

Abstract
AIMS
Ventricular arrhythmias are a common cause of death in patients with heart failure (HF). Structural and electrical abnormalities in the heart provide a substrate for such arrhythmias. Canine tachypacing-induced HF models of 4–6 weeks duration are often used to study pathophysiology and therapies for HF. We hypothesized that a chronic canine model of HF would result in greater electrical and structural remodeling than a short term model, leading to a more arrhythmogenic substrate.
MAIN METHODS
HF was induced by ventricular tachypacing for one (short-term) or four (chronic) months to study remodeling.
KEY FINDINGS
Left ventricular contractility was progressively reduced, while ventricular hypertrophy and interstitial fibrosis were evident at 4 month but not 1 month of HF. Left ventricular myocyte action potentials were prolonged after 4 (p<0.05) but not 1 month of HF. Repolarization instability and early afterdepolarizations were evident only after 4 months of HF (p<0.05), coinciding with a prolonged QTc interval (p<0.05). The transient outward potassium current was reduced in both HF groups (p<0.05). The outward component of the inward rectifier potassium current was reduced only in the 4 month HF group (p<0.05). The delayed rectifier potassium currents were reduced in 4 (p<0.05) but not 1 month of HF. Reactive oxygen species were increased at both 1 and 4 months of HF (p<0.05).
SIGNIFICANCE
Reduced Ito, outward IK1, IKs, and IKr in HF contribute to EAD formation. Chronic, but not short term canine HF, results in the altered electrophysiology and repolarization instability characteristic of end-stage human HF.
Keywords: heart failure, electrophysiology, fibrosis, hypertrophy
Introduction
Heart failure (HF) is a leading contributor to morbidity and mortality. In 2010, the number of deaths in the US attributed to HF was ~279,000, and HF was noted in 1 of 9 death certificates (Go et al. 2014). Sudden death due to lethal ventricular arrhythmias is six- to ninefold higher in HF patients than in the general population (Tomaselli and Zipes 2004; Thom et al. 2006), and accounts for up to 50% of deaths in HF patients(Tomaselli and Zipes 2004).
HF is associated with both structural and electrical remodeling that transforms the normal myocardium into a substrate susceptible to arrhythmogenesis. Left ventricular dilation, hypertrophy, and fibrosis are all examples of compensatory changes that are initially adaptive in the failing heart (Cohn et al. 2000). These changes may progress to become maladaptive resulting in further deterioration of heart function, and have been linked to the development of arrhythmia and/or sudden death (Hsia and Marchlinski 2002; Iles et al. 2011;Haider et al. 1998). At the cellular level, cardiomyocytes of the failing heart display electrical remodeling, including a signature prolongation of the action potential (AP) (Beuckelmann et al. 1995;Akar and Rosenbaum 2003).
We previously reported that chronic (four or more months) canine tachypacing HF becomes irreversible and emulates multiple aspects of chronic human HF (Nishijima et al. 2005). In the present study, we tested the hypothesis that dogs paced into chronic HF (4 M HF) would demonstrate greater structural and electrophysiological remodeling than dogs paced into acute HF (1 M HF), providing a substrate for ventricular arrhythmias. We used serial echocardiography and electrocardiograms to assess ventricular function and electrical activity, respectively. Patch clamp recordings were used to measure action potentials and K+ currents in ventricular myocytes. Real-time polymerase chain reaction (RT-PCR) was used to quantify ion channel subunit mRNA.
Materials and Methods
Heart failure canine model
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Ohio State University. A total of 63 adult mixed breed dogs of either sex (2–5 years of age) weighing between 8 and 20 kg with normal cardiac function were used. Dogs were verified to have normal cardiac function by routine electrocardiograms and echocardiographic examinations during butorphanol tartrate (0.5 mg kg −1 intramuscularly) sedation. Dogs had a RV pacemaker lead implanted in the RV apex, and HF was induced (n=16) by tachypacing for four months as previously described (Sridhar et al. 2009). To assess time dependence during the progression of HF, echocardiograms and electrocardiogram were measured at baseline, after 1 month, and 4 months of pacing in the 4 M HF group. A second group of dogs was RV tachypaced for 1 month at 180 bpm (n=17) and echocardiograms were measured at baseline and at the end of the pacing protocol as previously reported. (Nishijima et al. 2007; Nishijima et al. 2005). An age matched group of 30 healthy dogs were used as controls and studied in parallel. Transmural samples of left ventricular tissue were formalin fixed and embedded in paraffin and sectioned to 5 μm thickness, using standard procedures. Tissue sections were stained with Masson’s Trichrome to define the percentage area of fibrosis, as previously described (Nishijima et al. 2007)
Myocyte Isolation
On the day of the terminal procedure, the dogs were anesthetized with pentobarbital sodium (50 mg/kg IV). The heart was rapidly removed and perfused with cold cardioplegia solution containing the following in mM: NaCl 110, CaCl2 1.2, KCl 16, MgCl2 16 and NaHCO3 10. Cannulation of the left circumflex artery was used to perfuse the left ventricle, as previously described. (Sridhar et al. 2009; Bonilla et al. 2013a) Adjacent tissue samples were collected and snap frozen for protein analyses. Tyrode’s solution (mM) containing NaCl 130, KCl 5.4, MgCl2 3.5, NaH2PO4 0.5, Glucose 10, HEPES 5 and taurine 20, was used as the initial perfusate. During the cell isolation process the heart was perfused with three different solutions (36°C). First the heart was perfused for 10 minutes with Tyrode’s solution with 0.1 mM EGTA; followed by perfusion with Tyrode’s solution containing 0.3 mM Calcium, 0.12 mg/ml of Trypsin Inhibitor (NIBCO) and 1.33mg/ml of collagenase (Type II, Worthington), for a maximum of 45 minutes. Following enzymatic digestion, the heart was perfused with normal Tyrode’s solution for five minutes to remove residual enzyme. After digestion, the cells were resuspended in incubation buffer. This isolation procedure typically yields 40–60% rod shaped ventricular myocytes. All myocyte electrophysiology experiments were conducted within 10 hours of isolation.
Electrophysiological recordings
To assess myocyte electrophysiology, Amphotericin-B perforated patch clamp techniques with a bath temperature of 36 ± 0.5°C were used. The myocytes were placed in a laminin coated cell chamber (Cell Microcontrols, Norfolk, VA) and superfused with bath solution containing (in mM): 135 NaCl, 5 MgCl2, 5 KCl, 10 glucose, 1.8 CaCl2, and 5 HEPES with pH adjusted to 7.40 with NaOH. For current measurements the calcium in the bath solution was reduced to 1.0mM in addition, 2 μM nifidepine was added to the bath solution to avoid contamination with L-type Ca2+ current. Borosilicate glass micropipettes with tip resistance of 1.5–4.5 MΩ, were filled with pipette solution containing the following (in mM): 100 K-aspartate, 40 KCl, 5 MgCl, 5 EGTA, 5 HEPES, pH adjusted to 7.2 with KOH.
Action potentials (AP) were recorded in a train of 25 traces at 0.5, 1 and 2 Hz. The average of the last 10 traces (i.e. from trace 16–25) was used to calculate the action potential duration (APD). APD was calculated at 50 and 90 percent of repolarization (APD50 and APD90). The standard deviation of the APD90 for the last 10 traces not exhibiting EADs was used to evaluate repolarization variability (Thomsen et al. 2004). EADs were defined as positive oscillations occurring in Phase 2 or Phase 3 of the APD.
