

Abstract
Winter dormancy is a well known mechanism adopted by temperate plants, to mitigate the chilling temperature of winters. However, acquisition of sufficient chilling during winter dormancy ensures the normal phenological traits in subsequent growing period. Thus, low temperature appears to play crucial roles in growth and development of temperate plants. Apple, being an important temperate fruit crop, also requires sufficient chilling to release winter dormancy and normal phenological traits, which are often associated with yield and quality of fruits. DNA cytosine methylation is one of the important epigenetic modifications which remarkably affect the gene expression during various developmental and adaptive processes. In present study, methylation sensitive amplified polymorphism was employed to assess the changes in cytosine methylation during dormancy, active growth and fruit set in apple, under differential chilling conditions. Under high chill conditions, total methylation was decreased from 27.2% in dormant bud to 21.0% in fruit set stage, while no significant reduction was found under low chill conditions. Moreover, the demethylation was found to be decreased, while methylation increased from dormant bud to fruit set stage under low chill as compared to high chill conditions. In addition, RNA-Seq analysis showed high expression of DNA methyltransferases and histone methyltransferases during dormancy and fruit set, and low expression of DNA glcosylases during active growth under low chill conditions, which was in accordance with changes in methylation patterns. The RNA-Seq data of 47 genes associated with MSAP fragments involved in cellular metabolism, stress response, antioxidant system and transcriptional regulation showed correlation between methylation and their expression. Similarly, bisulfite sequencing and qRT-PCR analysis of selected genes also showed correlation between gene body methylation and gene expression. Moreover, significant association between chilling and methylation changes was observed, which suggested that chilling acquisition during dormancy in apple is likely to affect the epigenetic regulation through DNA methylation.
Introduction
The epigenetic modifications including DNA methylation and histone methylation/acetylation, are the heritable information [1], which modulate the gene expression without changing the DNA sequence. Therefore, these modifications provide an additional regulatory mechanism to influence the genes expression in response to internal or external environmental factors. Epigenetic regulation involves the post-translational histone protein modifications and DNA methylation [2, 3]. Epigenetic mechanisms have been known to modulate plant growth and development to adapt under environmental stresses. The methylation at cytosine base is well known strategy employed by plants to adopt the new environmental conditions without altering its DNA sequence [4]. The role of cytosine based DNA methylation in epigenetic regulation has been established and extensively studied in various plant species. Change in methylation pattern and chromatin modification of genomic DNA has been reported in various developmental processes and environmental stresses in plants [5]. These changes in methylation pattern have also been associated with transcriptional regulation in various plants [6–9]. The cytosine methylation is mediated by cytosine methyltransferases and is inheritable across the generations, while the cytosine demethylation is actively catalyzed by DNA glycosylases, beside the passive removal by replication process [10]. The cytosine methylation in higher plants occur predominantly at CG or CHG sequences (where H represents A, C, or T) [11–14] and sometimes at CHH sequences [15]. Irrespective of these sequence contexts, DNA cytosine methyltransferase, DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) catalyzes the de novo cytosine DNA methylation in plants. While, DNA METHYLTRANSFERASE 1 (MET1) maintains CG methylation; plant-specific DNA methyltransferase, CHROMOMETHYLASE 3 (CMT3) maintains CHG methylation and DRM2 maintains CHH methylation [10]. Whereas, the active removal of cytosine methylation is catalyzed by DEMETER (DME) and REPRESSOR OF SILENCING 1 (ROS1), and their paralogs DEMETER-LIKE 2 (DML2) and DEMETER-LIKE 3 (DML3), which are the members of DNA glycosylase family [10, 16, 17].
Although, DNA methylation occurs mainly in the transposable elements, methylation within the promoter and transcribed regions of the genes is common and correlated with their transcriptional activity [18–21]. Thus, it is apparent that cytosine methylation regulates various developmental and adaptive processes. Among all the presently available techniques to determine the cytosine methylation in genomic DNA, Methylation sensitive amplified polymorphism (MSAP) is reliable, cost effective and most widely used technique [22]. MSAP is a variant of amplified fragment length polymorphism technique (AFLP), and is widely used to study the changes in methylation pattern of genomic DNA. MSAP profile is based on the differential sensitivity of two isoschizomers, HpaII and MspI to methylation of their recognition sequences (CCGG). Activity of both the enzymes, HpaII and MspI is affected by the methylation state of external and internal cytosine residues. The full methylation (both strand methylated) of cytosine makes the HpaII inactive to cleave, but hemi-methylation (single strand methylated) site has no effect on its activity. Whereas MspI cleaves hemi or fully methylated site when internal cytosine is methylated (C5mCGG), while methylation of external cytosine (5mCCGG) inhibits activity of MspI [23].
Apple (Malus x domestica Borkh.) is an important fruit tree and cash crop for most of the temperate regions of the world. The low temperature of winter season plays crucial role in phenological events of apple tree. As winter approaches, the plant undergoes dormancy in order to survive the chilling temperature of winters. After accumulation of sufficient chilling, the dormant buds resume active growth in response to warm temperature of spring season. This phenomenon of bud dormancy during winter season is well understood and divided into three phases known as paradormancy, endodormancy and ecodormancy [24, 25]. It is reported that changing climatic conditions with high winter temperature resulted in improper flower development in tree species with high chilling requirement for winter dormancy release, which affect the productivity and quality of the developing fruit [3, 26]. Thus, chilling temperature during winter dormancy appears to have remarkable effect on quality and productivity of fruits. However, the molecular mechanism of chilling mediated dormancy release is still obscure and is a matter of debate. The understanding of chilling mediated dormancy release at genetic level may have profound impact on development of climate resilient cultivars.
The low temperature associated changes in DNA methylation have been evident from the several previous studies [11, 27–30]. However, no study related to changes in DNA methylation during dormancy period and its release in apple has been performed, so far. Therefore, to study the cytosine methylation based epigenetic regulation of chilling mediated dormancy release in apple, MSAP approach was employed to analyze the changes in cytosine methylation pattern during dormancy break and subsequent fruit set. In order to elucidate the effect of chilling temperature on methylation, the samples of Royal delicious apple variety were collected from two different locations in Kullu valley of Himachal Pradesh, India, with more than 1000 m altitude difference. A recent study by Jangra and Sharma [31] reported rise in average temperature during winter season in low altitude regions of Kullu valley. Therefore, the selected location with low altitude was assumed to receive less chilling during winter seasons as compared to location at high altitude. The Royal delicious is a high chill variety with >1200 chilling units (one chilling unit is equal to temperature below 7°C for one hour) requirement during dormancy period to resume normal active growth. In addition, the expression level of differentially methylated genes as well as genes involved in de novo cytosine methylation, its maintenance and active removal of cytosine methylation was analyzed in various stages of bud dormancy, dormancy release and fruit set using RNA-seq data of apple generated through Illumina Genome Analyzer IIx.
Materials and Methods
Plant material
For sample collection, apple trees of Royal delicious cultivar were selected from two private apple orchards situated at Palchan (32° 18′ 36″ N, 77° 10′ 40 E″; altitude 2350 m) and Seobag (31° 58′ 57″ N, 77° 07′ 43 E″; altitude 1250 m) of Kullu district, Himachal Pradesh (HP), India with owner’s permission. The selected locations have differential chilling availability during the winter season, where Seobag has less chilling availability, while the Palchan has more chilling availability during winter season. The daily maximum and minimum temperature data was obtained from Hill Agricultural Research & Extension Centre, Kullu (HP) and DRDO-Snow and Avalanche Study Estt. (SASE) station Bahang, Manali (HP) for Seobag and Palchan sampling locations, respectively. The samples comprising of four developmental stages, namely dormant bud (DB), silver tip (ST), green tip (GT) and initial fruit set (FS) were collected during the January 8, 2013 to April 15, 2013 from Seobag and during January 8, 2013 to May 8, 2013 from Palchan (Table 1). Each sample was collected from three trees and immediately frozen in liquid nitrogen and stored at – 80°C for further use.
Table 1. The sample abbreviation and their collection intervals.
| Developmental stages | Tissue type | Low chill location (Seobag) | High chill location (Palchan) | ||
|---|---|---|---|---|---|
| Sample abbreviation | Collection intervals | Sample abbreviation | Collection intervals | ||
| Bud | Dormant bud | DBL | 0 day | DBH | 0 day |
| Silver tip | STL | 45 day | STH | 56 day | |
| Bud break | Green tip | GTL | 57 day | GTH | 73 day |
| Fruit | Initial fruit set | FSL | 96 day | FSH | 119 day |
DNA extraction
The DNA was extracted as describe by Xin and Chen [32], with minor modifications. Briefly, the frozen tissue was crushed to powder in liquid nitrogen with activated charcoal and DNA extraction buffer (100mM Tris, pH 8.0; 20mM EDTA; 2% CTAB; 1.4M NaCl; freshly added 0.2% β-mercaptoethanol) was added and incubated at 60°C for 1 hr. After incubation, chloroform:isoamyl alcohol (24:1) treatment was given and 2 volumes of dilution buffer (100mM Tris, pH 8.0; 20mM EDTA; 2% CTAB) was added to the aqueous phase. The diluted samples were incubated at 60°C until DNA-CTAB precipitate starts appearing. The precipitate was washed with 70% ethanol and re-suspended in high salt TE buffer supplemented with RNase A (10mM Tris, pH 8.0; 2mM EDTA; 1M NaCl; 50μg/ml RNase), and further incubated at 60°C for 30 min. The RNase treated DNA was ethanol precipitated and re-suspended in TE buffer. The DNA was treated with proteinase K to remove any protein impurities which may interfere with restriction digestion of genomic DNA.
