

Abstract
Study Objectives:
The nuclear receptor REV-ERBα is a potent, constitutive transcriptional repressor critical for the regulation of key circadian and metabolic genes. Recently, REV-ERBα's involvement in learning, neurogenesis, mood, and dopamine turnover was demonstrated suggesting a specific role in central nervous system functioning. We have previously shown that the brain expression of several core clock genes, including Rev-erbα, is modulated by sleep loss. We here test the consequences of a loss of REV-ERBα on the homeostatic regulation of sleep.
Methods:
EEG/EMG signals were recorded in Rev-erbα knockout (KO) mice and their wild type (WT) littermates during baseline, sleep deprivation, and recovery. Cortical gene expression measurements after sleep deprivation were contrasted to baseline.
Results:
Although baseline sleep/wake duration was remarkably similar, KO mice showed an advance of the sleep/wake distribution relative to the light-dark cycle. After sleep onset in baseline and after sleep deprivation, both EEG delta power (1–4 Hz) and sleep consolidation were reduced in KO mice indicating a slower increase of homeostatic sleep need during wakefulness. This slower increase might relate to the smaller increase in theta and gamma power observed in the waking EEG prior to sleep onset under both conditions. Indeed, the increased theta activity during wakefulness predicted delta power in subsequent NREM sleep. Lack of Rev-erbα increased Bmal1, Npas2, Clock, and Fabp7 expression, confirming the direct regulation of these genes by REV-ERBα also in the brain.
Conclusions:
Our results add further proof to the notion that clock genes are involved in sleep homeostasis. Because accumulating evidence directly links REV-ERBα to dopamine signaling the altered homeostatic regulation of sleep reported here are discussed in that context.
Citation:
Mang GM, La Spada F, Emmenegger Y, Chappuis S, Ripperger JA, Albrecht U, Franken P. Altered sleep homeostasis in Rev-erbα knockout mice. SLEEP 2016;39(3):589–601.
Keywords: clock genes, mood, depression, neurogenesis, Process S, slow wave activity.
Significance.
Although circadian clock genes are named for their role in driving circadian rhythms in gene expression, physiology, and behavior, they can fulfill other important functions. The clock gene and transcriptional repressor REV-ERBα plays a role in pathways affecting metabolism and central nervous functioning. Using mice lacking the gene encoding REV-ERBα, Nr1d1, we could extend these finding to include the homeostatic regulation of sleep. Because the activity of REV-ERBα is modulated by cellular redox state, we propose that this molecule can sense and respond to the metabolic imbalance imposed at the neuronal level by periods of extended wakefulness. Recently developed synthetic drugs targeting REV-ERBα could thus be useful in the treatment of both the circadian and homeostatic aspects of sleep-wake related disorders.
INTRODUCTION
The timing and quality of sleep are controlled by the interaction of a homeostatic process, that tracks sleep need as a function of the previous sleep/wake history, and a circadian process that ensures the appropriate timing of sleep relative to the daily light-dark alternation.1,2 Although these two processes seem functionally and neurophysiologically distinct, at the molecular level, several core components of the circadian timing system were found to also play a role in maintaining proper sleep homeostasis.3,4
The molecular circadian oscillator consists of positive and negative elements. In mammals the positive elements comprise of CLOCK, NPAS2, and BMAL1, with CLOCK/ NPAS2:BMAL1 heterodimers driving the transcription of many target genes including that of the Period (Per1, -2) and Cryptochrome (Cry1, -2) genes.5 PER:CRY protein complexes suppress CLOCK/NPAS2:BMAL1-mediated transcription, including their own, thereby constituting the negative elements in this core feedback loop. Additional interactions between these core clock genes at the level of transcription, translocation back into the nucleus, and post-translational modifications add further complexity and stability to the circuit. One important auxiliary feedback loop involves the orphan nuclear receptor REV-ERBα, which binds to specific sequences in the promoters of all three positive elements,6–8 and inhibits their transcription.9–11 In turn, Rev-erbα expression is directly regulated by CLOCK/NPAS2:BMAL1-mediated transcription.8,12 Given REV-ERBα's ability to reset the phase of the molecular clock oscillation and to entrain central and peripheral clocks, it has been suggested to function as a synchronizing “hinge” of the clock gene machinery13 enabling it to act as “gatekeeper” in coordinating the circadian metabolic response.14
The REV-ERBα protein, encoded by the Nr1d1 (nuclear receptor subfamily 1, group D, member 1) gene, regulates gene transcription through binding to specific DNA sequences comprising an A/T-rich flank followed by AGGTCA in the promoter region of target genes.15,16 REV-ERBα is part of the ligand-binding receptor family, but lacks the carboxy-terminal tail of the ligand-binding domain required for co-activation and thus necessary for transcriptional activation.17 As a consequence, upon activation by its ligand, heme, REV-ERBα is a constitutive repressor of gene expression.16,18–21
Beside its important role in the generation of circadian rhythms, REV-ERBα is implicated in the regulation of various metabolic pathways including adipocyte differentiation, gluconeogenesis, bile acid synthesis, and heme and cholesterol homeostasis.20,22–24 Consistent with its function, Rev-erbα is highly expressed in tissues with high rates of metabolism such as adipose tissue, liver, skeletal muscle, and brain.25,26 Rev-erbα deletion impacts the expression of many metabolic genes, in particular genes involved in lipid metabolic pathways.12 The administration of REV-ERBα agonists in mice decreased diet-induced obesity by reducing fat content and improving hyper-glycemia and dyslipidemia.27 The expression of Rev-erbα can be directly activated through the peroxisome proliferator-activated receptors (PPARs) that play essential roles in energy metabolism and are themselves expressed in a circadian manner.28 REV-ERBα is a heme sensor and crucial for heme homeostasis, thereby further tightening the link between metabolic and circadian physiology.16,19–21
In the context of sleep homeostasis, we have previously shown that the expression of Rev-erbα in the forebrain of mice is decreased after sleep deprivation.29 We hypothesize that Rev-erbα could act as an integrator of both energy demand and sleep pressure. With the aim to establish such role, we evaluated sleep, the EEG, and cortical gene expression under baseline and sleep deprivation conditions, in Rev-erbα knockout (KO) mice and their wild-type (WT) littermate controls.
METHODS
Animals and Housing Conditions
Rev-erbα KO mice were kindly provided by Ueli Schibler (University of Geneva, Geneva, Switzerland) and maintained on a mixed 129/Sv × C57BL6 background. In these mice, exons 3 and 4, encoding the DNA binding domain, and part of exons 2 and 5 of the Rev-erbα gene, were replaced by an in-frame lacZ allele and a PGK-neo gene by homologous recombination in 129/SV ES cells, resulting in the absence of the transcript and protein.8 Wild type (WT) littermates were used as control animals. Mice were individually housed in polycarbonate cages (31×18×18 cm) in a temperature and humidity controlled room (25°C, 50% to 60%, respectively) and a 12 h light/12 h dark cycle (lights on at 09:00, 70–90 lux). Animals had access to food and water ad libitum. All experiments were approved by the Ethical Committee of the State of Vaud Veterinary Office, Switzerland.
EEG/EMG Implantation
At the age of 9 to 14 weeks, 13 KO and 9 WT male mice were implanted with EEG and EMG electrodes under deep xylazine/ ketamine anesthesia as previously described.30 Briefly, 6 gold-plated screws (diameter 1.1 mm) were screwed bilaterally into the skull, over the frontal and parietal cortices. Two served as EEG electrodes, and the remaining 4 anchored the electrode connector assembly. As EMG electrodes 2 gold wires were inserted into the neck musculature. The EEG and EMG electrodes were soldered to a connector and cemented to the skull. Animals were allowed to recover from surgery during 5–7 days before they were connected to the recording cables in their home cage. A minimum of 6 days were allowed for habituation to the cable and the experimental room prior to the experiments. Mice were 11–16 weeks old at the time of experiment and age did not differ between genotypes (t-test, P = 0.33).
Experimental Protocols and Data Acquisition
EEG and EMG signals were recorded continuously for 72 h. The recording started at light onset; i.e., Zeitgeber Time (ZT) 0 or ZT0. During the first 48 h, mice were left undisturbed and these two days were considered as baseline. Starting at ZT0 of day 3, animals were sleep deprived by gentle handling as described previously31 during 6 hours (ZT0–6). The remaining 18 h of day 3 were considered as recovery.
