

Abstract
Early access programs, (EAPs) are adopted by an increasing number of pharma companies due to several benefits offered by these programs. EAPs offer ethical, compliant, and controlled mechanisms of access to investigational drugs outside of the clinical trial space and before the commercial launch of the drug, to patients with life-threatening diseases having no treatment options available. In addition to the development of positive relationships with key opinion leaders (KOL), patients, advocacy groups and regulators, the data captured from the implementation of EAPs supports in the formulation of global commercialization strategies. This white paper outlines various circumstances to be considered for the implementation of EAPs named patient programs, the regulatory landscape, the benefits and challenges associated with implementing these programs and the key considerations for their successful implementation.
Key words: Early access programs, named patient programs, patients with life-threatening diseases
INTRODUCTION
After the recent outbreak of the Ebola virus in West Africa, the World Health Organization issued a statement indicating that it is ethical to resort to provide patients in need access to investigational drugs that have not been approved by regulators or even tested on human beings, so as to restrain the epidemic and save the lives of patients.[1,2] Hence, an investigational drug, a monoclonal antibody called as ZMapp developed by Mapp Biopharmaceuticals was administered to six Ebola infected patients, out of which four patients improved and two patients died.[3] Another case which received huge media attention was that of an ailing 7-year-old boy named Josh Hardy, who was suffering from a life-threating adenovirus infection. Josh was left with no treatment option as his existing treatment had to be stopped due to nephrotoxicity. The family requested chimerix, a biopharmaceutical company for compassionate use, access of the investigational antiviral drug – brincidofovir for their son, which denied the same as it wanted to focus on the completion of the ongoing phase III trial. The family then launched an aggressive social media campaign called “Save Josh,” the success of which resulted in the company granting access of brincidofovir to Josh by enrolling him as a part of the open-label pilot trial.[4] Such patients who are terminally ill with grim prospects of long-term survival with no other treatment options available, and who cannot participate in clinical trials for various reasons, benefit from early access program (EAP). From the patient's perspective, the benefit expected from the investigational drug outweighs the risk associated with the investigational drug.[5]
EARLY ACCESS PROGRAMS: AN OVERVIEW
These programs are also known by various other names such as compassionate use, early access, special access, etc., These programs along with clinical trials provide prelaunch access to the investigational drugs. However, there is a difference between a clinical trial and access programs with regards to the entity driving the access and the modality of providing access to the patients.
A clinical trial is protocol driven, and historically it was the only way for the patients in many countries to obtain preapproved drugs. Table 1 provides the comparison of expanded access, compassionate use and named patient programs (NPPs).[6]
Table 1.
Comparison of EAPs in the US to CUP and NPP in the EU
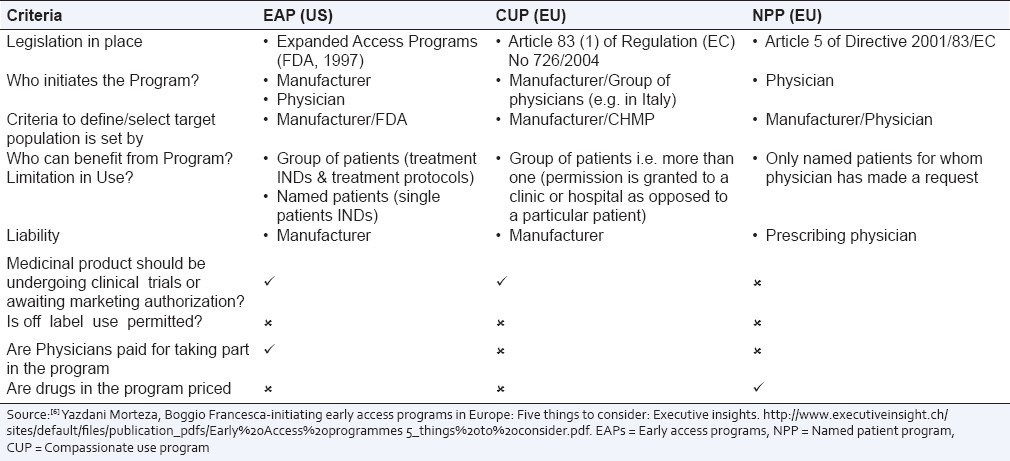
In addition there are various individual country specific variances of the access programs which are described in the regulatory section of this white paper.
