

Abstract
Study Objectives:
Optimal sleep is ensured by the interaction of circadian and homeostatic processes. Although synaptic plasticity seems to contribute to both processes, the specific players involved are not well understood. The EphA4 tyrosine kinase receptor is a cell adhesion protein regulating synaptic plasticity. We investigated the role of EphA4 in sleep regulation using electrocorticography in mice lacking EphA4 and gene expression measurements.
Methods:
EphA4 knockout (KO) mice, ClockΔ19/Δ19 mutant mice and littermates, C57BL/6J and CD-1 mice, and Sprague-Dawley rats were studied under a 12 h light: 12 h dark cycle, under undisturbed conditions or 6 h sleep deprivation (SLD), and submitted to a 48 h electrophysiological recording and/or brain sampling at different time of day.
Results:
EphA4 KO mice showed less rapid eye movement sleep (REMS), enhanced duration of individual bouts of wakefulness and nonrapid eye movement sleep (NREMS) during the light period, and a blunted daily rhythm of NREMS sigma activity. The NREMS delta activity response to SLD was unchanged in EphA4 KO mice. However, SLD increased EphA4 expression in the thalamic/hypothalamic region in C57BL/6J mice. We further show the presence of E-boxes in the promoter region of EphA4, a lower expression of EphA4 in Clock mutant mice, a rhythmic expression of EphA4 ligands in several brain areas, expression of EphA4 in the suprachiasmatic nuclei of the hypothalamus (SCN), and finally an unchanged number of cells expressing Vip, Grp and Avp in the SCN of EphA4 KO mice.
Conclusions:
Our results suggest that EphA4 is involved in circadian sleep regulation.
Citation:
Freyburger M, Pierre A, Paquette G, Bélanger-Nelson E, Bedont J, Gaudreault PO, Drolet G, Laforest S, Blackshaw S, Cermakian N, Doucet G, Mongrain V. EphA4 is involved in sleep regulation but not in the electrophysiological response to sleep deprivation. SLEEP 2016;39(3):613–624.
Keywords: delta activity, gene expression, rapid eye movement sleep, sigma activity, suprachiasmatic nuclei
Significance.
Our study provides the first findings supporting a role for the ephrin and Eph receptor cellular adhesion system in the regulation of sleep duration, sleep consolidation and synchronous activity of the cerebral cortex. It presents novel insight into molecular mechanisms involved in sleep regulation and more specifically on how molecules involved in synaptic plasticity participate in the control of wakefulness and sleep states. Given that the ephrin/Eph system has been linked to both neuronal development and neurodegeneration, our results highlight a new pathway, not only linking sleep to synaptic plasticity, but also connecting sleep alterations to neurodevelopmental and neurodegenerative diseases.
INTRODUCTION
Sleep is required for brain functioning and the maintenance of cognitive abilities,1,2 and alterations in sleep are observed in psychiatric and neurological conditions such as bipolar disorders and neurodegenerative diseases.2,3 Understanding cellular and molecular mechanisms of sleep regulation and their relationship to brain health is essential to define new therapeutic targets to alleviate sleep disturbances and cognitive impairments.
Sleep is tightly regulated by two main processes, a circadian and a homeostatic process, the interaction of which determines the quality of both wakefulness and sleep.4 The homeostatic pressure for sleep varies according to the prior duration of sleep and wakefulness and reflects sleep need. Sleep need rises during waking, and thus increases with prolonged wakefulness or sleep deprivation (SLD), and declines during sleep. The dynamics of specific markers such as electrocorticographic (ECoG) delta power (1–4 Hz) measured during nonrapid eye movement sleep (NREMS) indexes homeostatic sleep pressure.5,6 The circadian process modulates sleep timing, duration, and organization,4 and is controlled by the master clock located in the hypothalamic suprachiasmatic nuclei (SCN) in mammals. Circadian regulation depends on a molecular transcriptional-translational feedback loop that involves the core clock transcription factors CLOCK and BMAL1, which form a complex that activates gene expression via binding to specific DNA sequences (i.e., E-boxes).7
Both sleep regulatory processes seem to be linked to changes in synaptic functioning and therefore to mechanisms regulating synaptic plasticity. On the one hand, NREMS delta activity, the main marker of sleep homeostasis, is both affected by and affecting synaptic communication. For instance, altering the activity of plasticity mediators, such as N-methyl-d-aspartate glutamate receptors, with the use of antagonists modulates NREMS delta power,8,9 whereas delta oscillations during NREMS contribute to changes in synaptic strength through a glutamate receptor-dependent mechanism.10 On the other hand, synaptic plasticity events are also relevant to circadian regulation.11 Indeed, in the SCN, neuronal firing rate is lower during the active period than the inactive period,12 and the number of glutamatergic synapses on SCN vasoactive intestinal peptide (VIP) neurons is lower during the night than the day.13 In addition, molecular elements governing the circadian clock transcriptional-translational feedback loop, also called clock genes, appear to shape synaptic function outside the SCN, as mice carrying clock gene mutations show deficits in synaptic plasticity in the hippocampus.14,15 Consequently, although most underlying mechanisms remain to be defined, synaptic modifications play roles in the physiology of both sleep regulatory processes.
Our recent work on Neuroligin-1 (NLGN1) supports a role for synaptic adhesion systems regulating glutamate transmission in sleep regulation, and implicates clock genes in this pathway.16 In the adult brain, ephrins and their Eph receptors are also implicated in the regulation of neuronal and neuron-glia communications.17 EphA4, in particular, regulates synaptic strength by downregulating the expression of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) glutamate receptors,18 and mice lacking EphA4 exhibit impairments in both functional and morphological plasticity.19–21 These observations thus place EphA4 in a strategic position to act as a modulator of synaptic activity as a function of wakefulness and sleep.
Here, we verified the role of EphA4 in sleep regulation using ECoG recording in EphA4 knockout (KO) mice performed during baseline condition and after SLD, and using gene expression measurements. First, we measured vigilance state duration and ECoG spectral activity during wakefulness and sleep with a particular focus on electrophysiological markers of circadian and homeostatic sleep regulation (i.e., dynamics of NREMS delta and sigma activity).22,23 Because SLD extensively affects the brain transcriptome,24–27 we also assessed the gene expression response to SLD in EphA4 KO mice using quantitative polymerase chain reaction (qPCR) and microarray. In addition, we evaluated the effect of both SLD and time of day on the expression of elements of the ephrin/Eph system in three different brain regions in C57BL/6J mice. Last, the possibility that EphA4 represents an output from the circadian system was verified by measuring its expression in ClockΔ19 mutant mice and in the SCN, and by quantifying the expression of the major SCN neuropeptides in EphA4 KO mice. Our observations support a role for the ephrin/Eph system in sleep regulation.
METHODS
Animals and Protocols
Mice (all males) were maintained under standard housing conditions (food/water ad libitum, 22–25°C, 12 h light: 12 h dark [LD12:12]). EphA4 mutant mice28 were generously given by Keith K. Murai (McGill University) and bred on site. Homo-zygous EphA4 KO mice, and heterozygote (HET) and wild-type (WT) littermates were used for ECoG, qPCR, microarray, neurological assessment, and in situ hybridization. The ECoG of 44 mice (n = 13 KO [12.1 ± 0.3 w old, 21.1 ± 0.6 g], 16 HET [11.9 ± 0.2 w old, 23.1 ± 0.4 g], 15 WT [11.8 ± 0.2 w old, 24.4 ± 0.6 g]; significant genotype effect on weight F2,41 = 9.3, P < 0.01) was recorded during a 24-h baseline, during a 6-h SLD starting at light onset (Zeitgeber time 0: ZT0), and during 18 h of recovery. One week after the ECoG measurement, some of the same mice were sacrificed at ZT6 (6 h after light onset) under undisturbed condition (n = 4 KO, 4 WT) or after a second 6-h SLD (n = 5 KO, 6 WT), and forebrains (hindbrain excised) were used to measure the gene expression response to SLD. Neurological function was evaluated twice (ZT0 and ZT11) in a different cohort of mice (n = 7 KO [9.9 ± 0.2 w old, 20.5 ± 0.6 g], 7 HET [10.1 ± 0.2 w old, 22.3 ± 1.0 g], 10 WT [10.3 ± 0.3 w old, 24.2 ± 0.6 g]; significant genotype effect on weight F2,21 = 6.6, P < 0.01).