The analog EEG and EMG signals were amplified (2,000×) and filtered and then digitized at 2 kHz and subsequently down sampled to 200 Hz and stored. The EEG was subjected to a discrete Fourier transformation yielding power spectra (range: 0–100 Hz; frequency resolution: 0.25 Hz; time resolution: consecutive 4-sec epochs; window function: Hamming). Hardware (EMBLA) and software (Somnologica-3) were purchased from Medcare Flaga (EMBLA, Thornton, USA).
Determination of Behavioral States
Offline, the animal's behavior was visually classified as “Wakefulness,” “REM sleep,” or “NREM sleep” for consecutive 4-sec epochs based on the EEG and EMG signals as previously described.30 Wakefulness was characterized by EEG activity of mixed frequency and low amplitude. Muscle tone was present and variable. NREM sleep was defined by synchronous activity in the delta frequency (1–4 Hz) and low and stable muscle tone. REM sleep was characterized by regular theta oscillations (6–9 Hz) and muscle atonia with occasional twitches. Four-sec epochs containing EEG artifacts were marked according to the state in which they occurred and excluded from EEG spectral analysis. As the sleep/wake state of epochs in which EEG artifacts occurred could still be determined, they were included in the analysis of time-spent-asleep and -awake.
Data Analysis
Analysis of the time course of time spent in each sleep/wake state was performed on 1-, 12-, and 24-h values. For the 12-h and 24-h analysis, the 2 baseline days were averaged, as no significant differences between the 2 days were observed (3-way repeated measures analysis of the variance [rANOVA], factors “day,” “time,” and “genotype”; P > 0.12 for factor “day” and all its interactions). To further quantify the altered baseline distribution of sleep/wake states in KO mice, hourly values were accumulated and genotype differences calculated to assess the times at which the two distributions deviated.
Sleep/wake state quality was assessed by analyzing the spectral content of the EEG. To account for inter-individual differences in overall EEG power, EEG spectra of the 3 sleep/ wake states were expressed as a percentage of an individual reference value calculated as the total EEG power across all frequencies considered (0.75–45.0 Hz) and sleep/wake states. This reference value was weighted so that for all animals the relative contribution of the 3 sleep/wake states to this reference value was equal, according to.32 Theta peak frequency in wakefulness and REM sleep was calculated by determining the frequency at which maximum power density in the theta frequency range (5–10 Hz) was reached.
Effects of sleep deprivation were assessed by analyzing EEG delta power, sleep fragmentation, and time spent asleep. Time course analysis of EEG delta power (i.e., the mean EEG power density in the 1–4 Hz range in NREM sleep) during baseline and after sleep deprivation was performed as described previously.33 Briefly, the recording was divided into sections to which an equal number of 4-sec epochs scored as NREM sleep contributed (i.e., percentiles). The baseline light periods were divided into 12 such sections; the baseline and recovery dark periods into 6. The recovery light (ZT6–12) period was divided into 8 sections. The number of percentiles per recording period was chosen to assure that sufficient and similar numbers of NREM sleep epochs contributed to each interval necessary to obtain meaningful mean delta power values. Delta power values were normalized within each individual mouse by expressing all values relative to the mean value reached in the last 4 h of the main rest periods (i.e., ZT8–12) when delta power is minimal during baseline consistent with the fact that delta power is thought to reflect homeostatic sleep need which is lowest at the end of the major rest period. Besides its time course, delta power levels reached immediately after sleep onset in baseline and after sleep deprivation were determined separately. To assure that the same number of 4-sec epochs contributed, delta power was averaged over the first 20 min (300 4-sec epochs) scored as NREM sleep. Sleep onset was determined as the entry into the first episode of NREM sleep lasting > 1 min and not interrupted by 2 or more 4-sec epochs of wakefulness. This first episode was determined from light onset in the baseline days and from the end of the sleep deprivation for the recovery period.
The effect of 6 h sleep deprivation on time spent in and NREM and REM sleep was assessed by calculating the recovery-baseline difference in sleep time for 6-h intervals; i.e., the first 6 h of recovery still in the light period (ZT6–12) and two 6-h intervals in the dark (ZT12–18 and ZT18–24). The effect of sleep deprivation on NREM sleep spectra and sleep fragmentation was assessed by contrasting EEG spectra and the number of short awakenings (waking bouts lasting 4 consecutive 4-s epochs or less; i.e., < 16 s, expressed per hour of NREM sleep31) during the first 3 h of recovery sleep after sleep onset (ZT6–9) from the values observed during the last 4 h of the baseline light periods (ZT8–12), a period during which homeostatic sleep need (and EEG delta power) is lowest and sleep fragmentation highest. Similarly, NREM sleep spectra and the number of brief awakenings in the first 3 h after sleep onset (ZT0–3) in the 2 baseline days was calculated and contrasted to the same reference.
Cortical Gene Expression Analysis
Nine KO and 10 WT male mice were used to assess the effect of genotype and sleep deprivation on cortical gene expression. In each genotype, half the mice were submitted to a 6 h sleep deprivation (ZT0–6), and the other half was left undisturbed and used as control. At ZT6, both groups of mice were sacrificed, the cerebral cortex extracted, and immediately flash frozen in liquid nitrogen. Samples were stored at −80°C. RNA was extracted and purified using the RNeasy Lipid Tissue Mini Kit 50 (QIAGEN, Hombrechtikon, Switzerland) according to manufacturer's instructions. RNA quantity (NanoDrop ND-1000 spectrophotometer; Thermo Scientific, Wilmington, NC, USA) and quality (Agilent 2100 bioanalyzer chips; Agilent technologies, Basel, Switzerland) was measured and verified; 1,000 ng of purified RNA were reverse-transcribed in 20 μL using random hexamers and Superscript II reverse transcriptase (Invitrogen, Life Technologies, Europe, Zug, Switzerland) according to standard procedures. The cDNA was diluted 10 times and 2 μL were amplified in a 10 μL TaqMan reaction in technical triplicates on an ABI PRISM HT 7900 detection system (Applied Biosystems, Life Technologies, Europe, Zug, Switzerland). Cycler conditions were 50°C 2 min, 95°C 10 min, and 45 cycles at 95°C 15 s, and 60°C 1 min. To quantify the RNA expression level, specific forward and reverse primers and probes were used (Table S2, supplemental material). Gene expression levels were normalized to 4 reference genes (Gapdh, Tbp, Rsp9, and Eef1a1) using QbasePLUS software (Biogazelle, Zwijnaarde, Belgium) except for Fabp7 which was expressed relative to Gapdh only. The fold change indicative of the relative gene expression are based on the mean of three biological replicates in relation to control samples.
Statistics and Analysis Tools
TMT Pascal Multi-Target5 software (Framework Computers, Inc., Brighton, MA, USA) was used to manage the data, SigmaPlot V10.0 (Systat Software Inc., Chicago, IL, USA) for graphics, and SAS V9.2 (SAS Institute Software Inc., Cary, NC, USA) or Sigmastat V3.5. (Systat Software, Chicago, IL, USA) for statistical analysis. To assess the effect of genotype on the sleep/wake distribution, EEG power spectra, and time course of delta power, 2- or 3-way rANOVAs were performed. Significant effects and interactions were decomposed using post hoc Tukey HSD and t-tests. Genotype differences in the light/dark amount, accumulation, fragmentation of sleep/wake states, and in EEG delta power at sleep onset, as well as the cortical gene expression were evaluated using t-tests. Statistical significance was set to P = 0.05 and results are reported as mean ± standard error of the mean (SEM) or of the difference (SED).
RESULTS
Sleep/Wake Distribution Is Advanced in Rev-erbα Knockout Mice
Similar to WT mice, Rev-erbα KO mice were mostly asleep during the light period and mostly awake during the dark period (almost 2/3 of the time in the respective 12-h periods; Figure 1; Table S1, supplemental material). Nevertheless, the distribution of the 3 sleep/wake states over the 24-h day importantly differed between the 2 genotypes (Figure 1). Differences were observed mainly at the time encompassing the light-to-dark transition when KO mice spent more time awake than their WT littermates; i.e., in the 2 h prior and 3 h following this transition (ZT10–15; Figure 1A). The dynamics of the accumulation of time spent awake over the baseline days summarizes this effect and demonstrates that by ZT15 KO mice accrued approximately 1 h extra wakefulness compared to WT mice (Figure 1B). Interestingly, this gain in wakefulness was rapidly lost over the subsequent 3 h (ZT15–18) resulting in almost identical 24-h values for wakefulness (Figure 1B, Table S1). Similar, albeit opposite results were observed for NREM and REM sleep (analyses not shown).
Figure 1.

