

Abstract
Realizing the promise of precision medicine in cancer therapy depends on identifying and tracking of cancerous growths in order to maximize treatment options and improve patient outcomes. However, this goal of early detection remains unfulfilled by current clinical imaging techniques that fail to detect diseased lesions, due to their small size and sub-organ localization. With proper probes, optical imaging techniques can overcome this limitation by identifying the molecular phenotype of tumors at both macroscopic and microscopic scales. In this study, we propose the first use of nanophotonic short wave infrared technology to molecularly phenotype small sub-surface lesions for more sensitive detection and improved patient outcomes. To this end, we designed human serum albumin encapsulated rare-earth (RE) nanoparticles (ReANCs)[1, 2] with ligands for targeted lesion imaging. AMD3100, an antagonist to CXCR4 (a chemokine receptor involved in cell motility and a classic marker of cancer metastasis) was adsorbed onto ReANCs to form functionalized ReANCs (fReANCs). Functionalized nanoparticles were able to discriminate and preferentially accumulate in receptor positive lesions when injected intraperitoneally in a subcutaneous tumor model. Additionally, fReANCs, administered intravenously, were able to target sub-tissue tumor micro-lesions, at a maximum depth of 10.5 mm, in a lung metastatic model of breast cancer. Internal lesions identified with fReANCs were 2.25 times smaller than those detected with unfunctionalized ReANCs (p < .01) with the smallest tumor being 18.9 mm3. Thus, we present an integrated nanoprobe detection platform that allows target-specific identification of sub-tissue cancerous lesions.
Keywords: optical contrast agent, sub-tissue imaging, cancer nanoprobes, short-wave infrared imaging, molecular phenotyping
Introduction
Breast cancer, the most common form of cancer among women, is a heterogeneous disease with substantial inter-individual variability in the molecular phenotypic expression.[3.] Despite recent advances in treatment, 40% of breast cancer patients die as a result of distant site metastasis, commonly disseminated from the breast tissue to the patient’s lungs, bones or liver.[4] Early detection of small metastatic populations in these organs is critical to reducing the burden of metastatic disease. Development of sensitive and specific methods for non-invasively identifying these cancerous lesions can aid in determining optimal treatment regimens and improving patient outcomes. Unfortunately, current diagnostic methods lack the sensitivity and specificity to provide actionable biological readouts during cancer therapy.[5, 6] Techniques such as MRI, ultrasound and PET/SPECT, though capable of anatomical imaging and detection of breast tumors, are limited by their high cost, lack of targeted contrast agents (MRI) and use of radionuclides (PET/SPECT). Hence, these techniques are unable to provide molecular information about lesions in a low cost, high-resolution platform, which is pivotal to determining treatment regimen and patient response to therapy.
Optical imaging is a promising technique for high resolution detection that can facilitate molecular classification of disease lesions and avoid the hazard of radionuclides used in PET/SPECT imaging. Novel optical imaging tools have the potential to aid clinicians in detecting small metastatic tumors and tracking pharmacological agents, facilitating selection of appropriate therapeutic agents for nascent lesions and monitoring of molecular responses to therapy. However, current optical imaging modalities are faced with several limitations. One important limitation is the small number of clinically approved contrast agents. An additional limitation of widely used fluorescent organic dyes is their poor photostability and low quantum efficiency,[7–9] and their excitation and emission in visible light wavelengths. These wavelengths are absorbed and scattered by tissues and thus are limited in their ability to penetrate biological materials. Signal from these fluorophores is further attenuated by tissue absorption and autofluorescence in this visible wavelength range, limiting the utility and translational potential of the fluorophores. While the visible and near infrared wavelengths of light often used in biomedical optical imaging suffer from endogenous tissue autofluorescence, the short wave infrared range (1500–1700 nm) has less associated interference. This allows for improved detection of fluorescent moieties, greater sensitivity and improved penetration through biological tissue,.[10] Additionally, contrast agents with emissions in this range are frequently excited using NIR photons. Therefore, their excitation wavelength reduces associated tissue autofluorescence and scattering (as compared to UV and visible excitation wavelengths) leading to improved imaging.[11] Stable near-infrared emitting agents such as quantum dots and carbon nanotubes have shown promise for in vivo imaging, but do not achieve appreciable tissue penetration in vivo at biologically safe doses, limiting their clinical applicability.
To address limitations of existing technologies, in our previous work we have engineered optical nanoprobes as a new approach for detection of small sub-tissue cancerous lesions. These probes are based on ceramic nanoparticles doped with rare-earth (RE) cations that absorb near infrared (NIR) radiation to luminesce in the shortwave infrared (SWIR) spectrum (1000–3000 nm),[12, 13] emitting in the overlooked second “tissue transparent short-wave infrared window” of biological imaging.[14] Rare-Earth Albumin Nanocomposites (ReANCs), fabricated by encapsulating the RE nanoparticles within a human recombinant albumin shell, can be functionalized with ligands that complement cancer receptor markers. This functionalization results in imaging probes with selectivity to specific tumor molecular receptors.[1] Additionally, because of the hydrophobic nature of several binding pockets within the albumin shell, these probes exhibit high adsorptive capacity for small molecules pharmacologic drugs, and can be used for lesion-targeted delivery of antagonist molecules to cancers.[15]
In this study we investigated whether such functionalized nanoprobes could detect small cancerous lesions based on the combination of three key design features: the relative tissue transparency afforded by SWIR emitting nanoprobes, deeper tissue illumination potential of SWIR probes, and molecular targeting to cancerous lesion markers. Cancer targeting nanoprobes were generated by passive adsorption on albumin shells of AMD3100[16, 17] a hydrophobic small molecule inhibitor of the chemokine receptor CXCR4.[18–20] CXCR4 is strongly expressed on highly motile cancer cells, and has been previously used to target a PET imaging agent that localizes to metastatic lesions in the lungs.[21] CXCR4-targeted, AMD3100 functionalized nanoprobes preferentially accumulate withing receptor positive cells and tumors allowing for more sensitive detection of small-scale lung lesions in an in vivo metastatic breast cancer model (Figure 1). This is of particular clinical relevance as CXCR4 has previously been indicated in the site specific invasion and metastasis of breast cancer cells to the lungs, and is correlated to decreased survival time and poor prognosis.[19] Strikingly, the cancer targeted probes enabled imaging of microlesions (~25 mm3) in the lungs at a depth of approximately 1 cm while allowing for simultaneous molecular identification of the tumor population phenotype.
Figure 1. Design, fabrication, and preclinical efficacy of rare earth nanoprobes.