The effect of SLD on messenger RNA (mRNA) expression of targeted Eph receptors and ephrins was measured in the cerebral cortex, hippocampus and, to ensure rapid dissection, a region covering the thalamus and hypothalamus in C57BL/6J (B6) mice (12 w old) sacrificed at ZT6 after a 6-h SLD (n = 6–7) or under undisturbed condition (n = 5–7). SLD was performed by gentle handling.29 The effect of time of day on mRNA expression of the specific Eph receptors and ephrins in the same three brain regions was measured in B6 mice sacrificed at ZT0, ZT6, ZT12, and ZT18 under undisturbed condition (n = 4–5 per time, 10.5 w old). ClockΔ19/Δ19 mutant mice and WT littermates were the same animals as those used in a previous study,30 for which sacrifice was performed every 6 h under LD12:12 (ZT2 = 2 h after light onset, ZT8, ZT14, ZT20; n = 4–5 per genotype per time). Right forebrains were dissected from previously frozen whole brains. All mice used for qPCR gene expression measurements were sacrificed by cervical dislocation.
For in situ hybridization measuring EphA4 expression, two WT CD-1 mice (5 w old) and two male Sprague-Dawley rats (225–250 g; Charles River Laboratory) were injected with a lethal dose of ketamine/xylazine (80/10 mg/kg, intraperitoneally) around ZT6, and perfused through the heart with 4% paraformaldehyde in sodium tetraborate 0.1 M pH 9.5. For in situ hybridization measuring Vip, Grp, and Avp expression, three EphA4 KO mice (14.7 ± 2.8 w old) and three WT littermates (14.7 ± 2.8 w old) were sacrificed by cervical dislocation around ZT6, and brains were immediately embedded in O.C.T. Compound (Thermo Fisher Scientific, Waltham, MA, USA) and frozen. Experiments were approved by Animal Care and Use Committees (Hôpital du Sacré-Coeur de Montréal, Douglas Mental Health University Institute, Comité de déontologie de l'expérimentation animale Université de Montréal).
Vigilance State Analyses
EphA4 KO mice and littermates were implanted for ECoG and electromyography (EMG) as detailed previously.16 ECoG/ EMG was recorded 10 d postsurgery for 48 h (24 h baseline, 6 h SLD and 18 h recovery, as indicated in the previous section). Signals were recorded and processed as done previously.16 Wakefulness, NREMS, and rapid eye movement sleep (REMS) were visually assigned to each 4-sec epochs, and total durations were calculated for the 12 h light and dark periods, and expressed as a percent total recording time. Mean duration of vigilance state bouts was averaged per light and dark periods and per hour. The total number of REMS bouts, and number of short (32 sec) and long (4 min and 16 min) episodes of vigilance states were calculated for the 12-h light. Sleep latency after SLD was calculated as the time between the end of the SLD and the first sleep episode lasting ≥ 1 min, and not interrupted by more than two consecutive 4-sec epochs scored as wakefulness. NREMS lost was calculated as the difference between NREMS during SLD and the corresponding 6-h interval during baseline. The effect of SLD and recovery on NREMS and REMS duration was assessed using accumulated differences from the corresponding baseline values. ECoG spectral analysis was performed using Fast Fourier transform (FFT) to calculate 24-h mean power between 0.75 and 30 Hz per 0.25 Hz for the three vigilance states. Two mice were removed from baseline spectral analyses and SLD sleep architecture analyses, and one additional mouse for SLD spectral analyses because of artifacts. Spectra were normalized to total activity of all Hz-bins in all states. The time course of delta (1–4 Hz) and sigma (10–13 Hz) activity during NREMS was calculated using averages per equal intervals as done previously,16 and expressed in percent of the 24-h mean baseline for each mouse.
EMG and Neurological Function
EphA4 KO mice have dysfunctions in motor coordination and locomotion.28 We verified the effect of these dysfunctions on EMG tone using spectral analysis. The bipolar EMG signal of artifact-free epochs was submitted to FFT to calculate the 24-h mean baseline power between 10 and 64 Hz during vigilance states. Spectral power was normalized by expressing the activity in each state in percent of the total power for all states (three mice were excluded because of artifacts). Neurological function was evaluated at the beginning and end of the rest phase (ZT0 and ZT11) in EphA4 KO mice and littermates using a standardized 10-point Neurological Severity Score (NSS) as previously described,16 which assesses motor function, physiological behavior, and alertness.
Quantitative Polymerase Chain Reaction
RNA was extracted using the RNeasy Lipid Tissue Mini kit or the RNeasy Plus Universal Midi kit (Qiagen, Toronto, ON, Canada), and quality was verified using agarose gel electrophoresis and a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA USA). For reverse transcription, 0.5 μg of RNA was used with random hexamers and Superscript II reverse transcriptase (Life Technologies, Burlington, ON, Canada), according to manufacturer's procedures. qPCR was performed using a ViiA7 real-time cycler (Life Technologies). Complementary DNA (cDNA) were diluted and used in 10 μL reaction with Standard or Fast TaqMan Master Mix reagent (Life Technologies). Primers were purchased from Invitrogen, Life Technologies, or Operon (Huntsville, AL), and probes from Eurogentec (Seraing, Belgium) or Operon. Sequences of designed oligos or Taqman Gene Expression Assay numbers are provided in Table 1. The most stable endogenous controls were selected among Actin, Tbp, GusB, and Rps9 using Expression Suite v1.0 (Life Technologies), and two or three controls were used to perform relative quantification using a modified ΔΔCt method (Expression Suite v1.0, Life Technologies).
Table 1.
Sequences of primers or probes and product numbers of probe sets used for quantitative polymerase chain reaction.
Microarray Analyses
Sense-strand cDNA synthesis, fragmentation, labeling, hybridization and microarray scanning were performed by McGill University and Génome Québec Innovation Centre (Montreal, QC, Canada). A synthesized sense-strand cDNA was generated from 250 ng of total RNA using the Ambion WT Expression kit according to the manufacturer's instructions (Life Technologies). Fragmentation and labeling of the cDNA were done with the Affymetrix GeneChip WT Terminal Labeling kit also according to the manufacturer's instruction (Affymetrix, Santa Clara, CA, USA). DNA was hybridized on Mouse Gene 2.0 ST Array (Affymetric) and incubated at 45°C in a Genechip Hybridization oven 640 (Affymetrix) for 17 h at 60 rpm. Arrays were then washed in a GeneChips Fluidics Station 450 (Affymetrix) using Affymetrix Hybridization Wash and Stain kit according to the manufacturer's instructions (Affymetrix). Last, microarrays were scanned on a GeneChip scanner 3000 (Affymetrix). All microarray analyses were performed using R (R Core, 2004, www.R-project.org) and Bioconductor packages (www.Bioconductor.org) similar to those described in our previous publication.27 Normalized expression signals were calculated from Affymetrix CEL files using the RMA function of the oligo Bioconductor package.31 Control and low intensity probesets were removed prior to the differential expression analysis, leaving 22,568 probes for statistical analysis. Differential expressions were computed using the Bioconductor package “limma”.32 A linear model was fit for each gene, and comparisons of interest were extracted as contrasts (KO-control, KO-SLD, WT-control, WT-SLD). P values were adjusted for multiple testing with the Benjamini and Hochberg method to control the false discovery rate (FDR).33 A gene was called differentially expressed if its FDR was lower than 0.05.