Rev-erbα knockout mice have an earlier increase in wakefulness relative to dark onset. (A) Mean (± 1 SEM) hourly values of Wakefulness, NREM sleep, and REM sleep over 48 h baseline. Genotype significantly affected the time course for all 3 states, while the 2 baseline days did not differ (3-way rANOVA, factors genotype [‘G’: KO, WT], day [‘D’: 1, 2], and hour [‘H’: 1–24]; Wakefulness: ‘G’ P = 0.94, ‘D’ P = 0.19, ‘H’ P < 0.001, ‘GxD’ P = 0.33, ‘GxH’ P < 0.001, ‘DxH’ P = 0.56; NREMS: ‘G’ P = 0.35, ‘D’ P = 0.12, ‘H’ P < 0.001, ‘GxD’ P = 0.19, ‘GxH’ P < 0.001, ‘DxH’ P = 0.46; REMS: ‘G’ P = 0.22, ‘D’ P = 0.13, ‘H’ P < 0.001, ‘GxD’ P = 0.18, ‘GxH’ P < 0.001, ‘DxH’ P = 0.87). (B) Upper curve: Mean (± 1 SEM) accumulation of time spent awake calculated at 1-h increments. The two baseline days were averaged. Lower curve: Mean differences (KO-WT; ± 1 SED) in accumulated values represented in the upper curve. (C) Amplitudes of the difference in time spent awake between the two 12-h periods. The start time of the 12-h periods was shifted at 1-min increments with 0 shift corresponding to the actual dark onset. For each 1min shift the amplitude was calculated. Circles with bi-directional error bars indicated the mean (± 1 SEM) of the individual amplitude with 0 shift (symbols labeled ‘1’; same values as in Table S1) or at the shift yielding maximal amplitude (symbols labeled ‘2’). All panels, knockout (KO, n = 13) and wild type (WT, n = 8) mice are indicated with black lines and symbols and gray line and open symbols, respectively. Gray areas represent 12 h dark periods. Red squares in A) and B) indicate hours with significant genotype differences (post hoc t-tests, P < 0.05).
Because of this redistribution, KO mice spent significantly more time awake in the 12-h light period compared to WT and more time asleep during the 12-h dark period, although the latter effect did not reach significance levels (P = 0.07; Table S1). As a result, the differences between time-spent-asleep (or awake) in the 12-h dark and 12-h light periods, sometimes used to estimate the amplitude of the diurnal sleep/wake distribution, was significantly reduced in KO mice for all 3 sleep/wake states (Table S1). To further quantify the effect of the earlier wake onset on this reduced diurnal amplitude, the 12-h periods over which amplitude was calculated were systematically shifted at 1-min increments (Figure 1C). Advancing the 12 h periods by 65 ± 14 min yielded the highest diurnal amplitude in KO mice while for WT mice maximal amplitude was already reached with a zero shift (+3 ± 8 min; Figure 1C). This suggests that in KO mice the 12 h dark period does not adequately cover the active period. Moreover, the shift with which a zero amplitude was obtained (i.e., the time of day that divides the 24-h day into halves with equal sleep time) was advanced by 1.8 ± 0.3 h relative to WT mice (Figure 1C). Nevertheless, the maximum dark-light amplitude was still significantly lower in KO mice (Figure 1C; P = 0.01, t-test) indicating that besides an earlier onset other aspects of the sleep/wake distribution, such as the less pronounced main waking bout (analysis not shown), differed between the two genotypes.
Evidence of Altered Sleep Homeostasis in Rev-erbα Knockout Mice
To assess the consequence of a lack of Rev-erbα on sleep homeostasis mice were challenged with 6-h sleep deprivation. We found that several aspects of the response to sleep deprivation were altered including time spent asleep, sleep continuity, and the levels of EEG delta power reached.
Both genotypes responded to the sleep deprivation by sleeping more than in the corresponding periods in baseline with state-specific recovery dynamics; i.e., whereas extra NREM sleep was already obtained within the first 6 h of recovery (i.e., during the last 6 h of the light period; ZT6–12), recovery of REM sleep was deferred to the following dark period (Figure 2). However, for both sleep states, Rev-erbα KO mice obtained less extra sleep in the first 6 h of the recovery dark period when WT mice accrued most of the extra time spent in NREM and REM sleep (ZT12–18; Figure 2). During this time interval, KO mice did not significantly gain any NREM sleep (3 ± 6 min) whereas WT mice obtained 35 ± 7 min extra NREM sleep compared to that expressed in corresponding baseline hours (Figure 2). KO mice also gained less extra REM sleep (7 ± 2 min vs. 13 ± 1 min in WT mice; Figure 2B). Despite these different recovery dynamics, at the end of the 18-h recovery period, the deficit in sleep gained in KO mice no longer significantly differed from WT (NREM sleep: 41 ± 10 min and 57 ± 10 min; REM sleep: 14 ± 3 and 20 ± 3 min, for KO and WT respectively; P = 0.24 and 0.11 for NREM and REM sleep, respectively, t-tests).
Figure 2.

Rev-erbα knockout mice recover less sleep between ZT12 and ZT18 in recovery from sleep deprivation compared to their wild type littermates. Recovery-baseline differences in NREM sleep and REM sleep time spent in three 6 h intervals (i.e., during the light from ZT6–12, during the dark from ZT12–18 and ZT18–24). Indicated are the mean (± 1 SEM) differences for KO (black bars, n = 13) and WT (white bars, n = 8) mice. Red stars indicate significant genotype differences (t-tests, P < 0.05); blue stars significant recovery-baseline differences (paired t-tests, P < 0.05).
The general time course of the changes in EEG delta power was similar in the 2 genotypes; i.e., delta power increased during periods when waking prevails (i.e., the dark or active period), was highest immediately after light onset and decreased over the remainder of the light (or rest) period, and sleep deprivation resulted in an increase in EEG delta power in subsequent NREM sleep (Figure 3A). This increase was, however, significantly smaller in KO mice. Likewise, EEG delta power at sleep onset in baseline was lower in KO mice compared to WT mice (Figure 3A, 3B). High relative levels of EEG delta power during NREM sleep are often accompanied by a lower number of brief awakenings indicating deeper and more consolidated sleep.34 Accordingly, sleep deprivation resulted in a pronounced reduction of the number of brief awakenings in both genotypes (Figure 3C) compared to the baseline reference. Consistent with the smaller increase in EEG delta power, the decrease in the number of brief awakenings interrupting sleep in the first 3 h after sleep onset in baseline and after sleep deprivation was smaller in KO mice (rANOVA, P < 0.01; Figure 3C). Post hoc testing revealed, however, that the genotype effect in recovery sleep did not reach the 5% significance level (P = 0.01 and 0.06, for baseline and recovery sleep, respectively). After sleep onset in baseline, WT mice showed a reduced number of brief awakenings to the number observed at the end of the baseline light periods (ZT8–12), while in KO mice no difference from this baseline reference was observed (Figure 3C). In keeping with the analysis of EEG delta power, as reference for the effects on sleep fragmentation the number of brief awakenings occurring at the end of the baseline light periods was used (see Methods). This reference did not differ between genotypes (Figure S1, supplemental material).
Figure 3.

EEG delta power after sleep onset in baseline and after sleep deprivation is lower in the absence of Rev-erbα. (A) Mean (± 1 SEM) EEG delta power (upper curves) and NREM sleep amount (lower curves) during 48-h baseline, 6-h sleep deprivation (SDep; gray shaded area), and 18-h recovery. The time course of the changes in EEG delta power during baseline differed significantly between KO and WT (2-way rANOVA, factors genotype (‘G’ P < 0.001) and hour (‘H’ P < 0.001), ‘GxH’ P < 0.001; analyses of average of 2 baselines). Also in recovery the time course of both EEG delta power (2-way rANOVA, ‘G’ P = 0.0012, ‘H’ P < 0.001, ‘GxH’ P < 0.001) and of the hourly NREM sleep values (2-way rANOVA, ‘G’ P = 0.69, ‘H’ P < 0.001, ‘GxH’ P < 0.001) were affected by genotype. Intervals in which EEG delta power and NREM sleep in KO mice significantly differed from WT are indicated by red squares (post hoc t-tests, P < 0.05). For baseline NREM sleep statistics see Figure 1A. (B) Mean (± 1 SEM) level of EEG delta power in the first 20 min of NREM sleep after light onset in baseline (average of 2 days) and after the SDep. Both KO and WT have a significant increase in EEG delta power after sleep onset in baseline and after sleep deprivation compared to their lowest baseline levels (= 100%; t-tests, P < 0.05) but the levels reached in KO mice were lower in both conditions (red stars; t-tests, P < 0.05). (C) Recovery-baseline differences (± 1 SEM) in the number of brief awakenings (i.e., waking bouts lasting 16 sec or less) interrupting sleep during the first 3 h of the light period in baseline (Baseline) and during the 3 first hours at sleep onset after sleep deprivation (Recovery), contrasted to the number obtained during the last 4 h of the baseline light period (= 0 level). The decrease in number of brief awakenings was smaller in KO mice (2-way rANOVA, factors genotype (P = 0.01) and day (P < 0.001), interaction P = 0.44). Red stars mark significant genotype differences (post hoc t-tests; P < 0.05). Note that in recovery, genotype difference reached P = 0.06. Also note that while brief awakenings after recovery sleep onset was significantly lower than in the last 4 h of the baseline light periods for both genotypes (post-hoc t-tests; P < 0.001), after baseline sleep onset, they were reduced in WT mice only (post-hoc t-tests, P < 0.001 and P = 0.98 for WT and KO, respectively).