ReANCs were synthesized by controlled coacervation of albumin monomers in solution and REs dissolved in a solvent. (A) AMD3100 was adsorbed onto the surface of fully formed ReANCs to generate AMD3100 functionalized ReANCs, or fReANCs. (B) Athymic nude mice were inoculated with human breast cancer cells through the tail vein. Animals were treated with either ReANCs or fReANCs and SWIR imaging performed to determine nanoprobe-tumor localization. fReANCS were imaged in tumors up to 1 cm into the animal (B). Microlesions as small as 18 mm3 were detected with targeted probes (C). Probe localization to receptor positive tumors enabled optical molecular phenotyping (D).
2. Results
2.1. Synthesis and characterization of nanoparticles
Rare earth nanoparticles were synthesized via burst nucleation as previously described. Nanoparticle structure consisted of a NaYF4 core doped with ytterbium (Yb) and erbium (Er) coated in an undoped shell. Dopants were found to comprise 30% of the total nanoparticle atomic weight. Lifetime emissions scans revealed that the calculated delay time for the particles 1530 emissions was found to be 3.55 ms (Supplementary Figure 1). Rare earths were incorporated into albumin nanocomposites (ReANCs) by solvent induced controlled coacervation of albumin.[1, 2] Hydrodynamic diameters of ReANCs were determined using dynamic light scattering (DLS) (Figure 2A), which revealed monodisperse particle populations. The yield of ReANCs was found to be approximately 70% by BCA protein assay.
Figure 2. Characterization of functionalized Rare Earth Nanoprobes.

(A) fReANCs were synthesized by adsorbing AMD3100 onto fully formed ReANCs. Both populations had narrow size distributions and low polydispersity index. The formulation used for in vivo studies is as indicated with asterisk. (B) Scanning election microscopy (SEM) of fReANCs revealed spherical particles. The addition of AMD3100 at concentrations of 12.5 µM (B), 1.25 µM (C), 125 nM (D) and 37.5 nM (E) yielded an increase in granulated surface texture directly proportional to the loading concentration. Drug loading efficiency (last column) was determined using HPLC as described in methods section. Cellular treatment with REANCs, fReANCs or free drug did not have an effect on cell metabolic activity (F), suggesting that the nanoprobes are non-cytotoxic. (F) Error bars denote standard deviation (n = 3), p < .05
2.2. Cancer-Targeting Probes: AMD3100 Functionalized ReANCs (fReANCs)
Functionalized ReANCs (fReANCs), namely, ReANCs targeting the CXCR4 receptor over-expressed on motile cancer cells, were generated by adsorbing AMD3100, a small molecule antagonist of CXCR4, onto the ReANC surface (Figure 1A) by exploiting the native drug binding pockets on human serum albumin.[22] The particles were characterized to determine the effect of functionalization on the physical properties of the probes. fReANCs were somewhat larger than unmodified ReANCs (5–35 nm increase in diameter depending on loading concentration), with marginally but not statistically significant greater polydispersity (Figure 2A). Emission spectra for albumin coated ReANCs have been previously reported, showing that albumin encapsulated rare earth particles exhibit detectable SWIR emissions.[1] The adsorption of AMD3100 onto the albumin surface had a slight dampening effect on SWIR emissions; nonetheless, fluorescence was strong enough to be resolved via the current imaging prototype (supplemental figure 1).
Scanning electron microscopy (SEM) of ReANCs and fReANCs revealed spherical populations indicating successful adsorption, as evidenced by a change in surface texture with varying concentrations of AMD3100 (Figure 2B–E; Supplementary Figure 1). Loading efficiency was validated by HPLC and was determined to be dependent on the amount of AMD3100 introduced, ranging up to 70% (Figure 2A). Scatchard analysis of the loading data revealed a binding affinity of Kd=1.8×10−7 M. This value is comparable with binding of drug moieties such as paclitaxel to albumin indicating that drug binding activity is retained by albumin in a nanoshell form.[23] Additionally, AMD3100 loading of ReANCs had no significant effect on the viability of MDA-MB-231 breast cancer cells (Figure 2F).
2.3. Active receptor targeting of fReANCs
Effective targeting of fReANCs was determined by cellular association of AMD3100 functionalized probes using three breast cancer cell lines with varying levels of CXCR4 expression: 1) 4175-TR (a highly aggressive, lung-tropic subpopulation of the MDA-MB-231 cancer cell line[24]) with low CXCR4 expression, 2) MDA-MB-231, with moderate levels of CXCR4 expression, and 3) MCF-7, with relatively high levels of CXCR4 expression,[25] We observed a 3-fold increase in cellular association of fReANCs when compared to ReANCs by both receptor positive cell lines as determined by flow cytometry, with no significant change in association with receptor negative 4175-TR cells (Supplementary Figure 2 and Figure 3A, 3B, and 3E). Based on cell surface binding and cytotoxicity studies, the optimal concentration for loading REANCs was determined to be 125 nM. Confocal microscopy showed that nanoprobe internalization of both ReANCs and fReANCs occurred within 24 hours, with nanoprobes distributed in a punctate pattern throughout the cell cytoplasm and the increased intracellular fluorescence signal leading to overall increased signal in each individual cell. (Figure 3C and 3D; Supplementary Figure 3).
Figure 3. Active Cancer Targeting of Rare Earth Nanoprobes.

Cellular uptake and association between ReANCs (A) and fReANCs (B) by MDA-MB-231 breast cancer cells was analyzed by flow cytometry for up to 24 hours. Functionalization increased particle uptake as determined by FACS. This was confirmed with confocal imaging of receptor positive cells treated with ReANCs (C) or fReANCs (D) for 24 hours. White arrows indicate nanoparticle fluorescence. While ReANCs showed some cellular association, the degree of nanoprobe association was greater with the functionalized constructs. Quantitative flow cytometry analysis revealed that nanoprobe association increased with greater expression of the targeted CXCR4 receptor (E). Error bars denote standard deviation for n = 3. * p < .05 (t test, comparing between ReANC and fReANCs groups)
2.4. In vivo SWIR imaging
2.4.1. Tumor-Specificity of Targeted Nanoprobes
The ability of the functionalized probes to discriminate between receptor-positive (MDA-MB-231) and receptor-negative (4175-TR) lesions in vivo was assessed using nude mice bearing palpable bilateral subcutaneous tumors. Animals were treated with a bolus dose of 200 µL (10 mg kg−1) of either unfunctionalized ReANCs or fReANCs into the intraperitoneal cavity (i.p. administration). Immediately following injection of the particles, SWIR emissions were visualized in the abdominal cavity around the site of injection. The fReANCs cleared much more rapidly from the site of injection than the ReANCs, which were retained in the peritoneal cavity up to 48 hours post administration (data not shown). SWIR fluorescence was detected 24 h after fReANCs administration at the receptor-positive, left dorsal tumor (Figure 4B and C). This was in contrast to little or no SWIR signal seen with ReANC administration (Figure 4A). Additionally, little SWIR signal was observed with fREANC administration from the receptor-negative (4175-TR derived) right dorsal tumor (Figure 4B and C). Ex-vivo SWIR imaging also revealed irregular fluorescence patterns surrounding the receptor positive tumor validating tumor-specificity of our nanoprobes in vivo, indicated by white arrows (Figure 4E). There was no fluorescence associated with the receptor negative tumors excised from the animals’ right flank (Figure 4F and 4G) or the receptor positive tumors treated with ReANCs (Figure 4D).