For current recordings, only recordings with an access resistance <20 MΩ were included in the analyses. Transient outward potassium current (Ito), potassium inward rectifying current (IK1), and delayed rectifying currents (IKr + IKs) were elicited as previously described. (Sridhar et al. 2008; Bonilla et al. 2013b) To assess steady state inactivation kinetics of Ito, a series of 500 ms prepulses were clamped to voltages between −80 mV and +10 mV (holding potential of −60 mV) followed by a 300 ms step to +50 mV (peak current) (Nabauer et al. 1993). Ito elicted during each prepulse was normalized to peak current and plotted against the respective prepulse potential. The AUC for the window current was defined as the area beneath the intersection of the normalized Ito activation and inactivation curves, in the voltage range between −50 to +10 mV. To investigate Ito recovery from inactivation, a two-step protocol was used in which two 200 ms pulses from a holding potential of −60 mV to + 40 mV were separated by a variable interpulse interval of 20 to 1000 ms (Nabauer et al. 1993). Rectification ratio was calculated as previously described (Carnes and Dech 2002; Lopatin et al. 2000).
Data was collected with a low noise data acquisition system Digidata 1440A (Molecular devices, Sunnyvale, CA), Clampex software and an Axopatch 200A amplifier (Axon Instruments, Sunnyvale, CA).
Immunoblots
Following protein quantification, tissue lysates were analyzed on Mini-PROTEAN tetra cell (BioRad) on a 4–15% precast TGX gel (BioRad) in Tris/Glycine/SDS Buffer (BioRad). Gels were transferred to a nitrocellulose membrane using the Mini-PROTEAN tetra cell (BioRad) in Tris/Glycine buffer with 20% methanol (v/v, BioRad). Membranes were blocked for 1 hour at room temperature using a 3% BSA solution and incubated with primary antibody overnight at 4°C. Antibodies were KChiP2 (Alomone, Santa Cruz), Kv4.3 (Covance), and GAPDH (Fitzgerald). Donkey anti-rabbit-HRP (Jackson Laboratories) was used as the secondary antibody. Densitometry was performed using Image lab software and all data was normalized to GAPDH levels present in each sample.
Real-time PCR for gene expression
Total RNA was extracted from the cardiac tissues with Trizol reagent (Invitrogen), and 2 μg RNA was then converted to cDNA by using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression was quantified by real-time RT-PCR (Light Cycler 96, Roche Applied Science) using SYBR green assay reagent and gene-specific primer listed in Table1. Relative amplification was quantified by normalizing the gene-specific amplification to that of 18s rRNA in each sample. Changes in mRNA abundance were calculated using 2(−ΔΔCT) method (Livak and Schmittgen 2001;Kumar et al. 2013). qPCR reactions were run in triplicates. Primers used were as follows (5′–3′): Kv11.1(forward: CCTGCTGCTGGTCATCTACA; reverse: TCCTCGTTGGCATTGACATA), KCNE2 (forward: GAACACGACAGCTGAGCAAG; reverse: ACTGGTGGTAGGGGTCATTG), KChiP2 (forward: GCTGGTTTGTCGGTGATTCT; reverse: AAGAAGCTCTCCACGTGCTC), KCNQ1 (forward: CTTTACCTGCCAGGGGTACA; reverse: ACCACATACTCCGTCCCAAA), DPP6 (forward: CCCATCGAGTGTCAGCACTA; reverse: GATGGATCGGTACAGGTGCT), Kv4.3 (forward: GTTTGAGCAGAACTGCATGG; reverse: GTGGATGGTGCTGAGCTCTT), KCNJ2 (forward: TATCAACGTTGGGTTCGACA; reverse: AAATCAGTTATGGTTCCTTTGGT); 18s (forward: GCTCTAGAATTACCACAGTTATC; reverse: AAATCAGTTATGGTTCCTTTGGT)
TABLE 1.
Echocardiographic and Electrocardiogram parameters in 4 M HF animals
| Baseline | 1 Month HF | 4 Month HF | |
|---|---|---|---|
| Fractional shortening (%) | 29.6 ± 1.49 | 15.2 ± 0.97* | 11.2 ± 0.84*, # |
| Left Ventricle Dimension (cm) | |||
| Diastole | 3.46 ± 0.13 | 4.33 ± 0.15* | 5.28 ± 0.21*, # |
| Systole | 2.41 ± 0.07 | 3.67 ± 0.12* | 4.68 ± 0.18*, # |
| Left Ventricle mass (g) | 91.6 ± 9.15 | 121.2 ± 11.0 | 157.7 ± 16.1*, # |
| ECG parameters | |||
| PR (ms) | 110.7 ± 5.24 | 108.1 ± 5.01 | 107.9 ± 3.85 |
| QRS (ms) | 45.8 ± 2.47 | 50.1 ± 3.03 | 54.2 ± 4.10* |
| RR (ms) | 578.4 ± 35.4 | 520.2 ± 53.3 | 511.2 ± 69.4 |
| QT (ms) | 201.1 ± 6.10 | 199.6 ± 7.40 | 198.9 ± 8.38 |
| QTcf (ms) | 242.5 ± 3.35 | 250.1 ± 4.59 | 252.4 ± 4.33* |
N=7–10 per observation;
p<0.05 vs baseline;
p<0.05 vs 1 M HF. Values are means ± SE.
Electron paramagnetic resonance (EPR) spectroscopy
The ventricular tissue samples were flash frozen and stored in liquid nitrogen prior to electron paramagnetic resonance (EPR) analysis to measure radical and paramagnetic species. Both semi-quinone radical and Fe-S centers were quantified using previously described methods (Nishijima et al. 2011; Zweier et al. 1989; Zweier et al. 1987) Each EPR sample was prepared by transferring the frozen heart tissue (235–570 mg) into a ceramic mortar pre-chilled with liquid nitrogen. The tissue was then crushed in liquid nitrogen using a pestle. The tissue in liquid N2 was then loaded into a finger Dewar containing liquid nitrogen. Low temperature, 77 K, EPR spectra were recorded with a Bruker ESP 300E spectrometer (Bruker BioSciences, Billerica, MA, USA) operating at X-band with 100 KHz modulation frequency and a TM110 cavity as described previously (Zweier et al. 1995). The finger Dewar containing heart tissue samples in liquid nitrogen was placed within the EPR spectrometer cavity. All spectra were recorded with the following parameters: receiver gain = 1 × 105, modulation amplitude = 2 G (4 G for Fe-S signals), time constant = 164 ms, scan time = 60 s, microwave power = 1 mW (20 mW for Fe-S signals), and number of scans = 10.
Data Analysis
Cellular electrophysiology data were analyzed using Clampfit 10.3 software (Axon Instruments) and Origin 9.0 software (OriginLab, Northampton, MA, USA). One way repeated measured ANOVA was used to analyze differences within groups, while comparison between groups was analyzed by one-way ANOVA with post hoc least significant difference testing or Students t-test (OriginPro 8.6, OriginLab). Normality was tested via Kolmogorov-Smirnov. Non parametric analysis was performed using Chi-Square. All data are presented as mean ± SE (or SD for PCR quantification) and p<0.05 was the criterion for statistical significance for all comparisons.
Chemicals
All chemicals used were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Fisher Scientific (Pittsburgh, PA, USA), unless otherwise noted. All buffers and solutions were prepared daily.
Results
Chronic tachypacing is accompanied by an increase in left ventricular mass, a prolonged QTc interval, and interstitial fibrosis
RV tachypacing resulted in significantly impaired contractility, LV chamber dilation and increased LV mass at 4 M HF compared to 1 month HF and baseline (TABLE 1). The corrected QT interval was significantly prolonged at 4 M HF versus baseline.