To further investigate whether the genotype differences in EEG delta power reported above where specific to the delta frequency range and to homeostatic sleep need we analyzed the spectral composition of the NREM sleep EEG. Spectra did not differ between genotypes when averaged over the entire 48-h baseline (Figure S2A, supplemental material). Similarly, in the last 4 h of the baseline light periods, when sleep need is considered to be lowest, no spectral differences were observed (Figure 4A and 4B, dashed lines labeled 5 and 6). In contrast, when sleep need was high; i.e., at the start of the baseline light periods and after sleep deprivation, NREM sleep spectra in Rev-erbα KO mice did deviate from WT and the frequency bins in which power density differed in baseline and after sleep deprivation largely overlapped (Figure 4). Thus under both conditions, NREM sleep EEG power density between 2.5 and 4.75 Hz was significantly lower in KO mice.
Figure 4.

Genotype differences in NREM sleep EEG spectra are specific to the delta frequencies and appear only when sleep need is high. (A) Mean EEG spectral profiles in NREM sleep at times when sleep need is assumed to be high; i.e., in the first 3 h after sleep onset in baseline (left panel, Baseline; lines labeled 3 and 4 for WT [gray] and KO [black], respectively); and after sleep deprivation (right panel Recovery; lines labeled ‘1’ and ‘2’ for WT [gray] and KO [blue], respectively.). For comparison, spectral profiles of NREM sleep in the last 4 h of baseline, which served as reference for the analysis of EEG delta power (see Figure 3), when sleep need is lowest, are depicted (left panel; dashed lines, labeled ‘5’ and ‘6’ for KO [back] and WT [gray], respectively.) (B) Genotype differences for the spectral profiles in panels A. Mean spectral KO/WT ratios for recovery (Rec; blue line labeled ‘2/1’) and baseline (Bsl; black line; ‘4/3’) were remarkably similar and significant differences concerned higher delta frequencies only for both conditions (2.50–4.75 Hz; post hoc t-tests, P < 0.05, horizontal lines at the bottom), while no significant genotype differences were observed when sleep need is low (dashed black line labeled ‘5/6’). (C) The effect of sleep deprivation (SDep) on NREM sleep spectra in Recovery (right) panel A. Analysis of spectral recovery-to-baseline (Rec/Bsl) ratios in KO (blue line labeled ‘2/5’) and WT (gray; ‘1/6’) mice demonstrated that sleep loss increased EEG power density in frequencies < 21 Hz, while suppressing EEG activity in the low gamma range (> 32 Hz) in both genotypes (blue and gray lines at bottom). Although shape and the affected frequency ranges were highly similar, both the increases and the decreases in EEG power density were smaller in KO mice. Significant genotype differences were, however, highly frequency specific and limited to the delta frequency range (1.0–3.75 Hz) and in one frequency bin within the low gamma band (42.5 Hz).
Although the largest effects of sleep deprivation on the NREM sleep EEG concern the delta frequencies, significant recovery-to-baseline differences can be observed over a larger frequency range.35,36 In the current experiment, power density in frequencies up to 20 Hz were increased compared to baseline (Figure 4C). In addition, we found evidence that EEG activity in the low gamma range (32–45 Hz) was decreased. Although, the shape of the differential spectral profiles as well as the frequency ranges that were significantly affected by the sleep deprivation were very similar for the two genotypes, the magnitude of these changes was smaller in Rev-erbα KO mice. The frequency range for which the sleep deprivation effect was significantly smaller was again limited to the delta frequencies (1.0–3.75 Hz; Figure 4C).
The results concerning the genotype difference in the increase in EEG delta power suggest that when awake, homeostatic sleep need accumulates at a slower rate in mice lacking Rev-erbα. Given the published relationship between; e.g., theta activity in the waking EEG and delta activity in subsequent NREM sleep,37,38 we also quantified the spectral composition of waking EEG. When analyzed over the 48-h baseline, EEG activity during wakefulness showed higher power in the higher delta frequencies (1.5–5.5 Hz), frequencies between 11 and 20 Hz, and the low gamma range (33–42 Hz; Figure S2A). The first 2 of these 3 frequency ranges were similarly affected in REM sleep (Figure S2A). Moreover, the prevailing theta frequency during wakefulness, but not that of REM sleep, was slower in Rev-erbα KO mice. Slower theta oscillations in the wakefulness EEG were also observed during the sleep deprivation (Figure S2B).
We next focused on the waking EEG in the 3 h immediately preceding sleep onset in baseline and during the last 3 h of the sleep deprivation in an attempt to identify those frequency components that could have contributed to the delta power differences observed in subsequent NREM sleep. In the last 3 h of both the baseline dark periods (ZT21–24) and of the sleep deprivation (ZT3–6) the waking EEG showed clear theta activity in WT mice (Figure 5A). The appearance of a distinct theta peak in the waking EEG resulted from the combination of a decrease in high delta activity (3–6 Hz) and an increase in theta activity (6.5–11 Hz; Figure 5B). In addition, in both conditions, EEG activity in the high gamma range (40–90 Hz) significantly increased compared to that observed in the last 4 h of the baseline light periods which served as reference (Figure 5B; Figure S4, supplemental material). Although also in KO mice activity in the theta and high gamma frequency ranges was increased both in the baseline dark period and the sleep deprivation, the peak in theta activity during the baseline dark was less distinct because high delta activity remained elevated (Figure 5A, 5B). Moreover, the increase in theta power was smaller in both baseline and sleep deprivation when mice lack Rev-erbα. Correlation analyses showed that this increase in theta power predicted the increase in EEG delta power in subsequent NREM sleep (Figure 5C). In the sleep deprivation condition also the increase in high gamma activity was smaller in the KO mice.
Figure 5.

Mean EEG spectral profiles in wakefulness preceding the times at which NREMS EEG delta power differed between genotypes. (A) Mean spectral profiles in the 3 h before sleep onset in baseline (ZT21–24; left panel) and the last 3 h of the sleep deprivation (SDep; ZT3–6; right panel; KO black lines, WT gray lines). EEG power density was expressed as a percentage of individual total EEG power in baseline (see Methods and Figure S1C). (B) Effects of prolonged wakefulness on the spectral composition of the waking EEG was assessed by comparing the EEG spectra in panels A to the waking spectra obtained in the last 4 h of baseline (ZT8–12) when sleep need is lowest (see Figure S5 for a genotype comparison of the spectral EEG profiles at this time). Mean profiles of spectral ratios (last 3 h/baseline ZT8–12 reference) reached in last 3 h of the baseline dark periods (left panel) and the last 3 h of the sleep deprivation (right panel) were similar and significant differences concerned a decrease in EEG power in high delta frequencies (3–6 Hz) and increases in the theta (6.5–11 Hz) and gamma (40–90 Hz) frequency ranges (horizontal lines at bottom of panel; WT gray, KO black; post hoc paired t-tests, P < 0.05). These differences were, however, larger in sleep deprivation than in baseline (analyses not shown) and larger in WT than KO mice (red horizontal line at bottom of panel, post hoc t-tests P < 0.05). (C) The increase in EEG theta power in wakefulness as quantified in panel B, predicted the level of EEG delta power reached in NREM sleep after sleep onset as quantified in Figure 2B. The changes in theta power were averaged over the 2-Hz frequency range for which significant genotype differences were observed (8.5–10.5 Hz). For calculation of EEG delta power values see Figure 2B. Lines represent the linear correlation through all KO (black line: delta = 38 ± 9 × theta + 125 ± 8; R2 = 0.45, P = 0.0002, n = 26) and WT (gray line: delta = 50 ± 15 × theta + 133 ± 18; R2 = 0.45, P = 0.0047, n = 16) data points (i.e., Bsl + Rec). Slope and offset did not differ between genotypes. Black symbols: KO (n = 13); gray symbols WT (n = 8); circles baseline (Bsl); triangles recovery (Rec).