Figure 4. Functionalized nanoprobes are capable of molecular discrimination of tumors in vivo.

The ability of targeted constructs to molecularly target in vivo lesions was evaluated using nude mice bearing bilateral, subcutaneous, dorsal tumors derived from human breast cancer cells. Receptor positive MDA-MB-231 cells (Red open circle) were injected into the left dorsal flank, while receptor negative 4175-TR cells (Blue Open circle) were injected on the right. Unfunctionalized ReANCs (A) showed little associated red SWIR fluorescence at either lesion sites, while fReANCs (B) had red SWIR fluorescence associated with only the receptor positive tumor. The maximum SWIR values for each region of interest (C) show that fReANCs accumulated preferentially to the receptor positive left tumor. n = 2. (D–G insets) Ex vivo SWIR imaging revealed SWIR fluorescence associated with the receptor positive tumor treated with fReANCs. Excised tumors treated with (D, F) ReANC and (E, G) fReANC were sectioned and imaged to determine nanoprobe localization (green fluorescence) within the tumor mass. Scale bars represent 500 µm. Representative images from week 1.
Strikingly, the receptor-positive tumors were 4× smaller than the receptor-negative tumors (Figure 4D–G insets). This indicated that fReANCs possess the ability to detect and target small, poorly vascularized tumor microlesions. The maximum SWIR signal elicited with fReANCs was observed at 1–2 weeks of tumor growth and then declined, which may correlate with a decrease in CXCR4 expression as the metastasis develops into a mature tumor. These results validated the ability of targeted fReANCs to molecularly phenotype tumors in vivo.
Additionally, there was no overt toxicity from injection of the particles, and the particles were cleared 72–96 h post injection to non-detectable levels in the mouse (data not shown). Repeated injections of REANCs and fReANCs over time did not lead to toxicity as evidenced by uniform body weight over the course of the 11 week study (Figure 5A) and by lack of distress signals exhibited by the animals, as determined by the IACUC monitoring.
Figure 5. Rare-Earth nanoprobes proved safe for long-term use with no significant impact on animals’ weight or damage to organs of accumulation.
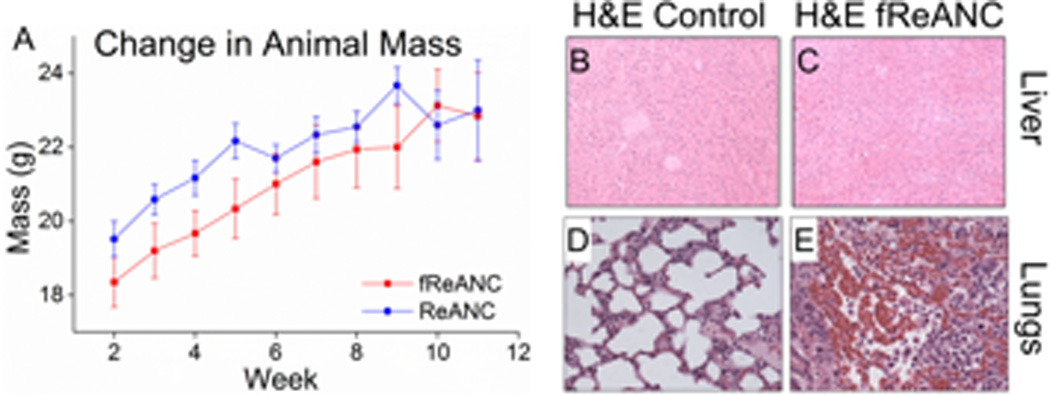
Animal’s body weight was recorded weekly over the course of the experiment (A) and animals were closely monitored for any signs of distress. Repeated injections were well tolerated by animals. Animals’ livers were excised and examined to determine changes in the organ’s structure with no (B) and fReANC (C) treatment. Analysis showed no change in the liver’s integrity with dosing. Lung sections, from fREANC treated animals showed tumor infiltration in tumor bearing animals (E) and no morphological change in the control healthy animals (D) confirming the safety of the nanoprobes. n = 6.
2.4.2.Histopathological analysis of Nanoparticle Biocompatibility
At experimental endpoints, animals were sacrificed and organs of clearance (liver and spleen) were analyzed for toxicity due to the administered ReANCs. Analysis of H&E sections revealed no alterations suggestive of toxicity when compared to untreated controls (Figure 5B and C). Lung sections, obtained from lesions from the lung tumor model described in later sections, revealed a high degree of tumor burden (Figure 5E). Notably, administration of ReANCs to control animals did not show overt toxicity or changes in lung architecture on H&E staining (Figure 5D). Taken together these results support the biocompatibility of the targeted-nanoprobes and their utility to detect microlesions and enable real-time imaging of tumors.
2.4.3. Ex vivo SWIR Imaging
Following necropsy, ex vivo SWIR imaging of tumors and organs revealed SWIR fluorescence associated with the livers of animals given i.v. injections of both ReANCs and fReANCs. Ex vivo analysis of animals that received intra-peritoneal injections of the particles showed SWIR fluorescence in the major organs of clearance (spleen and liver) for both ReANC and fReANC treatment groups (Supplementary Figure 4).
Excised tumors were sectioned and imaged with confocal fluorescence microscopy to determine the localization of the nanoprobes within the tumor interstitium. Analysis of receptor positive tumors treated with fReANCs revealed a punctate pattern of nanoparticles distributed throughout the tumor mass (Figure 4E and Supplementary Figure 5). Ex vivo results thus far confirmed that the targeted fReANCs are able to molecularly phenotype lesions in vivo.