In situ Hybridization
In situ hybridization for EphA4 was done as described elsewhere.34 Briefly, fixed brains were postfixed, cryopreserved, and sectioned in 30 μm-thick sections. A 313 bp fragment of the EphA4 gene (NM_007936.3) was used for probe synthesis (construct generously provided by Elena B. Pasquale), and inserted in pGEM-7Z(+) plasmids in opposite orientations to produce 35S-labeled sense and antisense riboprobes (T7 promoter). Probes were synthetized from 250 ng of linearized plasmids in transcription buffer 5X, 10 mM dithiothreitol, 0,2 mM R-ATP/ GTP/CTP, 100 μCi {α-35S} UTP, 40 U RNasin, and 20 U of RNA polymerase, and purified on mini Quick-Spin RNA columns (Roche Diagnostics, Mississauga, ON, Canada). Dehydrated slides were hybridized with 107 cpm of riboprobe in 1 mL of hybridization buffer. Sections were treated with RNase A (10 mg/mL), defatted in xylene, and slides were exposed on autoradiography film (KODAK BioMax, MR) during 15 d or dipped in liquid emulsion (Kodak NTB2) for light microscopy after 30 d.
In situ hybridization for Vip, Grp, and Avp was performed largely as described previously35 for EphA4 KO mice and WT littermates. In brief, 25 μm fresh-frozen brain slices were hybridized overnight at ∼67°C with digoxigenin (DIG)-labeled riboprobes. They were then incubated over-night in 1:5000 anti-DIG-alkaline phosphatase conjugated antibody (Roche) at 4°C and colorimetrically visualized the next day. Brightfield images were taken and stitched on an Axioskop 2 Mot Plus (Zeiss Microscopy). After blinded cell counts, total number of cells per hemisphere were estimated for each mouse (n = 3 per genotype).
Statistics
Vigilance state variables, spectral analysis data, NSS and gene expression measurements were compared using one-way or two-way analyses of variance (ANOVAs) with a repeated-measure design when appropriate. Effects were decomposed using post hoc Tukey tests, t tests, or planned comparisons. Significance level for repeated-measure analyses was adjusted using Huynh–Feldt correction. SCN cell counts were compared between genotypes using paired two-tailed t tests. Threshold for statistical significance was set to 0.05 and data are reported as mean ± standard error of the mean.
RESULTS
Altered REMS and Sleep Consolidation in EphA4 KO Mice
During the 24-h baseline, the duration of wakefulness and NREMS did not significantly differ between EphA4 KO mice and WT littermates. However, REMS duration was significantly lower in KO than WT mice during the light period (Figure 1A). This likely results from a lower number of REMS bouts (KO 75.5 ± 3.5, WT 87.2 ± 4.7, P = 0.08). The consolidation of vigilance state alternation, particularly that of wakefulness and NREMS, was also altered in EphA4 KO mice. KO mice showed longer bouts of wakefulness and NREMS than WT mice specifically during the light period (Figure 1B). The time course of the mean duration of NREMS bouts differed between geno-types with longer bouts in KO compared to WT mice found at the beginning and end of the light period (Figure 1C). In parallel, the number of short wakefulness and NREMS bouts (32 sec) was lower in KO than in WT mice, whereas the number of long bouts of NREMS (4 min) and wakefulness (16 min) was higher in KO mice compared to WT or HET (Figure 1D). HET mice did not significantly differ from WT or KO mice, except for three variables for which a significant difference from KO was observed (wakefulness bout mean duration during light, number of 16-min bouts of wakefulness and of 4-min bouts of NREMS). In summary, the absence of EphA4 impaired REMS occurrence during the light period, which was accompanied by more consolidated episodes of wakefulness and NREMS.
Figure 1.

(A) Mean duration of vigilance states expressed in percent total recording time calculated for baseline light and dark periods in EphA4 knockout (KO) mice and littermates. A significant genotype effect was observed for rapid eye movement sleep (REMS) during the light period (F2,41 = 3.3, P < 0.05). Stars show significant differences (P < 0.05) between indicated points (also for panels B and D). (B) Mean duration of vigilance state bouts during light and dark periods. A significant genotype effect was found for wakefulness (F2,41 = 4.3, P = 0.02) and nonrapid eye movement sleep (NREMS) (F2,41 = 3.9, P = 0.02) during the light period. (C) 24-h time course of vigilance state bout duration. A significant genotype-by-time interaction was found for NREMS (F46,943 = 1.9, P = 0.001). Red symbols show significant differences compared to WT. (D) Number of vigilance state bouts of different durations calculated over the light period. A significant genotype effect was observed for short (32 sec) and long (16 min) bouts of wakefulness (F2,41 > 3.9, P < 0.03) and short (32 sec) and long (4 min) bouts of NREMS (F2,41 > 4.2, P < 0.05). (E) Relative electromyographic (EMG) spectral power between 10 and 64 Hz averaged for 24 h in EphA4 KO mice and littermates. No significant genotype effect was found (wakefulness: F2,38 = 0.5, P = 0.5; NREMS: F 2,38 = 1.4, P = 0.3; REMS: F2,38 = 2.2, P = 0.1). (F) Neurological severity score (NSS) measured in EphA4 KO mice and littermates at ZT0 and ZT11. A significant genotype effect was found (F2,21 = 11.8, P < 0.01). NSS was significantly higher in KO mice than in wild-type (WT) and heterozygote (HET) mice (P < 0.003; genotype effect indicated by stars).
Neurological Function Deficit in EphA4 KO Mice
To verify that changes in wakefulness/sleep architecture in EphA4 KO mice did not result from a modified state identification caused by state-specific alterations in muscle tone, total EMG activity was compared between genotypes for each vigilance state. Although EMG activity seemed higher in KO mice in all vigilance states (Figure 1E), no significant difference was observed between genotypes. Nevertheless, a significant deficit in neurological function was found (Figure 1F), which was not influenced by time of day, and which adds to the literature showing dys-functions in motor control and locomotion in these mice.28
Blunted Dynamics of NREMS Sigma Activity in EphA4 KO Mice
Power spectra during wakefulness and REMS were not significantly affected by the mutation (Figure 2A). During NREMS, KO mice showed less delta/low theta activity than WT mice (Figure 2A). However, the time course of delta activity during NREMS measured under undisturbed conditions did not significantly differ with genotype (Figure 2B, upper panel). In contrast, the daily dynamics of NREMS sigma activity was significantly blunted in EphA4 KO mice compared to WT mice (Figure 2B, lower panel), with KO mice showing lower sigma activity when WT expressed their maximum (end of light period) and higher sigma when WT mice expressed low activity (mid-dark period). Given that the time course of individual sigma frequency bins in humans was shown to differentially index circadian and homeostatic influences,36 the time course of individual bins between 10 and 13 Hz was compared between genotypes (Figure 2C). Blunted dynamics were significant specifically for the 10 to 11 and 11 to 12 Hz bins, whereas the dynamics of 12 to 13 Hz activity, which seemed more strongly influenced by homeostatic sleep pressure, was not significantly affected by genotype.
Figure 2.

(A) Normalized spectral power between 0.75 and 30 Hz in EphA4 knockout (KO) mice and littermates for the three vigilance states. Significant genotype effect was found for nonrapid eye movement sleep (NREMS) low frequencies (F2,39 > 3.5, P < 0.05). Red symbols show significant differences compared to wild-type (WT) mice (also for panels B and C). (B) 24-h dynamics of relative NREMS delta and sigma activity in EphA4 KO mice and littermates. A significant genotype-by-interval interaction was found for sigma activity (F34,663 = 2.0, P = 0.01). (C) Dynamics of NREMS sigma activity for individual 1-Hz bins. Significant interactions were found for 10–11 Hz and 11–12 Hz (F34,663 ≥ 1.9, P ≤ 0.01).
Preserved Response to SLD in EphA4 KO Mice
Electrocorticography
To further explore if EphA4 has a role in sleep homeostasis, we measured the electrophysiological response to SLD in EphA4 KO mice. During the light period (first 6 h of recovery after SLD because no REMS was observed during SLD), REMS duration in KO mice was significantly lower than in WT and HET mice (Figure 3A), as it was observed during baseline. Here, reduced REMS duration seemed to result from shorter individual REMS bouts (Figure 3B). Sleep latency after SLD, NREMS occurring during SLD and NREMS loss during SLD did not significantly differ between genotypes (Figure 3C). Moreover, the three genotypes not only lost a similar amount of NREMS during SLD, but also expressed a similar rebound of NREMS duration during recovery (Figure 3D). The time course of REMS loss and recovery also did not significantly differ with genotype (Figure 3D). Furthermore, the time course of delta activity during NREMS after SLD and of theta activity during wakefulness measured during and after SLD did not significantly differ with genotype (Figure 3E). Nevertheless, the dynamics of NREMS sigma activity (10–13 Hz) and specifically of the 12 to 13 Hz bin was affected by genotype, with HET mice showing higher activity in the first recovery hours after SLD (Figure 3E and 3F).