The Molecular Consequences of Lack of Rev-erbα in the Cerebral Cortex
To test whether the lack of Rev-erbα also affects molecular markers of sleep need, we investigated the cortical expression of genes that are known to be reliably modulated by sleep deprivation; i.e., Homer1a, Sgk1, Dbp, and Per2.29,39 Homer1a, Sgk1, and Per2 were up-regulated by sleep deprivation and Dbp expression down-regulated although post-hoc testing showed that this decrease reached significance in WT mice only (Figure 6). We previously found that also Npas2 expression increases after sleep deprivation,29 but in the current experiment this increase was significant only in KO mice. These sleep-deprivation induced changes in cortical gene expression did, however, not statistically differ between the two genotypes (Figure 6).
Figure 6.
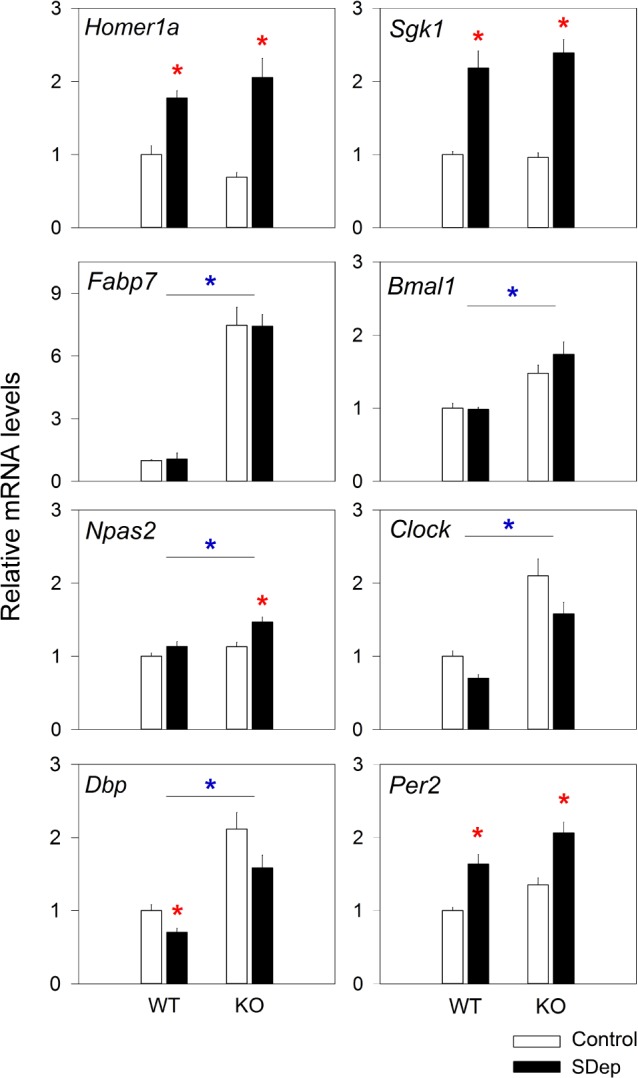
Cortical expression of several genes is affected by the lack of REV-ERBα and sleep deprivation. Mean (1 SEM) levels of mRNA expression in the cortex of WT (n = 10) and KO (n = 9) mice, submitted to either a 6 h sleep deprivation from ZT0–6 (SDep, black bars) or allowed to sleep ad lib (control, white bars). Normalized expression levels (see Methods) are expressed relative to the WT control group (set to 1.0). Red stars indicate a significant effect of the sleep deprivation within genotypes (P < 0.05; post hoc t-tests), while blue stars indicate genotype differences (2-way ANOVA, factors genotype [‘G’] and sleep condition [‘SDep’]; Homer1a: ‘G’ P = 0.99, ‘SDep’ P < 0.001; Sgk1: ‘G’ P = 0.77, ‘SDep’ P < 0.001; Fabp7: ‘G’ P < 0.001; ‘SDep’ P = 0.98; Bmal1: ‘G’ P < 0.001, ‘SDep’ P = 0.42; Npas2: ‘G’ P = 0.004, ‘SDep’ P = 0.003; Clock: ‘G’ P = 0.02, ‘SDep’ P = 0.18; Dbp: ‘G’ < 0.001, ‘SDep’ P = 0.02, Per2: ‘G’ P = 0.06; ‘SDep’ P = 0.003; For none of the transcripts did the interaction term reach significance; ‘GxSDep’ P > 0.13).
Next we assessed the expression of genes known to be directly regulated by Rev-erbα, to test whether Rev-erbα has a similar role on these targets in the cerebral cortex. The expression levels of Bmal1, Npas2, Clock, and the REV-ERBα target Fabp7 (fatty acid binding protein 7)40 were all significantly up-regulated in the cortex of Rev-erbα KO mice compared to WT (Figure 6); that of Fabp7 more than 7-fold. The overall level of Dbp and Per2 in the KO was also increased, although for Per2 it did not reach significance levels (P < 0.06, t-test). These results confirm that Rev-erbα directly, or indirectly in the case of Dbp and Per2, controls the expression of clock genes also in the cortex.
DISCUSSION
REV-ERBα Impacts Sleep Homeostasis
In the current study we identified a robust sleep homeostatic phenotype in mice lacking Rev-erbα. Significantly lower values for the electrophysiological correlate of homeostatic sleep need, EEG delta power, were reached after periods of prolonged wakefulness in KO mice compared to their WT littermates. This phenotype was observed both after enforced and spontaneous periods of wakefulness arguing against the possible influence of other variables than extended wakefulness that usually accompany a sleep deprivation protocol such as; e.g., increased stimulation, locomotion, and stress. This genotype difference in the NREM sleep EEG was specific to the delta frequencies and appeared only at times when sleep need was high. The observed differences do therefore not represent a general EEG phenomena but concern only those frequencies thought to reflect homeostatic sleep need. Moreover, the less pronounced increase in sleep consolidation, another, non-EEG variable reflecting homeostatic sleep need,33 confirmed that sleep immediately after sleep onset was less “deep.”
Together these findings argue for a slower accumulation of sleep need when Rev-erbα KO mice are awake. Correlates of the homeostatic sleep process can also be quantified in the waking EEG, the theta content of which is known to increase over the course of extended waking periods.34,38,41 Moreover, in the rat, levels of theta activity during wakefulness and/or time spent exploring, during which theta activity is prominent, predict delta power in subsequent NREM sleep,37,38 suggesting that these two EEG activities not only gauge the same underlying homeostatic process but that wakefulness with higher theta activity causes delta power to be higher during the sleep that follows. We here confirmed this relationship in the mouse. Rev-erbα KO mice displayed a smaller increase in theta activity during wakefulness in the 3 h preceding sleep onset that predicted the lower delta power immediately after sleep onset. We extended these theta findings to the 35–90 Hz or gamma frequencies, the activity in which is closely coupled to theta oscillations in the hippocampal formation.42,43 In both genotypes EEG activity in the gamma band was increased relative to the values reached when sleep need is lowest but, like for theta activity, this increase was less pronounced in the KO mice although the frequencies in which significant differences were observed concerned the 71–78 Hz range only. Lack of Reverbα may thus lead to deficits in either engaging in waking behaviors rich in theta activity such as exploratory behavior, and/or in the recruitment of neuronal populations contributing to these oscillations. The significantly slower theta frequency we observed during wakefulness constitutes additional evidence for a role of Rev-erbα in modulating theta oscillations of hippocampal origin. Of interest in this context is the fact that lack of Rev-erbα leads to increased adult neurogenesis in the hippocampus,40 which may affect the network properties contributing to theta oscillations.44
REV-ERBα, Sleep Homeostasis, and Dopamine
Impaired hippocampal function in Rev-erbα KO mice has been demonstrated for a number of hippocampal-dependent behaviors.40,45 This impairment was accompanied by increased dopamine turnover.45,46 The slower hippocampal theta oscillations we observed in Rev-erbα KO mice are consistent with the theta slowing observed when dopamine tone is increased as observed in Dopamine transporter (Dat) KO mice.47 In Rev-erbα KO mice the increased dopamine turnover was due to an up-regulation of Tyrosine hydroxylase (Th), the rate-limiting enzyme in dopamine production.45,46 Rev-erbα can directly repress Th expression in competition with the Nuclear receptor-related 1 protein (Nurr1), another nuclear receptor and key transcriptional activator of Th and other elements of the dopaminergic system.46,48 Of immediate interest for our current study is the observation that altered dopamine levels, resulting from altered Dat activity, were found to be associated with a change in the homeostatic sleep rebound both in flies and humans.49–51
Other clock genes have been implicated in the regulation of dopamine levels in the brain.46,52,53 Such role might be direct, like REV-ERBα‘s repression of Th expression discussed above and NPAS2:BMAL1's transcriptional activation of the dopa-mine degrading enzyme monoamine oxidase A (Maoa),53 or indirect through the effects of other clock genes on the expression of e.g., Rev-erbα. For instance, CLOCK/NPAS2:BMAL1 heterodimers induce Rev-erbα transcription while REV-ERBα, in turn, represses the expression of these three transcription factors.6–8,11 We here confirm that also in the cerebral cortex removing the repression provided by REV-ERBα increases the expression of Bmal1, Npas2, and Clock. Consistent with this up-regulation we found the CLOCK/NPAS2:BMAL1 targets Dbp and Per2 to be upregulated as well. PER2, in turn, not only provides negative feedback to CLOCK/NPAS2:BMAL1 induced transcription but, in addition, coordinates the action of various nuclear receptors, including REV-ERBα and NURR1, via protein-protein interactions.54 Thus REV-ERBα as part of a complex network of interacting transcriptional regulators, seems central in changing dopamine turnover.