2.4.4. In vivo detection of tumors in internal organs with SWIR imaging
We assessed the ability of the SWIR nanoprobe imaging approach to locate tumors in sub-tissue organs using female athymic nude mice bearing lung tumors in an established model of metastatic breast cancer.[26–28] One of the most significant observations was that the nanoprobe-related SWIR signal was detectable up to 10.5 mm into the animal. Animals were treated with 200 µL of unfunctionalized ReANCs or fReANCs (10 mg kg−1) via i.v. administration and imaged up to 24h post-injection and yet no toxicity was associated with repeated use of probes. SWIR fluorescence was distinguishable in animal’s livers immediately after injection. Notably, we obtained discernible SWIR signal using fReANCs in the lungs as early as three weeks following the inoculation of tumor cells (Figure 6D), compared to unfunctionalized ReANCs (Figure 6B and 6C) where signal was observed approximately 7 weeks post-inoculation. Preferential accumulation of fReANCs in tumors within lungs is indicated by the combination of enhanced lung-related and depressed liver-related signal in fReANC treated animals as compared to unfunctionalized, ReANC treated animals (Figure 6D and 6E). In particular, as shown in Figure 6D, fReANC accumulation in lungs increased significantly from week 2 after inoculation to week 4 after inoculation, which was in correlation with the increase in tumor volume.
Figure 6. Targeted fReANCs allow for detection of microscale lung lesions.

Nude mice were inoculated i.v. with CXCR4 expressing MBA-MB-231 cells. Cells colonized in the lungs and were monitored using MRI (A). Xenograft mice were administered either ReANCs (B) or fReANCs (C) at early stages (< 6 weeks) after inoculation. SWIR signal from the lung region was quantified at 24 h (D) post-injection. The fReANC associated SWIR signal was greater than the ReANC signal for early time-points, correlating to the presence of microlesions quantified by volumetric reconstruction of the MRIs. The SWIR signal from the liver was quantified for 0 h post injection (E) and showed a 2 fold increase in ReANC accumulation in the organs of clearance. Representative images (n=6) shown in all instances from week 3 of the study. The presence of tumors in the lungs was confirmed with vimentin staining of healthy (F) and tumor bearing (G) lungs, with the red fluorescence marked by arrows indicating tumor cells. (D) Error bars indicate SEM for n = 6. * indicates p < .01 (Wilcoxson Rank-Sum test) (E) * = p < .05 (Wilcoxson Rank-Sum test)
Tumor burden and depth of lesions relative to the surface of the animal were determined by bi-weekly MRI from time of inoculation of tumor cells (Figure 6A; Representative MRI). An especially significant outcome was that the tumors detected using fReANCs were found to be 27.8 mm3 in volume on average with the lowest tumor volume detectable being 18.9 mm3 (Figure 7B and C). In contrast, tumors detected with ReANCs were an average of 62.6 mm3 (2.25 times larger than those resolved with the functionalized particles) with a tumor minimum volume of 55.6 mm3 (Figure 7A and C). Tumors were located 7–10 mm from the surface of the animal. SWIR fluorescence and MRI images were overlaid (data not shown) to determine particle accumulation at lesion sites, confirming that the SWIR signal was associated with the lesions. The volumetric analysis reveals that fReANCs, in addition to detecting tumor lesions in sub-tissue earlier than ReANCs, are also able to resolve micro-lesions approximately 2.25 times smaller than those detected by ReANCs (Figure 7). Ex vivo analysis of excised organs revealed SWIR signal from multiple lesions in the lungs and chest wall (Supplementary Figure 4), thus establishing the tumor-specific targeting ability of fReANCs in the lung model.
Figure 7. Longitudinal tracking of internal lesions with SWIR imaging.

SWIR fluorescence images were taken each week for animals treated with both ReANC (A) and fReANC (B). Tumor burden was determined each week through MRI and compared to SWIR signal. Mean and minimum volume (C) of tumors detectable via SWIR imaging was calculated to determine the smallest tumors capable of resolution using the InGaAs camera. Treatment with fReANCs enabled detection of tumors on average 2.25 times smaller than those detected using ReANCs. (n=6)
Ex vivo tumors were analyzed for presence of infiltrating tumor cells by immunohistochemical staining for vimentin, a tumor-cell specific marker (Figure 6F and 6G). Taken together, these results confirm the ability of fReANCs to detect small (< 30 mm3) tumors up to 1 cm into biological tissue.
3. Discussion
Breast cancer, one of the most common causes of cancer death among women, is difficult to treat due to the frequency of micrometastatic populations that are phenotypically distinct from the parent tumor. It is therefore of critical importance to develop technologies capable of safe and sensitive detection and characterization of small sub-tissue tumors, based on their distinct molecular phenotype, with accurately defined margins. This study provides the first proof of concept model for optical-probe based molecular detection of sub-tissue micro-lesions. Here we demonstrate cancer-targeted SWIR emitting fREANCs target CXCR4-positive tumors in a bilateral tumor model of receptor-positive and negative tumors. Specifically, fREANCs are also shown to molecularly target CXCR4-positive tumors that are 4× smaller than receptor-negative tumors. The most significant observation from this study was the ability of cancer-targeted nanoprobes to detect sub-tissue lesions at a depth of approximately 1 cm and also to resolve micro-scale lesions. fREANCs were able to resolve tumors as small as 18.8 mm3 compared to lesions that were 62.6 mm3 detected by control REANCs.
SWIR emitting rare-earth albumin nanocomposites have previously been shown to be effective in vivo contrast agents that passively accumulate at the site of malignant lesions, possibly due to the enhanced permeability and retention (EPR) effect, allowing for imaging in vivo.[2] However, imaging of metastatic micro-lesions present additional challenges as the malignancy has not yet established its own vasculature to enable passive targeting. Prior work has shown that active targeting of nanoparticle based contrast agents to a specific disease marker can greatly improve their localization to targets of interest.[29, 30] Active targeting of contrast agents has been shown to improve particle biodistribution, improve signal-to-noise ratio, provide molecular information about a region of interest, and increase the optical signal allowing for more sensitive imaging.[31–37] Here, we have designed and characterized a novel targeted imaging probe consisting of rare-earth doped phosphors and human serum albumin onto which is adsorbed AMD3100 as the targeting ligand.
The approach to targeting of the ReANCs was designed based on the native drug binding properties of human serum albumin.[22, 38] HSA has been shown to simultaneously bind multiple distinct moieties at various ‘drug binding pockets’ across its surface.[22] Albumin nanocarriers retain the ability to strongly bind multiple therapeutic agents with little loss of efficacy.[15, 38] We utilized this property to adsorb AMD3100 as a targeting ligand onto the surface of the particles for tumor cell targeting with a high (up to 70%) efficiency of loading and affinity (Kd=1.8×10−7 M) owing to the several albumin drug binding sites within a nanoshell. The loading efficiency and binding affinity were found to be similar to reported values for albumin loading with other small molecule therapeutics such as paclitaxel and doxorubicin.[23] The resulting nanoprobe localization, combined with the sensitivity of an in-house SWIR imaging system, enables detection of small internal tumors with high specificity.
