc. 2008;130:6060. doi: 10.1021/ja8003528. [DOI] [PubMed] [Google Scholar]; (e) Kolonko KJ, Wherritt DJ, Reich HJ. J Am Chem Soc. 2011;133:16774. doi: 10.1021/ja207218f. [DOI] [PubMed] [Google Scholar]; (f) Casy BM, Flowers RA. J Am Chem Soc. 2011;133:11492. doi: 10.1021/ja205017e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Y, Hoepker AC, Gupta L, Faggin MF, Collum DB. J Am Chem Soc. 2010;132:15610. doi: 10.1021/ja105855v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The concentration of the lithium salts, although expressed in units of molarity, refer to the concentration of the monomer unit (normality).
- 13.Simple alkoxides in THF solution appear as broad mounds in the 6Li NMR spectra consistent the degrees of oligomerization.
- 14.Mulvey RE. Chem Soc Rev. 1991;20:167. [Google Scholar]
- 15.Collum DB. Acc Chem Res. 1993;26:227. [Google Scholar]
- 16.Zhao P, Condo A, Keresztes I, Collum DB. J Am Chem Soc. 2004;126:3113. doi: 10.1021/ja030582v. [DOI] [PubMed] [Google Scholar]
- 17.Evans DA, Britton TC, Ellman JA. Tetrahedron Lett. 1987;28:6141. [Google Scholar]
- 18.Crimmins MT, King BW, Tabet EA, Chaudhary K. J Org Chem. 2001;66:894. doi: 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]
- 19.Gage JR, Evans DA. Org Synth. 1990;68:83. [Google Scholar]
- 20.(a) Dias LC, Steil LJ, Vasconcelos VA. Tetrahedron: Asymmetry. 2004;15:147. [Google Scholar]; (b) May AE, Connell NT, Dahlmann HA, Hoye TR. Synlett. 2010;13:1984. doi: 10.1055/s-0030-1258480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans DA, Allison BD, Yang MG, Masse CE. J Am Chem Soc. 2001;123:10840. doi: 10.1021/ja011337j. [DOI] [PubMed] [Google Scholar]
-
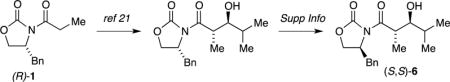
22.An authentic sample of (S,S)-6 was prepared by synthesizing the antipode of (R,R)-6 using (R)-5 via the literature preparation21 and then swapping the S configuration auxiliary as illustrated below (Supporting Information):
- 23.Aldol adduct 7 was characterized by spectroscopic and x-ray crystallographic studies.
- 24.As expected, the intermediate aldolates once again proved intractable by 6Li NMR spectroscopy.)
- 25.Fractional orders are problematic in that the curvature over a tenfold range of concentrations is difficult to detect within the noise. What distinguishes a fractional order from a first order is that the curvature is apparent when the fit is forced to a 0,0 origin. An analogous first-order dependence with a non-zero intercept can be dismissed deductively in some systems. Of course, a log-log plot will eliminate this annoyance from our perception without eliminating it in reality. Thus, reference to the idealized rate law27 takes on added meaning.
- 26.The rate law provides the stoichiometry of the transition structure relative to that of the reactants:; (a) Edwards JO, Greene EF, Ross J. J Chem Educ. 1968;45:381. [Google Scholar]; (b) Collum DB, McNeil AJ, Ramírez A. Angew Chem, Int Ed. 2007;46:3002. doi: 10.1002/anie.200603038. [DOI] [PubMed] [Google Scholar]
- 27.We define the idealized rate law as that obtained by rounding the observed reaction orders to the nearest rational order.