WHEN SHOULD AN EARLY ACCESS PROGRAM BE IMPLEMENTED?
The implementation of EAPs can be considered in a variety of stages and situations during the life cycle of the drug. Access programs can be considered in the early stages of product development where there is a likelihood of the product showing promising results for patients who cannot take part in clinical trials as they do not fulfill the eligibility criteria of the protocol or as they stay at a great distance from the trial site.[7] For terminally ill patients, access to the investigational drug is the only treatment option available and can be lifesaving in many cases. Many pharma companies implement open-label extension study (OLE), trials to ensure continued access of the study drug to patients who might have benefited from the investigational drug during participation in a clinical trial until the investigational drug is approved by regulators and is commercially available in the specified country. However when the company wants to implement extended access with the primary motive of providing continued access and not data collection, instead of an OLE, EAPs offer an efficient and a cost effective option.[8] These access programs are also implemented when the drug is approved in one country but not in another country where it is needed and requested by terminally ill patients. Followed by the approval of the drug by the regulators, there may be a considerable gap from the time the drug is approved and is commercially available to patients. For example in Europe, even after the drug is approved centrally by European Medicines Agency (EMA), there is a delay in the commercial launch of the drug in member countries, depending on local regulations, as the reimbursement process is not centralized, and the decision on the reimbursement or pricing is taken by the respective EU Country. The European Federation of Pharmaceutical Industries and Association had prepared a report known as “patients patients waiting to access innovative therapies” indicator for analyzing the delay in commercial launch and finalization of the reimbursement process from the time of marketing approval or authorization in 14 EU Countries.[9] It was seen that the mean time from EMA approval to commercial availability varied from 88 days to 392 days. It is under such situations when marketing authorizations are received in a staggered manner, that EAPs can be implemented.[10] EAPs can be also implemented for rare and orphan diseases like acute myeloid leukemia.[11] For pharma companies developing orphan drugs, EAP is a preferred option, instead of full-scale regulatory approval, for the countries or regions where it is not economical for the company to seek regulatory approval of an orphan drug.[12] Finally, when the drug is being discontinued from development or commercialization by a company in a specific country or region, NPP is the only channel through which patients who do not have any other treatment options available to get access to the drug.
REGULATORY SCENARIO
As per the US Food and Drug Administration (FDA) regulations,[13] there are three types of basic mechanisms that are available for patient access.
Access to individual patients which is considered on a case to case basis and which also includes access for emergency use (single patient/individual patient investigational new drug [IND])
Access to a medium group of patients
Access to a large group of patients (treatment IND or treatment protocol).
Based on different types of access, the FDA has subcategorized expanded access submissions into 8 types:
Individual patient IND
Individual patient protocol
Emergency IND
Emergency protocol
Intermediate-size patient population IND
Intermediate-size patient population protocol
Treatment IND
Treatment protocol.
The general criteria which must be met are:
There should be a legitimate expectation of worthwhile benefit from the investigational drug even though there is an absence of definite clinical trial data
The patient should be suffering from a life-threatening disease or chronic condition
Adequate treatment options and clinical trials for the patient should not be available. US regulations also stipulate that for all the expanded access types, investigators are required to obtain Ethics Committee (EC)/Institutional Review Board approval.
The regulation which enacts compassionate use and prelaunch accesses are Article 83[1] of regulation (EC) 726/2004.[14] The EMA has also specified that the implementation of compassionate use programs should be done by the EU Member States, which are known to vary significantly. Each member state has its own national regulations based on which compassionate use or NPPs are implemented, and EMA recommendations assist as guidelines.[15] The early access to medicine scheme (EAMS) which was launched in April 2014 in the UK is a three step process. In the first step, companies can submit a phase I/II data to the Medicines and Healthcare products Regulatory Agency (MHRA) and apply for a promising innovative medicine (PIM) designation. Upon receipt of the positive PIM designation, in stage 2, the MHRA reviews the data. If the EAMS scientific opinion is positive, then in the third stage is the commissioning of the drug in the National Health Service. Fees are levied at each stage of the application and contrary to other countries such as the USA and France, the early access provided by the company through EAMS is free of cost.[16] Other countries where early access to medicines is provided free of cost include Austria, Germany, Greece, and Spain.