Figure 3.

(A) Mean duration of vigilance states expressed in percent total recording time calculated for light and dark periods in EphA4 knockout (KO) mice and littermates during and after a 6-h sleep deprivation (SLD). A significant genotype effect was observed for rapid eye movement sleep (REMS) during the light period (F2,39 = 5.2, P < 0.01). Stars show significant differences (*P < 0.05, **P < 0.01) between indicated points (also for panel B). (B) Mean duration of vigilance state bouts during light and dark periods calculated during and after a 6-h SLD. A significant genotype effect was found for REMS (F1,26 = 6.3, P = 0.001) during the light period. (C) Sleep onset latency after SLD, nonrapid eye movement sleep (NREMS) measured during SLD and NREMS lost during SLD did not significantly differ between genotypes (F2,39 ≥ 1.7, P ≥ 0.2). (D) Time course of accumulated differences between the 6-h SLD followed by 18 h of recovery and baseline conditions for NREMS and REMS. No genotype effect or interaction was observed (respectively, F 2,39 ≤ 0.3, P ≥ 0.7; F46,897 ≤ 1.1, P ≥ 0.3). (E) 24-h dynamics of NREMS delta and sigma activity after SLD and of wakefulness theta activity during and after SLD in EphA4 KO mice and littermates. Significant interaction was found for sigma activity (F26,481 = 1.7, P = 0.04). (F) Dynamics of NREMS sigma activity for individual Hz-bins. A significant interaction was found for 12–13 Hz (F26,481 = 1.7, P < 0.05).
Gene Expression
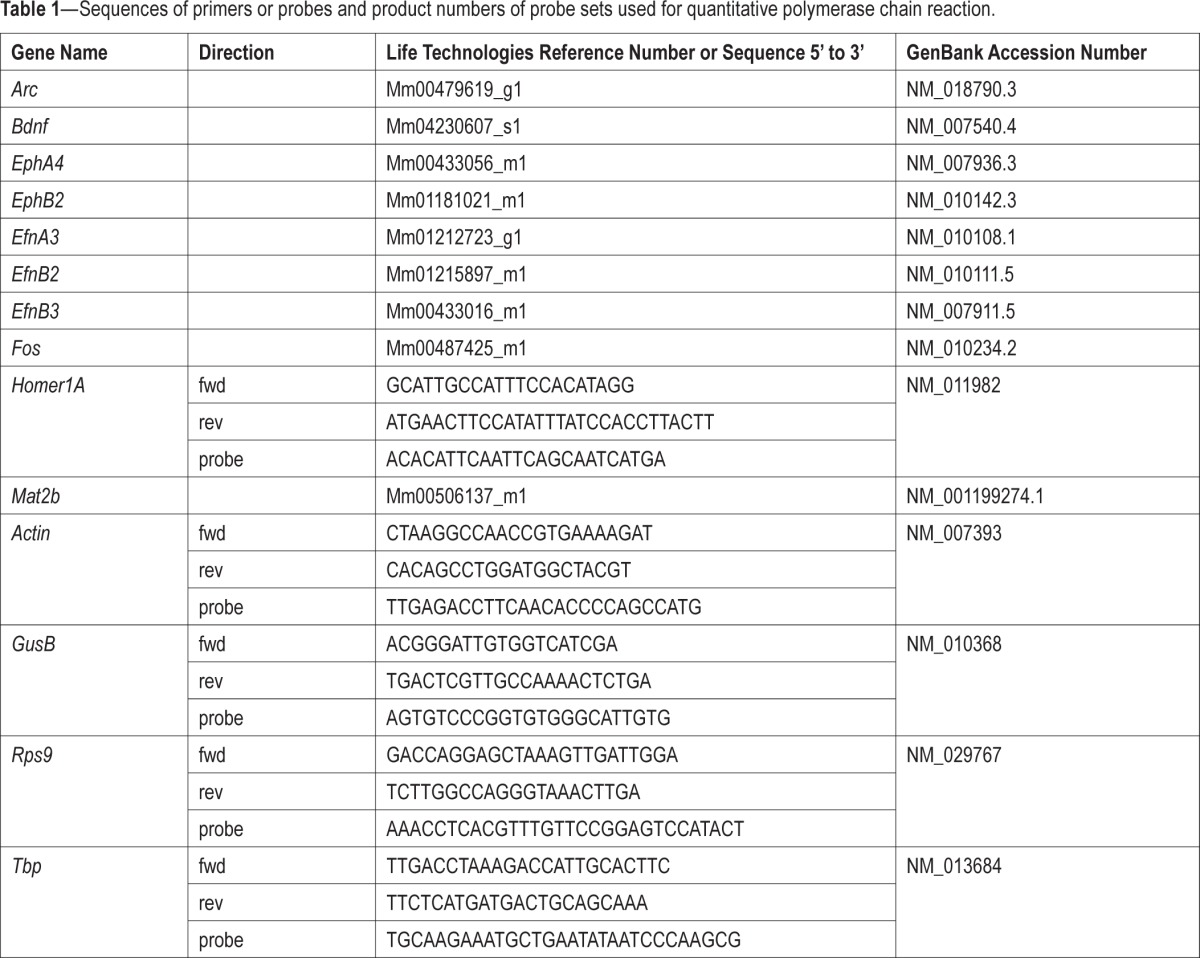
We also investigated the role of EphA4 in the response to SLD by measuring forebrain gene expression in EphA4 KO mice, with an initial focus on genes reported to be consistently increased by SLD (i.e., Arc, Bdnf, Fos, and Homer1A).26,27 The expression of EphA4, of two recognized ligands of EphA4, EfnB3, and EfnB2, and of a gene linked to DNA methylation that is modified by SLD (i.e., Mat2b)37 was also measured. SLD significantly increased the expression of Arc, Bdnf, Fos, and Homer1A in both WT and KO mice (Figure 4A). The expression of EphA4 was not detectable in EphA4 KO mice, which confirmed the absence of the transcript. In WT mice, its expression tended to be increased by SLD (P = 0.1). EfnB2 expression was significantly higher in EphA4 KO than in WT mice, but no interaction with SLD was found. The effect of SLD on the expression of EfnB3 tended to differ between geno-types, whereas the effect of SLD on Mat2b was significantly affected by genotype (Figure 4A). This differential effect of SLD resulted from a higher expression in KO than in WT mice under undisturbed/nonsleep deprived condition.
Figure 4.
(A) Relative messenger RNA expression of target genes in the forebrain of EphA4 knockout (KO) and wild-type (WT) mice measured in control condition or after a 6-h sleep deprivation (SLD). A significant SLD effect was found for Arc, Bdnf, Fos, and Homer1A (F1,15 ≥ 11.6, P < 0.001). A tendency for a SLD effect was found for EphA4 (t = −1.7, P = 0.1). A significant genotype effect was found for EfnB2 (F1,15 = 6.9, P < 0.02). A significant genotype-by-SLD interaction was found for Mat2b (F1,15 = 4.9, P < 0.05), and a trend for such interaction was found for EfnB3 (F1,15 = 3.1, P = 0.09). *P < 0.05, **P < 0.01, ***P < 0.001 between indicated points. (B) Venn diagram representing the number of probes significantly (false discovery rate [FDR] < 0.05) changed by SLD specifically in KO (gray circle) and in WT (blue circle) mice, and their overlap. (C) Heat maps of the 457 probes displaying an effect of SLD with FDR < 0.05. Columns refer to individual microarray data (WT Control n = 4; WT SLD n = 6; KO Control n = 4; KO SLD n = 5). Transcripts were ordered by hierarchical clustering (complete linkage).