- 28.Frisch MJ, et al. GaussianVersion 3.09; revision A.1. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 29.For a discussion of intrinisic reaction coordinate (IRC) calculations, see:; Foresman JB, Frisch AE. Exploring Chemistry with Electronic Structure Methods. 2nd. Gaussian, Inc; Pittsburgh: 1993. [Google Scholar]
- 30.(a) Larranaga O, de Cozar A, Bickelhaupt FM, Zangi R, Cossio FP. Chem Eur J. 2013;19:13761. doi: 10.1002/chem.201301597. [DOI] [PubMed] [Google Scholar]; (b) Li Y, Paddon-Row MN, Houk KN. J Am Chem Soc. 1988;110:3684. [Google Scholar]; (c) Li Y, Paddon-Row MN, Houk KN. J Org Chem. 1990;55:481. [Google Scholar]; (d) Li Y, Paddon-Row MN, Houk KN. J Am Chem Soc. 1988;110:7260. [Google Scholar]; (e) Pratt LM, Nguyen NV, Ramachandran B. J Org Chem. 2005;70:4279. doi: 10.1021/jo0503409. [DOI] [PubMed] [Google Scholar]
- 31.Zimmerman H, Traxler M. J Am Chem Soc. 1957;79:1920. [Google Scholar]
- 32.Kolonko KJ, Wherritt DJ, Reich HJ. J Am Chem Soc. 2011;133:16774. doi: 10.1021/ja207218f. [DOI] [PubMed] [Google Scholar]
- 33.Wanat RA, Collum DB. J Am Chem Soc. 1985;107:2078. [Google Scholar]
- 34.(a) Amstutz R, Schweizer WB, Seebach D, Dunitz JD. Helv Chim Acta. 1981;64:2617. [Google Scholar]; (b) Seebach D, Amstutz R, Dunitz JD. Helv Chim Acta. 1981;64:2622. [Google Scholar]; (c) Williard PG, Liu Q-Y. J Am Chem Soc. 1993;115:3380. [Google Scholar]; (d) House HO, Gall M, Olmstead HD. J Org Chem. 1971;36:2361. [Google Scholar]; (e) Horner JH, Vera M, Grutzner JB. J Org Chem. 1986;51:4212. [Google Scholar]; (f) Jackman LM, Petrei MM, Smith BD. J Am Chem Soc. 1991;113:3451. [Google Scholar]; (g) See ref 6c.
- 35.From Wikipedia, an isodesmic reaction is a chemical reaction in which the type of chemical bonds broken in the reactant are the same as the type of bonds formed in the reaction product.
- 36.(a) Lucht BL, Collum DB. J Am Chem Soc. 1995;117:9863. [Google Scholar]; (b) Scheschkewitz D. Angew Chem, Int Ed Engl. 2004;43:2965. doi: 10.1002/anie.200353347. [DOI] [PubMed] [Google Scholar]; (c) Niecke E, Nieger M, Schmidt O, Gudat D, Schoeller WW. J Am Chem Soc. 1999;121:519. [Google Scholar]; (d) Becker G, Eschbach B, Mundt O, Reti M, Niecke E, Issberner K, Nieger M, Thelen V, Noth H, Waldhor R, Schmidt M. Z Anorg Allg Chem. 1998;624:469. [Google Scholar]; (e) Becker G, Schwarz W, Seidler N, Westerhausen M. Z Anorg Allg Chem. 1992;612:72. [Google Scholar]; (f) Wang H, Wang H, Li H-W, Xie Z. Organometallics. 2004;23:875. [Google Scholar]; (g) Xu X, Zhang Z, Yao Y, Zhang Y, Shen Q. Inorg Chem. 2007;46:9379. doi: 10.1021/ic701148z. [DOI] [PubMed] [Google Scholar]; (h) Thiele K, Goris H, Imhof W, Seide W. Z Anorg Allg Chem. 2002;628:107. [Google Scholar]; (i) Ramirez A, Lobkovsky E, Collum DB. J Am Chem Soc. 2003;125:15376. doi: 10.1021/ja030322d. [DOI] [PubMed] [Google Scholar]; (j) Buchalski P, Grabowska I, Kaminska E, Suwinska K. Organometallics. 2008;27:2346. [Google Scholar]