In India, as per the Drugs and Cosmetic Act 1940 and Rules 1945, provisions are in place for the personal import of unapproved drugs in the country by a patient (Rule 36) and by a hospital or institution (Rule 34). A patient can apply via Form 12-A, after obtaining a prescription from his/her physician. The Drug Controller General of India (DCGI), after satisfactory review of the application, grants the import permit via form 12-B, which authorizes the patient to import the drug.[17] Pharma companies can apply to the DCGI to seek an no objections certificate to provide the drug for compassionate use (free of cost) to patients participating in global clinical trials, for which regulatory approval is pending.
Benefits
EAPs offer a number of benefits.[7,18] EAPs, allow the pharma company and physicians to meet the needs of patients suffering from serious life-threatening or rare diseases, by providing potentially life-saving drugs in an ethical and a compliant manner. Early adopting physicians could become brand advocates of the company's products and the programs help to build physician KOL relationships. KOLs may assist in providing their opinion about the investigational drug to regulatory agencies.[18] The programs also aid in establishing loyalty and positive relationships with patients and patient advocacy groups. The feedback on the use of an investigational drug obtained from early adopters/KOLs and their patients, is real-life safety data and is more varied clinically as well as ethnically as compared to patients fulfilling eligibility criteria in clinical trials. The information can provide insights on the wider use of the drug by different patient subtypes.[7,18] EAPs can form an essential component of the company's global market access plans and strategies. As EAPs are governed by the national regulations, these programs ensure the controlled and compliant supply of these preapproved drugs thereby reducing counterfeit opportunities. These programs also help in aligning key cross-functional team members of a company at a much earlier stage ensuring their preparedness for the global launch of the product. With respect to the financial benefits, EAPs can earn early revenues in the countries where early access through NPPs can be charged by the companies. EAPs can be immensely useful in early market penetration leading to increased acceptance and uptake by physicians and patients after the commercial launch of the product. In a study conducted to determine the financial impact of the implementation of NPPs, it was found that the NPP had a considerable effect on the market share of the product during the 1st year after the launch. The study showed that the probability of spending $1 on the drug was 1.362 times greater for the drug supplied when NPPs were implemented prior to the commercial launch.[19]
Challenges
There are numerous challenges faced in the implementation of EAPs.[18,19] Differences in national regulations pertaining to the implementation of EAPs pose a significant challenge to pharma companies. Pharma companies are concerned about the possible adverse effects that may be reported during the conduct of an NPP, which might be due to the investigational drug being used inappropriately by physicians, in the absence of a protocol like in case of NPP, thereby impacting the chances of obtaining marketing approval from regulators.[18] As the promotion of EAPs is prohibited, there is a fear that it may be perceived as illegal and as an attempt to promote unlicensed medicines.[18,20] In countries where the company can charge for EAPs, the challenge posed is with regards to frame the right pricing policy as it is expected that the price finalized during the EAP stage can be used as the benchmark price while launching the product. Uncertainty of the demand makes it difficult for the company to determine the quantity, production planning and supply of an investigational drug for the conduct of an EAP. There has been lot of debate and discussion in the media around the need of an investigational drug as the only treatment option for a terminally ill patient and a company's ethical obligation to complete the trial expediently so as to bring the investigational drug to the market as early as possible for the benefit of the wider patient population. It has also brought to light the issue of equitable access to patients, as only those patients who have access to social media are able to reach out to a pharma company faster, but those who do not have access to the same should be equally eligible to have access to the investigational drug.[21] Pharma companies believe that for implementing EAPs, they have to allocate resources which might impact ongoing developmental work and thus delay marketing approval and commercialization of the drug. The supply of the investigational drug to support an EAP is challenging for companies having lesser quantities of investigation drugs, such as biological.[20]