Methylation sensitive amplified polymorphism assay
The MSAP protocol was followed as described by Portis et al. [33] with minor modification. Briefly, 500 ng genomic DNA was digested with EcoRI and divided in to two equal halves for digestion with HpaII and MspI. The double digested DNA was subjected to proteinase K treatment and purified through ethanol precipitation. The quantification of restriction digested DNA was done using fluorescence based broad range Qubit assay (Invitrogen, USA). The 100 ng DNA of each digestion reaction was ligated to EcoRI and HpaII/MspI specific adaptors (S1 Table). The adaptor ligated DNA was 1/10 diluted and used as a template in pre-amplification PCR. The 1/20 diluted pre-amplification product was used as template for selective amplification PCR using 16 combinations of HpaII/MspI and EcoRI adaptor specific primers (S1 Table). Selectively amplified PCR products were mixed with equal amount of formamide dye (98% formamide, 10mM EDTA, 0.01% bromophenol blue w/v and 0.01% w/v xylene cyanol) and denatured at 95°C for 5 min. The denatured samples were separated on 6% denaturing sequencing gel and silver stained to visualize the DNA fragments.
Scoring and analysis of methylation patterns
The bands appeared on sequencing gel were scored according to Karan et al. [34]. Briefly, four types of bands were scored: type I fragments represent unmethylated and were present in both the EcoRI/MspI and EcoRI/HpaII lanes; type II fragments represent hemi-methylated loci and were present only in EcoRI/HpaII lane; type III fragments represent fully methylated loci and were present only in EcoRI/MspI lane; the type IV fragments represent the methylated/demethylated loci in two different samples which is indicated by absence of band in either enzyme combination in one of the samples.
Sequencing and analysis of polymorphic fragments
The randomly selected polymorphic fragments were excised from the gel, crushed in 20μl TE buffer and incubated at 94°C for five min. The eluted fragments were re-amplified with the same primer set and PCR thermal conditions as used for the corresponding selective amplification. The PCR products were cloned in pTZ57R/T TA cloning vector (Thermo Fisher Scientific). The plasmids were isolated from positive recombinant clones and sequenced using Big Dye Terminator v3.1 (Applied Biosystems, USA) with M13 forward and reverse primers. The resulted sequences were searched against phytozome (www.phytozome.net) as well as NCBI non-redundant database using BLASTN to find the genes and their flanking regions (within 1Kb) associated with methylation as identified through MSAP.
RNA sequencing and data analysis
Total RNA was extracted from 100 mg of plant material using iRIS solution [35], and quantified using NanoDrop UV-VIS spectrophotometer. The integrity of RNA was further checked on denaturing formaldehyde agarose gel. Eight RNA-Seq libraries, including DBL, STL, GTL, FSL and DBH, STH, GTH, FSH stages under low and high chill conditions, respectively, were prepared, using TruSeq RNA sample preparation kit v2 as per manufacturer’s instructions (Illumina Inc., USA). The quantification and insert size validation of libraries was done using Qubit dsDNA BR assay kit (Life technologies, USA) and Agilent Bioanalyzer DNA 1000 chip (Agilent Technologies), respectively. The paired-end (2×76) sequencing of RNA-Seq libraries was performed on Genome Analyzer IIx (Illumina) after cluster generation using cluster station (Illumina Inc., USA). The generated reads were de novo assembled on CLC Genomics Workbench 6.5 (http://www.clcbio.com) after quality filtering using NGS QC toolkit [36]. The assembled contig file was used as database to identify the MSAP loci associated transcripts using BLASTN with e-value cutoff 1e-05. The raw reads obtained from Illumina GAIIx of all the analyzed samples were submitted as BioProject (PRJNA306594) to NCBI Sequence Read Archive under accession number SRP067619.
Bisulfite sequencing
Two microgram of high quality genomic DNA from each of the eight samples corresponding to four developmental stages under differential chilling conditions was used for bisulfite treatment using Epitect Bisulfite Kit (Qiagen, USA) following the manufacturer’s instructions. The bisulfite converted DNA samples were used to amplify the four randomly selected polymorphic MSAP fragments; MDC013945.282, MDC019410.118, MDC011209.233 and MDC044052.10. Briefly, 2μl of bisulfite converted genomic DNA was used as template in 25μl of PCR reaction using hot start Advantage 2 polymerase mix (Clontech, USA) with touch-down thermal PCR profile. The primers corresponding to bisulfite converted polymorphic loci were designed using Methprimer (S2 Table), an online tool for designing bisulfite-conversion based Methylation PCR Primers [37]. The PCR amplified products were gel eluted using QIAEX II gel extraction kit (Qiagen, USA) and cloned in pTZ57R/T TA cloning vector. The transformants were confirmed by colony PCR and further sequencing of plasmid was performed using Big Dye Terminator v3.1 (Applied Biosystem, USA) with M13 forward and reverse primers. The sequence of unconverted DNA was used to compare the methylation status of selected polymorphic loci, where change from C (cytosine) in unconverted DNA to T (thymine) in bisulfite converted DNA represents non-methylated C, while no change from C to T represents the methylated C. The percentage of methylated cytosines was calculated using formula: [number of methylated cytosine/total number of cytosine present in different contexts] x 100.
Quantitative real-time PCR (qRT-PCR) analysis
For qRT-PCR analysis, 1μg of total RNA was converted into first strand cDNA using RevertAid RNAse H minus cDNA synthesis kit as per manufacturer’s instructions (Fermentas Life Sciences, USA). The primers (S3 Table) were designed using Primer Express software version 3.0.1 (Invitrogen). The 10 randomly selected genes associated with polymorphic fragments were analyzed for their expression levels using qRT-PCR based method as described earlier by Arya et al. [38]. The qRT-PCR data was normalized using GAPDH gene as internal control [39]. The relative expression of candidate target genes was calculated using REST software (Qiagen) considering high chill conditions as control.
Results
Methylation profiling of apple genomic DNA during chilling acquisition and fruit set
The MSAP profiling of apple genomic DNA in four developmental stages viz, dormant bud (DB), silver tip (ST), green tip (GT) and initial fruit set (FS) under differential chilling conditions was performed using 16 primer combinations. The temperature map for both the sampling locations showed difference in minimum and maximum temperature during the winters and onset of spring season (Fig 1). The scoring of fragments (ranging between 100bp to 1Kb) resulted in identification of 2306 fragments in four stages under low chill conditions, while, 2414 fragments were observed under high chill conditions (Fig 2, Table 2). Under high chill conditions, 1810 non-methylated fragments present in both EcoRI/HpaII and EcoRI/MspI lanes (type I), were identified, while under low chill conditions, 1740 non-methylated fragments were identified. Under low chill conditions, higher number (166) of hemi-methylated fragments, present only in EcoRI/HpaII lane (type II) were identified as compared to high chill conditions (132). Whereas, fully methylated fragments, present only in EcoRI/MspI lane (type III) were found to be almost equal under low chill (232) and high chill (234) conditions. The fragments, absent in both, EcoRI/MspI and EcoRI/HpaII lanes (type IV), were found to be more under high chill conditions (238) as compared to low chill conditions (168) (Table 2). The total methylation percentage was found to be 24.5% and 25% under low and high chill conditions, respectively. However, percentage of the fully methylated fragments (type III and IV) was slightly higher (19.6%) under high chill conditions than under low chill conditions (17.3%). In contrast, percentage of hemi-methylated fragments (type II bands) was found to be more (7.2%) under low chill conditions than under high chill conditions (5.5%). However, the percentage of fully methylated fragments was found to be gradually decreased along with the dormancy break and fruit set under high chill conditions. The chi-square test of independence between chilling availability during dormancy period and methylation patterns suggested association between chilling and methylation level (Table 2).
Fig 1. The graph showing the average of minimum and maximum daily temperature during November to April for three consecutive years (2011–2014) at both the sampling locations.
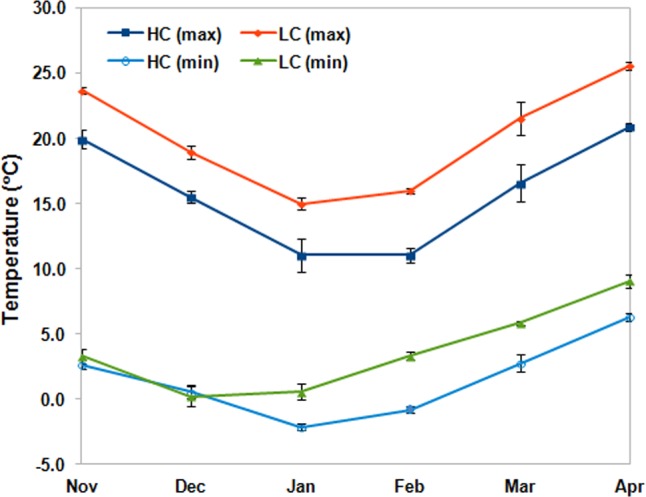
The HC and LC represent the high chill condition (Palchan) and low chill condition (Seobag), respectively. The min. and max. represent the minimum and maximum temperature, respectively.
Fig 2. A representative gel image of MSAP assay showing polymorphic fragments in different samples.
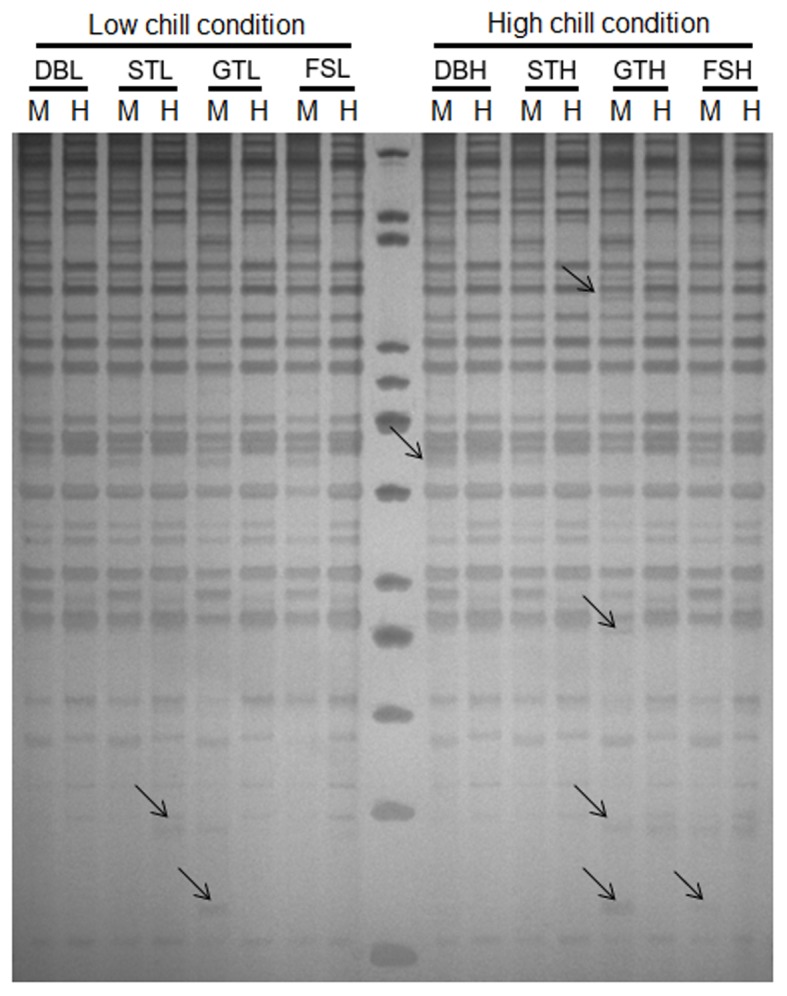
lane H: DNA digested with EcoRI–HpaII; lane M: DNA digested with EcoRI–MspI. The four developmental stages viz. Dormant bud (DB), Silver tip (ST), Green tip (GT) and Initial fruit set (FS) in apple, under low (L) chill and high (H) chill conditions, were used for MSAP analysis. The arrow marks show presence of polymorphic fragments.