The fReANCs were synthesized and loaded with varying concentrations of the small molecule antagonist, AMD3100. fReANCs not only showed improved association with receptor positive MDA-MB-231 and MCF7 cells in vitro but also exhibited a lack of association with receptor negative MDA-4175 cells, with no appreciable effect on cell viability. This effect was largely independent of bound AMD3100 concentration on fReANCs (Supplementary Figure 2), suggesting that the particles with modest net loading and a maximum loading efficiency (approximately 70% by HPLC) could provide efficient targeting selectivity. Confocal fluorescence microscopy of fReANCs’ association with malignant cells confirmed visually the increased association of fReANCs when compared to unfunctionalized ReANCs.
Proof of concept studies in a mouse xenograft breast cancer model showed tumor-specific binding of fReANCs to receptor-positive tumors and successful targeting of tumors that were 4× smaller than the corresponding receptor negative tumors. This clearly highlights the ability of fReANCs to accurately detect and molecularly phenotype early tumor lesions. Future longitudinal studies will address the ability of molecularly targeted ReANCs in tumor-dynamic tracking. It is important to note that fReANCs were able to accumulate in tumors enabling detection in contrast to unfunctionalized ReANCs that post-injection cleared immediately to the liver.
Thus far, one of the hurdles faced by optical imaging agents has been their inability to be resolved deeper for detection of lesions in internal organs.[39–41] Notably, targeted fReANCs were able to resolve lesions as small as 18.9 mm3 in the lungs of tumor bearing animals, with a depth of penetration of 10.5 mm through the tissue. Furthermore, the preferential accumulation in lung lesions was seen up to 24 h post-injection with little loss in SWIR signal. Future studies using large animal models will need to address improvement in depth of penetration using these probes to clinically acceptable depths. A limitation of near infrared excitation is localized photothermal effects.[42] Due to strong absorption of 980nm photons by water molecules, prolonged exposure of tissue to NIR wavelengths can induce non-localized temperature fluctuations.[43, 44] This effect can be mitigated by limiting the exposure of near infrared light to the skin. Thermal heating effects are typically generated after minutes of exposure at a high laser power (from 3–6 W cm−2).[45, 46] In contrast, the system presented here requires an intensity of only 1.7 W cm−2 with seconds of exposure. By limiting laser power and duration it is possible to circumvent detrimental effects on biological tissues post exposure.
Of significance is the fact that we were able to perform real-time imaging with no observed toxicity for a period of 11 weeks, as shown by consistent body weight and the lack of pathological evidence of toxicity in major organs of clearance such as the liver and spleen. Both nanoparticle formulations cleared completely from the animal’s body within 7 days. There was no apparent difference in organ accumulation between the functionalized and unfunctionalized particles (Supplementary figure 4). Since athymic nude mice used in this study lack a robust immune system, studies to validate lack of immunogenicity and toxicity to establish safety profile of the probes will be done in immunocompetent mice.
Emergent patient therapy towards metastatic disease is currently centered on a molecularly targeted therapeutic strategy.[47] Several recent preclinical studies have provided proof of concept that early targeting of metastasis-specific signaling pathways can result in enhanced therapeutic responses.[26, 27, 31, 48, 49] Implicit in the precision medicine approach is the need for cost-effective, safe and easy to use molecular diagnostic agents. Targeted nanoprobes from this study exhibit translational potential to monitor tumor progression, molecularly phenotype tumors and provide physicians with a diagnostic tool to make improved choices for tailored therapy and monitor in real-time response to therapy.
4. Conclusion
The albumin nanoshells utilized in this work show a high affinity and capacity for ligands and upon functionalization are capable of cellular discrimination based on receptor phenotype in vitro. Functionalized nanoprobes are able to preferentially localize to receptor positive tumors in mice, allowing for detection of positive tumors 4× smaller than receptor negative tumors. The most significant observation of clinical importance from this study was the ability of functionalized nanoprobes a) to detect sub-tissue microlesions approximately 1 cm from the imaging surface and b) to resolve microlesions as small as 18.9 mm3 when compared to 2.25 times larger lesions detected by ReANCs. The safety profile of both fReANCs and ReANCs, as indicated by no overt toxicity during the course of the study, is an added benefit for plausible clinical applicability of these agents.
5. Experimental Section
Reagents
AMD3100 was purchased from EMD Millipore (Billerica, MA, USA) and used without further modification.
Cell Culture
MDA-MB-231, MCF7 (ATCC, Manassas, VA) and MDA-MB-231 derived 4175-TR cells (kind gift from Dr. Yibin Kang, Princeton University, USA) were cultured in DMEM (ATTC, Manassas, VA) supplemented with 10% FBS and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA) at 37°C in an atmosphere of 5% CO2.
ReANC Fabrication
ReANCs were synthesized as previously described.[1, 2] Briefly, 2% (w/v) solution of human serum albumin (Sigma-Aldrich, St. Louis, Mo) was dissolved in NaCl (2 mM) and the pH was adjusted to 8.50 ± 0.05 with NaOH (.1 M). Under continuous stirring at 700 rpm at room temperature, ethanol (2 mL) sonicated with rare-earth nanoprobes (0.2 mg mL−1) was added at 1.5 mL min−1 to the HSA solution (500 µL) with a syringe pump (Harvard Apparatus PHD 2000, Holliston, MA). Subsequently glutaraldehyde (2.34 µL) (Sigma-Aldrich, St Louis, Mo) was added to the suspension as a cross-linker following ethanol addition and particles were allowed to crosslink overnight. Particles were then purified by 3 cycles of centrifugation at 48,400 g. Particles were characterized by BCA assay (Pierce Biotechnology, Rockford, Il) to determine % yield. DLS was performed to determine particle size and polydispersity.
Preparation of functionalized nanoparticles (fReANCs)
AMD3100 (EMD Millipore, Darmstadt, Germany) was dissolved in sterile water (1 mg ml−1) and was added into a suspension of ReANCs in PBS+1 mM EDTA to yield final concentrations ranging from 0.0125–12.5 µM. AMD3100 adsorption was achieved by constant agitation of samples at room temperature for 3h followed by purification through centrifugation. AMD3100 functionalized ReANCs (fReANCs) were characterized as described above.