On the basis of rules and regulations of expanded access, in August 2009, FDA mentioned that the sponsor companies conducting expanded access must provide information on adverse events. The agency has also noted that this information must be incorporated in IND annual reports and safety reports and that the new drug application must at least cover the summary of the expanded access exposure to the patients. With regards to the use of the data obtained from the expanded access, FDA clearly mentions that the data can be useful in assessing drugs safety profile. For example, the information on a rare adverse event which is observed during the expanded access might be useful in assessing the safety profile of the drug to the patients which were not part of clinical trials. The agency has also cited that in certain cases the safety information obtained from expanded access was included in the approved product labeling.[22]
Similarly for EU, among the 49 orphan drugs approved from 2005 to 2012, 7 (14%) applications incorporated safety data from the compassionate use programs. There are a number of studies published which indicate the usefulness of the data for patient subtypes not included in the clinical trials. For example, clinical trial data of Celgene's Vidaza (azacitidine) show the effectives of the drug to the patient with high risk of myelodisplastic syndrome (MDS). However, data collected from NPP indicate that the drug is effective to the patients with low risk of MDS whereas the clinical trial included very fewer patients with low risk of MDS. Finally, data collected from the access programs can be used to formulate patient-centric approaches to the treatment. For example, as per the study published in British Journal of Urology International, the data generated from access programs in the US and the UK was used to decide treatment approach to various patient subtypes suffering from renal cell carcinoma.[23]
Key considerations for the successful implementation of early access program
Early planning (6–12 months prior to the expected demand) to ascertain whether an EAP should be implemented and if so, at what stage of clinical development should it be implemented is beneficial.[6] This would depend on the presentation of the results, the launch of the product in the first country or the news of submission of the data to regulators, the availability of the investigational drug, drug labeling requirements, supply chain considerations, the duration and objective of the program, the number of countries involved, country-specific access regulations, provisions for collecting safety data, and ensuring that there is no impact on ongoing clinical trials are some of the factors to be considered.[7,12] The investigational drug may either be provided free of charge to ex-trial patients until the commercial launch while others are charged; free of charge before the first market launch to all and then chargeable to all; or free of charge in some of the countries and chargeable in others.
CONCLUSION
The demand for EAPs has been increasing due to patients and physicians having online access to information on clinical trials and investigational drugs from websites and blogs. There are huge benefits of EAPs not only to patients but also to pharma companies having drugs in the development stage which can generate early demand from patients and physicians. In addition to other challenges, pharma companies now also need to deal with patient advocacy groups and social media campaigns, as a result of an increased proportion of an informed and vocal patient population.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Geneva: World Health Organization; 2014. [Last accessed on 2015 Apr 30]. World Health Organization. Ethical Considerations for Use of Unregistered Interventions for Ebola Viral Diseases: Report of an Advisory Panel to WHO. Available from: http://www.who.int/mediacentre/news/statement/2014/ebola-ethical-review-summary/en/ [Google Scholar]
- 2.Hantel A, Olopade CO. Drug and vaccine access in the Ebola epidemic: Advising caution in compassionate use. Ann Intern Med. 2015;162:141–2. doi: 10.7326/M14-2002. [DOI] [PubMed] [Google Scholar]
- 3.Gallagher J. Ebola: Experimental drug ZMapp is ‘100% effective’ in animal trials: BBC News; 29 August, 2014. [Last accessed on 2015 Apr 30]. Available from: http://www.bbc.com/news/health-28980153 .
- 4.Arthur C, Kenneth M. Rescue Me: The Challenge of Compassionate Use in the Social Media Era: Health Affairs Blog; 27 August, 2014. [Last accessed on 2015 Apr 30]. Available from: http://www.healthaffairs.org/blog/2014/08/27/rescue-me-the-challenge-ofcompassionate-use-in-the-social-media-era/
- 5.Life Science Update – US Edition. [Last accessed on 2015 jun 15]. Available from: https://www.lifescicompliance.com/wp-content/uploads/2015/05/LSCUApril2015v4.pdf .
- 6.Morteza Y, Francesca B. Initiating Early Access Programs in Europe: Five Things to Consider: Executive Insights. [Last accessed on 2015 jun 15]. Available from: http://www.executiveinsight.ch/sites/default/files/publication_pdfs/Early%20Access%20programmes_5_things%20to%20consider.pdf .