Table 2. Changes in DNA methylation under high and low chill conditions during various stages of dormancy, dormancy release and fruit set.
| MSAP band type | Dormant bud | Silver tip | Green tip | Initial fruit set | Total methylation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DBH | DBL | STH | STL | GTH | GTL | FSH | FSL | High chill condition | Low chill condition | ||
| Fragments type I | 444 | 458 | 446 | 438 | 466 | 434 | 454 | 410 | 1810 | 1740 | |
| Fragments type II | 33 | 32 | 37 | 50 | 30 | 35 | 32 | 49 | 132 | 166 | |
| Fragments type III | 50 | 50 | 59 | 54 | 62 | 76 | 63 | 52 | 234 | 232 | |
| Fragments type IV | 83 | 62 | 70 | 43 | 59 | 34 | 26 | 29 | 238 | 168 | |
| Total fragments | 610 | 602 | 612 | 585 | 617 | 579 | 575 | 540 | 2414 | 2306 | |
| Chi-square test statistics | χ2 | 3.22 | 8.08 | 8.46 | 5.93 | 14.87 | |||||
| P-value | 0.358 | 0.044 | 0.03 | 0.115 | 0.002 | ||||||
| Methylation percentage | |||||||||||
| Total methylation (%) | 27.2 | 23.9 | 27.1 | 25.1 | 24.5 | 25.0 | 21.0 | 24.1 | 25.0 | 24.5 | |
| Fully methylated fragments (%) | 21.8 | 18.6 | 21.1 | 16.6 | 19.6 | 19.0 | 15.5 | 15.0 | 19.6 | 17.3 | |
| Hemi-methylated fragments (%) | 5.4 | 5.3 | 6.0 | 8.5 | 4.9 | 6.0 | 5.6 | 9.1 | 5.5 | 7.2 | |
Dynamics of methylation/demethylation events in relation to chilling availability
The apple cultivar Royal Delicious, taken for study is a high chill variety and requires high chilling (>1200 CU) during dormancy period for subsequent quality fruit development. Therefore, high chill condition was considered as control to study the methylation/demethylation events in four different comparative conditions, including DBL-vs-DBH, STL-vs-STH, GTL-vs-GTH and FSL-vs-FSH during dormancy break to fruit set. The comparative MSAP profiling under differential chilling conditions showed changes in methylation and demethylation events. The differential banding patterns were considered to analyze the methylation and demethylation status of 5'-CCGG-3' sites in apple genome (Table 3). The comparative analysis revealed that methylation events under low chill conditions were increased from 0.3% in DBL-vs-DBH to 3.3% in FSL-vs-FSH, while the demethylation events were decreased from 6.5% in DBL-vs-DBH to 2.8% in FSL-vs-FSH (Table 3). The change in methylation and demethylation events in DBL-vs-DBH to GTL-vs-GTH and DBL-vs-DBH to FSL-vs-FSH comparisons were also found to be statistically significant with chi-square test of independence (S4 Table). These changes in methylation and demethylation events suggested that availability of high chilling during dormancy period was associated with progressive demethylation events during dormancy break and fruit set.
Table 3. DNA methylation under low chill conditions with respect to high chill conditions in various developmental stages during dormancy, dormancy release and fruit set.
| Band Pattern type | *Class of banding pattern | Banding pattern | Developmental stages | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | Treatment | ||||||||
| No change | MspI | HpaII | MspI | HpaII | DBL-vs-DBH | STL-vs-STH | GTL-vs-GTH | FSL-vs-FSH | |
| A | 1 | 1 | 1 | 1 | 432 | 420 | 420 | 402 | |
| B | 0 | 1 | 0 | 1 | 23 | 30 | 28 | 27 | |
| C | 1 | 0 | 1 | 0 | 44 | 50 | 56 | 49 | |
| D | 0 | 0 | 0 | 0 | 62 | 43 | 34 | 29 | |
| Total | 561 | 543 | 538 | 507 | |||||
| Percentage | 93.2 | 92.8 | 92.9 | 93.9 | |||||
| Demethylation | |||||||||
| E | 0 | 1 | 1 | 1 | 10 | 2 | 0 | 0 | |
| F | 1 | 0 | 1 | 1 | 8 | 6 | 8 | 2 | |
| G | 0 | 0 | 1 | 1 | 8 | 10 | 6 | 6 | |
| H | 1 | 0 | 0 | 1 | 0 | 2 | 0 | 1 | |
| I | 0 | 0 | 0 | 1 | 7 | 11 | 5 | 4 | |
| J | 0 | 0 | 1 | 0 | 6 | 3 | 8 | 2 | |
| Total | 39 | 34 | 27 | 15 | |||||
| Percentage | 6.5 | 5.8 | 4.7 | 2.8 | |||||
| Methylation | |||||||||
| K | 1 | 1 | 0 | 1 | 2 | 7 | 2 | 17 | |
| L | 1 | 1 | 1 | 0 | 0 | 1 | 12 | 1 | |
| M | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| N | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| O | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| P | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Total | 2 | 8 | 14 | 18 | |||||
| Percentage | 0.3 | 1.4 | 2.4 | 3.3 | |||||
*Alphabets A-P represent classes of banding patterns.
Analysis of MSAP polymorphic fragments
The polymorphic fragments representing methylation/demethylation events in different samples during DB to FS under differential chilling conditions were randomly selected and sequenced. The size of polymorphic DNA fragments ranged between 62 to 307 bp. From the total sequenced fragments, 47 fragments were found to be either within the gene body or within the 1Kb upstream/downstream regions of protein coding genes (Table 4). The BLASTN analysis of sequenced fragments showed that these were related to AUX/IAA27 B, pentatricopeptide repeat domain containing protein, NAC domain containing protein, DTT domain containing protein (Homeodomain-like), molecular chaperone (dnaJ), histone acetyltransferase HAC1-like, exonuclease, leucine-rich repeat receptor-like protein kinase, cytochrome P450, pectinesterase 3 (involved in cell wall modification), solute carrier family protein, ribosomal protein kinase G11A-like, acid phosphatase1-like, thioredoxin-like protein, cation efflux protein, serine/threonine-protein phosphatase, SNARE protein, tRNA-nucleotidyltransferase (ACT domain-containing protein), sodium/hydrogen exchanger family, NADH-cytochrome B5 reductase, etc. (Table 4). Most of these genes were related to cellular metabolism, DNA/RNA processing, stress response, antioxidant system and transcriptional regulation. Therefore, annotation of polymorphic fragments suggests that methylation might regulate the wide range of molecular and cellular functions during dormancy release and fruit set in apple.
Table 4. List of selected MSAP polymorphic fragments, their annotation, their location in apple genome and their corresponding contigs identified in RNA-Seq data.