Quantification of loading
The concentration of AMD3100 loaded on ReANCs was determined via high-performance liquid chromatography (HPLC, Beckman Coulter Model 166, Brea, CA) used with a Luna C18 column (3 µm, 100 × 4.6 mm i.d., Phenomenex, Torrance, CA). Briefly, the functionalized fReANCs were flash frozen and lyophilized for 48 hours. The powders were re-suspended in water and assayed on HPLC to determine AMD3100 concentrations. For chromatographic elution the flow rate was 1 mL minute−1. The mobile phase consisted of 0.1% TFA in de-ionized water and acetonitrile. AMD3100 was detected at 212 nm. Binding affinity between albumin nanoparticles and AMD3100 was determined via scatchard analysis: The ratio of bound/free ligand was determined for various concentrations of ligand from the HPLC data and plotted against the amount of ligand loaded onto the particles. Slope of the resulting best fit line was used to determine the particle dissociation constant.
In Vitro cellular uptake of fReANCs
MDA-MB-231, MCF-7, and 4175-TR cells expressing different relative amounts of CXCR4 were seeded onto 96-well microtiter plates at a density of 5×105 cells/well and incubated for 24 hours at 37 °C. To quantify the amount of nanoparticle uptake, cells were treated with 10% w/v of either ReANCs or differently functionalized fReANCs for up to 48 hours. Cells were subsequently trypsinized, washed and fixed in 1% PFA prior to being analyzed with fluorescent flow cytometry (FACscalibur, BD Biosciences, San Jose, CA) using the autofluorescence of the albumin shell to determine uptake as described previously. Data was collected for 10,000 cells.
Cell uptake and confocal imaging
To further visualize and confirm active targeting of receptor + cells, MDA-MB-231 and MCF7 cells (with elevated expression of CXCR4) and 4175-TR cells (with low expression of the receptor) were seeded on borosilicate Lab-Tek chambers (Nuc, Rochester, NY) and incubated overnight at 37 °C. Cells were then treated with either unfunctionalized ReANCs or AMD3100 loaded fReANCs for 24 hours. Cells were then washed, fixed, and stained with DAPI. NIR imaging was performed using a titanium:sapphire laser on a Leica TCS SP2 fluorescence microscope (Leica Microsystems, Exton, PA) to confirm cellular uptake.
Cell viability assay
The metabolic activity of cells treated with ARAs was assessed using the CellTiter96 Aqueous One Solution Reagent (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, MTS) (Promega, Madison, WI). MDA-MB-231 and 4175-TR cells were cultured on 96 well microtiter plates at a density of 5000 cells/well and treated with 10% w/v ReANCs or fReANCs suspended in PBS for 24 hours. After 24 hours culture media was changed and MTS reagent was added to each well, and the plate was allowed to incubate for an additional 4 hours at 37°C. Absorbance was measured at 450 nm using a microplate reader (Bio-Rad Model 680, Hercules, CA). Cell viability was calculated relative to the absorbance of the untreated cell population.
In Vivo imaging
Imaging studies were conducted using female homozygous nude mice (Taconic, Hudson, NY). For subcutaneous tumor imaging studies, MDA-MB-231 and 4175-TR human breast cancer cells were injected into the dorsal area at 107 cells per site. Animals were used for imaging studies once tumors became palpable. For imaging of internal organs at a sub-tissue level, MDA-MB-231 cells were injected via the tail vein at 3×105 cells per animal. All animal studies were approved by the Institutional Review Board for the Animal Care and Facilities Committee of Rutgers University and performed in accordance with institutional guidelines on animal handling.
SWIR-imaging
Animals were imaged using a previously described in-house small animal SWIR imaging prototype for real time non-invasive optical imaging. Mice were fully anesthetized using 2% isoflurane (Butler-Schein, Dublin, OH) and were continuously scanned with a 1.7 W collimated 980 nm laser in various positions to excite nanoparticles. SWIR fluorescence was detected with an IR sensitive InGaAs camera (Sensors Unlimited, Princeton, NJ) equipped with long-pass 1000 nm and band-pass 1550 nm filters (Semrock, Rochester, NY) and a 25 mm SWIR lens (StingRay Optics, Keene, NH. This system is capable of real-time live animal imaging with an exposure time of ~50 ms frame−1.[2] Images were acquired as .bin video files and were processed using custom Matlab scripts (Supplementary Figure 6).
MRI Imaging
MR images are acquired using 1 tesla M2-High Performance MRI System (Aspect Magnet Technologies Ltd, Netanya, Israel). All imaging procedures were performed under inhalation anesthesia with isoflurane at a concentration of 4% for induction of anesthesia and 1–2% for maintenance. MRI images were analyzed using VivoQuant software (Aspect Magnet Technologies Ltd, Netanya, Israel).
Detection of subcutaneous cancer lesions
MDA-MB-231 (CXCR4+) and 4175-TR (CXCR4 −) cells were cultured in DMEM supplemented with 10% FBS (Invitrogen, Carlsbad, CA) and 1% penicillin-streptomycin. Female athymic nude mice were purchased from Taconic laboratories at 3–4 weeks of age and inoculated with bilateral dorsal injections of MDA-MB-231 and 4175-TR cells at 107 cells per site and allowed to grow until palpable. Animals were treated with weekly i.p. injections of ReANCs or fReANCs at a dose of 10 mg kg−1 and imaged with SWIR imaging at 24 h post injection to determine particle accumulation in the tumor space. Animals were sacrificed once tumor volume reached 500 mm3 and organs were excised and imaged ex vivo to determine particle distribution.
Ex vivo analysis of subcutaneous tumors
At experimental endpoints animals were euthanized and subcutaneous tumors were excised and flash frozen. Tumors were cut to 25 µM sections with a cryotome (Thermo Fisher, Waltham, MA) and counterstained with Hoechst to detect cell nuclei. Sections were imaged using confocal microscopy to determine particle accumulation in the tumor mass.
Detection of lung metastatic lesions
Female homozygous athymic nude mice were obtained from Taconic Laboratories (Hudson, NY) at 3–4 weeks of age and were injected through the tail vein with MDA-MB-231 cells in an established model of lung metastasis. Tumor burden and location was evaluated using twice-monthly MRI (Aspect Imaging, Shoam, Israel) as described above. Animals were treated with weekly i.v. injections of either ReANCs or fReANCs at a dose of 10mg kg−1 body weight and imaged at 24h post treatment to determine the ability of functionalized nanoprobes to resolve changes in the tumor burden over time. SWIR signal was correlated to tumor size and location as determined by MRI. Animals were sacrificed upon weight loss and organs were excised and imaged with SWIR imaging ex vivo to determine particle biodistribution and localization to lesion sites.