Ventricular tissue samples (control, 1 M HF, and 4 M HF) had significantly increased fibrosis in the 4 M HF group (p<0.05 vs control) (Figure 1). Collectively, these results suggest that time-dependent decreases in cardiac function with continued RV tachypacing are associated with the development of structural remodeling (cardiac hypertrophy and fibrosis) in chronic (4 M) HF.
Figure 1. Interstitial Fibrosis is Increased in Chronic HF.
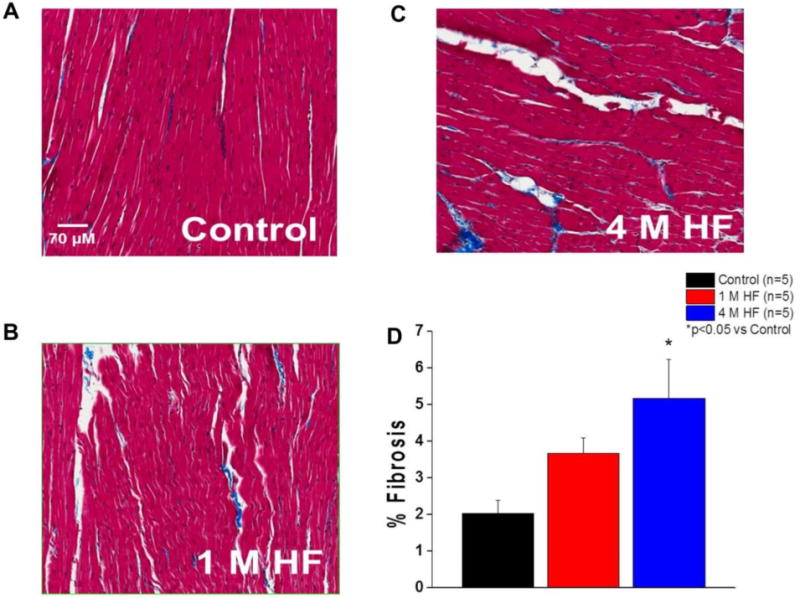
Representative Masson’s Trichrome staining of LV tissue. A. Control. B. 1 M HF. C. 4 M HF. D. Summary data (*p<0.05 vs control).
Chronic tachypacing results in a prolonged AP associated with early afterdepolarizations, increased myocyte size, and downregulation of repolarizing K+ currents
Heart failure prolonged ventricular cardiomyocyte action potentials (Figure 2). In addition to AP prolongation, a disappearance of the prominent phase one “notch” occurred with HF. APD50 was significantly prolonged at all rates tested (0.5, 1, and 2 Hz) in both 1 M HF and 4 M HF groups compared with control myocytes (p<0.05) (Figure 2B). APD90 was significantly increased in 4 M HF compared to both baseline and 1 M HF at all rates (p<0.05) (Figure 2C). Rate-adaptation of the APD was maintained in each group (p<0.05). The beat-to-beat variability of APD90, a marker of proarrhythmic potential (Oosterhoff et al. 2007) (Figure 2D) was significantly increased in the 4 M HF group at all rates versus control (p<0.05), and at 1 Hz versus 1 M HF (p<0.05). Consistent with these findings, EADs were significantly more frequent in the 4 M HF group compared to both control and 1 M HF (p<0.05) (Figure 2E). No change in the resting membrane potential was found between the groups. Collectively, this data suggests that HF duration mediates action potential prolongation and the development of a proarrhythmic ventricular substrate.
Figure 2. Progressive action potential prolongation and cellular arrhythmias during heart failure.
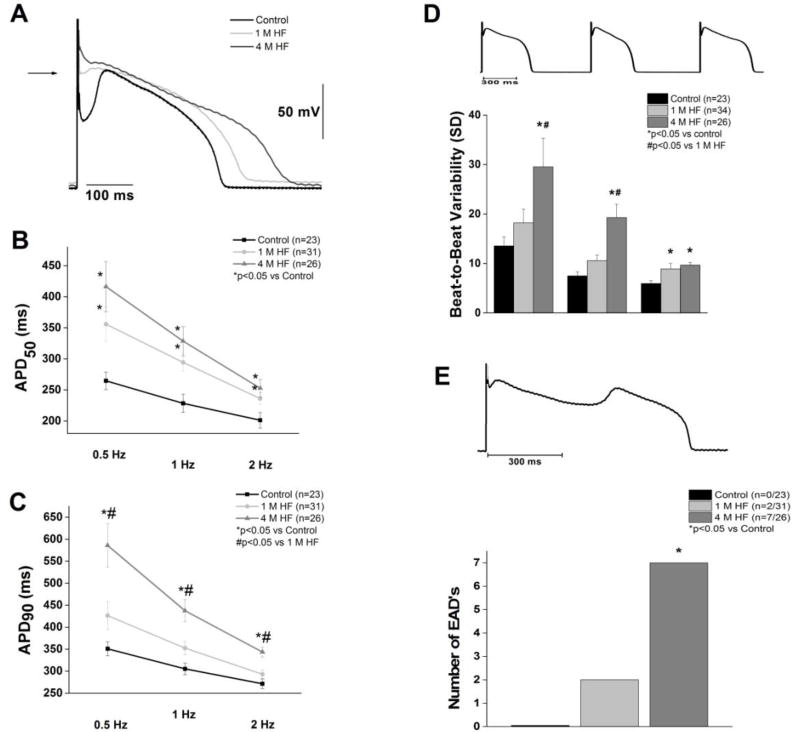
A. Representative action potentials at 1 Hz. B. APD50 is prolonged at 1 and 4 months of HF (p<0.05). C. APD90 is significantly prolonged in 4 M HF compared to 1 M HF and control (p<0.05). D. Beat-to-beat variability (representative, top) was significantly increased in 4 M HF (*p<0.05 vs. control, #p<0.05 vs. 1M HF). E. Early afterdepolarizations (representative, top) were more frequent in 4 M HF (*p<0.05 vs control).
Membrane capacitance was used as a measure of ventricular cell hypertrophy. Consistent with the LV mass findings, 4 M HF ventricular myocytes had a significantly larger capacitance (239.5 ± 16.6, n=23, p<0.05) compared to controls (159.0 ± 6.6 pF, (n=52) and 1 M HF (163.7 ± 9.4 pF, n=29)
Transient outward current (Ito) density and the corresponding slope conductance was significantly decreased in both 1 M and 4 M HF groups (p<0.05 vs Control) (Figure 3B and 3C). Inactivation of Ito was best fitted as the sum of two exponentials, a rapidly inactivating Ito,fast (τ1) and a slowly inactivating Ito,slow (τ1). There was no significant difference in either time constant between groups. Mean τ1 and τ2 at +50 mV was 18.4 ± 1.9 ms and 87.3 ± 21.1 ms, 11.3 ± 0.80 ms and 43.1 ± 10.2 ms, and 15.8 ± 1.5 ms and 68.9 ± 21.5 ms for control (n=23), 1 M HF (n=5), and 4 M HF (n=10), respectively. The rapidly inactivating component comprised 80.6 ± 2.6% of the decay current in the control group, 73.7 ± 6.2% in 1 M HF, and 74.8± 7.1% in 4 M HF (p=NS). A plot of the steady state inactivation of Ito against current activation revealed no voltage shift in kinetics (Figure 4A). However, there was a decrease in the overall current available (AUC) in both 1 and 4 M HF compared to controls (AUC values were reduced by 97% for 1 M HF and 63% for 4 M HF). This “window” current peaked around −35 mV, representing approximately 5% of peak Ito (Figure 4B). The time for Ito recovery from activation was assessed using a two-step protocol (Figure 4C). The time for 50% recovery from inactivation was prolonged in the 4 M HF group vs. controls (185.5±16.9 ms vs. 107.0±17.3 ms, p<0.05).