| Sl. No | Methylation status | Tissue type | Gene annotation | E-value | Associated apple protein | Fragment position | MDC ID | RNA-seq contig |
|---|---|---|---|---|---|---|---|---|
| 1 | Demethylated | DB | PREDICTED: wall-associated receptor kinase 2-like | 1.4E-39 | MDP0000216164 | exon | MDC022332.50 | C_47891 |
| 2 | Demethylated | DB | DNA-directed RNA polymerases II and IV subunit 5A | 5E-30 | MDP0000305507 | 100bp 3'UTR | MDC017731.272 | C_10585 |
| 3 | Demthylated | DB | NAC domain-containing protein 90-like | 6E-79 | MDP0000130797 | exon | MDC018994.343 | C_56817 |
| 4 | Demthylated | DB | Homeodomain-like transcriptional regulator isoform 2 | 1.1E-87 | MDP0000194500 | exon and 3'UTR | MDC009126.325 | C_38969 |
| 5 | Methylated | DB | Hydroxymethylglutaryl-CoA lyase | 8.3E-70 | MDP0000168020 | 700 bp 3'UTR | MDC004192.624 | C_15455 |
| 6 | Methylated | DB | probable alpha,alpha-trehalose-phosphate synthase | 3.3E-62 | MDP0000257194 | intron | MDC004525.265 | C_12925 |
| 7 | Methylated | DB | quinone oxidoreductase PIG3 | 2.9E-41 | MDP0000303908 | 800bp 5'UTR | MDC016620.364 | C_8592 |
| 8 | Methylated | DB | VAM6/VPS39-like protein | 9.7E-28 | MDP0000312750 | exon | MDC019094.96 | No hit |
| 9 | Methylated | DB | AUX/IAA27 B | 7.6E-84 | MDP0000361838 | 100 bp 5'UTR | MDC003296.431 | C_7332 |
| 10 | Methylated | DB | U3 small nucleolar ribonucleoprotein protein IMP3 | 8.3E-57 | MDP0000365539 | exon and intron | MDC022311.89 | C_8153 |
| 11 | Demethylated | ST | ACT domain-containing protein ACR8-like | 9.5E-39 | MDP0000162605 | exon and intron | MDC000413.350 | C_5857 |
| 12 | Demethylated | ST | dnaJ homolog subfamily A member 3, mitochondrial-like | 2.8E-49 | MDP0000200636 | exon and intron | MDC012610.126 | C_24702 |
| 13 | Demethylated | ST | pentatricopeptide repeat-containing protein | 7E-23 | MDP0000269242 | exon | MDC001244.150 | C_14588 |
| 14 | Demethylated | ST | Embryo defective 2016, putative | 4.7E-40 | MDP0000299377 | exon | MDC011779.270 | C_9444 |
| 15 | Demethylated | ST | TMV resistance protein N-like isoform X1 (NBS-LRR) | 1.1E-19 | MDP0000378930 | exon and intron | MDC000285.554 | C_33868 |
| 16 | Demethylated | ST | Nuclear transcription factor Y subunit B-3-like | 8.3E-70 | MDP0000818967 | 150 5'UTR | MDC000337.410 | C_34825 |
| 17 | Methylated | ST | UDP-galactose transporter 2-like | 1.5E-52 | MDP0000198172 | exon and 3'UTR | MDC013793.94 | C_33785 |
| 18 | Methylated | ST | Werner Syndrome-like exonuclease isoform X2 | 3.2E-96 | MDP0000226301 | 200bp 3'UTR | MDC027549.17 | C_9043 |
| 19 | Methylated | ST | galactose oxidase-like | 1.1E-68 | MDP0000896660 | exon | MDC044052.10 | C_30737 |
| 20 | Demethylated | GT | uncharacterized ATP-dependent helicase C29A10.10c-like | 1.3E-46 | MDP0000296615 | exon and intron | MDC013945.282 | C_32594 |
| 21 | Demethylated | GT | Ribosomal protein kinase G11A-like | 1.9E-25 | MDP0000153928 | intron | MDC019410.118 | C_15847 |
| 22 | Demethylated | GT | acid phosphatase 1-like | 1.1E-33 | MDP0000186556 | exon and 3'UTR | MDC011209.233 | C_10934 |
| 23 | Demethylated | GT | Cation efflux protein/ zinc transporter | 6.7E-37 | MDP0000230536 | exon and intron | MDC003233.264 | C_45074 |
| 24 | Demethylated | GT | Histone acetyltransferase HAC1-like | 2.1E-29 | MDP0000272113 | exon | MDC003556.304 | C_21725 |
| 25 | Demethylated | GT | probable salt tolerance-like protein | 1.3E-39 | MDP0000551876 | exon | MDC015976.77 | C_4845 |
| 26 | Demethylated | GT | Pectinesterase 3 | 8.7E-22 | MDP0000621927 | exon | MDC004699.471 | C_25510 |
| 27 | Demethylated | GT | galactose oxidase-like | 5.7E-66 | MDP0000646516 | exon | MDC003162.287 | No hit |
| 28 | Methylated | GT | serine/threonine-protein phosphatase 7 inactive homolog | 8.4E-28 | MDP0000158250 | intron | MDC000502.364 | C_29245 |
| 29 | Methylated | GT | UDP-galactose/UDP-glucose transporter 3 | 2E-78 | MDP0000165524 | exon and intron | MDC000968.94 | C_25294 |
| 30 | Methylated | GT | E3 ubiquitin-protein ligase RING1 | 3.6E-26 | MDP0000189022 | exon | MDC011159.221 | C_450 |
| 31 | Methylated | GT | vesicle-associated membrane protein 726 (SNARE protein) | 2.2E-8 | MDP0000231646 | exon | MDC011573.204 | C_14669 |
| 32 | Methylated | GT | Transcriptional corepressor SEUSS isoform X1 | 8.4E-21 | MDP0000278350 | exon | MDC007401.275 | C_1813 |
| 33 | Methylated | GT | Cytochrome P450 77A3 | 2.3E-79 | MDP0000322366 | exon and intron | MDC017031.336 | C_14881 |
| 34 | Methylated | GT | LRR receptor-like protein 2 | 1.7E-37 | MDP0000600580 | exon and intron | MDC040598.17 | C_32440 |
| 35 | Methylated | GT | aldehyde dehydrogenase family 2 member C4-like | 9.4E-41 | MDP0000662387 | 380 bp 3'UTR | MDC018307.94 | C_63120 |
| 36 | Methylated | GT | thioredoxin-like protein YLS8 isoform X1 | 1.1E-27 | MDP0000748886 | 200bp 5'UTR | MDC022326.61 | C_7466 |
| 37 | Methylated | GT | protein Iojap-related, mitochondrial | 4.1E-15 | MDP0000750612 | exon and intron | MDC010033.126 | C_10847 |
| 38 | Methylated | GT | cytochrome P450 71A25-like | 1.2E-26 | MDP0000843359 | exon and down | MDC012381.537 | No hit |
| 39 | Methylated | GT | IQ-DOMAIN 14-like protein | 2.7E-35 | MDP0000890508 | exon | MDC018371.124 | C_27091 |
| 40 | Demethylated | FS | vam6/Vps39-like protein | 1.2E-32 | MDP0000134240 | exon | MDC003486.520 | C_30705 |
| 41 | Demethylated | FS | NADH-cytochrome b5 reductase 1 | 1.3E-25 | MDP0000164574 | exon | MDC002037.232 | C_9830 |
| 42 | Demethylated | FS | DNA mismatch repair protein MSH7-like | 2.8E-42 | MDP0000306261 | exon and intron | MDC017615.201 | C_130 |
| 43 | Demethylated | FS | cation/H(+) antiporter 24-like | 2.1E-14 | MDP0000310134 | exon | MDC020062.119 | No hit |
| 44 | Demethylated | FS | LRR receptor-like serine/threonine-protein kinase EFR | 6.8E-58 | MDP0000712707 | intron | MDC007491.483 | C_32170 |
| 45 | Methylated | FS | metal tolerance protein 10-like | 2.3E-57 | MDP0000130310 | 200bp 5'UTR | MDC018194.83 | No hit |
| 46 | Methylated | FS | cysteine proteinase | 2.2E-9 | MDP0000189200 | exon and 3'UTR | MDC009544.135 | C_2542 |
| 47 | Methylated | FS | ATP-dependent Clp protease ATP-binding subunit clpX-like | 1.8E-8 | MDP0000719271 | 150bp 5'UTR | MDC014085.237 | C_15418 |
Global expression analysis of MSAP fragments associated genes
In order to determine the effect of methylation change on the expression of MSAP fragments associated genes in various developmental stages during dormancy and fruit set, RNA-seq analysis of eight samples representing 2 dormant stages of floral spur bud (DB and ST), one stage of actively growing floral bud (GT) and one stage of fruit initiation (FS) under high and low chill conditions was performed (Table 1). The paired-end sequencing generated 97,960,812 raw reads of which 7,080,502 low quality reads were filtered out. The de novo assembly of 90,880,310 high quality reads resulted in 68,455 contigs with N50 of 766 bp. Out of 47 randomly selected MSAP fragments, expression of 42 was found in all the analyzed tissues, while remaining 5 genes exhibit either very low or no expression in any of the tissues. From the 42 MSAP fragments, the expression of majority of demethylated fragments associated genes was observed to exhibit high expression while majority of methylated contigs were either down-regulated or moderately expressed (Fig 3), which suggest that expression of these genes is affected by their DNA methylation. Overall, this analysis shows the moderate correlation between methylation status and level of gene expression.
Fig 3. Heat map representation of relative expression based on FPKM values of differentially methylated fragments in various developmental tissues as identified in MSAP analysis.

The majority of demethylated fragments showed upregulation under low chill conditions as compared to high chill conditions. The relative expression of majority of methylated fragments was downregulated. The relative FPKM values were used to generate the heat-map using MeV4. The color scale at the bottom represents the expression level, where red, green and black colors indicate upregulation, downregulation and unaltered expression, respectively. Contig number (starting with C_) and their annotation are given on the left and right side of the heat map, respectively.
The expression of genes involved in DNA and histone methylation/demethylation was also analyzed in various developmental stages under differential chilling conditions (Fig 4). In our RNA-seq data, twelve transcripts were annotated as DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) and DRM2-like genes, which are involved in de novo DNA methylation. In DBL-vs-DBH comparison, only four transcripts (C_6670, C_1114, C_17545 and C_18318) of DRM2 and DRM2-like were found to be upregulated (>1.5 fold change). However, in FSL-vs-FSH comparison, expression of nine DRM2 and DRM2-like transcripts was found to be upregulated, while expression of remaining three transcripts (C_1114, C_33214 and C_47355) was found to be unaltered. Although, most of the DRM2 and DRM2-like transcripts (six out of twelve) were found to exhibit downregulation (<0.75 fold change) in GTL-vs-GTH, the expression of all the transcripts (except, C_8277 and C_40483 with un-altered expression) of DNA METHYLTRANSFERASE1 (MET1) and MET1-like transcripts, which maintain CG methylation, was found to be upregulated in GTL-vs-GTH comparative condition (Fig 4). In addition, six out of eleven MET1 and MET1-like transcripts were also found to be upregulated in STL-vs-STH (C_35761, C_8276, C_15157, C_40483, C_8277 and C_43388) and FSL-vs-FSH (C_20719, C_32224, C_15157, C_40483, C_8277 and C_43388) comparisons. Similarly, out of seven transcripts of CHROMOMETHYLASE 3 (CMT3), which maintain CHG methylation, five were found to be upregulated in DBL-vs-DBH (C_5233, C_16270, C_23926, C_24032 and C_26644) and GTL-vs-GTH (C_1013, C_1627, C_5233, C_23926 and C_26644) comparisons (Fig 4). In addition, expression of DNA glycosylases, DEMETER (DME) and REPRESSOR OF SILENCING1 (ROS1), which are involved in active demethylation of cytosine, was also analyzed. The expression of five out of six DME-like transcripts was upregulated in FSL-vs-FSH, while only two exhibited upregulation in DBL-vs-DBH (C_13671 and C_33814) and STL-vs-STH (C_12667 and C_33814). Whereas, in GTL-vs-GTH comparison, most of the DME-like transcripts were either downregulated or have unaltered expression. The high relative expression of majority of ROS1-like transcripts was observed to exhibit upregulation in various comparative conditions. Out of eleven ROS1-like transcripts, expression of five, eight, four and eight transcripts was found to be upregulated DBL-vs-DBH, STL-vs-STH, GTL-vs-GTH and FSL-vs-FSH comparisons, respectively (Fig 4). These results suggest that, the de novo DNA methylation by DRM2 during dormant bud (DB) and fruit set (FS) stages and its maintenance by MET1 and CMT3 during bud break (GT) and fruit set with concomitant downregulation of DME-like and ROS1-like transcripts during bud break might be responsible for high percentage of cytosine methylation under low chill conditions even after dormancy release (Table 2).