Ex vivo analysis of metastatic tumors
At experimental endpoints, animals were euthanized and tumors were excised, and fixed in formalin. Paraffin-embedded sections were imaged using confocal microscopy to determine particle accumulation in the tumor microenvironment. Slides were stained with Haemotoxylin and Eosin (Sigma-Aldrich, St. Louis, MO) to determine changes in microlesion architecture and photographed using (Nikon, Chiyoda, Japan). For staining with Vimentin, paraffin-embedded sections were warmed at 40°C for 1h and deparaffination procedure performed. Antigen retrieval using citrate buffer at pH 6.0 was performed by microwave heating method. Following staining with anti-human Alexa647 conjugated Vimentin antibody (Santacruz Biotechnology, St.Louis,MO, USA) sections were counterstained with Hoechst 3342 to stain nuclei. Vimentin staining was visualized by fluorescence microscopy. Isotype matched control antibody staining was performed for non-specific binding.
Supplementary Material
Acknowledgments
The authors are grateful to access to the Rutgers Molecular Imaging Core, and funding from the NIH NIBIB EB018378-01 and EB015169-02. We would also like to thank D. Naczynski for the foundational research that inspired this study, S. Chen of the Pharmacology-Laboratory for Cancer Research and S. Ganesan of Rutgers-CINJ for their insights with the clinical and preclinical models, Y. Kang for the 4175-TR cells, N. Stebbins and J. Faig of the Uhrich laboratory at Rutgers for their help with the analysis of drug loading and L. Higgins for assistance with microscopy.
Contributor Information
Margot Zevon, Department of Biomedical Engineering, Rutgers University, 599 Taylor Road, Piscataway, NJ 08854.
Vidya Ganapathy, Department of Biomedical Engineering, Rutgers University, 599 Taylor Road, Piscataway, NJ 08854.
Harini Kantamneni, Department of Chemical and Biochemical Engineering, Rutgers University, Piscataway, NJ 08854, USA.
Marco Mingozzi, Department of Biomedical Engineering, Rutgers University, 599 Taylor Road, Piscataway, NJ 08854.
Paul Kim, Department of Materials Science and Engineering, Rutgers University, 607 Taylor Road, Piscataway, NJ 08855.
Derek Adler, Email: Derek.adler@rutgers.edu, Molecular Imaging Center, 41 Gordon Road (Suite D), Piscataway NJ 08854.
Yang Sheng, Engineering Product Development, Singapore University of Technology and Design, 20 Dover Drive, 138682, Singapore.
Mei Chee Tan, Email: meichee.tan@sutd.edu.sg, Engineering Product Development, Singapore University of Technology and Design, 20 Dover Drive, 138682, Singapore.
Mark Pierce, Department of Biomedical Engineering, Rutgers University, 599 Taylor Road, Piscataway, NJ 08854.
Richard E. Riman, Email: riman@rci.rutgers.edu, Department of Materials Science and Engineering, Rutgers University, 607 Taylor Road, Piscataway, NJ 08855.
Charles M. Roth, Email: cmroth@rci.rutgers.edu, Department of Biomedical Engineering, Rutgers University, 599 Taylor Road, Piscataway, NJ 08854; Department of Chemical and Biochemical Engineering, Rutgers University, Piscataway, NJ 08854, USA.
Prabhas V. Moghe, Email: moghe@rci.rutgers.edu, Department of Biomedical Engineering, Rutgers University, 599 Taylor Road, Piscataway, NJ 08854; Department of Chemical and Biochemical Engineering, Rutgers University, Piscataway, NJ 08854, USA.
References
- 1.Naczynski DJ, Andelman T, Pal D, Chen S, Riman RE, Roth CM, Moghe PV. Small. 2010;6(15):1631–1640. doi: 10.1002/smll.200902403. [DOI] [PubMed] [Google Scholar]
- 2.Cui M, Naczynski DJ, Zevon M, Griffith CK, Sheihet L, Poventud-Fuentes I, Chen S, Roth CM, Moghe PV. Adv Healthc Mater. 2013;2(9):1236–1245. doi: 10.1002/adhm.201200467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clemons M, Danson S, Hamilton T, Goss P. Cancer Treat Rev. 2001;27(2):67–82. doi: 10.1053/ctrv.2000.0204. [DOI] [PubMed] [Google Scholar]
- 4.Weigelt B, Peterse JL, van 't Veer LJ. Nat Rev Cancer. 2005;5(8):591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 5.Seemann MD. Technol Cancer Res Treat. 2005;4(5):577–582. doi: 10.1177/153303460500400512. [DOI] [PubMed] [Google Scholar]
- 6.Antoch G, Bockisch A. Eur J Nucl Med Mol Imaging. 2009;36(Suppl 1):S113–S120. doi: 10.1007/s00259-008-0951-6. [DOI] [PubMed] [Google Scholar]
- 7.Hwang JY, Park J, Kang BJ, Lubow DJ, Chu D, Farkas DL, Shung KK, Medina-Kauwe LK. PLoS One. 2012;7(4):e34463. doi: 10.1371/journal.pone.0034463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Methods. 2008;5(9):763–775. doi: 10.1038/nmeth.1248. DOI nmeth.1248 [pii] 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 9.He H, Xie C, Ren J. Anal Chem. 2008;80(15):5951–5957. doi: 10.1021/ac8005796. [DOI] [PubMed] [Google Scholar]
- 10.Diao Shuo, Hong, Guosong, Antaris, Alexander, Blackburn, Jeffrey, Cheng, Kai, Cheng, Zhen, Dai, Hongjie Nano Research. 2015;8(9):3027–3034. [Google Scholar]
- 11.Villa I, Vedda A, Cantarelli IX, Pedroni M, Piccinelli F, Bettinelli M, Speghini A, Quintanilla M, Vetrone F, Rocha U, Jacinto C, Carrasco E, Rodriguez FS, Juarranz A, del Rosal B, Ortgies DH, Gonzalez PH, Sole JG, Garcia DJ. Nano Research. 2015;8(2):649–665. [Google Scholar]
- 12.Riman Richard E, Kumar GA, Atakan V, Brennan John G, Ballato J. Proc. SPIE-Int. Soc. Opt. Eng. 2007;6707 (Copyright (C) 2013 American Chemical Society (ACS). All Rights Reserved.), 670707/1-670707/11. [Google Scholar]
- 13.Riman RE, Tan MC, Gangadharan KA. Near Infrared-Emitting ER and Yb/Er doped CeF3 Nanoparticulates with no visible upconversion. 2012 doi: 10.1364/OE.17.015904. [DOI] [PubMed] [Google Scholar]