Figure 3. HF decreases Ito.
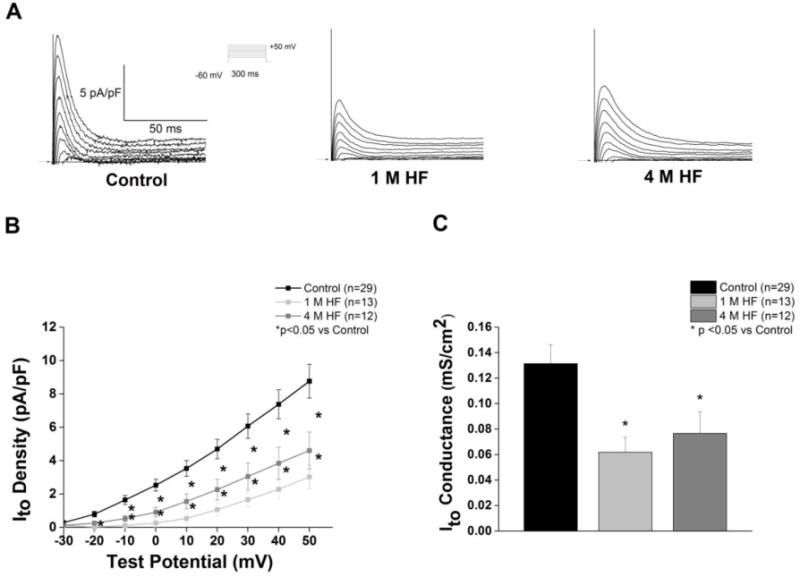
A. Representative Ito current tracings from each group; voltage protocol shown in the inset. B. I–V curves (*p<0.05 vs. control) C. Ito slope conductance is decreased in HF (*p<0.05).
Figure 4. Ito kinetics are altered in chronic HF.
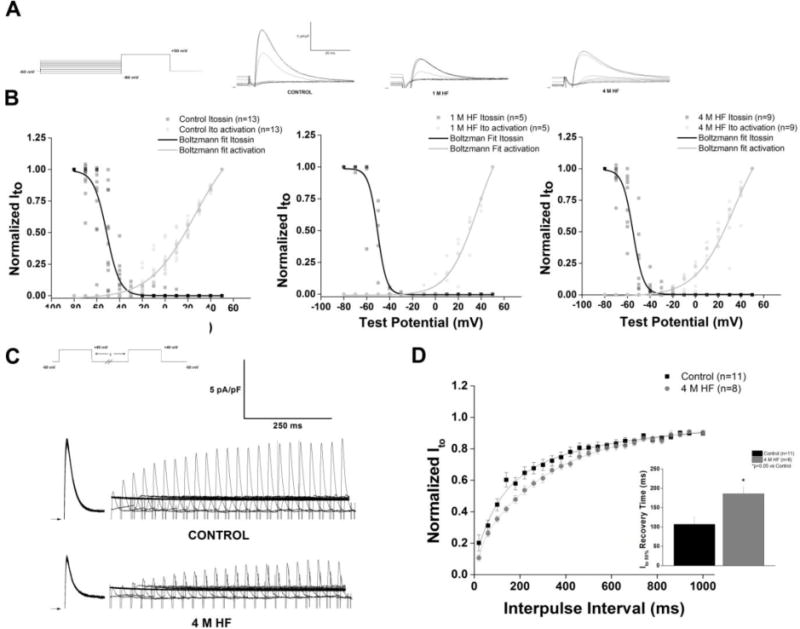
A. Representative steady state inactivation traces of Ito recorded with the voltage protocol displayed in the inset. B. Steady state inactivation and activation curves of Ito fit to Boltzmann functions, demonstrates a “window” current, that is reduced as the heart fails C. Representative traces elicited by two-step protocol in control and 4 M HF; voltage protocol in inset. D. Summary data of recovery from inactivation; HF significantly prolongs recovery to 50% of total Ito current (p<0.05 vs control).
HF-dependent changes in the inward rectifier current, IK1 are shown in Figure 5. There were no significant differences in inward slope conductance between groups. Peak IK1 outward current was significantly decreased in 4 M HF compared to controls (p<0.05, Figure 5C). The rectification ratio was significantly increased in 4 M HF (p<0.05 vs control, Figure 5D).
Figure 5. Outward IK1, but not inward IK1, is reduced in chronic but not short-term HF.
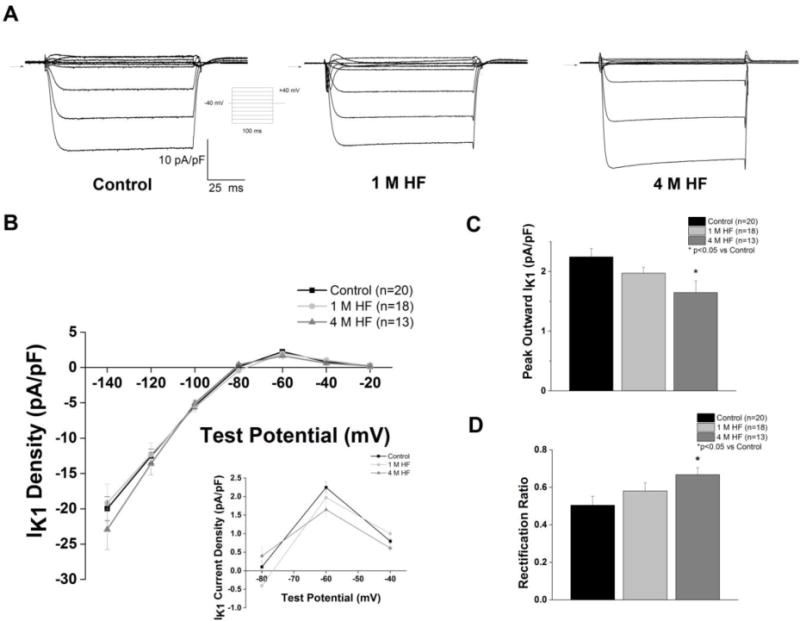
A. Representative IK1 current tracing, voltage protocol displayed in the inset. B. I–V curves; inset shows expanded I–V curve of outward IK1 C. Peak outward IK1 is significantly reduced in 4 M HF vs. control (p<0.05). D. Rectification ratio is significantly increased in 4 M HF vs control (p<0.05).
HF-induced alterations in the delayed rectifier currents, IKr and IKs are shown in Figure 6. IKs amplitude was significantly decreased in 4 M HF at all test potentials compared to both 1 M HF and controls (Figure 6B), and IKs slope conductance was decreased in 4 M HF (p<0.05 vs control) (Figure 6C). IKr was significantly decreased in 4 M HF compared to 1 M HF and control (p<0.05) (Figure 6D).
Figure 6. IKs and IKr are reduced in chronic HF.