Fig 4. Heat map representation of relative expression based on FPKM values of DNA methyltransferases, DNA glycosylases and histone methyltransferases in various developmental tissues.

The relative FPKM values were used to generate the heat-map using MeV4. The color scale at the bottom represents the expression level, where red, green and black colors indicate upregulation, downregulation and unaltered expression, respectively. Contig number (starting with C_) and their annotation are given on the left and right side of the heat map, respectively.
Since, histone methylation marks are observed to be linked with DNA methylation, expression of histone lysine methyltransferase (HMTase), including SUVH-like and SUVR-like transcripts was also analyzed. In total, 33 transcripts were found to be annotated as SUVH-like, which belong to different classes including, SUVH1-like (10 transcripts), SUVH4-like (8 transcripts), SUVH5-like (4 transcripts), SUVH6-like (5 transcripts) and SUVH9 (6 transcripts). The relative expression of seven out of ten SUVH1-like transcripts was found to be upregulated in each STL-vs-STH and FSL-vs-FSH comparisons. Similarly, majority of SUVH4-like (five out of eight), SUVH5-like (three out of four) and SUVH9 (five out of six) transcripts were found to be upregulated in FSL-vs-FSH. In addition, five transcripts of SUVH9 were also found to be upregulated in STL-vs-STH, while majority of SUVH6-like transcripts were found to be upregulated in all the comparative conditions, except in GTL-vs-GTH, where only two SUVH6-like transcripts were upregulated. Similarly, majority of SUVR2-like transcripts were also found to be upregulated in DBL-VS-DBH and FSL-VS-FSH comparative conditions, while majority of SUVR5-like transcripts were found to be upregulated in all the comparative conditions, except GTL-VS-GTH. Only two transcripts were annotated as SUVR4-like, both of them exhibit upregulation in FSL-VS-FSH comparisons (Fig 4).
Quantitative-real time PCR analysis of genes associated with methylation changes
The FPKM based expression obtained through RNA-Seq for genes associated with MSAP loci was validated using qRT-PCR (Fig 5). In total, eight genes, which have methylation change either within the gene body (intron/exon) or upstream/downstream regions were selected for qRT-PCR analysis. Out of these, six genes viz. MDP0000153928, MDP0000162605, MDP0000186556, MDP0000296615, MDP0000299377 and MDP0000378980 were demethylated, while, two genes viz. MDP0000198172, and MDP0000896660 were methylated in different developmental stages (Table 4). The FPKM based expression values were found to be in accordance with qRT-PCR expression values. Among six demethylated genes, expression of four (MDP0000162605, MDP0000186556, MDP0000299377 and MDP0000378930) was found to be up-regulated (>1.5 fold change) in samples of low chill conditions where demethylated fragments of these genes were observed in MSAP analysis (Fig 5). While the expression of other two demethylated genes, MDP0000153928 and MDP0000162605, was observed to be unaltered in corresponding samples. Similarly, from the two methylated genes, only one (MDP0000896660) was found to be downregulated in corresponding samples under low chill conditions as compared to high chill conditions, while the expression of MDP0000198172 was either up-regulated or remained unaltered (Fig 5). From the qRT-PCR analysis, it was observed that MSAP methylation status of genes was highly correlated with their expression induction or inhibition. For instance, four out of six demethylated genes (MDP0000162605, MDP0000186556, MDP0000299377 and MDP0000378930), were upregulated in samples where demethylation of these genes were observed in MSAP analysis. Similarly, the correlation between methylation and suppression of expression was observed for methylated gene, MDP0000896660, which showed downregulated expression in corresponding sample where methylated fragment of this gene was observed.
Fig 5.

Relative expression of randomly selected (A) demethylated and (B) methylated genes analyzed through qRT-PCR. Pearson correlation coefficient between fold change of FPKM based expression and qRT-PCR was 0.431 (p-value<0.0005).
Bisulfite sequencing of polymorphic fragments
In order to accurately determine the level of cytosine methylation of polymorphic MSAP loci and to correlate with their expression levels, four loci; MDC013945.282 (MDP0000296615), MDC019410.118 (MDP0000153928), MDC011209.233 (MDP0000186556) and MDC044052.10 (MDP0000896660) were randomly selected for bisulfite sequencing. The methylation pattern of three types of cytosine sequence contexts CG, CHH and CHG (where H represents the A, T or C), were analyzed. In all the four bisulfite sequenced loci, maximum methylated cytosines were found to be of CG type (15), followed by CHH (13) and CHG (5) (S1, S2, S3 and S4 Figs). Interestingly, under differential chilling conditions, the total methylation events were found to be higher under low chill conditions with 20 methylated cytosines, while under high chill conditions, only 13 methylated cytosines were observed in any of the sequence context. Irrespective of chilling conditions, the number of methylated cytosines during the dormant period (DBL, DBH, STL and STH) was relatively higher as compared to the active growth period (GTL, GTH, FSL and FSH). However, among dormant stages, thirteen methylated cytosines were identified under low chill conditions (DBL and STL) as compared to only seven methylated cytosines under high chill conditions (DBH and STH). However, during the active growth period (GT and FS), the number of methylated cytosine were comparable under both the chilling conditions. The methylation status of all the bisulfite sequenced loci, except MDP0000896660, indicates that low chill conditions have comparatively high cytosine methylation even after the dormancy release, which is in agreement with the MSAP scoring. The MDP0000296615 gene (MDC013945.282 loci), was found to have seven methylated cytosines in different sequence contexts, where most of them were present in DBL, STL, and FSL, while, methylated cytosines were found at positions 61 in STH and 233 in DBH, GTH and FSH (S1 Fig). Moreover, the percentage of methylated cytosines was observed to be high under low chill conditions with 8.57% in each DBL and STL and 5.71% in FSL, except in GTL, where it was only 2.85%. Whereas, constantly low (2.85%) percentage of methylated cytosines was observed in all the stages under high chill conditions. Therefore, it appeared that the gene body of MDP0000296615 is hyper-methylated under low chill conditions as compared to high chill conditions. In case of MDP0000153928 gene (MDC019410.118 loci), methylated cytosines were observed at position 6 in DBL and GTL, and at position 29 in STL with constant percentage of methylated cytosines (5%) in these samples, while no methylated cytosine was observed in FSL, DBH, STH, GTH and FSH (S2 Fig). This observation indicates that MDP0000153928 gene body was also hyper-methylated under low chill conditions. The MDP0000186556 gene (MDC011209.233 loci), was found to have methylated cytosines at positions 52 and 58 in DBL and at position 81 and 159 in STL and GTL, respectively. While, the cytosines at positions 126 and 131 in DBH, and at position 128 and 159 in STH were observed to be methylated (S3 Fig). The percentage of methylated cytosines under high chill conditions was observed to be constant with 3.7% in both DBH and STH, thereafter, no methylated cytosine was observed during active growth phase. While, under low chill conditions, the percentage of methylated cytosine was found to be 3.7% during DBL, thereafter, decreased towards the dormancy release, with 1.88% methylated cytosine in STL and GTL. In case of MDP0000896660 gene (MDC044052.10 loci), under low chill conditions, the methylated cytosines were observed at position 88 and 272 in STL and at position 164 and 272 in GTL and FSL, respectively. While under high chill conditions, the nucleotide positions at 150 in DBH, 78 in GTH and nucleotide positions 45, 200 and 272 in FSH were found to have methylated cytosines (S4 Fig). Although, similar to other bisulfite sequenced genes under low chill conditions, gene body of MDP0000896660 was found to be hyper-methylated during dormant period in STL with 3.27% methylated cytosines, thereafter decreased towards active growth (GTL and FSL) with 1.63% methylated cytosine, increases in cytosine methylation under high chill conditions was observed during fruit set (FSH) with 4.91% of methylated cytosines. From the qRT-PCR analysis and bisulfite sequencing of these four MSAP fragments, it is observed that methylation level and gene expression is moderately correlated.
Discussion
In temperate fruit crops, the low temperature during winter dormancy period is crucial for the normal phenological traits and quality and yield of the crop. Most of the previous studies related to dormancy release in various plants species have been based on the gene expression using global transcriptome analysis [40–43]. However, the regulatory network behind chilling requirement in relation to fruit development is highly complex. The epigenetic processes, like DNA methylation/demethylation, have been shown to play important roles in regulating gene expression in response to diverse environmental signals [21, 44]. Apple, being a temperate fruit crop, also requires adequate chilling temperature during dormancy period to resume normal active growth in response to warm temperature. While, lack of sufficient chilling leads to irregular bud break and delayed flowering, which consequently have detrimental effects on yield and quality of fruits. Therefore, in order to study the consequences of differential chilling availability on epigenetic regulation during winter dormancy, MSAP and RNA-Seq approaches have been employed.