Because Mat2b codes for a protein regulating the synthesis of S-adenosylmethionine,38 a methyl donor involved in DNA methylation reactions, the observation of an increase in KO mice could suggest alterations in transcriptional regulation. Therefore, we measured the effect of the mutation and of SLD on genome-wide forebrain gene expression using microarray. SLD significantly changed the expression of 457 probes, including many of the transcripts known to be consistently modified by SLD (e.g., Arc, Bdnf, Hspa5, Sgk1, Nr4a1, Dnajb5, Cdkn1, Dio2, Vip). When analyzing the effect of SLD separately in KO and WT mice, 166 probes were significantly changed in KO mice and 131 probes in WT mice, with an overlap of 67 probes (Figure 4B). However, except for EphA4, no genome-wide significant geno-type effect was found. Moreover, only two probes showed a significant genotype-by-SLD interaction, one predicted gene (Gm7008), and Olfr661 (olfactory receptor 661). Overall, forebrain gene expression changes after SLD were thus similar in EphA4 KO and WT mice (Figure 4C).
SLD and Time of Day Affect the Expression of EphA4 and Related Elements
Given our finding of alterations in sleep macroarchitecture and microarchitecture in EphA4 KO mice, we investigated if homeostatic sleep pressure and time of day modulate the expression of specific elements of the ephrin/Eph system in B6 mice. This was specifically verified for two brain regions involved in sleep regulation, the cerebral cortex and a region covering the thalamus and hypothalamus, as well as for the hippocampus, which expresses high levels of EphA421 and is particularly sensitive to SLD.39 Fos expression was used as a positive control of the effect of SLD, and was significantly increased by SLD in all regions (Figure 5A). Interestingly, only EphA4 showed a significant increase after SLD and only in the thalamic/hypothalamic region (Figure 5A). In all three brain areas, the expression of EphA4 and EfnB2 was not significantly modified by time of day (Figure 5B). However, in the cerebral cortex, a rhythmic expression was found for EphB2 and EfnB3 showing peak expression at ZT18 and ZT12, respectively. In the hippocampus, EfnA3 expression was significantly lower at ZT12 than at other times. In the thalamic/ hypothalamic region, opposite rhythms were found for EfnA3 and EfnB3 expression which, respectively, showed lower and higher expression at ZT12 than at ZT0 (Figure 5B). Overall, the expression of EphA4 and of targeted members of the same system responded to SLD or time of day in a brain area-dependent manner.
Figure 5.

Relative messenger RNA expression of EphA4, EphB2, EfnA3, EfnB2, and EfnB3 measured in the cerebral cortex, hippocampus and a region covering the thalamus and hypothalamus in B6 mice. (A) Expression measured under undisturbed condition or after sleep deprivation (SLD). A significant effect of SLD was found for the positive control gene Fos in each region (t ≥ −3.2, P < 0.02) and for EphA4 in the thalamic/hypothalamic region (t = −2.5, P = 0.03). *P < 0.05, ***P < 0.001 in comparison to the control condition. (B) Expression measured at four different Zeitgeber times (ZT0, 6, 12, and 18). A significant time of day effect was found for EfnB3 and EphB2 in the cortex (F3,15 > 4.1, P < 0.05), for EfnA3 in the hippocampus (F3,15 = 5.2, P = 0.01), and for EfnA3 and EfnB3 in the thalamic/hypothalamic region (F3,15 > 3.7, P < 0.05). Stars illustrate significant differences between indicated points (P < 0.05).
EphA4/Ephrins Expression is Reduced in Clock Mutant Mice
Because the sleep phenotype in EphA4 KO mice included variables under circadian control (REMS, sleep consolidation, sigma dynamics),23,40 and because EphA4 expression was affected by SLD in the thalamic/ hypothalamic region, we suspected that EphA4 might represent an output from the circadian system and that its expression could be controlled by clock elements. In support of this, we observed that the putative promoter region of EphA4 and one of its ligands, EfnB3, contains many predicted E-box sequences (CANNTG) in both the mouse and rat genomes (Figure 6A). Given that the core clock transcription factors CLOCK and BMAL1 are known to bind to E-boxes in their target genes,7 we evaluated if the expression of EphA4 and other ephrin/Eph is controlled by CLOCK. To do this, we measured the expression of ephrin/Eph-coding genes in Clock mutant mice at four different times. We found that the expression of EphA4, EfnA3, and EfnB2 was significantly decreased in the fore-brain of Clock mutant mice (Figure 6B). In addition, we tested whether EphA4 is expressed in the central circadian clock using in situ hybridization on mouse and rat brains. We indeed found EphA4 expression in the SCN (Figure 6C and 6D), in addition to high expression in the cerebral cortex and striatum and modest expression in other hypothalamic and thalamic nuclei (Figure 6D).
Figure 6.

(A) Diagram of the mouse and rat EphA4 and EfnB3 genes showing the position of E-boxes relative to the transcription start sites (arrows). Blue boxes represent E-boxes with the sequence CANNTG and red boxes, CACGTG (sequence with the highest affinity for CLOCK/BMAL1). Not to scale. (B) Expression of EphA4, EphB2, EfnA3, EfnB2, and EfnB3 measured in the right forebrain of ClockΔ19/Δ19 mutant mice at four different Zeitgeber times (ZT2, 8, 14, and 20). A significant genotype effect was found for EphA4, EfnA3, and EfnB2 (F1,31 ≥ 4.7, P ≤ 0.03). (C) Liquid emulsion autoradiography of EphA4 in situ hybridization on brain sections covering the hypothalamic SCN of two different mice. Scale bar = 100 μm. (D) Film autoradiographs following in situ hybridization of EphA4 messenger RNA on brain sections of two different rats. Red arrows indicate the suprachiasmatic nuclei of the hypothalamus.
Preserved Cell Number Expressing SCN Neuropeptides in EphA4 KO Mice
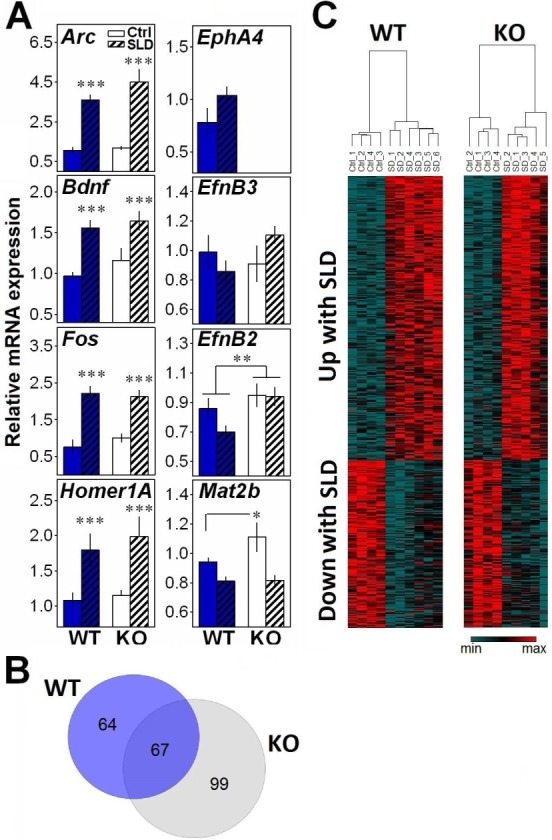
Given our observation of EphA4 expression in the SCN, and given that Vip KO mice exhibit a decrease in REMS specifically in the light period,41 and that another adhesion molecule, PSA-NCAM, regulates VIP neuron number in the SCN,42 a developmental change in SCN neuropeptide disposition could represent a mechanism underlying the sleep phenotype observed in EphA4 KO mice. As a first attempt to evaluate the role of EphA4 in SCN development, we quantified the expression of Vip, Grp, and Avp in EphA4 KO mice. However, the number of SCN neuropeptide-positive cells was not significantly different in EphA4 KO mice compared to WT littermates for all three neuropeptides studied (Figure 7). In addition, the overall morphology of SCN subdomains demarcated by these neuropeptides appeared to be intact. Thus, these observations support that a developmental change in the pattern of expression of SCN neuropeptides is unlikely to contribute to the sleep alterations observed in EphA4 KO mice.