- 7.Simon E. Integrating managed access programs: Global considerations. Applied Clinical Trials. [Last accessed on 2015 Apr 30];23(2) Available from http://www.appliedclinicaltrialsonline.com/integrating-managed-access-programs-global-considerations . [Google Scholar]
- 8.Nicky W. Managed Access Programs: Maintaining Treatment Options Post-trial: Pharmaphorum; 6th October, 2014. [Last accessed on 2015 Apr 30]. Available from: http://www.pharmaphorum.com/articles/managed-accessprogrammes-maintaining-treatment-access-post-trial .
- 9.EFPIA Patients W.A.I.T Indicator-2010 Report-based on EFPIA's Database (first EU Marketing Authorization in the Period 2007-09), EFPIA-MAD WG. 22.09.10-28.11.10 Final. [Last accessed on 2015 Apr 30]. Available from: http://www.efpia.eu/documents/22/48/Patients-39-W-A-I-T-Indicator-Report-2010 .
- 10.Ways to Optimize Patient Access Through the Product Life Cycle with Managed Access Programs – A Practical Guide by Idis. [Last accessed on 2015 Apr 30]. Available from: http://www.digitaleditions.pmlive.com//launch.aspx?eid=3eb4e601-0158-4f8e-bb17-14a57a8d5fc9 .
- 11. [Last accessed on 2015 Apr 30]. Available from: http://www.pmlive.com/pharma_intelligenceorphan_drugs_the_early_access_regulatory_environment_ 511024 .
- 12.Simon E. Managing access to the drugs prior to approval and launch. Life Science Leader May 2010. [Last accessed on 30 April 2015]. Available from: http://www.idispharma.com/sites/default/files/uploads/Life-Science-Leader.pdf .
- 13.Guidance for Industry Expanded Access to Investigational Drugs for Treatment Use-Qs and As: U. S. Dept. of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER)-May, 2013 Procedural [Google Scholar]
- 14.European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP): Guideline on Compassionate Use of Medicinal Products, Pursuant to Article 83 of Regulation (EC) no 726/2004. European Medicines Agency. 2007. [Last accessed on 2015 Apr 30]. Available from: http://www. ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004075.pdf .
- 15.London, UK: European Medicines Agency; 2010. European Medicines Agency. Questions and Answers on the Compassionate use of Medicines in the European Union. EMEA/72144/2006 (rev) [Google Scholar]
- 16.MHRA-Early Access to Medicines Scheme (EAMS) [Last accessed 2015 Apr 30]. Available from: http://www.mhra.gov.uk/Howweregulate/Innovation/EarlyaccesstomedicinesschemeEAMS/index.htm .
- 17.Central Drugs Standard Control Organization. Procedure for permission to Import Small Quantities of Drugs for Personal Use. [Last accessed 2015 Apr 30]. Available from: http://www.cdsco.nic.in/forms/list.aspx?lid=1852and Id=30 .
- 18.Early Access Programs in Europe: A Regulatory Tool with Pre-marketing Impact-the Pharma Letter; 29 September. 2008. [Last accessed 2015 Apr 30]. Available from: http://www.thepharmaletter.com/article/early-access- programs-in-europe-a-regulatory-tool-with-pre-marketing-impact .
- 19.Andree B. Marketing briefs: Implementing a pre-launch named patient program: Evidence of increased market share. J Med Mark. 2008;8:319–324. [Google Scholar]
- 20. [Last accessed on 2015 Jun 15]. Available from: http://www.pharmaphorum.com/articles/earlyaccess-the-ground-rules-for-communicating-with-physiciansand-patient .
- 21.Steve U, Washington Editor, Viral Crossroads: Hardy Case Shows Flaws in Compassionate Use System, Provides Catalyst for Change. [Published on 2014 Mar 31]. Available from: http://www. biocentury.com/biotech-pharma-news/coverstory/2014-03-31/hardy-case-shows-flaws-in-compassionateuse-system-providescatalyst-for-change-a1 .
- 22.Rules and Regulations. Federal Register. Vol. 74. Thursday, August 13. 2009. [Last accessed 2015 Jun 15]. Available from: http://www.gpo.gov /fdsys/pkg/FR-2009-08-13/pdf/E9-19005.pdf .
- 23. [Last accessed 2015 Jun 15]. Available from: http://www.idispharma.com/sites/default/filesuploads/Pharmaceutical%20Executive_November%202012%20Reprint.pdf .