In the present study, the temporal changes in methylation status of 5'-CCGG-3' tetra-nucleotide in apple genome were analyzed in four developmental stages from dormant bud to fruit set (DB, ST, GT and FS) under differential availability of chilling using MSAP technique. The MSAP profiles reveal the significant changes in methylation status during different temporal development stages of apple bud and fruit set under differential chilling availability (Table 2, Table 3). The results indicate the biological importance of epigenetic modifications in response to chilling during dormancy release and subsequent fruit set in apple. The methylation patterns of comparable stages, showed decrease in demethylation events under low chill condition as compared to high chill conditions, which suggests that re-initiation of active growth (bud break) and fruit set is linked with progressive demethylation events under ideal high chill conditions. In Arabidopsis, early flowering has been found to be associated with reduced 5-methyl cytosine level in vernalized (prolonged cold treatment) and 5-azacytidine (a DNA demethylating agent) treated plants (Burn et al. 1983). The decrease in DNA methylation at 5'-CCGG-3' sites with dormancy release was also reported in potato tubers and chestnut [45–46]. The previous study in pepper reported increase in demethylation during seed germination [33]. The low temperature associated demethylation in subsequent reproductive growth was also reported in wheat, rapeseed and Arabidopsis [11, 27–30]. Thus, it is evident from the previous studies, that the acquisition of low temperature has been associated with decrease in methylation and/or increase in demethylation during active growth phase of the plant. Interestingly, in the present study, availability of less chilling during dormancy period was found to be associated with less demethylation of 5'-CCGG-3' sequences in apple genome. Moreover, the methylation was also observed to be progressively decreased along the chilling mediated dormancy release and subsequent fruit set in apple (Table 2). The similar developmental stage specific changes in drought stress induced DNA methylation were also observed in rice [47].
The sequencing and gene ontology (GO) annotation of 47 MSAP fragments reveal that, the genes which undergo methylation changes are involved in diverse physiological and regulatory pathways (Table 4). These genes are broadly related to GO terms, such as response to stimulus, transcriptional regulation, catalytic activity, regulation of biological processes, cellular and metabolic processes, etc. The genes encoding pentatricopeptide repeat protein (PPR) and thioredoxin-like were previously reported to be involved in transcriptional regulation of gene expression in plants cell organelles [48–50]. In our analysis, the PPR gene was found to be demethylated in silver tip (ST) stage, where plant is committed to resume active growth. While, the thioredoxin-like gene was found to be methylated in green tip (GT) stage. The gene annotated as transcriptional corepressor SEUSS isoform X1 (SEU), was found to be methylated in GT stage, where floral meristem development is initiated. The SEU along with LEUNIG (LUG) was previously reported to be involved in repression of AGAMOUS (AG), a C class floral homeotic MADS gene [51]. Therefore, our results suggest that transcriptional SEU might be epigenetically regulated through cytosine methylation to remove repressive effect of SEU-LUG repressor complex on AG expression to initiate the flower development in apple. In Arabidopsis, HAC1 mutants exhibited late-flowering phenotype and therefore implicated to be involved in flowering time regulation [52]. The increase in histone acetyltransferase activity was previous reported in germinating Zea mays embryos [53]. Interestingly in our study, the histone acetyltransferase HAC1-like gene was found to be demethylated in GT stage, where plant resumes its active growth after dormant period. Moreover, a homeodomain containing Arabidopsis genes, FWA and BEL1 was reported to be involved in reproductive tissue development, where expression of FWA was epigenetically regulated through hypo-methylation of its promoter and coding region [54, 55]. In our analysis, homeodomain-like gene was also found to be demethylated at DB stage, however the gene identified in present analysis was similar to Arabidopsis homeodomain-like transcription regulator which was found to be expressed mainly during the reproductive organ development [56]. The auxin responsive gene has also been found to have altered expression during dormancy release in plants [3, 57]. In present analysis, the gene AUX/IAA27 B, of AUX/IAA family, was found to be methylated. Gene encoding NAC domain containing protein was found to be demethylated during the chilling acquisition that might have some role in flowering regulation. Previously LOV1, a NAC transcription factor was also found to be involved in flowering time regulation by negatively regulating the expression of CONSTANS (CO) and enhancing cold tolerance in transgenic Arabidopsis [58]. Recently, Arabidopsis NAC050 and NAC052 have been shown to regulate flowering time by associating with histone demethylase, JMJ14 [59].
In addition, the genes involved in RNA/DNA processing and transcriptional regulation, including RNA polymerase, U3 small nucleolar ribonucleoprotein, exonuclease, nuclear transcription factor Y, IQ-DOMAIN 14-like, DNA mismatch repair protein MSH7-like and E3 ubiquitin-protein ligase RING1 were also found to be differentially methylated in present analysis. Similarly, the gene involved in DNA/RNA processing and transcriptional regulation has been found to be differentially methylated in ryegrass [60] and maize [61]. Previous studies in poplar, kiwifruit and raspberry have reported the differential expression of stress related genes during the dormancy release [62–64]. In the present analysis, the genes involved in stress response, including CYP71A25-like, cation efflux protein, salt tolerance-like protein and quinone oxidoreductase PIG3 homolog were also found to be either methylated or demethylated, and exhibit differential expression (Table 4). In addition, wall-associated receptor kinase2-like, pectinesterase3, NADH-cytochrome b5 reductase1, aldehyde dehydrogenase family2 member C4-like, cysteine proteinase and SNARE protein involved in cellular and metabolic processes were also found to have altered methylation patterns (Table 4).
In Arabidopsis, the transcriptional regulation of genes was found to be determined by methylation levels within their gene bodies. Interestingly, these genes were found to be functionally more important as compared to the genes which are lacking methylation sites within their gene bodies [65, 66]. In addition, gene body methylation was also reported in other plants, like rice and sorghum [34, 47, 67]. The methylation changes at 5' and 3' regions of gene bodies and their flanking regions, was found to have regulatory effect on gene expression [68]. Similarly, in the present analysis, all the MSAP fragments reveal methylation in either gene body or 5' and 3' flanking regions (Table 4), which suggest that methylation may provide an additional regulatory mechanism to alter the transcription of these genes. In qRT-PCR analysis, expression of five out of eight genes associated with the methylated and demethylated fragments, was observed in accordance with their DNA methylation status (Fig 5). Therefore, our results suggest that changes in methylation status within the gene bodies are correlated with their gene expression patterns. These genes are involved in different biological processes and molecular functions. For instance, gene encoding UDP-galactose transporter (MDP0000198172) is involved in transport of UDP-galactose in golgi lumen for galactosylation of lipids, proteins and cell wall matrix polysaccharide [69, 70]. The embryo defective gene (MDP0000299377), that might have some role in embryo development [71], was found to be demethylated at ST stage, where plant was committed to grow in response to warm temperature. Similarly, the gene encoding ACT domain containing protein, previously reported to act as metabolism regulatory or sensory proteins in plants [72], was also found to be demethylated in ST stage. Moreover, the MSAP analysis represents only the 5'-CCGG-3' methylation, the cytosine methylation changes at other sequence contexts may also regulate the gene expression. Therefore, to accurately correlate the gene expression with gene body methylation percentage, bisulfite sequencing of randomly selected four MSAP fragments was performed. The ATP-dependent helicase belonging to DNA2/NAM7 helicase family (MDP0000296615), involved in replication/transcription, was found to be hyper-methylated, however, its expression pattern was moderately correlated with methylation level (Fig 5, S1 Fig). In contrast, high correlation between methylation level and expression pattern was observed for hyper-methylated MDP0000153928 gene (ribosomal protein kinase G11A-like) involved in signal transduction and metabolic regulation (Fig 5, S2 Fig). Similarly, the acid phosphatase 1-like gene (MDP0000186556), involved in phosphate energy metabolism [73], has low methylation level under low chill condition, which correlates with its expression pattern (Fig 5, S3 Fig). The hyper-methylation of galactose oxidase-like gene (MDP0000896660) under high chill condition was also found to be correlated with its expression pattern (Fig 5, S4 Fig). The galactose oxidase gene is involved in the production of reactive oxygen species, particularly H2O2 that also acts as signaling molecule [74]. Moreover, bisulfite sequencing of these genes suggests that low chilling might be associated with hyper-methylation of gene body that might result into suppression of their expression. Therefore, these results indicate that gene body methylation is a determining factor in regulating the gene expression. Moreover, FPKM based expression profiles of other genes associated with MSAP fragments also suggest that methylation status of genes was also correlated with their transcriptional activation or inhibition (Fig 3). The similar association between DNA methylation and transcriptional regulation of genes has also been reported in Arabidopsis [65].
In addition, the expression analysis of DNA methyltransferases, DNA gylcosylases and histone methyltransferases showed that chilling availability during winter dormancy has profound impact on the expression of genes encoding enzymes which actively catalyze the DNA methylation/demethylation and histone methylation. For instance, under low chill conditions, high percentage of fully methylated MSAP fragments was in agreement with high expression of MET1 and CMT3 methyltransferases in addition to low expression of DME-like and ROS1-like DNA glycosylases during active growth (GTL). Whereas, under high chill conditions, the gradual decrease in methylated fragments along the dormancy release and fruit set, was consistent with down-regulated expression of MET1 and CMT3 transcripts and high expression of DNA glycosylases. It has been previously reported that, some of the histone methylation marks are correlated with DNA cytosine methylation, which suggests cross talk between DNA methylation and histone methylation marks [75–78]. The H3K9, histone methylation mark was reported to be associated with DNA cytosine methylation at CHG sequence context [79], while, H3K4me1 mark has been reported to be associated with DNA methylation at CG sequence context [80]. In MSAP, based on methylation-sensitivity of the restriction enzymes (MspI/HpaII), it can be assumed that CHG (5mCCGG) and CG (C5mCGG) sequence contexts are mainly represented by hemi-methylated and fully methylated fragments, respectively. However, this assumption is inexact, since the different cytosine methylation combinations at 5'-CCGG-3' restriction site cannot be detected by activity of MspI/HpaII [22]. In the present analysis, the high percentage of hemi-methylated fragments in STL and FSL was in correlation with concomitant upregulation of SUVH4, SUVH6 and SUVH9 transcripts, involved in methylation of H3K9 mark [79,81–83], which further reinforce the presence of higher CHG methylation during STL and FSL, under low chill condition. In addition, SUVR4 and SUVR5, which are also involved in methylation of H3K9 histone mark [84, 85], were observed to be upregulated under low chill conditions in most of the comparative conditions. Conclusively, these findings suggest that the availability of chilling modulates the complex interplay between active and passive demethylation (replication and base excision), and associated de novo methylation and its maintenance by altering the gene expression of participating enzymes which are responsible for methylation changes.