Figure 7.
(A) In situ hybridization of suprachiasmatic nuclei of the hypothalamus (SCN) using probes for Vip, Grp, and Avp shown for a representative EphA4 knockout (KO) mouse and wild-type (WT) littermate. Scale bar = 100 μm. (B) Group mean of estimated cell number per hemisphere expressing these main SCN neuropeptides did not significantly differ between EphA4 KO and WT mice (n = 3 per genotype; Vip t = 0.30, P = 0.78; Grp t = −0.02, P = 0.98; Avp t = 2.36, P = 0.14).
DISCUSSION
Our findings point to an implication of the ephrin/Eph system, and particularly of EphA4, in sleep regulation. We first report that, under baseline condition, mice not expressing EphA4 have less REMS and longer bouts of wakefulness and NREMS during the rest period. This was accompanied by a blunted dynamics of sigma activity during NREMS. Nevertheless, the electro-physiological and molecular response to SLD in KO mice was similar to that of WT mice, except for the expression of Mat2b, which was significantly higher in KO mice under control conditions. Furthermore, we highlighted that the expression of EphA4 and members of its system is modulated by homeostatic sleep pressure and by daytime in different brain areas linked to sleep, and that some of these genes contain cis clock regulatory elements and have their expression modified in Clock mutant mice. Overall, these findings and the observation that EphA4 mRNA is expressed in the SCN support an implication of this specific adhesion protein in circadian sleep regulation.
The sleep phenotype of animals lacking EphA4 is reminiscent of a preserved sleep homeostat but of an altered circadian regulation of sleep. Indeed, although NREMS delta activity was generally decreased in EphA4 KO mice, its dynamics during baseline and after SLD, which indexes sleep homeostasis, were indistinguishable from those of WT mice. Conversely, we observed that several sleep variables under circadian control were altered in EphA4 KO mice. First, the daily dynamics of NREMS sigma activity, which has been linked to circadian regulation,23,36 was blunted. Moreover, EphA4 KO mice showed decreased REMS duration specifically during the light period in baseline, and the distribution of REMS is another sleep parameter under the influence of the circadian clock.40 The decrease in REMS duration in the light period only is still present after SLD, indicating a robust phenotype, whereas changes in wakefulness and NREMS bout duration and of sigma dynamics after SLD appear to override the between-genotype difference for these variables. These results could thus suggest that the loss of EphA4 affects the circadian regulation of sleep.
Interestingly, under baseline conditions, decreased REMS duration was accompanied by more consolidated wakefulness and NREMS, as reflected by longer individual bouts. This could suggest that a lowered pressure for REMS might be permissive to enhance consolidation of other states in mice. Reduced REMS duration accompanied by longer wakefulness and NREMS bouts has also been reported in Vip KO mice.41 Of note is that enhanced wakefulness and NREMS consolidation in EphA4 KO mice do not occur in combination with better cognitive functions. On the contrary, EphA4 KO mice express spatial memory deficits,43 in addition to impaired locomotion and neurological function. Memory deficits could be related to decreased REMS duration and altered sigma activity because REMS and sleep spindles (associated with NREMS sigma activity) have been linked to hippocampus-dependent and independent memory.1,44–47 Nevertheless, memory deficits of EphA4 KO mice could be independent from sleep alterations and rather result from other abnormalities in brain circuitry. Accordingly, future work should assess if sleep alterations contribute to memory impairments in EphA4 KO mice.
In addition to the electrophysiological response to SLD, the gene expression response to SLD was also highly similar in EphA4 KO and WT mice, further supporting a minor role, if any, for EphA4 in sleep homeostasis. The expression of targeted immediate early genes was increased by SLD independent of genotype, and in a manner similar to previous studies.48,49 Also, the genome-wide gene expression response to SLD was not significantly affected by genotype, and resembled that previously reported for the forebrain using microarray,26,27 even if the absence of EphA4 differentially affected the expression of Mat2b, a transcriptional regulator.50 Nevertheless, the expression of EphA4 itself responded to SLD in the thalamus/hypothalamus, potentially suggesting a modulatory role under elevated sleep pressure. Our future work will aim at defining the functional significance of this change in normally developed animals.
Importantly, our results reveal a pathway by which clock elements can be linked to synaptic activity in various brain areas. We indeed report that ephrin/ Eph elements (1) are expressed in a rhythmic manner, (2) present cis regulatory elements in their genes that might mediate regulation by clock transcription factors, and (3) have an altered expression in Clock mutant mice. The presence of E-boxes in EphA4 and EfnB3 genes does not necessarily implicate transcriptional regulation by core clock factors because E-box elements are not all functional. The functionality of identified elements will thus need to be verified using in vitro mutagenesis as-says.30,51 Nevertheless, this observation in combination with findings of rhythmic expression of specific ephrin/Eph in different brain regions and of decreased EphA4, EfnA3, and EfnB2 expression in Clock mutant mice suggest regulation by core clock transcription factors. This last observation was likely enabled by the absence of compensation by other partners of BMAL1, such as NPAS2, given the dominant negative nature of the ClockΔ19 mutation.52 Yet, regional differences in the rhythmic expression of the different ephrin/Eph may originate from differential transcriptional regulation by different clock elements (e.g., NPAS2 versus CLOCK). Indeed, CLOCK and BMAL1 may regulate the rhythmic expression of some ephrin/Eph elements in certain brain areas, whereas NPAS2 and BMAL1 may regulate the expression of the same or different ephrin/Eph in other regions. This is in agreement with the reported brain area-specificity in ephrin/Eph expression and with their various roles in neuronal function.17 In the hippocampus specifically, given the high expression of EphA4,21 and our finding of rhythmic expression of its ligand EfnA3, control of ephrin/Eph by clock elements may represent a route by which synaptic plasticity varies with time of day.53 Such a pathway could contribute to deficits in hippocampal long-term potentiation (LTP) in animals with clock gene mutations.14,15 In the cerebral cortex, rhythmic expression of EphB2 and EfnB3 may also impact on synaptic activity as well as plasticity. Last, rhythmic expression of EphA4 ligands (EfnA3, EfnB3) in the thalamus/hypothalamus could contribute to the circadian sleep phenotype observed here given the numerous thalamic/hypothalamic nuclei involved in sleep regulation.
Considering that EphA4 is expressed in the SCN, it is also tempting to speculate that its modulatory effects on circa-dian sleep variables originate from the SCN. The absence of EphA4 was shown to impair synaptic potentiation as measured by long-term potentiation,54 which was linked to the role of EphA4 in neuron-astrocyte communication.17,20,21 Interestingly, astrocytic coverage in the SCN varies with time of day,13 which represents a form of structural plasticity that modulates neurotransmission.55 This is presumably dependent on astrocytes cytoskeletal rearrangement, which was shown to be attenuated in EphA4 KO astrocytes in culture.56 A role of EphA4 in SCN structural plasticity could contribute to rhythmic changes of the SCN output signal, without necessarily altering SCN neuropeptide expression. Co-localization experiments assessing the SCN cell type(s) where EphA4 is expressed according to time and measuring circadian behavior in constant darkness in EphA4 KO mice will be required to define the contribution of this receptor to the circadian timing system.
Alternatively, developmental effects of the absence of EphA4 on sleep-relevant circuits could underlie the sleep phenotypes observed in EphA4 KO mice. Preservation of the number of cells expressing the main SCN neuropeptides in EphA4 KO mice suggests that developmental effects on SCN cell fate selection are not responsible. However, this does not rule out other possible developmental contributions. For example, EphA4 KO mice were shown to have a loss of the anterior commissure,28 which mediates interhemispheric communication of numerous brain regions. Furthermore, EphA4 KO mice have a thinner cerebral cortex,57 and an altered development of ventral trigeminal axons innerving the somatosensory cortex.58 These or other structural/circuit abnormalities, related to the role of EphA4 in axon guidance,59 could thus specifically affect REMS duration, sleep consolidation variables, and ECoG activity. However, circuit abnormalities in most regions cannot fully explain why EphA4 KO mice are showing altered sleep architecture variables only at certain times of the day (e.g., less REMS and longer NREMS bouts during the light period) and opposed changes in ECoG activity at opposite times of the day under baseline conditions (e.g., lower sigma in KO than WT during the light period but higher in KO than WT during the dark period). Aside from the SCN, these changes could originate from abnormalities in brain circuits controlling the daily pattern of, for instance, REMS such as the circuit encompassing the sublaterodorsal tegmental nucleus, which mainly expresses glutamate.60 In the KO mice studied here, a minimal effect of developmental compensation by other molecules is expected on the sleep phenotype observed, because changes in dendritic spine morphology in EphA4 KO mice do not seem to be compensated for by other ephrins/Eph.19 Nevertheless, we have observed a higher EfnB2 expression in EphA4 KO mice than in WT mice. Consequently, replication of our findings using targeted downregulation (or overexpression) strategies will be required to identify the precise brain circuits by which EphA4 (and ligands) regulate sleep architecture as well as ECoG activity.