The link between dopamine levels and sleep homeostasis is intriguing and could also have contributed to the profound sleep homeostatic phenotype we observed in Rev-erbα KO mice. This relationship is, however, not straightforward; the mutations in Dat activity referred to above led to increased dopamine post-synaptically due to compromised dopamine re-uptake which was associated with increased homeostatic sleep rebounds,49–51 while in Rev-erbα KO mice the reduced inhibition of Th expression led to increased dopamine levels pre-synaptically and, as we show here, was associated with a decreased homeostatic sleep rebound. Further illustrating this complexity is the fact that REV-ERBα can directly inhibit the expression of the Dopamine D3 receptor.55 The activation of this receptor is thought to be inhibitory, reducing novelty seeking behaviors, and to reduce dopamine through post-synaptic negative feedback.56
REV-ERBα and Circadian Organization of Overt Behavior
Although Rev-erbα is important for the circadian molecular circuitry and for setting the phase of circadian rhythms in peripheral tissues,12 its lack only modestly affects rhythms in overt behaviors. Rev-erbα KO mice do maintain circadian organization of locomotor activity under constant conditions albeit with a significantly shorter free-running period.9,12 The earlier onset of the main waking period under the entrained conditions of our experiment is consistent with a shorter endogenous period although the approximate 24 min shortening of the period seems insufficient to account for the > 1 h advance of sleep/ wake distribution. The inducible depletion of both Rev-erbα and its homolog Rev-erbβ does lead to a profound disruption of circadian behavior12 pointing to a functional redundancy between the two at least for this phenotype. Whether also the sleep homeostatic phenotype becomes more pronounced in double KO mice remains to be determined.
In addition to altered EEG delta power and sleep fragmentation after sleep deprivation, we also observed a distinctly different pattern for the recovery of sleep time lost during the sleep deprivation to which the advance of the sleep/wake distribution might have contributed. In both genotypes, maximal levels of wakefulness are reached within the first 6 h of the dark period under baseline conditions. After the sleep deprivation, it was during these 6 h that WT mice adapt their behavior to allow for recovery sleep gaining 35 min of NREM sleep. KO mice conspicuously maintain their baseline sleep/ wake pattern and did not increase NREM sleep time above basal levels. As a consequence of the extra NREM sleep in the recovery dark period, EEG delta power in WT mice, exceptionally, reached lower levels than KO mice. The inflexibility of Rev-erbα KO mice to mount the appropriate behavioral sleep response is reminiscent to that observed in mice lacking the clock gene Npas2.57 Also Npas2 KO mice have intact circadian behavior but a poor homeostatic response to food or sleep deprivation.58
Sleep Deprivation, Mood, Metabolism, and Clock Genes
Strong, bidirectional links exist between sleep and mood disorders, perhaps best illustrated by the amazingly rapid, albeit short lasting, antidepressant effects of sleep deprivation in depression.59 These beneficial effects of sleep deprivation combined with the observation made in some, but not all studies, that EEG delta power is reduced in major depressive disorder,60 led to the hypothesis that sleep need accumulates at a slower rate during wakefulness and is causally involved in the dysregulation of mood.61 Abnormal dynamics in waking theta activity over the course of a sleep deprivation further suggest abnormal sleep homeostasis in depressed patients.62 Both the altered theta activity in wakefulness and the altered delta activity in NREM sleep we here describe for the KO mice are reminiscent of these EEG changes associated with mood disorders.
Sleep homeostasis and mood disorders could also be linked at the molecular level. Many studies reported on the role of clock genes in mood regulation63,64 and our own work revealed a bidirectional relationship between clock genes and sleep homeostasis,3 further illustrated here by the altered homeostatic regulation of sleep in Rev-erbα KO mice and by the effects of sleep deprivation on the cortical expression of the clock genes Npas2 and Per2, and the clock-controlled transcription factor Dbp. We previously showed that like Dbp, Rev-erbα was decreased by sleep deprivation.29 We also quantified two transcripts known to be reliably upregulated sleep deprivation; i.e., Homer1a and Sgk1. Although the expression of both was indeed increased in the current study, their increase did not differ between genotypes pointing to a dissociation between EEG and molecular markers of sleep need.
Among the clock genes, REV-ERBα seems to play a central role in the regulation of mood as evidenced by; e.g., a mania-like behavior in KO mice,40,46,65 and, perhaps, the altered sleep homeostasis in the current study. The involvement of REV-ERBα in mood might, in part, relate to its role in dopaminergic signaling discussed above. Other likely candidate pathways concern the role of REV-ERBα in adult hippocampal neurogenesis,40 which has also been linked to mood disorders,66 and its well-established role in metabolism and circadian rhythms.64 One important mediator of the increased adult hippocampal neurogenesis is FABP7,40,67 a direct target of REV-ERBα.40 Because of the large, 7.5-fold over-expression of Fabp7 in the brain that we observed (Figure 6),40 FABP7 might also be involved in mediating other phenotypes observed in Rev-erbα KO mice such as anxiety and memory deficits.67–69 Concerning the sleep homeostatic phenotype, overexpression of the mouse Fabp7 or its fly homologue Fabp-B in the fruit fly Drosophila melanogaster resulted in decreased sleep duration and consolidation,68 the Drosophila correlates of reduced homeostatic sleep need.50,70 Whether FABP7 plays a role in the homeostatic regulation of sleep in mammals has yet to be determined.
Several groups have investigated the role of the core clock genes in sleep homeostasis by studying the effects of sleep deprivation on EEG delta power in mice carrying targeted disruptions of single or a combination of clock genes.3,4 The first core clock genes for which altered sleep-wake dependent dynamics of EEG delta power was demonstrated were the Cryptochromes.71 Cry1,2 double KO mice displayed a more rapid build-up of homeostatic sleep need during wakefulness resulting in overall higher levels of EEG delta power during baseline, explaining the smaller relative increase in EEG delta power after sleep deprivation. In contrast, as in our Rev-erbα KO mice, evidence for a slower build rate was obtained in Bmal1 and Npas2 knock-out mice,57,72 while in Clock-mutant mice no differences were reported.73 Sleep homeostasis was also assessed in Per1 and Per2 single and double mutant KO mice but with inconsistent outcomes.74,75 These disparate results do not support a simple, unifying mechanism through which the clock gene circuitry, as a whole, alters the dynamics of the sleep homeostat but rather suggest that factors such as tissue/cell type-specificity and yet to be identified modifiers associated with differences in genetic background play important roles as has been demonstrated for circadian-related phenotypes.5
CONCLUSION
The role of REV-ERBα in the control of sleep homeostasis is of particular interest because both the molecule and the process are tightly linked to metabolism as well as circadian rhythms.3,14,76,77 The activity of REV-ERBα as a transcriptional repressor is modulated by cellular redox state through altered binding of its endogenous ligand, heme.78,79 In the context of our sleep homeostatic phenotype, Rev-erbα could thus act as a sensor of the metabolic imbalance imposed at the neuronal level by periods of extended wakefulness in keeping with our proposal that clock genes not only set time-of-day, but in the cerebral cortex, can also be used to keep track of and respond to time-spent-awake.3,77 A recent study demonstrated that synthetic agonists targeting both REV-ERB proteins leads to a strong, immediate reduction in sleep time when administrated at ZT6,80 implying that pharmacologically targeting of REVERBα could be useful in the treatment sleep disorders.