Conclusion
To best of our knowledge, this is the first report of changes in methylation patterns during dormancy release and fruit set in apple, under differential chilling availability. The analysis of developmental stage specific changes in methylation level suggested that adequate (high) chilling availability during dormancy period is associated with reduced methylation of apple genome along the dormancy release and fruit set. Moreover, the chilling availability appears to modulate the expression of genes involved in active DNA methylation and demethylation. In addition, the genes encoding proteins involved in various biological processes like transcriptional regulation, catalysis, transport, defense, response to stimulus etc. were found to have altered methylation status under differential chilling. Thus, our results demonstrate that epigenetic modification through cytosine methylation is an important regulatory mechanism to control the dormancy release in apple and is likely to be modulated by chilling acquisition during dormancy period.
Supporting Information
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We gratefully acknowledge Director, CSIR-Institute of Himalayan Bioresource Technology, Palampur for the constant support. GK acknowledges senior research fellowship received from University Grants Commission, India. We kindly acknowledge Associate Director, Hill Agricultural Research & Extension Centre, Bajaura, Kullu (HP) and Director, SASE (DRDO) station Bahang, Manali (HP) for providing the temperature data. This manuscript represents CSIR-IHBT communication number 3952.
Data Availability
The reads obtained from Illumina GAIIx of all the analyzed samples were submitted as BioProject (PRJNA306594) to NCBI Sequence Read Archive under accession number SRP067619.
Funding Statement
This study was supported by the grants from Council of Scientific and Industrial Research, New Delhi, in the form of Network Project PlaGen (BSC0107) to AKS at CSIR-IHBT, Palampur.
References
- 1. Bond DM, Finnegan EJ. Passing the message on: inheritance of epigenetic traits. Trends Plant Sci. 2007; 12:211–216. [DOI] [PubMed] [Google Scholar]
- 2.Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008; 659:40–8. 10.1016/j.mrrev.2008.02.004 [DOI] [PubMed] [Google Scholar]
- 3.Shim D, Ko JH, Kim WC, Wang Q, Keathley DE, Han KH. A molecular framework for seasonal growth-dormancy regulation in perennial plants. Hortic Res. 2014; 1:14059 10.1038/hortres.2014.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Causevic A, Delaunay A, Ounnar S, Righezza M, Delmotte F, Brignolas F, et al. DNA methylating and demethylating treatments modify phenotype and cell wall differentiation state in sugarbeet cell lines. Plant Physiol Biochem. 2005; 43:681–691. [DOI] [PubMed] [Google Scholar]
- 5.Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature. 2007; 447:418–424. [DOI] [PubMed] [Google Scholar]
- 6.Messeguer R, Ganal MW, Steffens JC, Tanksley SD. Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear DNA. Plant Mol Biol. 1991; 16:753–770. [DOI] [PubMed] [Google Scholar]
- 7.Lund G, Messing J, Viotti A. Endosperm-specific demethylation and activation of specific alleles of alpha-tubulin genes of Zea mays L. Mol Gen Genet. 1995; 246:716–722. [DOI] [PubMed] [Google Scholar]
- 8.Ulian EC, Magill JM, Magill CW, Smith RH. DNA methylation and expression of NPT II in transgenic petunias and progeny. Theor Appl Genet. 1996; 92:976–981. 10.1007/BF00224037 [DOI] [PubMed] [Google Scholar]
- 9.Hashida SN, Uchiyama T, Martin C, Kishima Y, Sano Y, Mikami T. The temperature-dependent change in methylation of the Antirrhinum transposon Tam3 is controlled by the activity of its transposase. Plant Cell. 2006; 18:104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010; 11:204–220. 10.1038/nrg2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finnegan EJ, Genger RK, Kovac K, Peacock WJ, Dennis ES. DNA methylation and the promotion of flowering by vernalization. Proc Natl Acad Sci USA. 1998; 95:5824–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pradhan S, Adams RLP. Distinct CG and CNG DNA methyltransferases in Pisum sativum. Plant J. 1995; 7:471–481. [DOI] [PubMed] [Google Scholar]
- 13.Kovarik A, Matyasek R, Leitch A, Gazdova B, Fulnecek J, Bezdek M. Variability in CpNpG methylation in higher plant genomes. Gene. 1997; 204:25–33. [DOI] [PubMed] [Google Scholar]
- 14.Jeddeloh JA, Richards EJ. mCCG methylation in angiosperms. Plant J. 1996; 9:579–586. [DOI] [PubMed] [Google Scholar]
- 15.Steward N, Ito M, Yamakuchi Y, Koizumi N, Sano H. Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J Biol Chem. 2002; 277:37741–37746. [DOI] [PubMed] [Google Scholar]
- 16.Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, Fischer RL. DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci USA. 2007; 104:6752–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ortega-Galisteo AP, Morales-Ruiz T, Ariza RR, Roldán-Arjona T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol Biol. 2008; 67:671–681. 10.1007/s11103-008-9346-0 [DOI] [PubMed] [Google Scholar]
- 18.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphate sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008; 452:215–219. 10.1038/nature06745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang XY. The epigenetic landscape of plants. Science. 2008; 320:489–492. 10.1126/science.1153996 [DOI] [PubMed] [Google Scholar]
- 20.Tran RK, Henikoff J, Zilberman D, Ditt R, Jacobsen S, Henikoff S. DNA methylation profiling identifies CpG methylation clusters in Arabidopsis genes. Curr Biol. 2005; 15:154–159. [DOI] [PubMed] [Google Scholar]
- 21.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002; 16:6–21. [DOI] [PubMed] [Google Scholar]
- 22.Fulneček J, Kovařík A. How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet. 2014; 15:2 10.1186/1471-2156-15-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McClelland M, Nelson M, Raschke E. Effect of site-specific modification on restriction endonucleases and DNA modification methyl-transferases. Nucleic Acid Res. 1994; 22:3640–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson JV, Gesch RW, Jia Y, Chao WS, Horvath DP. Seasonal shifts in dormancy status, carbohydrate metabolism, and related gene expression in crown buds of leafy spurge. Plant Cell Environ. 2005; 28:1567–1578. [Google Scholar]
- 25.Lang GA. Dormancy: a new universal terminology. HortScience. 1987; 22:817–820. [Google Scholar]
- 26.Sugiura T, Sumida H, Yokoyama S, Ono H. Overview of recent effects of global warming on agricultural production in Japan. JARQ. 2012; 46:7–13. [Google Scholar]
- 27.Burn JE, Bagnall DJ, Metzger JD, Dennis ES, Peacock WJ. DNA methylation, vernalization, and the initiation of flowering. Proc Natl Acad Sci USA. 1993; 90:287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lizal P, Relichova J. The effect of day length, vernalization and DNA demethylation on the flowering time in Arabidopsis thaliana. Physiol Plant. 2001; 113:121–127. [Google Scholar]
- 29.Sherman JD, Talbert LE. Vernalization-induced changes of the DNA methylation pattern in winter wheat. Genome. 2002; 45:253–260. [DOI] [PubMed] [Google Scholar]
- 30.Wrobelska JG, Filek M, Kaliciak A, Szarejko I, Machackova I, Krekule J, et al. Vernalization and photoperiod-related changes in the DNA methylation state in winter and spring rapeseed. Acta Physiol Plant. 2013; 35:817–827. [Google Scholar]
- 31.Jangra MS, Sharma JP. Climate resilient apple production in Kullu valley of Himachal Pradesh. Intl J Farm Sci. 2013; 3:91–98. [Google Scholar]
- 32.Xin Z, Chen J. A high throughput DNA extraction method with high yield and quality. Plant Methods. 2012; 8:26 10.1186/1746-4811-8-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Portis E, Acquadro A, Comino C, Lanteri S. Analysis of DNA methylation during germination of pepper (Capsicum annuum L.) seeds using methylation-sensitive amplification polymorphism (MSAP). Plant Sci. 2004; 166:169–178. [Google Scholar]
- 34.Karan R, DeLeon T, Biradar H, Subudhi PK. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS One. 2012; 7:e40203 10.1371/journal.pone.0040203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muoki RC, Paul A, Kumari A, Singh K, Kumar S. An improved protocol for the isolation of RNA from roots of tea (Camellia sinensis (L.) O. Kuntze). Mol Biotechnol. 2012; 52:82–88. 10.1007/s12033-011-9476-5 [DOI] [PubMed] [Google Scholar]
- 36.Patel RK, Jain M. NGS QC toolkit: A toolkit for quality control of next generation sequencing data. PLoS One. 2012; 7:e30619 10.1371/journal.pone.0030619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 18:1427–31. [DOI] [PubMed] [Google Scholar]
- 38.Arya P, Kumar G, Acharya V, Singh AK. Genome-wide identification and expression analysis of NBS-encoding genes in Malus x domestica and expansion of NBS genes family in rosaceae. PLoS One, 2014; 9:e107987 10.1371/journal.pone.0107987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar G, Singh AK. Reference Gene validation for qRT-PCR based gene expression studies in different developmental stages and under biotic stress in apple. Sci Hortic. 2015; 10.1016/j.scienta.2015.10.025 [DOI] [Google Scholar]
- 40.Rowland LJ, Alkharouf N, Darwish O, Ogden EL, Polashock JJ, Bassil NV, et al. Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 2012; 12:46 10.1186/1471-2229-12-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedley PE, Russell JR, Jorgensen L, Gordon S, Morris JA, Hackett CA, et al. Candidate genes associated with bud dormancy release in blackcurrant (Ribes nigrum L.). BMC Plant Biol. 2010; 10:202 10.1186/1471-2229-10-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leida C, Conejero A, Arbona V, Gomez-Cadenas A, Llacer G, Badenes ML, et al. Chilling-dependent release of seed and bud dormancy in peach associates to common changes in gene expression. PLoS One. 2012; 7:e35777 10.1371/journal.pone.0035777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luca M, Hancock RD, Haupt S, Walker PG, Pont SDA, McNicol J, et al. Co-ordinated gene expression during phases of dormancy release in raspberry (Rubus idaeus L.) buds. J Exp Bot. 2007; 58:1035–1045. [DOI] [PubMed] [Google Scholar]