To our knowledge, this is the first study implicating the ephrin/Eph system in sleep regulation. Importantly, in light of the role of EphA4 in synaptic plasticity, an implication of EphA4 in neurodegenerative diseases has been described. In fact, patients with Alzheimer's disease (AD) exhibit a twofold increase in EphA4 mRNA in synaptoneurosomes,61 and the intracellular signaling of EphA4 is involved in synaptic transmission impairment in animal models of AD.62,63 Interestingly, AD patients and also animal models for AD have disturbances in the sleep-wake cycle and circadian rhythms.64 Accordingly, our findings support the hypothesis that the same molecular elements are simultaneously regulating cognitive functioning and sleep quality and quantity, and thus that dysfunctions in neuronal circuitry or plasticity originating from the absence of such elements are responsible for both memory and sleep alterations.
DISCLOSURE STATEMENT
This was not an industry supported study. Research conducted thanks to a J.A. De Sève fellowship (MF), a Université de Montréal COPSE fellowship (GP), a Université de Montréal graduate studies fellowship (MF), NSERC discovery grants (NC, VM), a NSF predoctoral award (JB), and FRQS salary awards (VM, NC). The content is solely the responsibility of the authors and does not represent the official views of the aforementioned funding agencies. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank Chloé Provost, Caroline Bouchard, Hélène Blais, Alexis Blanchet-Cohen, Gaétan Tremblay, Renaud Massart, Abhijith Bathini, and Gaétan Poirier for technical help; Paul Franken, Samer Hattar, and J-Martin Beaulieu for helpful discussions and advice; Keith K. Murai for providing animals for initial breeding; and Elena B. Pasquale for providing the EphA4 construct for in situ hybridization
REFERENCES
- 1.Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–26. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- 2.Jagannath A, Peirson SN, Foster RG. Sleep and circadian rhythm disruption in neuropsychiatric illness. Curr Opin Neurobiol. 2013;23:888–94. doi: 10.1016/j.conb.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Hatfield CF, Herbert J, van Someren EJ, Hodges JR, Hastings MH. Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer's dementia. Brain. 2004;127:1061–74. doi: 10.1093/brain/awh129. [DOI] [PubMed] [Google Scholar]
- 4.Dijk DJ, Lockley SW. Integration of human sleep-wake regulation and circadian rhythmicity. J Appl Physiol. 2002;92:852–62. doi: 10.1152/japplphysiol.00924.2001. [DOI] [PubMed] [Google Scholar]
- 5.Achermann P, Dijk DJ, Brunner DP, Borbely AA. A model of human sleep homeostasis based on EEG slow-wave activity: quantitative comparison of data and simulations. Brain Res Bull. 1993;31:97–113. doi: 10.1016/0361-9230(93)90016-5. [DOI] [PubMed] [Google Scholar]
- 6.Borbely AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14:557–68. doi: 10.1177/074873099129000894. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–75. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feinberg I, Campbell IG. Ketamine administration during waking increases delta EEG intensity in rat sleep. Neuropsychopharmacology. 1993;9:41–8. doi: 10.1038/npp.1993.41. [DOI] [PubMed] [Google Scholar]
- 9.Campbell IG, Feinberg I. Comparison of MK-801 and sleep deprivation effects on NREM, REM, and waking spectra in the rat. Sleep. 1999;22:423–32. doi: 10.1093/sleep/22.4.423. [DOI] [PubMed] [Google Scholar]
- 10.Chauvette S, Seigneur J, Timofeev I. Sleep oscillations in the thalamocortical system induce long-term neuronal plasticity. Neuron. 2012;75:1105–13. doi: 10.1016/j.neuron.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank MG, Cantera R. Sleep, clocks, and synaptic plasticity. Trends Neurosci. 2014;37:491–501. doi: 10.1016/j.tins.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deboer T, Vansteensel MJ, Detari L, Meijer JH. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat Neurosci. 2003;6:1086–90. doi: 10.1038/nn1122. [DOI] [PubMed] [Google Scholar]
- 13.Girardet C, Blanchard MP, Ferracci G, et al. Daily changes in synaptic innervation of VIP neurons in the rat suprachiasmatic nucleus: contribution of glutamatergic afferents. Eur J Neurosci. 2010;31:359–70. doi: 10.1111/j.1460-9568.2009.07071.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang LM, Dragich JM, Kudo T, et al. Expression of the circadian clock gene Period2 in the hippocampus: possible implications for synaptic plasticity and learned behaviour. ASN Neuro. 2009:1. doi: 10.1042/AN20090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rawashdeh O, Jilg A, Jedlicka P, et al. PERIOD1 coordinates hippocampal rhythms and memory processing with daytime. Hippocampus. 2014;24:712–23. doi: 10.1002/hipo.22262. [DOI] [PubMed] [Google Scholar]
- 16.El Helou J, Belanger-Nelson E, Freyburger M, et al. Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc Natl Acad Sci U S A. 2013;110:9974–9. doi: 10.1073/pnas.1221381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murai KK, Pasquale EB. Eph receptors, ephrins, and synaptic function. Neuroscientist. 2004;10:304–14. doi: 10.1177/1073858403262221. [DOI] [PubMed] [Google Scholar]
- 18.Fu AK, Hung KW, Fu WY, et al. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat Neurosci. 2011;14:181–9. doi: 10.1038/nn.2715. [DOI] [PubMed] [Google Scholar]
- 19.Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci. 2003;6:153–60. doi: 10.1038/nn994. [DOI] [PubMed] [Google Scholar]
- 20.Carmona MA, Murai KK, Wang L, Roberts AJ, Pasquale EB. Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc Natl Acad Sci U S A. 2009;106:12524–9. doi: 10.1073/pnas.0903328106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filosa A, Paixao S, Honsek SD, et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci. 2009;12:1285–92. doi: 10.1038/nn.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franken P, Dudley CA, Estill SJ, et al. NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: genotype and sex interactions. Proc Natl Acad Sci U S A. 2006;103:7118–23. doi: 10.1073/pnas.0602006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yasenkov R, Deboer T. Interrelations and circadian changes of electroencephalogram frequencies under baseline conditions and constant sleep pressure in the rat. Neuroscience. 2011;180:212–21. doi: 10.1016/j.neuroscience.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 24.Cirelli C, Tononi G. Gene expression in the brain across the sleep-waking cycle. Brain Res. 2000;885:303–21. doi: 10.1016/s0006-8993(00)03008-0. [DOI] [PubMed] [Google Scholar]
- 25.Mackiewicz M, Shockley KR, Romer MA, et al. Macromolecule biosynthesis: a key function of sleep. Physiol Genomics. 2007;31:441–57. doi: 10.1152/physiolgenomics.00275.2006. [DOI] [PubMed] [Google Scholar]
- 26.Maret S, Dorsaz S, Gurcel L, et al. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A. 2007;104:20090–5. doi: 10.1073/pnas.0710131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mongrain V, Hernandez SA, Pradervand S, et al. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep. 2010;33:1147–57. doi: 10.1093/sleep/33.9.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dottori M, Hartley L, Galea M, et al. EphA4 (Sek1) receptor tyrosine kinase is required for the development of the corticospinal tract. Proc Natl Acad Sci U S A. 1998;95:13248–53. doi: 10.1073/pnas.95.22.13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franken P, Dijk DJ, Tobler I, Borbély AA. Sleep deprivation in rats: effects on EEG power spectra, vigilance states, and cortical temperature. Am J Physiol. 1991;261:R198–208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- 30.Mongrain V, Ruan X, Dardente H, Fortier EE, Cermakian N. Clock-dependent and independent transcriptional control of the two isoforms from the mouse Rorgamma gene. Genes Cells. 2008;13:1197–210. doi: 10.1111/j.1365-2443.2008.01237.x. [DOI] [PubMed] [Google Scholar]