DISCLOSURE STATEMENT
This was not an industry supported study. This study was performed at the Universities of Lausanne and Fribourg, Switzerland, and supported by the Swiss National Science Foundation (SNF n°130825 and 136201 to PF and SNF n°146265 to UA) and the states of Vaud supporting GMM, YE, and PF and Fribourg supporting JAR and UA. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors are grateful for the expert technical support provided by Ashot Sargsyan and Antoinette Hayoz and all colleagues who helped with the sleep deprivations.
REFERENCES
- 1.Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- 2.Daan S, Beersma DG, Borbely AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246(2 Pt 2):R161–83. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- 3.Franken P. A role for clock genes in sleep homeostasis. Curr Opin Neurobiol. 2013;23:864–72. doi: 10.1016/j.conb.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Mang GM, Franken P. Genetic dissection of sleep homeostasis. Curr Top Behav Neurosci. 2015;25:25–63. doi: 10.1007/7854_2013_270. [DOI] [PubMed] [Google Scholar]
- 5.Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Adv Genet. 2011;74:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crumbley C, Burris TP. Direct regulation of CLOCK expression by REV-ERB. PLoS One. 2011;6:e17290. doi: 10.1371/journal.pone.0017290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crumbley C, Wang Y, Kojetin DJ, Burris TP. Characterization of the core mammalian clock component, NPAS2, as a REV-ERBalpha/RORalpha target gene. J Biol Chem. 2010;285:35386–92. doi: 10.1074/jbc.M110.129288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akashi M, Takumi T. The orphan nuclear receptor RORalpha regulates circadian transcription of the mammalian core-clock Bmal1. Nat Struct Mol Biol. 2005;12:441–8. doi: 10.1038/nsmb925. [DOI] [PubMed] [Google Scholar]
- 9.Preitner N, Damiola F, Lopez-Molina L, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–60. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 10.Ueda HR, Chen W, Adachi A, et al. A transcription factor response element for gene expression during circadian night. Nature. 2002;418:534–9. doi: 10.1038/nature00906. [DOI] [PubMed] [Google Scholar]
- 11.Sato TK, Panda S, Miraglia LJ, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–37. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 12.Cho H, Zhao X, Hatori M, et al. Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature. 2012;485:123–7. doi: 10.1038/nature11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazzoccoli G, Cai Y, Liu S, et al. REV-ERBalpha and the clock gene machinery in mouse peripheral tissues: a possible role as a synchronizing hinge. J Biol Regul Homeost Agents. 2012;26:265–76. [PubMed] [Google Scholar]
- 14.Duez H, Staels B. Rev-erb-alpha: an integrator of circadian rhythms and metabolism. J Appl Physiol. 2009;107:1972–80. doi: 10.1152/japplphysiol.00570.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harding HP, Lazar MA. The monomer-binding orphan receptor Rev-Erb represses transcription as a dimer on a novel direct repeat. Mol Cell Biol. 1995;15:4791–802. doi: 10.1128/mcb.15.9.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burris TP. Nuclear hormone receptors for heme: REV-ERBalpha and REV-ERBbeta are ligand-regulated components of the mammalian clock. Mol Endocrinol. 2008;22:1509–20. doi: 10.1210/me.2007-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renaud JP, Harris JM, Downes M, Burke LJ, Muscat GE. Structure-function analysis of the Rev-erbA and RVR ligand-binding domains reveals a large hydrophobic surface that mediates corepressor binding and a ligand cavity occupied by side chains. Mol Endocrinol. 2000;14:700–17. doi: 10.1210/mend.14.5.0444. [DOI] [PubMed] [Google Scholar]
- 18.Yin L, Lazar MA. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol Endocrinol. 2005;19:1452–9. doi: 10.1210/me.2005-0057. [DOI] [PubMed] [Google Scholar]
- 19.Raghuram S, Stayrook KR, Huang P, et al. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REVERBbeta. Nat Struct Mol Biol. 2007;14:1207–13. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin L, Wu N, Curtin JC, et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–9. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 21.Estall JL, Ruas JL, Choi CS, et al. PGC-1alpha negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erb(alpha) axis. Proc Natl Acad Sci U S A. 2009;106:22510–5. doi: 10.1073/pnas.0912533106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duez H, van der Veen JN, Duhem C, et al. Regulation of bile acid synthesis by the nuclear receptor Rev-erbalpha. Gastroenterology. 2008;135:689–98. doi: 10.1053/j.gastro.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 23.Le Martelot G, Claudel T, Gatfield D, et al. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. 2009;7:e1000181. doi: 10.1371/journal.pbio.1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delezie J, Dumont S, Dardente H, et al. The nuclear receptor REVERBalpha is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. 2012;26:3321–35. doi: 10.1096/fj.12-208751. [DOI] [PubMed] [Google Scholar]
- 25.Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–37. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 26.Torra IP, Tsibulsky V, Delaunay F, et al. Circadian and glucocorticoid regulation of Rev-erbalpha expression in liver. Endocrinology. 2000;141:3799–806. doi: 10.1210/endo.141.10.7708. [DOI] [PubMed] [Google Scholar]
- 27.Solt LA, Wang Y, Banerjee S, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–8. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L, Yang G. PPARs integrate the mammalian clock and energy metabolism. PPAR Res. 2014;2014:653017. doi: 10.1155/2014/653017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mongrain V, Hernandez SA, Pradervand S, et al. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep. 2010;33:1147–57. doi: 10.1093/sleep/33.9.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mang GM, Franken P. Sleep and EEG Phenotyping in Mice. Curr Protoc Mouse Biol. 2012;2:55–74. doi: 10.1002/9780470942390.mo110126. [DOI] [PubMed] [Google Scholar]
- 31.Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–69. [PubMed] [Google Scholar]
- 32.Franken P, Malafosse A, Tafti M. Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol. 1998;275:R1127–37. doi: 10.1152/ajpregu.1998.275.4.R1127. [DOI] [PubMed] [Google Scholar]
- 33.Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–21. doi: 10.1523/JNEUROSCI.21-08-02610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franken P, Dijk DJ, Tobler I, Borbély AA. Sleep deprivation in rats: effects on EEG power spectra, vigilance states, and cortical temperature. Am J Physiol. 1991;261:R198–208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- 35.Borbely AA, Baumann F, Brandeis D, Strauch I, Lehmann D. Sleep deprivation: effect on sleep stages and EEG power density in man. Electroencephalogr Clin Neurophysiol. 1981;51:483–95. doi: 10.1016/0013-4694(81)90225-x. [DOI] [PubMed] [Google Scholar]
- 36.Kopp C, Petit JM, Magistretti P, Borbely AA, Tobler I. Comparison of the effects of modafinil and sleep deprivation on sleep and cortical EEG spectra in mice. Neuropharmacology. 2002;43:110–8. doi: 10.1016/s0028-3908(02)00070-9. [DOI] [PubMed] [Google Scholar]
- 37.Huber R, Tononi G, Cirelli C. Exploratory behavior, cortical BDNF expression, and sleep homeostasis. Sleep. 2007;30:129–39. doi: 10.1093/sleep/30.2.129. [DOI] [PubMed] [Google Scholar]
- 38.Vyazovskiy VV, Tobler I. Theta activity in the waking EEG is a marker of sleep propensity in the rat. Brain Res. 2005;1050:64–71. doi: 10.1016/j.brainres.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 39.Curie T, Mongrain V, Dorsaz S, Mang GM, Emmenegger Y, Franken P. Homeostatic and circadian contribution to EEG and molecular state variables of sleep regulation. Sleep. 2013;36:311–23. doi: 10.5665/sleep.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schnell A, Chappuis S, Schmutz I, et al. The nuclear receptor REV-ERBalpha regulates Fabp7 and modulates adult hippocampal neurogenesis. PLoS One. 2014;9:e99883. doi: 10.1371/journal.pone.0099883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cajochen C, Brunner DP, Krauchi K, Graw P, Wirz-Justice A. Power density in theta/alpha frequencies of the waking EEG progressively increases during sustained wakefulness. Sleep. 1995;18:890–4. doi: 10.1093/sleep/18.10.890. [DOI] [PubMed] [Google Scholar]