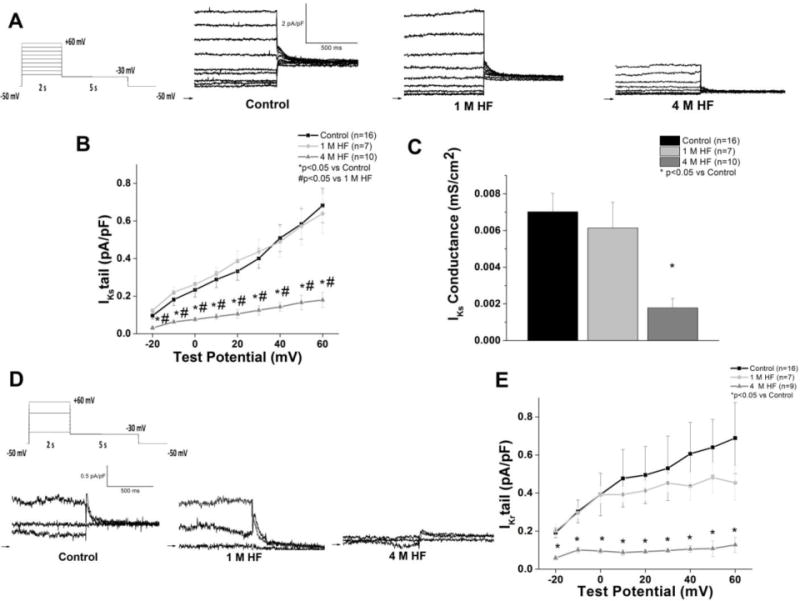
A. Representative IKs tail currents (defined as the sotalol-insensitive current) from each group; inset: voltage protocol B. I–V curves (*p<0.05 vs. control; #p<0.05 vs. 1 M HF) C. IKs slope conductance is reduced in 4 M HF (p<0.05 vs control). D. Representative IKr tail currents (defined as the sotalol-sensitive current) recorded with the same voltage protocol in (A). For ease of viewing, only traces recorded −20 mV, +30 mV, and +60 mV are depicted. E. IKr I–V curves (*p<0.05 vs control).
Protein and mRNA expression of K Channel Subunits does not correlate with function
Ito in the canine ventricle is conducted through channel pore-forming subunits Kv4.3 and Kv1.4, with Kv4.3 being the main alpha subunit contributing to the current (Akar et al. 2004a) Kv4.3 can assemble with multiple accessory proteins including K+ channel interacting protein 2 (KChiP2) which can modulate Kv4.3 gating and function (Deschenes et al. 2002). Dipeptidyl-aminopeptidase-like protein 6 (DPP6) also coassembles with Kv4.3, and has been shown to alter Ito kinetics (Radicke et al. 2005). Biochemical analysis showed no significant difference between protein expression of Kv4.3 and KChiP2 in the control, 1 M HF, and 4 M HF group (Figure 7A and 7B). In contrast, notable HF-dependent changes were observed in transmural gene expression of K+ channel subunits (Figure 7C). The mRNA of the Kv4.3 and Kv1.4 subunits demonstrated no change at 1 M HF, but increased gene expression at 4 M HF (p<0.05 vs control and 1 M HF). Similarly the mRNA levels for KChiP2 and DPP6 increased at 4 M HF. KCNJ2 encodes the Kir2.1 channel, the pore-forming subunit of IK1. At 1 M HF, KCNJ2 gene expression was significantly elevated (p<0.05 vs control), and continued to increase at 4 M HF (p<0.05 vs control, p<0.05 vs 1 M HF). IKs is carried via a complex which includes the pore-forming KvLTQ1 (KCNQ1) and minK (KCNE1). Both KCNQ1 and KCNE1 mRNA are significantly upregulated at 4 M HF compared to both control (p<0.05) and 1 M HF (p<0.05). IKr is carried through Kv11.1 channels, and no differences in Kv11.1 mRNA were found between the three groups.
Figure 7. Protein expression and mRNA levels of K+ channel subunits.
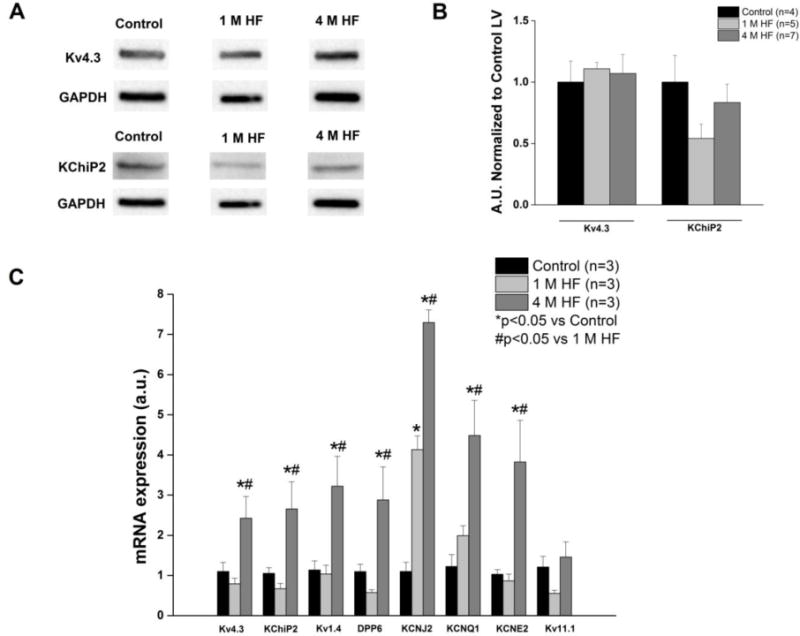
A. Representative Western Blots of Ito subunits Kv4.3 and KChiP2. B. Summary data of Kv4.3 and KChIP2 protein expression (p=NS) C. mRNA for K+ channel subunits. (*p<0.05 vs. control; #p<0.05 vs. 1 M HF).
EPR spectroscopy
EPR spectroscopy revealed a modest increase in semiquinone radicals that did not reach significance in left ventricular tissue among either of the HF groups compared to control (P=NS) (Figure 8B). However, Fe-S center signals were significantly increased in both 1 M HF and 4 M HF (p<0.05 vs control) (Figure 8C).
Figure 8. Heart failure increases ventricular oxidative stress.
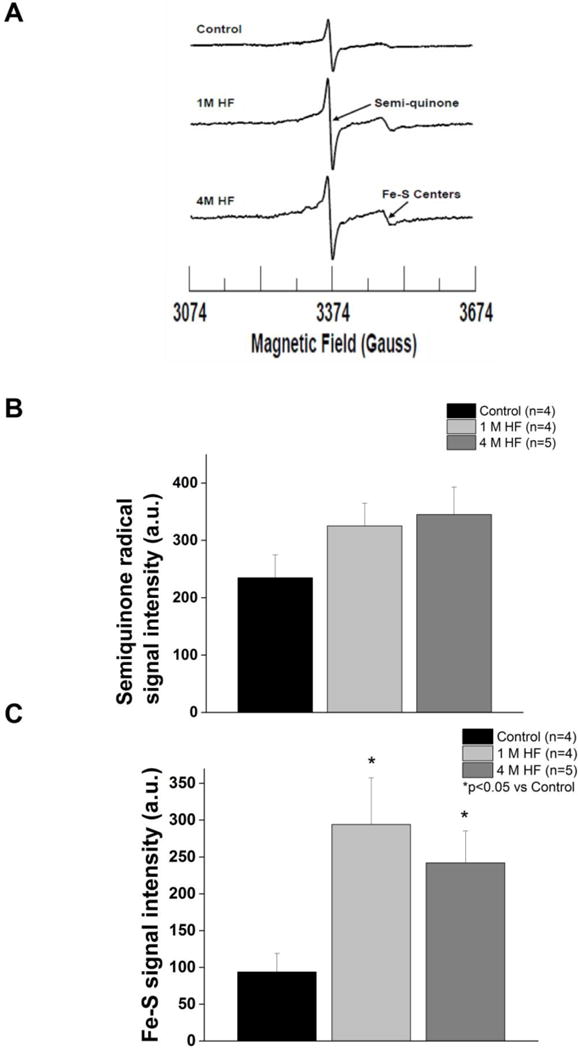
A. Representative EPR spectra of tissue homogenates measured at 77 K in control, 1 M HF, and 4 M HF. B. Summary data of semi-quinone radical centeres. (C) Summary data of Fe-S centers. (p< 0.05 vs. control).