- 37.(a) Comins DL, Brown JD, Mantlo NB. Tetrahedron Lett. 1982;23:3979. [Google Scholar]; (b) Larsen RD, Corley EG, King AO, Carroll JD, Davis P, Verhoeven TR, Reider PJ, Labelle M, Gauthier JY, Xiang YB, Zamboni RJ. J Org Chem. 1996;61:3398. [Google Scholar]; (c) Comins DL, Killpack MO. J Org Chem. 1990;55:69. [Google Scholar]
- 38.Ager DJ. Org React. 1990;38:1. [Google Scholar]
- 39.(a) Adler M, Marsch M, Nudelman NS, Boche G. Angew Chem, Int Ed Engl. 1999;38:1261. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1261::AID-ANIE1261>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]; (b) Kaiser F, Schwink L, Velder J, Schmalz H-G. J Org Chem. 2002;67:9248. doi: 10.1021/jo026232t. [DOI] [PubMed] [Google Scholar]; (c) Roschangar F, Brown JC, Cooley BE, Sharp MJ, Matsuoka RT. Tetrahedron. 2002;58:1657. [Google Scholar]; (d) Juaristi E, Beck AK, Hansen J, Matt T, Mukhopadhyay T, Simson M, Seebach D. Synthesis. 1993;1271 [Google Scholar]; (e) Hart DJ, Kanai K, Thomas DG, Yang T-K. J Org Chem. 1983;48:289. [Google Scholar]; (f) Lebegue N, Flouquet N, Berthelot P, Pfeiffer B, Renard P. Synth Commun. 2002;32:2877. [Google Scholar]; (g) Koide H, Uemura M. Chirality. 2000;12:352. doi: 10.1002/(SICI)1520-636X(2000)12:5/6<352::AID-CHIR9>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (h) Nudelman NS, García-Linares GE. J Org Chem. 2000;65:1629. doi: 10.1021/jo9908445. [DOI] [PubMed] [Google Scholar]; (i) Matsumoto T, Hamura T, Kuriyama Y, Suzuki K. Tetrahedron Lett. 1997;38:8985. [Google Scholar]; (j) Armstrong DR, Davies RP, Raithby PR, Snaith R, Wheatley AEH. New J Chem. 1999;23:499. [PubMed] [Google Scholar]; (k) Corruble A, Valnot J-Y, Maddaluno J, Duhamel P. J Org Chem. 1998;63:8266. [Google Scholar]; (l) Corruble A, Valnot J-Y, Maddaluno J, Duhamel P. Tetrahedron: Asymmetry. 1997;8:1519. [Google Scholar]; (m) Reynolds KA, Finn MG. J Org Chem. 1997;62:2574. doi: 10.1021/jo9610016. [DOI] [PubMed] [Google Scholar]; (n) Nudelman NS, García-Linares GE. J Org Chem. 2000;65:1629. doi: 10.1021/jo9908445. [DOI] [PubMed] [Google Scholar]; (o) Fressigné C, Maddaluno J, Marquez A, Giessner-Prettre C. J Org Chem. 2000;65:8899. doi: 10.1021/jo000648u. [DOI] [PubMed] [Google Scholar]; (p) Davies JE, Raithby PR, Snaith R, Wheatley AEH. J Chem Soc, Chem Commun. 1997:1721. [Google Scholar]; (q) Armstrong DR, Davies JE, Davies RP, Raithby PR, Snaith R, Wheatley AEH. New J Chem. 1999;23:35. [Google Scholar]; (r) See ref 14.
- 40.Under a specific set of conditions, in situ IR spectroscopic monitoring of the enolization of cyclohexanone by LiHMDS showed the disappearance of ketone before the appearance of the enolate. Gruver, J. B. and Collum, D. B., unpublished.
- 41.Kofron WG, Baclawski LM. J Org Chem. 1976;41:1879. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.