- 42.Penley SC, Hinman JR, Sabolek HR, Escabi MA, Markus EJ, Chrobak JJ. Theta and gamma coherence across the septotemporal axis during distinct behavioral states. Hippocampus. 2012;22:1164–75. doi: 10.1002/hipo.20962. [DOI] [PubMed] [Google Scholar]
- 43.Sirota A, Montgomery S, Fujisawa S, Isomura Y, Zugaro M, Buzsaki G. Entrainment of neocortical neurons and gamma oscillations by the hippocampal theta rhythm. Neuron. 2008;60:683–97. doi: 10.1016/j.neuron.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rangel LM, Quinn LK, Chiba AA, Gage FH, Aimone JB. A hypothesis for temporal coding of young and mature granule cells. Front Neurosci. 2013;7:75. doi: 10.3389/fnins.2013.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jager J, O'Brien WT, Manlove J, et al. Behavioral changes and dopaminergic dysregulation in mice lacking the nuclear receptor Reverbalpha. Mol Endocrinol. 2014;28:490–8. doi: 10.1210/me.2013-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung S, Lee EJ, Yun S, et al. Impact of circadian nuclear receptor REV-ERBalpha on midbrain dopamine production and mood regulation. Cell. 2014;157:858–68. doi: 10.1016/j.cell.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 47.Dzirasa K, Santos LM, Ribeiro S, et al. Persistent hyperdopaminergia decreases the peak frequency of hippocampal theta oscillations during quiet waking and REM sleep. PLoS One. 2009;4:e5238. doi: 10.1371/journal.pone.0005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–50. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- 49.Greenspan RJ, Tononi G, Cirelli C, Shaw PJ. Sleep and the fruit fly. Trends Neurosci. 2001;24:142–5. doi: 10.1016/s0166-2236(00)01719-7. [DOI] [PubMed] [Google Scholar]
- 50.Pfeiffenberger C, Allada R. Cul3 and the BTB adaptor insomniac are key regulators of sleep homeostasis and a dopamine arousal pathway in Drosophila. PLoS Genet. 2012;8:e1003003. doi: 10.1371/journal.pgen.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holst SC, Bersagliere A, Bachmann V, Berger W, Achermann P, Landolt HP. Dopaminergic role in regulating neurophysiological markers of sleep homeostasis in humans. J Neurosci. 2014;34:566–73. doi: 10.1523/JNEUROSCI.4128-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parekh PK, Ozburn AR, McClung CA. Circadian clock genes: effects on dopamine, reward and addiction. Alcohol. 2015;49:341–9. doi: 10.1016/j.alcohol.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hampp G, Ripperger JA, Houben T, et al. Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr Biol. 2008;18:678–83. doi: 10.1016/j.cub.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 54.Schmutz I, Ripperger JA, Baeriswyl-Aebischer S, Albrecht U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Gene Dev. 2010;24:345–57. doi: 10.1101/gad.564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ikeda E, Matsunaga N, Kakimoto K, et al. Molecular mechanism regulating 24-hour rhythm of dopamine D3 receptor expression in mouse ventral striatum. Mol Pharmacol. 2013;83:959–67. doi: 10.1124/mol.112.083535. [DOI] [PubMed] [Google Scholar]
- 56.Holmes A, Lachowicz JE, Sibley DR. Phenotypic analysis of dopamine receptor knockout mice; recent insights into the functional specificity of dopamine receptor subtypes. Neuropharmacology. 2004;47:1117–34. doi: 10.1016/j.neuropharm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 57.Franken P, Dudley CA, Estill SJ, et al. NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: genotype and sex interactions. Proc Natl Acad Sci U S A. 2006;103:7118–23. doi: 10.1073/pnas.0602006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dudley CA, Erbel-Sieler C, Estill SJ, et al. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science. 2003;301:379–83. doi: 10.1126/science.1082795. [DOI] [PubMed] [Google Scholar]
- 59.Benedetti F. Antidepressant chronotherapeutics for bipolar depression. Dialogues Clin Neurosci. 2012;14:401–11. doi: 10.31887/DCNS.2012.14.4/fbenedetti. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frey S, Birchler-Pedross A, Hofstetter M, et al. Challenging the sleep homeostat: sleep in depression is not premature aging. Sleep Med. 2012;13:933–45. doi: 10.1016/j.sleep.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 61.Borbely AA, Wirz-Justice A. Sleep, sleep deprivation and depression. A hypothesis derived from a model of sleep regulation. Hum Neurobiol. 1982;1:205–10. [PubMed] [Google Scholar]
- 62.Plante DT, Goldstein MR, Landsness EC, et al. Altered overnight modulation of spontaneous waking EEG reflects altered sleep homeostasis in major depressive disorder: a high-density EEG investigation. J Affect Disord. 2013;150:1167–73. doi: 10.1016/j.jad.2013.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McClung CA. Circadian genes, rhythms and the biology of mood disorders. Pharmacol Ther. 2007;114:222–32. doi: 10.1016/j.pharmthera.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Albrecht U. Circadian clocks and mood-related behaviors. Handb Exp Pharmacol. 2013:227–39. doi: 10.1007/978-3-642-25950-0_9. [DOI] [PubMed] [Google Scholar]
- 65.Yin L, Wang J, Klein PS, Lazar MA. Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science. 2006;311:1002–5. doi: 10.1126/science.1121613. [DOI] [PubMed] [Google Scholar]
- 66.Samuels BA, Hen R. Neurogenesis and affective disorders. Eur J Neurosci. 2011;33:1152–9. doi: 10.1111/j.1460-9568.2011.07614.x. [DOI] [PubMed] [Google Scholar]
- 67.Matsumata M, Inada H, Osumi N. Fatty acid binding proteins and the nervous system: their impact on mental conditions. Neurosci Res. 2014 Sep 6; doi: 10.1016/j.neures.2014.08.012. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 68.Gerstner JR, Vanderheyden WM, Shaw PJ, Landry CF, Yin JC. Fatty-acid binding proteins modulate sleep and enhance long-term memory consolidation in Drosophila. PLoS One. 2011;6:e15890. doi: 10.1371/journal.pone.0015890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimamoto C, Ohnishi T, Maekawa M, et al. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum Mol Genet. 2014;23:6495–511. doi: 10.1093/hmg/ddu369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andretic R, Shaw PJ. Essentials of sleep recordings in Drosophila: moving beyond sleep time. Methods Enzymol. 2005;393:759–72. doi: 10.1016/S0076-6879(05)93040-1. [DOI] [PubMed] [Google Scholar]
- 71.Wisor JP, O'Hara BF, Terao A, et al. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002;3:20. doi: 10.1186/1471-2202-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laposky A, Easton A, Dugovic C, Walisser J, Bradfield C, Turek F. Deletion of the mammalian circadian clock gene BMAL1/Mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep. 2005;28:395–409. doi: 10.1093/sleep/28.4.395. [DOI] [PubMed] [Google Scholar]
- 73.Naylor E, Bergmann BM, Krauski K, et al. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci. 2000;20:8138–43. doi: 10.1523/JNEUROSCI.20-21-08138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kopp C, Albrecht U, Zheng B, Tobler I. Homeostatic sleep regulation is preserved in mPer1 and mPer2 mutant mice. Eur J Neurosci. 2002;16:1099–106. doi: 10.1046/j.1460-9568.2002.02156.x. [DOI] [PubMed] [Google Scholar]
- 75.Shiromani PJ, Xu M, Winston EM, Shiromani SN, Gerashchenko D, Weaver DR. Sleep rhythmicity and homeostasis in mice with targeted disruption of mPeriod genes. Am J Physiol Regul Integr Comp Physiol. 2004;287:R47–57. doi: 10.1152/ajpregu.00138.2004. [DOI] [PubMed] [Google Scholar]
- 76.Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–60. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- 77.Albrecht U. Circadian rhythms and sleep--the metabolic connection. Pflugers Arch. 2012;463:23–30. doi: 10.1007/s00424-011-0986-6. [DOI] [PubMed] [Google Scholar]
- 78.Gupta N, Ragsdale SW. Thiol-disulfide redox dependence of heme binding and heme ligand switching in nuclear hormone receptor reverb{beta} J Biol Chem. 2011;286:4392–403. doi: 10.1074/jbc.M110.193466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carter EL, Ragsdale SW. Modulation of nuclear receptor function by cellular redox poise. J Inorg Biochem. 2014;133:92–103. doi: 10.1016/j.jinorgbio.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Banerjee S, Wang Y, Solt LA, et al. Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour. Nat Comm. 2014;5:5759. doi: 10.1038/ncomms6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.