- 14.Smith AM, Mancini MC, Nie S. Nat Nanotechnol. 2009;4(11):710–711. doi: 10.1038/nnano.2009.326. DOI nnano.2009.326 [pii] 10.1038/nnano.2009.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui M, Naczynski DJ, Zevon M, Griffith CK, Sheihet L, Poventud-Fuentes I, Chen S, Roth CM, Moghe PV. Adv Healthc Mater. 2013 doi: 10.1002/adhm.201200467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, Bridger G, Henson GW. Antimicrob Agents Chemother. 2000;44(6):1667–1673. doi: 10.1128/aac.44.6.1667-1673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Bon B, Van Craynest N, Daoudi JM, Di Giorgio C, Domb AJ, Vierling P. Bioconjug Chem. 2004;15(2):413–423. doi: 10.1021/bc034220o. [DOI] [PubMed] [Google Scholar]
- 18.Hinton CV, Avraham S, Avraham HK. Clin Exp Metastasis. 2010;27(2):97–105. doi: 10.1007/s10585-008-9210-2. [DOI] [PubMed] [Google Scholar]
- 19.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410(6824):50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 20.Zlotnik A. Contrib Microbiol. 2006;13:191–199. doi: 10.1159/000092973. [DOI] [PubMed] [Google Scholar]
- 21.Nimmagadda S, Pullambhatla M, Stone K, Green G, Bhujwalla ZM, Pomper MG. Cancer Res. 2010;70(10):3935–3944. doi: 10.1158/0008-5472.CAN-09-4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kratz F. J Control Release. 2008;132(3):171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Paal K, Muller J, Hegedus L. Eur J Biochem. 2001;268(7):2187–2191. doi: 10.1046/j.1432-1327.2001.02107.x. [DOI] [PubMed] [Google Scholar]
- 24.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. Cancer Cell. 2003;3(6):537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y, Mao X, Fan C, Liu C, Guo A, Guan S, Jin Q, Li B, Yao F, Jin F. Tumour Biol. 2014;35(8):7765–7773. doi: 10.1007/s13277-014-1816-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, Kang Y, Lonning S, McPherson J, Yingling JM, Biswas S, Mundy GR, Reiss M. Mol Cancer. 2010;9:122. doi: 10.1186/1476-4598-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massague J. J Clin Invest. 2005;115(1):44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly KA, Bardeesy N, Anbazhagan R, Gurumurthy S, Berger J, Alencar H, DePinho RA, Mahmood U, Weissleder R. Plos Medicine. 2008;5(4):657–668. doi: 10.1371/journal.pmed.0050085. DOI ARTN e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao ZY, Levy-Nissenbaum E, Alexis F, Luptak A, Teply BA, Chan JM, Shi JJ, Digga E, Cheng J, Langer R, Farokhzad OC. Acs Nano. 2012;6(1):696–704. doi: 10.1021/nn204165v. DOI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anhorn MG, Wagner S, Kreuter J, Langer K, von Briesen H. Bioconjug Chem. 2008;19(12):2321–2331. doi: 10.1021/bc8002452. [DOI] [PubMed] [Google Scholar]
- 32.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Proc Natl Acad Sci U S A. 2007;104(39):15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang MJ, Park SH, Kang MH, Park MJ, Choi YW. Int J Nanomedicine. 2013;8:1155–1165. doi: 10.2147/IJN.S39491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Low K, Wacker M, Wagner S, Langer K, von Briesen H. Nanomedicine-Nanotechnology Biology and Medicine. 2011;7(4):454–463. doi: 10.1016/j.nano.2010.12.003. DOI. [DOI] [PubMed] [Google Scholar]
- 35.Ming X, Carver K, Wu L. Biomaterials. 2013;34(32):7939–7949. doi: 10.1016/j.biomaterials.2013.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vlashi E, Kelderhouse LE, Sturgis JE, Low PS. Acs Nano. 2013;7(10):8573–8582. doi: 10.1021/nn402644g. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Yu Y, Dai W, Lu J, Cui J, Wu H, Yuan L, Zhang H, Wang X, Wang J, Zhang X, Zhang Q. Biomaterials. 2012;33(33):8451–8460. doi: 10.1016/j.biomaterials.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 38.Kratz F, Elsadek B. J Control Release. 2012;161(2):429–445. doi: 10.1016/j.jconrel.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 39.Hellebust A, Richards-Kortum R. Nanomedicine (Lond) 2012;7(3):429–445. doi: 10.2217/nnm.12.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chinnathambi S, Chen S, Ganesan S, Hanagata N. Adv Healthc Mater. 2014;3(1):10–29. doi: 10.1002/adhm.201300157. [DOI] [PubMed] [Google Scholar]
- 41.Biju V, Itoh T, Anas A, Sujith A, Ishikawa M. Anal Bioanal Chem. 2008;391(7):2469–2495. doi: 10.1007/s00216-008-2185-7. [DOI] [PubMed] [Google Scholar]
- 42.Jayakumar MK, Idris NM, Huang K, Zhang Y. Nanoscale. 2014;6(15):8441–8443. doi: 10.1039/c4nr02422b. [DOI] [PubMed] [Google Scholar]
- 43.Wang YF, Liu GY, Sun LD, Xiao JW, Zhou JC, Yan CH. ACS Nano. 2013;7(8):7200–7206. doi: 10.1021/nn402601d. [DOI] [PubMed] [Google Scholar]
- 44.Haro-Gonzalez P, Ramsay WT, Martinez Maestro L, del Rosal B, Santacruz-Gomez K, Iglesias-de la Cruz Mdel C, Sanz-Rodriguez F, Chooi JY, Rodriguez Sevilla P, Bettinelli M, Choudhury D, Kar AK, Sole JG, Jaque D, Paterson L. Small. 2013;9(12):2162–2170. doi: 10.1002/smll.201201740. [DOI] [PubMed] [Google Scholar]
- 45.Maestro LM, Haro-Gonzalez P, del Rosal B, Ramiro J, Caamano AJ, Carrasco E, Juarranz A, Sanz-Rodriguez F, Sole JG, Jaque D. Nanoscale. 2013;5(17):7882–7889. doi: 10.1039/c3nr01398g. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Wang R, Zhang F, Zhou L, Shen D, Yao C, Zhao D. Sci Rep. 2013;3(3536) doi: 10.1038/srep03536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kummar S, Gutierrez M, Doroshow JH, Murgo AJ. Br J Clin Pharmacol. 2006;62(1):15–26. doi: 10.1111/j.1365-2125.2006.02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen TM. Nature Reviews Cancer. 2002;2(10):750–763. doi: 10.1038/nrc903. DOI. [DOI] [PubMed] [Google Scholar]
- 49.Horm TM, Schroeder JA. Cell Adh Migr. 2013;7(2):187–198. doi: 10.4161/cam.23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.