Discussion
Large animal models of dilated cardiomyopathy are accepted as a surrogate of the pathology of human HF (Houser et al. 2012). In the present study, we found that the 4 month duration of HF overcomes a limitation of the short-term canine tachypacing model - the lack of structural remodeling. We observed an increase in LV mass at 4 M HF (≈1.7 fold), coinciding with an increase in myocyte membrane capacitance (≈1.5 fold) – both indicators of hypertrophy. iThese results are consistent with a previous report of increased LV mass after 7 to 10 months of chronic RV pacing (Nishijima et al. 2005). We also report an increase in interstitial fibrosis at 4 M HF. The appearance of increased fibrosis in 4 to 6 week canine HF models varies between studies (Burashnikov et al. 2014; Akar et al. 2004b; Hanna et al. 2004). A key difference between the current model and previous reports is that our chronic pacing rates are not as rapid (180 bpm vs 240 bpm) resulting in a sustained hypertrophic response not seen in more rapid, short-term pacing models. This notion is supported by our previous work, where we observed increases in LV mass after 10 months of pacing, 6 months of which was maintained at 160 bpm (Nishijima et al. 2005). It is well known that hypertrophy induces the structural remodeling of the collagen matrix necessary for fibrosis. (Weber et al. 1988). The lack of fibrosis in rapid pacing models may be responsible for their “reversible” nature.
A hallmark of HF is downregulation of repolarizing K+ currents and subsequent AP prolongation. The most studied and consistently downregulated K+ current in both canine HF models and human HF is Ito (Akar et al. 2004a; Beuckelmann et al. 1993; Li et al. 2002), and we found reductions in both 1 M HF and 4 M HF groups. The role of Ito in early repolarization is evident as the loss of the prominent phase one “notch” during HF. Ito may contribute to the prolonged APD50 in both HF groups, as altered Ito can alter ICa (Sah et al. 2002). The role of Ito in late repolarization (APD90) is less clear. The peak of the “window current” we observed is in agreement with findings from other groups (Virag et al., 2011), and suggests Ito may contribute repolarizing current during the plateau of the AP.
The molecular mechanisms for downregulation of Ito in HF are still not fully elucidated. In both short-term canine tachypacing models and limited human HF studies, reductions in Kv4.3 protein have been reported (Akar et al. 2005; Zicha et al. 2004). The relatively consistent finding of reduced Kv4.3 mRNA in canine HF models is a plausible mechanism for decreased Ito (Akar et al. 2005; Zicha et al. 2004; Kaab et al. 1998). However, gene expression studies in human HF are conflicting, demonstrating decreases (Borlak and Thum 2003) or increases (Soltysinska et al. 2009) in Kv4.3 mRNA. Here, we report an increase in Kv4.3 mRNA at 4 M HF with no change in its protein expression. Consistent with our findings, other animal models of tachypacing-induced HF have reported no change in total Kv4.3 mRNA or protein, yet have also found reduced Ito (Rose et al. 2005). One potential explanation for the reduced current could a change in the accessory subunit, KChiP2. However, we found no change in KChIP2 protein expression despite a significant increase in KChiP2 mRNA at 4 M HF. This finding is contradictory to reports of KChIP2 mRNA being downregulated in failing human hearts (Radicke et al. 2006; Soltysinska et al. 2009)
Interestingly, we report a prolonged time of Ito recovery from inactivation in 4 M HF compared to controls. Such results were not seen in other canine HF studies using midmyocardial cells (Kaab et al., 1996), but were reported in epicardial cells (Cordeiro et al., 2012). DPP6, an ancillary subunit of Kv4.3, has been shown to slow recovery from inactivation in CHO cells when expressed with Kv4.3 (Radicke et al. 2005). Here, we report that gene expression of DPP6 is increased in 4 M HF. More studies will be needed to determine the precise role of DPP6 in modulating cardiac Ito, as well as whether Ito inactivation contributes to APD90 prolongation at faster heart rates (cycle length<500 ms) due to the slow recovery from inactivation.
Outward IK1 is a modulator of terminal repolarization (Lopatin and Nichols 2001). We observed unchanged inward current with a decrease in the peak outward current (−60 mV) reflecting altered IK1 rectification (Lopatin et al. 2000; Carnes and Dech 2002). Because rectification factors, such as polyamines and intracellular magnesium, are involved in the normal gating of IK1 (Lopatin et al. 1994), our data suggests a possible role for these factors in decreasing the outward portion of this current in HF. Given the role of IK1 in the pathogenesis of arrhythmias (Dhamoon and Jalife 2005), additional study of IK1 in HF will be required to clarify the variable results.
The delayed rectifier currents, IKr and IKs, play a prominent role in phase 2 and phase 3 repolarization (Nerbonne and Kass 2005). Studies in both humans and canines have suggested that IKs reduction only prolongs the APD in the setting of downregulation of other K+ currents (reduced repolarization reserve) (Jost et al. 2013; Roden 1998;Varro et al. 2000). Consistent with a previous 4 to 6 week canine model of HF (Li et al. 2002), we found a reduction in IKs at 4 M HF. IKr is a dominant modulator of ventricular repolarization, with reductions evident as both AP and QT prolongation (Tseng 2001). We found a reduction in IKr in 4 M HF, which does not occur in 4–6 week tachypacing models (Li et al. 2002) or in 1 month HF in the present study. The HF duration-dependent reduction may be explained by hypertrophic modulation of IKr as this was only observed in the 4 M HF group. This is consistent with reduced IKr observed in a proarrhythmic chronic AV nodal block canine model of biventricular hypertrophy (Volders et al. 1999). While not measuring IKr directly, human studies using E-4031 (a specific IKr blocker) have demonstrated reduced response to IKr blockade in failing human hearts compared to controls. This study also found Kv11.1 hERG 1a protein expression was reduced compared to controls(Holzem KM et al. 2011), suggesting reduced functional expression of IKr may be one mechanism contributing to HF-induced IKr downregulation.
Since we found reductions in multiple repolarizing currents, this suggests a role for reduced repolarization reserve (Roden 1998) as the basis of EAD formation. Reduction of IKr, specifically, has been demonstrated to produce repolarization instability, which in itself can predict proarrhythmia (Thomsen et al. 2006). Our study showed that chronic HF results in both AP and QTc prolongation with concurrent increased beat-to-beat variability of cellular repolarization and the appearance of EADs. In normal canine ventricle, combined inhibition of IKr, IKs, and Ito block can induce EADs (Virag et al. 2011), suggesting these reductions as a mechanism for our observed EADs in HF.
A recent report suggests a role for reactive oxygen species (ROS) in the regulation of IKr function (Shu et al., 2013). Increased oxidative stress has been implicated in the pathophysiology of several forms of HF, included non-ischemic dilated cardiomyopathy (Sam et al. 2005). While we found no increases in superoxide anion formation, we had previously demonstrated a generalized increase in ventricular ROS in this 4 M HF model (Terentyev et al. 2008). Nitric oxide synthase-dependent ROS are suggested to have a role in the regulation of several repolarizing K+ currents (Bonilla et al. 2012). In this study, we found increases in Fe-S signal intensity in both 1 M HF and 4 M HF, consistent with increased reduction of these centers that could arise secondary to an increase in nitric oxide (NO) that can block distal electron transport (Zweier et al. 1995;Cleeter et al. 1994). NO has been found to reduce Ito in human cardiomyocytes (Gomez et al. 2008) and therefore the observed HF-induced current decreases may be in part attributable to altered NO signaling.