- 31.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–7. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth G. Limma: linear models for microarray data. In: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S, editors. Bioinformatics and computational biology solutions using R and bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 33.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B. 1995;57:289–300. [Google Scholar]
- 34.Poulin JF, Chevalier B, Laforest S, Drolet G. Enkephalinergic afferents of the centromedial amygdala in the rat. J Comp Neurol. 2006;496:859–76. doi: 10.1002/cne.20956. [DOI] [PubMed] [Google Scholar]
- 35.Bedont JL, LeGates TA, Slat EA, et al. Lhx1 controls terminal differentiation and circadian function of the suprachiasmatic nucleus. Cell Rep. 2014;7:609–22. doi: 10.1016/j.celrep.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dijk DJ, Shanahan TL, Duffy JF, Ronda JM, Czeisler CA. Variation of electroencephalographic activity during non-rapid eye movement and rapid eye movement sleep with phase of circadian melatonin rhythm in humans. J Physiol. 1997;505:851–8. doi: 10.1111/j.1469-7793.1997.851ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massart R, Freyburger M, Suderman M, et al. The genome-wide landscape of DNA methylation and hydroxymethylation in response to sleep deprivation impacts on synaptic plasticity genes. Transl Psychiatry. 2014;4:e347. doi: 10.1038/tp.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murray B, Antonyuk SV, Marina A, et al. Structure and function study of the complex that synthesizes S-adenosylmethionine. IUCrJ. 2014;1:240–9. doi: 10.1107/S2052252514012585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen C, Hardy M, Zhang J, LaHoste GJ, Bazan NG. Altered NMDA receptor trafficking contributes to sleep deprivation-induced hippocampal synaptic and cognitive impairments. Biochem Biophys Res Commun. 2006;340:435–40. doi: 10.1016/j.bbrc.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 40.Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15:3526–38. doi: 10.1523/JNEUROSCI.15-05-03526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu WP, Li JD, Colwell CS, Zhou QY. Decreased REM sleep and altered circadian sleep regulation in mice lacking vasoactive intestinal polypeptide. Sleep. 2011;34:49–56. doi: 10.1093/sleep/34.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen H, Watanabe M, Tomasiewicz H, Rutishauser U, Magnuson T, Glass JD. Role of neural cell adhesion molecule and polysialic acid in mouse circadian clock function. J Neurosci. 1997;17:5221–9. doi: 10.1523/JNEUROSCI.17-13-05221.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willi R, Winter C, Wieske F, et al. Loss of EphA4 impairs short-term spatial recognition memory performance and locomotor habituation. Genes Brain Behav. 2012;11:1020–31. doi: 10.1111/j.1601-183X.2012.00842.x. [DOI] [PubMed] [Google Scholar]
- 44.Smith C, Rose GM. Evidence for a paradoxical sleep window for place learning in the Morris water maze. Physiol Behav. 1996;59:93–7. doi: 10.1016/0031-9384(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 45.Luo J, Phan TX, Yang Y, Garelick MG, Storm DR. Increases in cAMP, MAPK activity, and CREB phosphorylation during REM sleep: implications for REM sleep and memory consolidation. J Neurosci. 2013;33:6460–8. doi: 10.1523/JNEUROSCI.5018-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuentemilla L, Miró J, Ripollés P, et al. Hippocampus-dependent strengthening of targeted memories via reactivation during sleep in humans. Curr Biol. 2013;23:1769–75. doi: 10.1016/j.cub.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 47.Fogel SM, Albouy G, Vien C, et al. fMRI and sleep correlates of the age-related impairment in motor memory consolidation. Hum Brain Mapp. 2014;35:3625–45. doi: 10.1002/hbm.22426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terao A, Wisor JP, Peyron C, et al. Gene expression in the rat brain during sleep deprivation and recovery sleep: an Affymetrix GeneChip study. Neuroscience. 2006;137:593–605. doi: 10.1016/j.neuroscience.2005.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson CL, Wisor JP, Lee CK, et al. Molecular and anatomical signatures of sleep deprivation in the mouse brain. Front Neurosci. 2010;4:165. doi: 10.3389/fnins.2010.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Igarashi K, Katoh Y. Metabolic aspects of epigenome: coupling of S-adenosylmethionine synthesis and gene regulation on chromatin by SAMIT module. Subcell Biochem. 2013;61:105–18. doi: 10.1007/978-94-007-4525-4_5. [DOI] [PubMed] [Google Scholar]
- 51.Kiyohara YB, Nishii K, Ukai-Tadenuma M, Ueda HR, Uchiyama Y, Yagita K. Detection of a circadian enhancer in the mDbp promoter using prokaryotic transposon vector-based strategy. Nucleic Acids Res. 2008;36:e23. doi: 10.1093/nar/gkn018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gekakis N, Staknis D, Nguyen HB, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–9. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 53.Raghavan AV, Horowitz JM, Fuller CA. Diurnal modulation of long-term potentiation in the hamster hippocampal slice. Brain Res. 1999;833:311–4. doi: 10.1016/s0006-8993(99)01523-1. [DOI] [PubMed] [Google Scholar]
- 54.Zhuang Z, Yang B, Theus MH, et al. EphrinBs regulate D-serine synthesis and release in astrocytes. J Neurosci. 2010;30:16015–24. doi: 10.1523/JNEUROSCI.0481-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev. 2008;88:983–1008. doi: 10.1152/physrev.00036.2007. [DOI] [PubMed] [Google Scholar]
- 56.Puschmann TB, Turnley AM. Eph receptor tyrosine kinases regulate astrocyte cytoskeletal rearrangement and focal adhesion formation. J Neurochem. 2010;113:881–94. doi: 10.1111/j.1471-4159.2010.06655.x. [DOI] [PubMed] [Google Scholar]
- 57.North HA, Zhao X, Kolk SM, Clifford MA, Ziskind DM, Donoghue MJ. Promotion of proliferation in the developing cerebral cortex by EphA4 forward signaling. Development. 2009;136:2467–76. doi: 10.1242/dev.034405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.North HA, Karim A, Jacquin MF, Donoghue MJ. EphA4 is necessary for spatially selective peripheral somatosensory topography. Dev Dyn. 2010;239:630–8. doi: 10.1002/dvdy.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wegmeyer H, Egea J, Rabe N, et al. EphA4-dependent axon guidance is mediated by the RacGAP alpha2-chimaerin. Neuron. 2007;55:756–67. doi: 10.1016/j.neuron.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 60.Luppi PH, Clément O, Sapin E, et al. The neuronal network responsible for paradoxical sleep and its dysfunctions causing narcolepsy and rapid eye movement (REM) behavior disorder. Sleep Med Rev. 2011;15:153–63. doi: 10.1016/j.smrv.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 61.Williams C, Mehrian Shai R, Wu Y, et al. Transcriptome analysis of synaptoneurosomes identifies neuroplasticity genes overexpressed in incipient Alzheimer's disease. PLoS One. 2009;4:e4936. doi: 10.1371/journal.pone.0004936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu AK, Hung KW, Huang H, et al. Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer's disease. Proc Natl Acad Sci USA. 2014;111:9959–64. doi: 10.1073/pnas.1405803111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vargas LM, Leal N, Estrada LD, et al. EphA4 activation of c-Abl mediates synaptic loss and LTP blockade caused by amyloid-beta oligomers. PLoS One. 2014;9:e92309. doi: 10.1371/journal.pone.0092309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Musiek ES, Xiong DD, Holtzman DM. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med. 2015;47:e148. doi: 10.1038/emm.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]