- 44.Angers B, Castonguay E, Massicotte R. Environmentally induced phenotypes and DNA methylation: how to deal with unpredictable conditions until the next generation and after. Mol Ecol. 2010; 19:1283–1295. 10.1111/j.1365-294X.2010.04580.x [DOI] [PubMed] [Google Scholar]
- 45.Law RD, Suttle JC. Transient decreases in methylation at 5'-CCGG-3' sequences in potato (Solanum tuberosum L.) meristem DNA during progression of tubers through dormancy precede the resumption of sprout growth. Plant Mol Biol. 2003; 51: 437–447. [DOI] [PubMed] [Google Scholar]
- 46.Santamaria ME, Rodriguez R, Canal MJ, Toorop PE. Transcriptome analysis of chestnut (Castanea sativa) tree buds suggests a putative role for epigenetic control of bud dormancy. Ann Bot. 2011; 108:485–498. 10.1093/aob/mcr185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang WS, Pan YJ, Zhao XQ, Dwivedi D, Zhu LH, Ali J, et al. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot. 2011; 62:1951–1960. 10.1093/jxb/erq391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okuda K, Myouga F, Motohashi R, Shinozaki K, Shikanai T. Conserved domain structure of pentatricopeptide repeat proteins involved in chloroplast RNA editing. Proc Natl Acad Sci USA. 2007; 104: 8178–8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barkan A, Small I. Pentatricopeptide repeat proteins in plants. Annu Rev Plant Biol. 2014; 65:415–442. 10.1146/annurev-arplant-050213-040159 [DOI] [PubMed] [Google Scholar]
- 50.Yu QB, Ma Q, Kong MM, Zhao TT, Zhang XL, Zhou Q, et al. AtECB1/MRL7, a thioredoxin-like fold protein with disulfide reductase activity, regulates chloroplast gene expression and chloroplast biogenesis in Arabidopsis thaliana. Mol Plant. 2014; 7:206–217. 10.1093/mp/sst092 [DOI] [PubMed] [Google Scholar]
- 51.Sridhar VV, Surendrarao A, Gonzalez D, Conlan RS, Liu Z. Transcriptional repression of target genes by LEUNIG and SEUSS, two interacting regulatory proteins for Arabidopsis flower development. Proc Natl Acad Sci USA. 2004; 101:11494–11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deng WW, Liu CY, Pei YX, Deng X, Niu LF, Cao XF. Involvement of the histone acetyltransferase AtHAC1 in the regulation of flowering time via repression of FLOWERING LOCUS C in Arabidopsis. Plant Physiol. 2007; 143:1660–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Georgieva EI, Lopez-Rodas G, Sendra R, Grobner P, Loidl P. Histone acetylation in Zea mays. II. Biological significance of post-translational histone acetylation during embryo germination. J Biol Chem. 1991; 266:18751–18760. [PubMed] [Google Scholar]
- 54.Reiser L, Modrusan Z, Margossian L, Samach A, Ohad N, Haughn GW, et al. The BELL1 gene encodes a homeodomain protein involved in pattern formation in the Arabidopsis ovule primordium. Cell. 1995; 83:735–742. [DOI] [PubMed] [Google Scholar]
- 55.Soppe WJJ, Jacobsen SE, Alonso-Blanco C, Jackson JP, Kakutani T, Koornneef M, et al. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell. 2000; 6:791–802. [DOI] [PubMed] [Google Scholar]
- 56.Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005; 37:501–506. [DOI] [PubMed] [Google Scholar]
- 57.Bassa C, Mila I, Bouzayen M, Audran-Delalande C. Phenotypes associated with down-regulation of Sl-IAA27 support functional diversity among Aux/IAA family members in tomato. Plant Cell Physiol. 2012; 53:1583–1595. 10.1093/pcp/pcs101 [DOI] [PubMed] [Google Scholar]
- 58.Yoo SY, Kim Y, Kim SY, Lee JS, Ahn JH. Control of flowering time and cold response by a NAC-domain protein in Arabidopsis. PLoS One. 2007; 2: e642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ning YQ, Ma ZY, Huang HW, Mo H, Zhao TT, Li L, et al. Two novel NAC transcription factors regulate gene expression and flowering time by associating with the histone demethylase JMJ14. Nucleic Acids Res. 2015; 43:1469–1484. 10.1093/nar/gku1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang XM, Tao X, Wang Y, Ma DW, Li D, Yang H, et al. Analysis of DNA methylation of perennial ryegrass under drought using the methylation sensitive amplification polymorphism (MSAP) technique. Mol Genet Genomics. 2014; 289:1075–1084. 10.1007/s00438-014-0869-6 [DOI] [PubMed] [Google Scholar]
- 61.Shan X, Wang X, Yang G, Wu Y, Su S, Li S, et al. Analysis of the DNA methylation of maize (Zea mays L.) in response to cold stress based on methylation-sensitive amplified polymorphisms. J Plant Biol. 2013; 56:32–38. [Google Scholar]
- 62.Mazzitelli L, Hancock RD, Haupt S, Walker PG, Pont SD, McNicol J, et al. Co-ordinated gene expression during phases of dormancy release in raspberry (Rubus idaeus L.) buds. J Exp Bot. 2007; 58:1035–1045. [DOI] [PubMed] [Google Scholar]
- 63.Walton EF, Wu RM, Richardson AC, Davy M, Hellens RP, Thodey K, et al. A rapid transcriptional activation is induced by the dormancy-breaking chemical hydrogen cyanamide in kiwifruit (Actinidia deliciosa) buds. J Exp Bot. 2009; 60:3835–3848. 10.1093/jxb/erp231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ko JH, Prassinos C, Keathley D Han KH. Novel aspects of transcriptional regulation in the winter survival and maintenance mechanism of poplar. Tree physiol. 2011; 31:208–225. 10.1093/treephys/tpq109 [DOI] [PubMed] [Google Scholar]
- 65.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007; 39:61–69. [DOI] [PubMed] [Google Scholar]
- 66.Takuno S, Gaut BS. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol. 2012; 29: 219–227. 10.1093/molbev/msr188 [DOI] [PubMed] [Google Scholar]
- 67.Zhang M, Xu C, Wettstein DV, Liu B. Tissue-specific differences in cytosine methylation and their association with differential gene expression in sorghum. Plant Physiol. 2011; 156:1955–1966. 10.1104/pp.111.176842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gehring M, Henikoff S. DNA methylation dynamics in plant genomes. Biochim Biophys Acta. 2007; 1769:276–286. [DOI] [PubMed] [Google Scholar]
- 69.Rautengarten C, Ebert B, Moreno I, Temple H, Herter T, Link B, et al. The Golgi localized bifunctional UDP-rhamnose/UDP-galactose transporter family of Arabidopsis. Proc Natl Acad Sci U S A. 2014; 111:11563–11568. 10.1073/pnas.1406073111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abedi T, Khalil MF, Asai T, Ishihara N, Kitamura K, Ishida N, et al. UDP-galactose transporter gene hUGT1 expression in tobacco plants leads to hyper-galactosylated cell wall components. J Biosci Bioeng. 2015; 10.1016/j.jbiosc.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 71.Tzafrir I, Pena-Muralla R, Dickerman A, Berg M, Rogers R, Hutchens S, et al. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004; 135:1206–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hsieh MH, Goodman HM. Molecular characterization of a novel gene family encoding ACT domain repeat proteins in Arabidopsis. Plant Physiol. 2002; 130:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duff SMG, Sarath G, Plaxton WC. The role of acid phosphatases in plant phosphorus metabolism. Physiol Plant. 1994; 90:791–800. [Google Scholar]
- 74.Phan HA, Iacuone S, Li SF, Parish RW. The MYB80 transcription factor is required for pollen development and the regulation of tapetal programmed cell death in Arabidopsis thaliana. Plant Cell. 2011; 23:2209–2224. 10.1105/tpc.110.082651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Callis J, et al. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol. 2007; 17:379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Woo HR, Dittmer TA, Richards EJ. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet. 2008; 4:e1000156 10.1371/journal.pgen.1000156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou J, Wang X, He K, Charron JB, Elling AA, Deng XW. Genome-wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol Biol. 2010; 72:585–595. 10.1007/s11103-009-9594-7 [DOI] [PubMed] [Google Scholar]
- 78.Du J, Zhong X, Bernatavichute YV, Stroud H, Feng S, Caro E, et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell. 2012; 151: 167–180. 10.1016/j.cell.2012.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bernatavichute YV, Zhang X, Cokus S, Pellegrini M, Jacobsen SE. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS One. 2008; 3:e3156 10.1371/journal.pone.0003156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang X, Bernatavichute YV, Cokus S, Pellegrini M, Jacobsen SE. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009; 10:R62 10.1186/gb-2009-10-6-r62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ebbs ML, Bartee L, Bender J. H3 lysine 9 methylation is maintained on a transcribed inverted repeat by combined action of SUVH6 and SUVH4 methyltransferases. Mol Cell Biol. 2005; 25:10507–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ebbs ML, Bender J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell. 2006; 18:1166–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu ZW, Shao CR, Zhang CJ, Zhou JX, Zhang SW, Li L, et al. The SET domain proteins SUVH2 and SUVH9 are required for Pol V occupancy at RNA-directed DNA methylation loci. PLoS Genet. 2014; 10:e1003948 10.1371/journal.pgen.1003948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thorstensen T, Fischer A, Sandvik SV, Johnsen SS, Grini PE, Reuter G, et al. The Arabidopsis SUVR4 protein is a nucleolar histone methyltransferase with preference for monomethylated H3K9. Nucleic Acids Res. 2006; 34:5461–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Caro E, Stroud H, Greenberg MV, Bernatavichute YV, Feng S, Groth M, et al. The SET-domain protein SUVR5 mediates H3K9me2 deposition and silencing at stimulus response genes in a DNA methylation-independent manner. PLoS Genet. 2012; 8:e1002995 10.1371/journal.pgen.1002995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
Sequence of top strand (ORG) represents the non-bisulfite converted locus and was cloned from the genomic DNA of bud samples.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
The reads obtained from Illumina GAIIx of all the analyzed samples were submitted as BioProject (PRJNA306594) to NCBI Sequence Read Archive under accession number SRP067619.