Limitations
Our model is a model of non-ischemic dilated cardiomyopathy, and does not necessarily reflect all etiologies of HF. We also limited our study to ventricular myocytes from the midmyocardial layer which may not fully reflect all transmural changes. We did not assess in vivo arrhythmias, but have previously reported increased premature ventricular contractions in chronic canine HF (Kubalova et al. 2005). The present study did not evaluate the contribution of other arrhythmogenic mechanisms; previous work in this model has shown that changes in calcium handling attributable in part to posttranslational modifications of ryanodine receptors by ROS occur (Terentyev et al. 2008) and may also contribute to arrhythmogenesis. Our study also did not evaluate other potentially affected ion currents (e.g. late sodium or sodium-calcium exchanger) which may also contribute to arrhythmogenesis (Valdivia et al. 2005; Sipido et al. 2007).
We did not assess protein expression of all K+ subunits. Furthermore, differences in mRNA expression also exist between this model and previous reports (Akar et al. 2005; Zicha et al. 2004). The literature on ion channel gene and protein expression in heart failure is highly variable and sometimes contradictory (Nattel et al. 2007; Soltysinska et al. 2009). The variability may reflect the complexity of the system which is affected by modulation downstream of mRNA transcription, such as micro-RNA targeted degradation, post-translational modifications, and/or changes in protein trafficking can determine ion channel function. Therefore, alterations in gene expression may be d ynamic and not always translate to equivalent protein expression.
Conclusion
Electrophysiological modeling during human HF is poorly defined due to the reliance on end-stage HF (explanted hearts from transplant recipients) who are treated with multiple drugs which can elicit their own electrophysiologic effects (Haverkamp et al. 2000)and limited access to true normal controls (Hearse and Sutherland 2000). In this paper, we present a chronic canine HF model which emulates many of the alterations seen in human HF more accurately than other short-term canine tachypacing models. The downregulation of IKr, not seen in other pacing models, along with other K+ currents provides a rational mechanism for EAD formation. We present data suggesting that duration of HF produces progressive electrical remodeling, resulting in proarrhythmic potential at the cellular and organ level. Further studies are warranted to elucidate the relationships between ion channel subunit gene, protein and function during heart failure.
Acknowledgments
The authors thank Jeanne Green, RVT for technical assistance and Vadim V. Fedorov, PhD for helpful discussion of the manuscript. Pacing devices and leads provided as a gift of St. Jude Medical.
Grants:
This work was supported by the National Institutes of Health [HL115580, HL089836 to CAC; HL074045 to SG; HL084583, HL083422 to PJM; HL63744, HL65608, HL38324 to JLZ; CA178443 to NL]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
None
Reference List
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014 Jan 21;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004 Oct 15;95(8):754–63. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, et al. Heart disease and stroke statistics–2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006 Feb 14;113(6):e85–151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000 Mar 1;35(3):569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- Hsia HH, Marchlinski FE. Electrophysiology studies in patients with dilated cardiomyopathies. Card Electrophysiol Rev. 2002 Dec;6(4):472–81. doi: 10.1023/a:1021109130276. [DOI] [PubMed] [Google Scholar]
- Iles L, Pfluger H, Lefkovits L, Butler MJ, Kistler PM, Kaye DM, et al. Myocardial fibrosis predicts appropriate device therapy in patients with implantable cardioverter-defibrillators for primary prevention of sudden cardiac death. J Am Coll Cardiol. 2011 Feb 15;57(7):821–8. doi: 10.1016/j.jacc.2010.06.062. [DOI] [PubMed] [Google Scholar]
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998 Nov;32(5):1454–9. doi: 10.1016/s0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Kruger C, Erdmann E. Altered diastolic [Ca2+]i handling in human ventricular myocytes from patients with terminal heart failure. Am Heart J. 1995 Apr;129(4):684–9. doi: 10.1016/0002-8703(95)90316-x. [DOI] [PubMed] [Google Scholar]
- Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003 Oct 3;93(7):638–45. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- Nishijima Y, Feldman DS, Bonagura JD, Ozkanlar Y, Jenkins PJ, Lacombe VA, et al. Canine nonischemic left ventricular dysfunction: a model of chronic human cardiomyopathy. J Card Fail. 2005 Oct;11(8):638–44. doi: 10.1016/j.cardfail.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Sridhar A, Nishijima Y, Terentyev D, Khan M, Terentyeva R, Hamlin RL, et al. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc Res. 2009 Nov 1;84(2):227–36. doi: 10.1093/cvr/cvp216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima Y, Sridhar A, Viatchenko-Karpinski S, Shaw C, Bonagura JD, Abraham WT, et al. Chronic cardiac resynchronization therapy and reverse ventricular remodeling in a model of nonischemic cardiomyopathy. Life Sci. 2007 Sep 15;81(14):1152–9. doi: 10.1016/j.lfs.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla IM, Sridhar A, Nishijima Y, Gyorke S, Cardounel AJ, Carnes CA. Differential effects of the peroxynitrite donor, SIN-1, on atrial and ventricular myocyte electrophysiology. J Cardiovasc Pharmacol. 2013 Jan 29a;61(5):401–7. doi: 10.1097/FJC.0b013e31828748ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de PG, van OJ, et al. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation. 2004 Oct 19;110(16):2453–9. doi: 10.1161/01.CIR.0000145162.64183.C8. [DOI] [PubMed] [Google Scholar]
- Sridhar A, Nishijima Y, Terentyev D, Terentyeva R, Uelmen R, Kukielka M, et al. Repolarization abnormalities and afterdepolarizations in a canine model of sudden cardiac death. Am J Physiol Regul Integr Comp Physiol. 2008 Nov;295(5):R1463–R1472. doi: 10.1152/ajpregu.90583.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla IM, Vargas-Pinto P, Nishijima Y, Pedraza-Toscano A, Ho HT, Long VP, III, et al. Ibandronate and Ventricular Arrhythmia Risk. J Cardiovasc Electrophysiol. 2013 Nov 20b; doi: 10.1111/jce.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabauer M, Beuckelmann DJ, Erdmann E. Characteristics of transient outward current in human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993 Aug;73(2):386–94. doi: 10.1161/01.res.73.2.386. [DOI] [PubMed] [Google Scholar]
- Carnes CA, Dech SJ. Effects of dihydrotestosterone on cardiac inward rectifier K(+) current. Int J Androl. 2002 Aug;25(4):210–4. doi: 10.1046/j.1365-2605.2002.00349.x. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Shantz LM, Mackintosh CA, Nichols CG, Pegg AE. Modulation of potassium channels in the hearts of transgenic and mutant mice with altered polyamine biosynthesis. J Mol Cell Cardiol. 2000 Nov;32(11):2007–24. doi: 10.1006/jmcc.2000.1232. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001 Dec;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Kumar S, Pan CC, Bloodworth JC, Nixon A, Theuer C, Hoyt DG, et al. Antibody-directed coupling of endoglin and MMP-14 is a key mechanism for endoglin shedding and deregulation of TGF-beta signaling. Oncogene. 2013 Sep 30; doi: 10.1038/onc.2013.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima Y, Sridhar A, Bonilla I, Velayutham M, Khan M, Terentyeva R, et al. Tetrahydrobiopterin depletion and NOS2 uncoupling contribute to heart failure-induced alterations in atrial electrophysiology. Cardiovasc Res. 2011 Jul 1;91(1):71–9. doi: 10.1093/cvr/cvr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier JL, Kuppusamy P, Williams R, Rayburn BK, Smith D, Weisfeldt ML, et al. Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J Biol Chem. 1989 Nov 15;264(32):18890–5. [PubMed] [Google Scholar]
- Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci U S A. 1987 Mar;84(5):1404–7. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier JL, Wang P, Kuppusamy P. Direct measurement of nitric oxide generation in the ischemic heart using electron paramagnetic resonance spectroscopy. J Biol Chem. 1995 Jan 6;270(1):304–7. doi: 10.1074/jbc.270.1.304. [DOI] [PubMed] [Google Scholar]
- Oosterhoff P, Oros A, Vos MA. Beat-to-beat variability of repolarization: a new parameter to determine arrhythmic risk of an individual or identify proarrhythmic drugs. Anadolu Kardiyol Derg. 2007 Jul;7(Suppl 1):73–8. [PubMed] [Google Scholar]
- Akar FG, Wu RC, Deschenes I, Armoundas AA, Piacentino V, III, Houser SR, et al. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: insights into molecular composition of ventricular Ito. Am J Physiol Heart Circ Physiol. 2004 Feba;286(2):H602–H609. doi: 10.1152/ajpheart.00673.2003. [DOI] [PubMed] [Google Scholar]
- Deschenes I, DiSilvestre D, Juang GJ, Wu RC, An WF, Tomaselli GF. Regulation of Kv4.3 current by KChIP2 splice variants: a component of native cardiac I(to)? Circulation. 2002 Jul 23;106(4):423–9. doi: 10.1161/01.cir.0000025417.65658.b6. [DOI] [PubMed] [Google Scholar]
- Radicke S, Cotella D, Graf EM, Ravens U, Wettwer E. Expression and function of dipeptidyl-aminopeptidase-like protein 6 as a putative beta-subunit of human cardiac transient outward current encoded by Kv4.3. J Physiol. 2005 Jun 15;565(Pt 3):751–6. doi: 10.1113/jphysiol.2005.087312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, et al. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012 Jun 22;111(1):131–50. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- Burashnikov A, Di Diego JM, Sicouri S, Doss MX, Sachinidis A, Barajas-Martinez H, et al. A temporal window of vulnerability for development of atrial fibrillation with advancing heart failure. Eur J Heart Fail. 2014 Mar;16(3):271–80. doi: 10.1002/ejhf.28. [DOI] [PubMed] [Google Scholar]
- Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004 Oct 1b;95(7):717–25. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- Hanna N, Cardin S, Leung TK, Nattel S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc Res. 2004 Aug 1;63(2):236–44. doi: 10.1016/j.cardiores.2004.03.026. [DOI] [PubMed] [Google Scholar]
- Weber KT, Janicki JS, Shroff SG, Pick R, Chen RM, Bashey RI. Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ Res. 1988 Apr;62(4):757–65. doi: 10.1161/01.res.62.4.757. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993 Aug;73(2):379–85. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002 Sep;283(3):H1031–H1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- Sah R, Ramirez RJ, Backx PH. Modulation of Ca(2+) release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res. 2002 Feb 8;90(2):165–73. doi: 10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- Akar FG, Wu RC, Juang GJ, Tian Y, Burysek M, Disilvestre D, et al. Molecular mechanisms underlying K+ current downregulation in canine tachycardia-induced heart failure. Am J Physiol Heart Circ Physiol. 2005 Jun;288(6):H2887–H2896. doi: 10.1152/ajpheart.00320.2004. [DOI] [PubMed] [Google Scholar]
- Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, et al. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004 Dec 15;561(Pt 3):735–48. doi: 10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, et al. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998 Oct 6;98(14):1383–93. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- Borlak J, Thum T. Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J. 2003 Sep;17(12):1592–608. doi: 10.1096/fj.02-0889com. [DOI] [PubMed] [Google Scholar]
- Soltysinska E, Olesen SP, Christ T, Wettwer E, Varro A, Grunnet M, et al. Transmural expression of ion channels and transporters in human nondiseased and end-stage failing hearts. Pflugers Arch. 2009 Nov;459(1):11–23. doi: 10.1007/s00424-009-0718-3. [DOI] [PubMed] [Google Scholar]
- Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, et al. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol. 2005 May;288(5):H2077–H2087. doi: 10.1152/ajpheart.00526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radicke S, Cotella D, Graf EM, Banse U, Jost N, Varro A, et al. Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc Res. 2006 Sep 1;71(4):695–703. doi: 10.1016/j.cardiores.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on I(K1) J Mol Cell Cardiol. 2001 Apr;33(4):625–38. doi: 10.1006/jmcc.2001.1344. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994 Nov 24;372(6504):366–9. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Dhamoon AS, Jalife J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm. 2005 Mar;2(3):316–24. doi: 10.1016/j.hrthm.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005 Oct;85(4):1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Jost N, Virag L, Comtois P, Ordog B, Szuts V, Seprenyi G, et al. Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J Physiol. 2013 Sep 1;591(Pt 17):4189–206. doi: 10.1113/jphysiol.2013.261198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998 May;21(5):1029–34. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- Varro A, Balati B, Iost N, Takacs J, Virag L, Lathrop DA, et al. The role of the delayed rectifier component IKs in dog ventricular muscle and Purkinje fibre repolarization. J Physiol. 2000 Feb 15;523(Pt 1):67–81. doi: 10.1111/j.1469-7793.2000.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GN. I(Kr): the hERG channel. J Mol Cell Cardiol. 2001 May;33(5):835–49. doi: 10.1006/jmcc.2000.1317. [DOI] [PubMed] [Google Scholar]
- Volders PG, Sipido KR, Vos MA, Spatjens RL, Leunissen JD, Carmeliet E, et al. Downregulation of delayed rectifier K(+) currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation. 1999 Dec 14;100(24):2455–61. doi: 10.1161/01.cir.100.24.2455. [DOI] [PubMed] [Google Scholar]
- Holzem KM, Glukhov AV, Efimov IR. The role of IKr in transmural repolarization abnormalities in human heart failure. Circulation. 2011;124 Ref Type: Generic. [Google Scholar]
- Thomsen MB, Volders PG, Beekman JD, Matz J, Vos MA. Beat-to-Beat variability of repolarization determines proarrhythmic outcome in dogs susceptible to drug-induced torsades de pointes. J Am Coll Cardiol. 2006 Sep 19;48(6):1268–76. doi: 10.1016/j.jacc.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Sam F, Kerstetter DL, Pimental DR, Mulukutla S, Tabaee A, Bristow MR, et al. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail. 2005 Aug;11(6):473–80. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008 Dec 5;103(12):1466–72. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla IM, Sridhar A, Gyorke S, Cardounel AJ, Carnes CA. Nitric oxide synthases and atrial fibrillation. Front Physiol. 2012;3:105. doi: 10.3389/fphys.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994 May 23;345(1):50–4. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- Gomez R, Nunez L, Vaquero M, Amoros I, Barana A, de PT, et al. Nitric oxide inhibits Kv4.3 and human cardiac transient outward potassium current (Ito1) Cardiovasc Res. 2008 Dec 1;80(3):375–84. doi: 10.1093/cvr/cvn205. [DOI] [PubMed] [Google Scholar]
- Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, et al. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci U S A. 2005 Sep 27;102(39):14104–9. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005 Mar;38(3):475–83. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Bito V, Antoons G, Volders PG, Vos MA. Na/Ca exchange and cardiac ventricular arrhythmias. Ann N Y Acad Sci. 2007 Mar;1099:339–48. doi: 10.1196/annals.1387.066. [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le BS, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007 Apr;87(2):425–56. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, et al. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc Res. 2000 Aug;47(2):219–33. doi: 10.1016/s0008-6363(00)00119-x. [DOI] [PubMed] [Google Scholar]
- Hearse DJ, Sutherland FJ. Experimental models for the study of cardiovascular function and disease. Pharmacol Res. 2000 Jun;41(6):597–603. doi: 10.1006/phrs.1999.0651. [DOI] [PubMed] [Google Scholar]